
Ab initio coverage-dependent microkinetic modeling of benzene hydrogenation on Pd(111)†
 *a
Marie-Françoise
Reyniers
a
and
*a
Marie-Françoise
Reyniers
a
and
Abstract
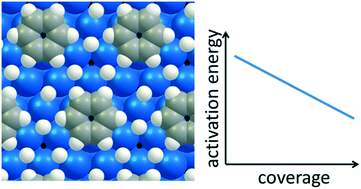
The effect of hydrogen coverage on the kinetics of benzene hydrogenation on Pd(111) has been investigated with optPBE-vdW density functional theory calculations and a coverage-dependent microkinetic model. The dominant reaction path consists of the consecutive hydrogenation of carbon atoms located in ortho positions relative to the previously hydrogenated carbon atom, independent of the hydrogen coverage. Increasing the hydrogen coverage destabilizes all surface species, which leads to weaker adsorption and increased rate coefficients for the hydrogenation steps due to stronger destabilization of reactants than transition states. The catalytic activities simulated using the constructed coverage-dependent microkinetic model exceed those obtained using a low-coverage microkinetic model by several orders of magnitude and are comparable to experimentally observed activities. The rate coefficients to which the global rate is most sensitive depend on the reaction conditions and differ from those calculated using low coverage kinetics. Therefore, properly accounting for coverage dependence on the kinetics and thermodynamics of catalytic hydrogenation reactions is not only required for an accurate DFT-based prediction of the catalytic activity but also for a correct understanding of the reaction mechanism.
- This article is part of the themed collections: 2017 Catalysis Science & Technology HOT Articles and Catalytic reactivity of surfaces: in recognition of François Gault
 Please wait while we load your content...
Please wait while we load your content...

