
Imaging and chemically probing catalytic processes using field emission techniques: a study of NO hydrogenation on Pd and Pd–Au catalysts
 *ab
Thierry
Visart de Bocarmé
*ab
*ab
Thierry
Visart de Bocarmé
*ab
Abstract
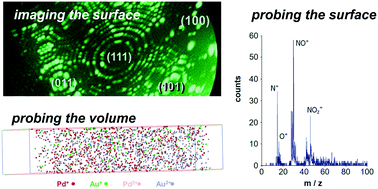
Nitric oxide hydrogenation is investigated on palladium and gold–palladium alloy crystallites, i.e. the extremity of sharp tip samples aimed at modelling a single catalytic grain. Field ion microscopy and field emission microscopy are used to monitor adsorption and reaction in real time. One-dimensional atom probe and atom probe tomography are used on the same samples to unravel the surface composition of the adsorbed layers and the composition of the very first atomic layers of the Pd–Au surface. At constant NO pressure and at 450 K, the surface composition of the adsorbed layer on Pd samples shows a strong hysteresis behavior when H2 gas is varied. Under oxidizing conditions, N2O is formed via the occurrence of surface (NO)2 dimers. In the presence of Pd–Au alloys, the NO–H2 interaction comprises a simple NO dissociation causing the formation of surface NO2 species. On Pd–Au tip samples, atom probe tomography proves the occurrence of significant surface enrichment of palladium atoms in the presence of NO gas, but it is not sufficient to drift the behavior of the surface to that of pure palladium. Accordingly, external control parameters could be changed to tune the surface composition of Pd–Au catalysts and thus their activity and/or selectivity.
- This article is part of the themed collections: 2017 Catalysis Science & Technology HOT Articles and Catalytic reactivity of surfaces: in recognition of François Gault
 Please wait while we load your content...
Please wait while we load your content...

