
Upgrading of oxygenated compounds present in aqueous biomass-derived feedstocks over NbOx-based catalysts†
M. E.
Domine  *a
and
*a
and
*a
and
Abstract
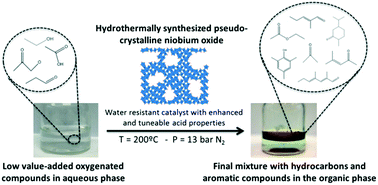
The influence of synthesis and post-synthesis procedures of different niobium oxides on their catalytic performance in the aqueous phase condensation of oxygenated compounds is studied. Hydrothermally synthesized niobium oxide with a pseudo-crystalline structure shows enhanced acid properties, surface area and consequently better catalytic activity than Nb2O5 prepared by other synthesis methods. The optimized NbOx-based catalyst also demonstrates higher stability after several reuses compared to the Ce–Zr mixed oxide reference catalyst.
- This article is part of the themed collection: 2017 Catalysis Science & Technology HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

