
Improved dispersion of transition metals in mesoporous materials through a polymer-assisted melt infiltration method†
A.
Ungureanu,  *a
S.
Royer
*bcd
*a
S.
Royer
*bcd
*a
S.
Royer
*bcd
Abstract
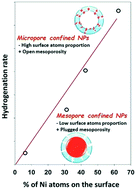
Melt infiltration (MI) has been described as an effective way to disperse transition metals (TM) in ordered mesoporous supports. The alternative method described here is based on the infiltration of molten precursors into the support pores in the presence of a support polymer porogen. The resulting materials contain metal oxide particles (metallic particles after reduction) smaller than 2 nm, dispersed within the support micropores, which prove to be stable upon reduction up to 900 °C and to exhibit improved catalytic performances for the hydrogenation of cinnamaldehyde due to the high fraction of the active metal exposed.
- This article is part of the themed collection: Catalytic reactivity of surfaces: in recognition of François Gault
 Please wait while we load your content...
Please wait while we load your content...

