
Fabrication, characterization, and stability of supported single-atom catalysts

Abstract
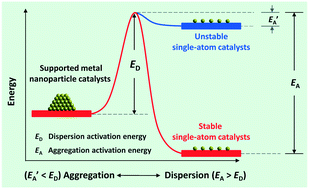
Supported single-atom catalysts (SACs) and their catalysis have become a hot topic recently, because the dispersion of isolated metal atoms on support surfaces can maximize the atomic efficiency/economy of noble metals and the resulting SACs often possess unprecedented catalytic activity. More importantly, with the development of SACs, it is relatively easy for us to identify the nature of catalytically active sites on these catalysts and establish intrinsic reaction mechanisms. However, it is still a great challenge to develop thermally and chemically stable SACs by a relatively easy method, which is also the case for the characterization of these catalysts because that would need higher resolution and precision. In this minireview, we generalize the advantages of SACs, outline the recent progress of the fabrication, characterization, and stability of SACs, and propose that electronic metal–support interactions are key to the development of stable SACs with pronounced catalytic activity. Some directions for future research are briefly discussed.
- This article is part of the themed collection: Single atom catalysis
 Please wait while we load your content...
Please wait while we load your content...

