
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reduction of carbon dioxide and organic carbonyls by hydrosilanes catalysed by the perrhenate anion†
Danny S.
Morris
a,
Catherine
Weetman
a,
Julian T. C.
Wennmacher
b,
Mirza
Cokoja
b,
Markus
Drees
 b,
Fritz E.
Kühn
b and
Jason B.
Love
*a
b,
Fritz E.
Kühn
b and
Jason B.
Love
*a
aEaStCHEM School of Chemistry, University of Edinburgh, David Brewster Road, Edinburgh EH9 3FJ, UK. E-mail: jason.love@ed.ac.uk; Tel: +44 (0)131 650 4762
bCatalysis Research Center, Technische Universität München, Ernst-Otto-Fischer-Straße 1, D-85747 Garching bei München, Germany
First published on 8th June 2017
Abstract
The simple perrhenate salt [N(hexyl)4][(ReO4)] acts as a catalyst for the reduction of organic carbonyls and carbon dioxide by primary and secondary hydrosilanes. In the case of CO2, this results in the formation of methanol equivalents via silylformate and silylacetal intermediates. Furthermore, the addition of alkylamines to the reaction mixture favours catalytic amine N-methylation over methanol production under certain conditions. DFT analysis of the mechanism of CO2 reduction shows that the perrhenate anion activates the silylhydride forming a hypervalent silicate transition state such that the CO2 can directly cleave a Si–H bond.
Introduction
The hydrosilylation of CO2 to silylformates and silylethers is an attractive route for CO2 utilisation as, unlike hydrogenation, this reaction is exergonic due to the comparative ease of Si–H bond activation and the strength of the Si–O bond, and the availability of relatively inexpensive and environmentally benign hydrosilanes.1–3 A variety of catalysts have been discovered and developed for this reaction, including complexes of Co,4 Cu,5–8 Ir,9–14 Ni,15–17 Pd/Pt,18 Rh,19 Ru,20–25 Sc,26 Zn,27–29 and Zr,30 frustrated Lewis pairs,31–36 organocatalysts,37–42 alkali metal carbonates,43 and even polar solvents such as DMF.38 These reactions result in various reduction products including silyl-formates, -acetals, and -ethers, CO, and methane. This reactivity has been further exploited in a ‘diagonal’ approach to CO2 reduction/functionalisation, for example in the use of CO2 as the C1 source for amine N-methylation.3,37,44–54 High oxidation state transition metal oxo complexes55 have been studied in CO2 reduction chemistry and have been shown to react to form metal carbonates where, in the case of molybdate, the resulting carbonate undergoes hydrosilylation to formate and silylated molybdate, albeit stoichiometrically.56–59 Furthermore, rhenium(V) mono and dioxo complexes act as catalysts for the hydrosilylation of organic carbonyls,60–65 but this activity is not generally seen for the parent perrhenate, ReO4−. Significantly, metal oxo complexes have the potential to overcome the oxygen and moisture sensitivity issues from which many current CO2 hydrosilylation catalysts suffer.It was shown previously that the pyridinium perrhenate salt [4,6-But2C5H2NH-2-CH{(CO)NH(C6H13)}2][ReO4] 1 acts as a catalyst for the epoxidation of alkenes by hydrogen peroxide under biphasic conditions.66 Important to this chemistry is the formation of a lipophilic ion pair in which electrostatic and hydrogen-bonding interactions are enhanced in the hydrophobic phase. This catalyst has the advantages of ease of synthesis and manipulation and lack of air-sensitivity, and the recovery of the precious metal is driven by straightforward reverse phase-transfer. Given the efficacy of this new catalyst system and the use of high oxidation state rhenium oxo complexes in reduction catalysis, it is shown here that perrhenate can act as a catalyst for the reduction of carbon dioxide to methanol equivalents by hydrosilanes, that the methylation of alkylamines using CO2 as the C1 source occurs under similar catalytic conditions, and that this method can be used to reduce aldehydes and ketones to silylalcohols.
Results and discussion
Reduction of carbon dioxide
Initially, the reaction between CO2 (2.5 bar), PhSiH3 (2.0 mmol), and pyridinium perrhenate 1 (2.0 mol%) at 80 °C in C6D6 in a Teflon-tapped NMR tube was monitored by 1H NMR spectroscopy. However, only 15% consumption of hydrosilane is seen under these conditions after 16 h (Fig. S2 and S3, ESI†). In contrast, when the reaction is carried out using the simple lipophilic quaternary ammonium salt [N(hexyl)4][ReO4] 2 (2.5 mol%) with PhSiH3 in C6D6 at 1 bar of CO2 and at 80 °C, complete consumption of the hydrosilane is observed in the 1H NMR spectrum after 12 h (Fig. S4†) along with the formation of silyformate (3), bis(silyl)acetal (4) and silylated methanol (5) products (Table 1, entry 1 and Scheme 1). Negligible depletion of hydrosilane (<3%) is seen when the reaction is carried out in the absence of CO2 (under air) for 1 h (Fig. S5 and S6†), confirming that CO2 is integral to the reaction. This air-stable perrhenate catalyst is also integral as no reaction is seen in the absence of 2 nor in the presence of 5 mol% of N(hexyl)4Br (Fig. S7†). Increased turnover and selectivity towards methanol equivalents is observed when switching solvents to the more polar CD3CN (Table 1, entries 3–6). In this case, complete consumption of hydrosilane is observed in 4 h after which the addition of water and heating at 80 °C for a further 2 h results in the formation of methanol, seen as a singlet at 3.28 ppm in the 1H NMR spectrum (e.g. Fig. S9†). Prolonged reaction times to ensure full consumption of all Si–H containing species, i.e. no mono/oligomeric siloxyhydride resonances remaining at ca. 5.5 ppm, result in an increase in yield of 5. GC-MS analysis of the reduction with Ph2SiH2 in CD3CN prior to quenching indicates the sole presence of Ph2Si(OMe)2 confirming that all silylhydrides are consumed in the reaction (Table 1, entry 6, Fig. S60†).| Silane | Time (h) | Solvent | Conversiona (%) | Ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:5b :4:5b |
TOFc (h−1) | |
|---|---|---|---|---|---|---|
| a Consumption of hydrosilane. b NMR ratio vs. Me3SiPh after water quench. c TOF = (conversion/catalyst loading)/time. d Prolonged reaction times to consume all Si–H containing species. e 150 °C. | ||||||
| 1 | PhSiH3 | 12 | C6D6 | 95 | 51:18:22 |
3 |
| 2 | Ph2SiH2 | 12 | C6D6 | 100 | 73:16:11 |
3 |
| 3 | PhSiH3 | 3 | CD3CN | 96 | 15:0:85 |
10 |
| 4 | Ph2SiH2 | 4 | CD3CN | 100 | 16:0:84 |
13 |
| 5 | PhSiH3d | 16 | CD3CN | 100 | 2:0:98 |
3 |
| 6 | Ph2SiH2d | 16 | CD3CN | 100 | 7:0:93 |
3 |
| 7 | Et3SiH | 24 | CD3CN | 0 | 0 | 0 |
| 8 | (EtO)3SiH | 24 | CD3CN | 15 | 99:0:1 |
0.3 |
| 9 | Ph2SiH2 | 1.5 | d5-py | 100 | 38:0:62 |
27 |
| 10 | PhSiH3 | 1 | d7-DMF | 100 | 10:0:90 |
40 |
| 11 | Ph2SiH2 | 1 | d7-DMF | 100 | 16:0:84 |
40 |
| 12 | Ph3SiHe | 8 | d7-DMF | 25 | 21:0:79 |
1 |
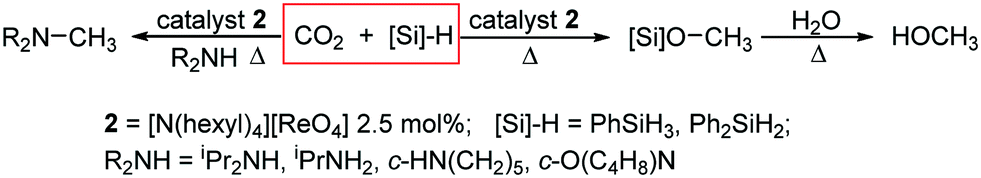 | ||
| Scheme 1 Reactions of carbon dioxide with hydrosilanes catalysed by the lipophilic perrhenate salt 2. | ||
The choice of hydrosilane is important to the reaction as Et3SiH (Table 1, entry 7) showed no conversion after 24 h at 80 °C, whereas (EtO)3SiH (Table 1, entry 8) is highly selective towards the formation of silylformate 3, and the use of more sterically demanding Ph3SiH (Table 1, entry 12) required elevated temperatures. This variation in reactivity is due to a combination of steric inaccessibility of the Si–H bond and the lower Lewis acidity of alkyl silanes.
Further solvent screening found the catalysis to be accelerated in pyridine, with complete hydrosilane consumption observed after 1.5 h (Table 1, entry 9). Using d7-DMF accelerates the reaction further, with complete hydrosilane depletion after 1 h and an increase in selectivity towards methanol formation compared to the reaction carried out in pyridine (Table 1, entries 10 and 11). Significantly however, reactions in DMF suffer from competitive solvent reduction by hydrosilane to (CD3)2NCDH2, consuming ca. 10% of the hydrosilane in this process (Fig. S21†). The variation of solvent relates to an increase in turnover frequencies (TOFs), determined from the time taken to consume the hydrosilane at 80 °C, of 3.3 h−1 for C6D6 to 40 h−1 for d7-DMF. The increase in rate is, in part, due to the increased solubility of CO2 in the more polar solvent; indeed, a reaction carried out in CD3CN under 2 bar of CO2 shows complete hydrosilane depletion within 1.5 h (Fig. S22†).
The longevity of the catalyst system was probed, and at the end of the first catalytic run with Ph2SiH2 in d7-DMF (TOF 40 h−1), the NMR tube was recharged with Ph2SiH2 and 1 bar of CO2 and heated again at 80 °C; this procedure was repeated 3 times. A marginal drop in TOF is observed on the third recharge cycle (36 h−1) after which the reaction was quenched with water and selective formation of methanol is seen (ratio 1:0:99, Fig. S23†). Whilst only a small drop in activity was noted after subsequent recharges, the perrhenate catalyst has significant advantages over current reports due to the ease in which the perrhenate anion can be recovered through simple reverse-phase-transfer techniques.
The faster rate of the reaction in d7-DMF allowed it's monitoring by 1H and 13C NMR spectroscopy at 300 K using 13CO2. On depletion of hydrosilane, broad resonances are seen after 3 h consistent with the formation of silylformate 3 at 8.65 ppm (d, JHC 219 Hz) and 162.6 ppm (d, JCH 219 Hz) in the 1H and 13C NMR spectra, respectively (Fig. S24–S29†). After 5 h, quartet resonances at 51.9 ppm (JCH 144 Hz) appear in the 13C NMR spectrum, consistent with [Si]OCH35 formation, along with some small triplet resonances at 82.8 ppm (JCH 171 Hz) for the bis(silyl)acetal [Si]OCH2O[Si] 4. In d7-DMF, the reaction between Ph2SiH2 and CO2 in the absence of perrhenate catalyst also leads to silylformate and silylether reduction products in a ratio of 38:62.38 However, this background reaction is very slow, taking 144 h to consume the Ph2SiH2 at 300 K compared with 10 h when using perrhenate (Fig. S30†).
A series of stoichiometric reactions were undertaken to probe the catalytic mechanism. It was first thought that the mechanism may follow a similar route to the molybdate dianion which reacts initially with CO2 to form the carbonate complex Mo(CO3)O32−;56 however, no reaction of 2 with 13CO2 in the absence of hydrosilane is seen, even after prolonged heating at 80 °C (Fig. S31†), thus ruling out initial carbonate formation in this catalytic cycle. Also, no intermediates are seen in stoichiometric reactions of 2 with varying equivalents of hydrosilane in the absence of CO2. It is of note, however, that upon extended heating (>16 h at 80 °C) decomposition of 2 occurs to form trihexylamine, siloxanes and a brown precipitate containing rhenium oxide clusters (Fig. S32–S34†). In light of these experiments, the mechanism of this reaction was evaluated computationally.
DFT modelling of the catalytic mechanism
To explain how the carbon dioxide reacts with the hydrosilane and the perrhenate catalyst, three different principle pathways have been elucidated (Scheme 2). | ||
| Scheme 2 The pathways considered for the DFT investigation of the reduction of CO2 by hydrosilanes catalysed by the perrhenate salt 2. | ||
Two pathways include activation of the H–Si bond of the relevant hydrosilane by either a [3 + 2] or [2 + 2] cycloaddition to the Re![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or OReO fragment of the perrhenate to form a siloxyhydride, with subsequent reaction of this intermediate with CO2 by hydride migratory insertion.63,65 If Si–H activation would occur to form a hydride, then the most probable mode would be [2 + 2] addition. However, the DFT results favour an alternative ΔG energy profile in the gas phase (Fig. 1) which includes the formation of a simple complex of the hydrosilane with perrhenate without preliminary Si–H activation, forming a five-coordinate hypervalent silicon centre.2,60,67–70 The CO2 activation occurs then directly at a Si–H bond to form a HCO2− species that adds to the silicon to form the observed silylformate (here as adduct of ReO4−). This adduct can be seen as a starting point of activating the next Si–H bond by a second CO2. In principle, CO2 can also undergo a cycloaddition to silyl perrhenate intermediates, but the barriers are higher in all pathways. Calculations incorporating a solvent continuum were carried out, with neither benzene nor DMF models significantly altering the preferred mechanism or its energies (see ESI†). Furthermore, alternative mechanisms involving the cycloaddition of CO2 to a rhenium silicate intermediate were explored, but were found to have higher energy barriers than the route described above (see ESI†).
O or OReO fragment of the perrhenate to form a siloxyhydride, with subsequent reaction of this intermediate with CO2 by hydride migratory insertion.63,65 If Si–H activation would occur to form a hydride, then the most probable mode would be [2 + 2] addition. However, the DFT results favour an alternative ΔG energy profile in the gas phase (Fig. 1) which includes the formation of a simple complex of the hydrosilane with perrhenate without preliminary Si–H activation, forming a five-coordinate hypervalent silicon centre.2,60,67–70 The CO2 activation occurs then directly at a Si–H bond to form a HCO2− species that adds to the silicon to form the observed silylformate (here as adduct of ReO4−). This adduct can be seen as a starting point of activating the next Si–H bond by a second CO2. In principle, CO2 can also undergo a cycloaddition to silyl perrhenate intermediates, but the barriers are higher in all pathways. Calculations incorporating a solvent continuum were carried out, with neither benzene nor DMF models significantly altering the preferred mechanism or its energies (see ESI†). Furthermore, alternative mechanisms involving the cycloaddition of CO2 to a rhenium silicate intermediate were explored, but were found to have higher energy barriers than the route described above (see ESI†).
 | ||
| Fig. 1 Calculated Gibbs free-energy profile of the three different pathways. | ||
N-Methylation of amines using carbon dioxide
The observation that silylformates are formed during the catalytic reaction and their implication as intermediates in the N-methylation of amines using CO2 led us to assess the use of 2 to catalyse this latter reaction. Accordingly, the reaction between CO2, HNiPr2, 4 eq. of Ph2SiH2, and 2.5 mol% of 2 at 80 °C in CD3CN was monitored by 1H NMR spectroscopy. After 2 h, complete loss of HNiPr2 is observed with the concomitant formation of MeNiPr2 in quantitative yields (Table 2, entry 1; Fig. S35†). The excess hydrosilane is then consumed in the formation of [Si]OCH3 (5) as noted by the characteristic resonances in the 1H NMR spectrum between 3.4–3.6 ppm. The scope of the amine substrate was evaluated, revealing that several aliphatic amines are N-methylated, including isopropylamine (to form Me2NiPr), piperidine, pyrrolidine and morpholine (Table 2, entries 2–5). The N-methylation of aromatic primary or secondary amines at this temperature does not occur (Table 2, entries 6 and 7), and instead the hydrosilane is consumed in competitive CO2 reduction. However, lowering the temperature of reaction negates the further reduction of silylformate by hydrosilane to favour reaction with the poorly nucleophilic N-methylaniline and, after 100 h at 20 °C, its conversion to N,N-dimethylaniline was seen in 40% yield. Interestingly, the major product of this reaction is the aminal CH2(NMePh)2 in 60% yield, seen by its characteristic resonances at 2.86 and 4.78 ppm in the 1H NMR spectrum (Fig. S41†). Similar aminal formation from CO2 has been seen previously in organocatalysed or Ru-catalysed reactions.47,54 As the aminal can be prepared by the condensation reaction of N-methylaniline and formaldehyde, its formation in this catalysed reaction is likely a result of transient formaldehyde production which can arise from the decomposition of CH2(OSiH2Ph)2.71It was reported recently that selective formylation of amines is possible using hydrosilanes and CO2 in polar solvents such as DMF and DMSO.72 In contrast, it was found that the simple aliphatic amine HNiPr2 underwent N-methylation in DMF at room temperature in the absence of 2, and subsequent in situ monitoring of this reaction by 1H NMR spectroscopy found the initial rates essentially identical for the catalysed and uncatalysed reaction (Fig. S40†). Reactions in DMF for the other amine substrates are also selective towards N-formylation, which was possible in the absence of 2; however, to achieve N-methylation catalyst 2 was required.
Reduction of aldehydes and ketones
Following on from the success of the CO2 reduction, attention was turned to CO reduction for a variety of carbonyl containing substrates.73–75 An initial trial NMR reaction with benzaldehyde was carried out using the standard reaction conditions of 1.2 eq. of PhSiH3, 0.5 mL of CD3CN and 2.5 mol% of 2 at 80 °C. After 1 h, the 1H NMR spectrum showed near complete consumption of the aldehyde through loss of the characteristic CHO resonance at 10.01 ppm and formation of approximately a 50:50 mixture of PhSi(H)(OCH2Ph)2 and PhSi(OCH2Ph)3; subsequent heating for a further 30 minutes to ensure full consumption of benzaldehyde resulted in no change to the product distribution. Simple hydrolysis through the addition of a small excess of water resulted in full conversion to the benzylalcohol product with concomitant formation of siloxanes (Fig. S42†). Following the same reaction protocol the substrate scope was expanded to other substituted arylaldehydes and alkylaldehydes (Table 3).
| Aldehyde | Time (h) | NMR yielda (%) | TOFb (h−1) | |
|---|---|---|---|---|
| a NMR ratio vs. Me3SiPh after water quench. b TOF = (conversion/catalyst loading)/time. | ||||
| 1 | Benzaldehyde | 1.5 | 86 | 23 |
| 2 | p-Bromobenzaldehyde | 1 | 92 | 37 |
| 3 | p-Tolualdehyde | 2.5 | 79 | 13 |
| 4 | p-Anisaldehyde | 2.5 | 89 | 14 |
| 5 | m-Anisaldehyde | 1.5 | 96 | 26 |
| 6 | Trimethylacetaldehyde | 4 | 91 | 2 |
| 7 | (1R)-(−)-Myrtenal | 4 | 47 | 4 |
| 8 | Crotonaldehyde | 2.5 | 48 | 8 |
| 9 | Butyraldehyde | 2 | 46 | 9 |
The arylaldehyde substrates provided efficient turnover to the corresponding benzylalcohol product upon quenching with water. Aryl substituents containing electron-withdrawing groups (Table 3, entry 2 and 5) proceed with higher TOFs compared to their electron-donating counterparts (Table 3, entry 3 and 4), in line with previous reports of rhenium catalysed hydrosilylation reactions of aldehydes.76 In contrast the alkylaldehydes containing more sterically demanding substituents require twice the reaction time in comparison with the linear butyl chain derivatives.
Further extension of this perrhenate hydrosilylation catalysis examined the reduction of ketones (Table 4). Using the same reaction protocol with acetophenone, and monitoring by 1H NMR spectroscopy, the loss of the singlet resonance for the CH3 protons at 2.56 ppm is seen after 21 h at 80 °C with three new doublet resonances appearing in the range 1.49–1.37 ppm that correspond to oligomeric PhCH2(O–[Si])CH3 products; upon quenching with water these resonances resolve into a single species at 1.42 ppm (JHH = 6.51 Hz). The same is also true for the CH(O[Si])CH3 proton, visible around 5.1 ppm, but due to the oligomeric nature of the silylether products and the overlapping remaining [Si]–H resonances this appears as a series of multiplets which resolve into the characteristic quartet at 4.85 ppm (JHH = 6.39 Hz) on quenching. Expanding the scope of this ketone reduction to include substituted acetophenones follows the same trend as observed with the benzaldehyde derivatives. In this case, however, prolonged reaction times are required for the electron-donating substituents, taking 40 h for only 50% consumption of the starting ketone (Table 4, entries 2 and 4 vs. 3 and 5). Reduction of benzophenone to the corresponding alcohol proceeds efficiently, and again the use of a more sterically demanding alkyl ketone requires prolonged reaction times. Interestingly, a small amount of H/D exchange was seen to take place during extended heating. Hydrogen formation occurs during the initial stages of the reaction where some of the hydrosilane is initially consumed in the reduction of trace water to form H2 and siloxanes and, in the case of the acetophenone, HD is seen in the 1H NMR spectrum as a characteristic 1:1:1 triplet at 4.56 ppm (JHD = 42.6 Hz) (Fig. S58†). Further H/D exchange was seen at the acetyl position of the acetophenone substrates and is thought to occur through keto–enol tautomerisation in the presence of the [Si]–D which results from the reaction of [Si]–H and D2O. The use of dry d-solvents negates this HD exchange.
| Ketone | Time (h) | NMR yielda (%) | TOFb (h−1) | |
|---|---|---|---|---|
| a NMR ratio vs. Me3SiPh after water quench; b TOF = (conversion/catalyst loading)/time. | ||||
| 1 | Acetophenone | 21 | 72 | 1 |
| 2 | p-Bromoacetophenone | 40 | 45 | 0.5 |
| 3 | p-Acetyltoluene | 21 | 66 | 1 |
| 4 | p-Acetanisole | 40 | 39 | 0.4 |
| 5 | m-Acetanisole | 20 | 81 | 2 |
| 6 | Benzophenone | 21 | 66 | 1 |
| 7 | Pinacolone | 37 | 22 | 0.2 |
Experimental
Synthesis of [N(hexyl)4][ReO4], 2
An aqueous solution of ammonium perrhenate (1.26 g, 4.7 mmol) was added to a chloroform solution of tetrahexylammonium bromide (2.00 g, 4.6 mmol) and stirred together for 6 hours at room temperature after which the two layers were separated and the aqueous layer extracted with chloroform (2 × 10 mL). The combined chloroform extracts were dried over magnesium sulphate and the solvent removed under reduced pressure to provide a colourless solid of tetrahexylammonium perrhenate (2.10 g, 75% yield).1H NMR (500 MHz; d3-MeCN, 300 K): δH (ppm) 3.07 (m, 8H, NCH2), 1.60 (m, 8H, CH2), 1.33 (m, 24H, CH2), 0.91 (t, 12H, CH3).
13C{1H} NMR (500 MHz; d3-MeCN, 300 K): δC (ppm) 59.48 (NCH2), 31.84 (CH2), 26.57 (CH2), 26.57 (CH2), 23.10 (CH2), 22.34 (CH2), 14.21 (CH3); FTIR (ATR): νmax/cm−1 902 (Re–O); ESI-MS: M+m/z found (calculated): 354.41068 (354.40943 C24H52N2), 959.77918 (959.75512 (C24H54N)2ReO4), 1565.16124 (1565.10132 (C24H54N)3(ReO4)2). M−m/z found (calculated): 250.93270 (250.93597 ReO4), 854.26749 (854.27860 (C24H52N)(ReO4)2), 1459.59185 (1459.62427 (C24H52N)2(ReO4)3) 2064.90770 (2064.97026 (C24H52N)3(ReO4)4); analysis: C24H52NO4Re found (calc.); C: 47.55% (47.65), H: 8.72% (8.67), N: 2.38% (2.32).
General procedure for catalytic reactions of hydrosilanes with CO2
[N(hexyl)4][ReO4] 2 (3 mg, 2.5 mol%) in 0.6 mL d-solvent, hydrosilane (0.2 mmol) and trimethylphenylsilane (2 μL) as an internal standard (unless otherwise stated) were added to a Teflon-tapped NMR tube. The solution was freeze–pump–thaw degassed three times before being refilled with 1 bar of CO2. The 1H NMR spectrum was recorded and then the tube was placed in a preheated oil bath (80 °C). The reactions were monitored by 1H NMR spectroscopy regularly until the hydrosilane was consumed. At this point the reaction was quenched with water (10 μL, 0.56 mmol), heated for 2 h and the 1H NMR spectrum recorded.General procedure for catalytic N-methylation of amines with CO2
[N(hexyl)4][ReO4] 2 (3 mg, 2.5 mol%) in 0.6 mL d-solvent, amine (0.2 mmol), silane (0.8 mmol) and trimethylphenylsilane (2 μL) as an internal standard (unless otherwise stated) were added to a Teflon-tapped NMR tube. The solution was freeze–pump–thaw degassed three times before being refilled with 2 bar of CO2. The 1H NMR spectrum was recorded and then the tube was placed in a preheated oil bath (80 °C) for 2 h, after which the 1H NMR spectrum was recorded.General procedure for catalytic hydrosilylation of aldehydes and ketones
[N(hexyl)4][ReO4] 2 (3 mg, 2.5 mol%) in 0.5 mL CD3CN, phenylsilane (0.24 mmol), carbonyl (0.2 mmol) and trimethylphenylsilane (2 μL) as an internal standard were added to a NMR tube. The 1H NMR spectrum was recorded and then the tube was placed in a preheated oil bath (80 °C). The reactions were monitored by 1H NMR spectroscopy regularly until the hydrosilane or carbonyl was consumed. At this point the reaction was quenched with water (10 μL, 0.56 mmol), heated for 15 min and the 1H NMR spectrum recorded.Computational details
All calculations have been performed with the software Gaussian09.77 The hybrid density functional B3LYP78–81 has been used together with dispersion correction GD3BJ82 and the triple zeta basis set 6-311++G**83,84 for all elements except Re. The Re atoms are described with the Stuttgart-Dresden-ECP.85 The energies reported are free energies in gas-phase or for the solvents benzene and DMF with SMD solvent calculations at 298.15 K.86Conclusions
We have shown that the simple, lipophilic perrhenate salt [N(hexyl)4][ReO4] 2 acts as a catalyst for the reduction of CO2 by hydrosilanes. This is the first time that perrhenate has acted as a catalyst for CO2 reduction, and is likely facilitated by dissolution of the lipophilic assembly into a hydrophobic, organic solvent. The calculated mechanism shows that direct attack of CO2 on a Si–H bond of a perrhenate hypervalent silicate occurs instead of alternative mechanisms through rhenium hydride formation. Furthermore, the observation that N-methylation of amines using CO2 and organic carbonyl reduction can also be undertaken using a perrhenate catalyst expands considerably the scope of this chemistry. The ease of transfer of perrhenate from an organic phase back into an aqueous phase by the addition of base (e.g. NaOH) should lead to straightforward rhenium recycling, an essential step if this precious metal is to be further exploited in catalytic chemistry.Acknowledgements
We thank the TUM Graduate School, the Johannes Hübner Stiftung (sponsorship of JTCW), the University of Edinburgh and the EPSRC (EP/J018090/1), and the Leibnitz Computing Centre Munich for support.Notes and references
- F. J. Fernández-Alvarez, A. M. Aitani and L. A. Oro, Catal. Sci. Technol., 2015, 4, 611–624 Search PubMed.
- M. Iglesias, F. J. Fernández-Alvarez and L. A. Oro, ChemCatChem, 2014, 6, 2486–2489 CrossRef CAS.
- C. Das Neves Gomes, O. Jacquet, C. Villiers, P. Thuéry, M. Ephritikhine and T. Cantat, Angew. Chem., Int. Ed., 2012, 51, 187–190 CrossRef CAS PubMed.
- M. L. Scheuermann, S. P. Semproni, I. Pappas and P. J. Chirik, Inorg. Chem., 2014, 53, 9463–9465 CrossRef CAS PubMed.
- Y. Tani, K. Kuga, T. Fujihara, J. Terao and Y. Tsuji, Chem. Commun., 2015, 51, 13020–13023 RSC.
- K. Motokura, D. Kashiwame, N. Takahashi, A. Miyaji and T. Baba, Chem. – Eur. J., 2013, 19, 10030–10037 CrossRef CAS PubMed.
- L. Zhang, J. H. Cheng and Z. M. Hou, Chem. Commun., 2013, 49, 4782–4784 RSC.
- K. Motokura, D. Kashiwame, A. Miyaji and T. Baba, Org. Lett., 2012, 14, 2642–2645 CrossRef CAS PubMed.
- A. Julian, E. A. Jaseer, K. Garces, F. J. Fernandez-Alvarez, P. Garcia-Orduna, F. J. Lahoz and L. A. Oro, Catal. Sci. Technol., 2016, 6, 4410–4417 CAS.
- A. Julian, V. Polo, E. A. Jaseer, F. J. Fernandez-Alvarez and L. A. Oro, ChemCatChem, 2015, 7, 3895–3902 CrossRef CAS.
- E. A. Jaseer, M. N. Akhtar, M. Osman, A. Al-Shammari, H. B. Oladipo, K. Garces, F. J. Fernandez-Alvarez, S. Al-Khattaf and L. A. Oro, Catal. Sci. Technol., 2015, 5, 274–279 CAS.
- H. B. Oladipo, E. A. Jaseer, A. Julian, F. J. Fernandez-Alvarez, S. Al-Khattaf and L. A. Oro, J. CO2 Util., 2015, 12, 21–26 CrossRef CAS.
- S. Park, D. Bézier and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 11404–11407 CrossRef CAS PubMed.
- T. C. Eisenschmid and R. Eisenberg, Organometallics, 1989, 8, 1822–1824 CrossRef CAS.
- P. Rios, N. Curado, J. Lopez-Serrano and A. Rodriguez, Chem. Commun., 2016, 52, 2114–2117 RSC.
- L. Gonzalez-Sebastian, M. Flores-Alamo and J. J. Garcia, Organometallics, 2013, 32, 7186–7194 CrossRef CAS.
- R. Lalrempuia, M. Iglesias, V. Polo, P. J. S. Miguel, F. J. Fernandez-Alvarez, J. J. Perez-Torrente and L. A. Oro, Angew. Chem., Int. Ed., 2012, 51, 12824–12827 CrossRef CAS PubMed.
- S. J. Mitton and L. Turculet, Chem. – Eur. J., 2012, 18, 15258–15262 CrossRef CAS PubMed.
- A. J. Huckaba, T. K. Hollis and S. W. Reilly, Organometallics, 2013, 32, 6248–6256 CrossRef CAS.
- T. T. Metsänen and M. Oestreich, Organometallics, 2015, 34, 543–546 CrossRef.
- P. Deglmann, E. Ember, P. Hofmann, S. Pitter and O. Walter, Chem. – Eur. J., 2007, 13, 2864–2879 CrossRef CAS PubMed.
- A. Jansen and S. Pitter, J. Mol. Catal. A: Chem., 2004, 217, 41–45 CrossRef CAS.
- A. Jansen, H. Gorls and S. Pitter, Organometallics, 2000, 19, 135–138 CrossRef CAS.
- H. Koinuma, F. Kawakami, H. Kato and H. Hirai, J. Chem. Soc., Chem. Commun., 1981, 213–214 RSC.
- G. Süss-Fink and J. Reiner, J. Organomet. Chem., 1981, 221, C36–C38 CrossRef.
- F. A. LeBlanc, W. E. Piers and M. Parvez, Angew. Chem., Int. Ed., 2014, 53, 789–792 CrossRef CAS PubMed.
- M. M. Deshmukh and S. Sakaki, Inorg. Chem., 2014, 53, 8485–8493 CrossRef CAS PubMed.
- A. Rit, A. Zanardi, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2014, 53, 13273–13277 CrossRef CAS PubMed.
- W. Sattler and G. Parkin, J. Am. Chem. Soc., 2012, 134, 17462–17465 CrossRef CAS PubMed.
- T. Matsuo and H. Kawaguchi, J. Am. Chem. Soc., 2006, 128, 12362–12363 CrossRef CAS PubMed.
- J. W. Chen, L. Falivene, L. Caporaso, L. Cavallo and E. Y. X. Chen, J. Am. Chem. Soc., 2016, 138, 5321–5333 CrossRef CAS PubMed.
- S. A. Weicker and D. W. Stephan, Chem. – Eur. J., 2015, 21, 13027–13034 CrossRef CAS PubMed.
- R. Dobrovetsky and D. W. Stephan, Isr. J. Chem., 2015, 55, 206–209 CrossRef CAS.
- Y. Jiang, O. Blacque, T. Fox and H. Berke, J. Am. Chem. Soc., 2013, 135, 7751–7760 CrossRef CAS PubMed.
- M. Khandelwal and R. J. Wehmschulte, Angew. Chem., Int. Ed., 2012, 51, 7323–7326 CrossRef CAS PubMed.
- A. Berkefeld, W. E. Piers and M. Parvez, J. Am. Chem. Soc., 2010, 132, 10660–10661 CrossRef CAS PubMed.
- D. Mukherjee, D. F. Sauer, A. Zanardi and J. Okuda, Chem. – Eur. J., 2016, 22, 7730–7733 CrossRef CAS PubMed.
- M. A. Courtemanche, M. A. Legare, E. Rochette and F. G. Fontaine, Chem. Commun., 2015, 51, 6858–6861 RSC.
- K. Motokura, M. Naijo, S. Yamaguchi, A. Miyaji and T. Baba, Chem. Lett., 2015, 44, 1464–1466 CrossRef CAS.
- C. Lescot, D. U. Nielsen, I. S. Makarov, A. T. Lindhardt, K. Daasbjerg and T. Skrydstrup, J. Am. Chem. Soc., 2014, 136, 6142–6147 CrossRef CAS PubMed.
- S. N. Riduan, J. Y. Ying and Y. G. Zhang, ChemCatChem, 2013, 5, 1490–1496 CrossRef CAS.
- S. N. Riduan, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2009, 48, 3322–3325 CrossRef CAS PubMed.
- C. Fang, C. Lu, M. Liu, Y. Zhu, Y. Fu and B.-L. Lin, ACS Catal., 2016, 6, 7876–7881 CrossRef CAS.
- S. Q. Zhang, Q. Q. Mei, H. Y. Liu, H. Z. Liu, Z. P. Zhang and B. X. Han, RSC Adv., 2016, 6, 32370–32373 RSC.
- T. V. Q. Nguyen, W. J. Yoo and S. Kobayashi, Adv. Synth. Catal., 2016, 358, 452–458 CrossRef CAS.
- T. V. Q. Nguyen, W. J. Yoo and S. Kobayashi, Angew. Chem., Int. Ed., 2015, 54, 9209–9212 CrossRef CAS PubMed.
- X. Frogneux, E. Blondiaux, P. Thuéry and T. Cantat, ACS Catal., 2015, 5, 3983–3987 CrossRef CAS.
- L. D. Hao, Y. F. Zhao, B. Yu, Z. Z. Yang, H. Y. Zhang, B. X. Han, X. Gao and Z. M. Liu, ACS Catal., 2015, 5, 4989–4993 CrossRef CAS.
- Z. Lu, H. Hausmann, S. Becker and H. A. Wegner, J. Am. Chem. Soc., 2015, 137, 5332–5335 CrossRef CAS PubMed.
- O. Santoro, F. Lazreg, Y. Minenkov, L. Cavallo and C. S. J. Cazin, Dalton Trans., 2015, 44, 18138–18144 RSC.
- X. Frogneux, O. Jacquet and T. Cantat, Catal. Sci. Technol., 2014, 4, 1529–1533 CAS.
- O. Jacquet, X. Frogneux, C. Das Neves Gomes and T. Cantat, Chem. Sci., 2013, 4, 2127–2131 RSC.
- O. Jacquet, C. D. Gomes, M. Ephritikhine and T. Cantat, ChemCatChem, 2013, 5, 117–120 CrossRef CAS.
- Y. Li, X. Fang, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 9568–9571 CrossRef CAS PubMed.
- R. G. de Noronha and A. C. Fernandes, Curr. Org. Chem., 2012, 16, 33–64 CrossRef CAS.
- I. Knopf, T. Ono, M. Temprado, D. Tofan and C. C. Cummins, Chem. Sci., 2014, 5, 1772–1776 RSC.
- J. P. Krogman, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2013, 52, 3022–3031 CrossRef CAS PubMed.
- J. S. Silvia and C. C. Cummins, Chem. Sci., 2011, 2, 1474–1479 RSC.
- N. P. Tsvetkov, J. G. Andino, H. Fan, A. Y. Verat and K. G. Caulton, Dalton Trans., 2013, 42, 6745–6755 RSC.
- L. Huang, W. Wang and H. Wei, J. Mol. Catal. A: Chem., 2015, 400, 31–41 CrossRef CAS.
- P. Gu, W. Wang, Y. Wang and H. Wei, Organometallics, 2013, 32, 47–51 CrossRef CAS.
- S. C. A. Sousa, I. Cabrita and A. C. Fernandes, Chem. Soc. Rev., 2012, 41, 5641–5653 RSC.
- K. A. Nolin, J. R. Krumper, M. D. Pluth, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 14684–14696 CrossRef CAS PubMed.
- G. Du, P. E. Fanwick and M. M. Abu-Omar, J. Am. Chem. Soc., 2007, 129, 5180–5187 CrossRef CAS PubMed.
- E. A. Ison, E. R. Trivedi, R. A. Corbin and M. M. Abu-Omar, J. Am. Chem. Soc., 2005, 127, 15374–15375 CrossRef CAS PubMed.
- M. Cokoja, I. I. E. Markovits, M. H. Anthofer, S. Poplata, A. Pothig, D. S. Morris, P. A. Tasker, W. A. Herrmann, F. E. Kühn and J. B. Love, Chem. Commun., 2015, 51, 3399–3402 RSC; M. D. Zhou, M. Liu, J. Huang, J. Zhang, J. Wang, X. Li, F. E. Kühn and S. L. Zang, Green Chem., 2015, 17, 1186–1193 RSC; I. I. E. Markovits, A. A. Eger, S. Yue, M. Cokoja, C. J. Münchmeyer, B. Zhang, M. D. Zhou, A. Genest, J. Mink, S. L. Zang, N. Rösch and F. E. Kühn, Chem. Eur. J., 2013, 19, 5972–5979 CrossRef CAS PubMed; B. Zhang, S. Li, S. Yue, M. Cokoja, M. D. Zhou, S. L. Zang and F. E. Kühn, J. Organomet. Chem., 2013, 744, 108–112 CrossRef.
- M. J. Bearpark, G. S. McGrady, P. D. Prince and J. W. Steed, J. Am. Chem. Soc., 2001, 123, 7736–7737 CrossRef CAS PubMed.
- R. Corriu, C. Guérin, B. Henner and Q. Wang, Inorg. Chim. Acta, 1992, 198, 705–713 CrossRef.
- Q. Zhou and Y. Li, J. Am. Chem. Soc., 2015, 137, 10182–10189 CrossRef CAS PubMed.
- M. Oestreich, Angew. Chem., Int. Ed., 2016, 55, 494–499 CrossRef CAS PubMed.
- F. Huang, G. Lu, L. Zhao, H. Li and Z.-X. Wang, J. Am. Chem. Soc., 2010, 132, 12388–12396 CrossRef CAS PubMed.
- H. Lv, Q. Xing, C. Yue, Z. Lei and F. Li, Chem. Commun., 2016, 52, 6545–6548 RSC.
- S. Abbina, S. Bian, C. Oian and G. Du, ACS Catal., 2013, 3, 678–684 CrossRef CAS.
- T. V. Truong, E. A. Kastl and G. Du, Tetrahedron Lett., 2011, 52, 1670–1672 CrossRef CAS.
- H. Dong and H. Berke, Adv. Synth. Catal., 2009, 351, 1783–1788 CrossRef CAS.
- D. E. Perez, J. L. Smeltz, R. D. Sommer, P. D. Boyle and E. A. Ison, Dalton Trans., 2017, 46, 4609–4616 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. R. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- K. Raghavachari, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef.
- A. Bergner, M. Dolg, W. Küchle, H. Stoll and H. Preuß, Mol. Phys., 1993, 80, 1431–1441 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and spectra. See DOI: 10.1039/c7cy00772h |
| This journal is © The Royal Society of Chemistry 2017 |