
Doping carbon nanotubes with N, S, and B for electrocatalytic oxygen reduction: a systematic investigation on single, double, and triple doped modes
 *ab
*ab
Abstract
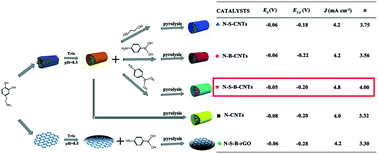
In this work, polydopamine (PDA)-coated multi-walled carbon nanotubes (CNTs) have been employed as reactive platforms for the anchoring of multiple heteroatom dopants. Single-, double-, and triple-doped CNTs with the elements N, S and B have been successfully prepared and systematically investigated as electrocatalysts for the oxygen reduction reaction (ORR). The obtained catalysts have been fully characterized by the nitrogen-adsorption technique, transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), and Raman spectroscopy. Combining with the physicochemical properties of different doped CNTs and their catalytic performance, various doping modes are compared and the mechanism of these processes has been investigated. The synergetic effects on double and triple doped CNTs with N, S, and B are found. The new non-metal catalyst N–S–B-CNTs exhibits high electrocatalytic activity and good stability for ORR.
 Please wait while we load your content...
Please wait while we load your content...

