
The effect of CeO2–ZrO2 structural differences on the origin and reactivity of carbon formed during methane dry reforming over NiCo/CeO2–ZrO2 catalysts studied by transient techniques†

Abstract
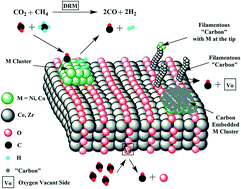
Nickel (1.2 wt%) and cobalt (1.8 wt%) were dispersed over Ce0.75Zr0.25O2−δ solid solution (3NiCo EG) or over a mixture of CeO2 and ZrO2 single phases (3NiCo HT) and tested for dry reforming of methane (DRM) at 750 °C. The structural, morphological, textural and redox differences between 3NiCo EG and 3NiCo HT catalysts were probed by powder XRD, HAADF/STEM and SAED, N2 adsorption/desorption at 77 K, H2-chemisorption, H2-TPR and H2 transient isothermal reduction (H2-TIR) techniques. The origin, concentration and reactivity of “carbon” deposits formed in DRM (via the CH4 and CO2 activation routes) towards gaseous H2 and O2 but also towards the support's labile oxygen species in the prepared catalysts were analyzed by a series of various transient and SSITKA experiments (use of 18O2 and 13CO2). Regardless of the EG or HT support, the %-contribution of CH4 and CO2 to the “carbon” deposition is very similar but the amount and reactivity towards oxidation are largely different. On the other hand, the concentration of active carbon formed in the carbon pathway of the CO2 activation route is very small (θC < 0.16% after 2 h in DRM). Participation of mobile lattice oxygen species in gasification of deposited “carbon” to CO(g) does occur to a large extent on both EG- and HT-supported NiCo catalysts. The catalyst deactivation rate during the first 5 h of DRM was found to depend on the structure of the support (EG vs. HT). The faster deactivation observed with the 3NiCo EG catalyst cannot be linked to the existing differences in the rates of inactive “carbon” formation and depletion or the concentration of active carbon but rather to the different rates of encapsulation of NiCo bimetallic particles in the carbon layers formed (∼30 nm thick) as revealed by HAADF/STEM.
- This article is part of the themed collection: Catalytic reactivity of surfaces: in recognition of François Gault
 Please wait while we load your content...
Please wait while we load your content...

