
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal dimer sites in ZSM-5 zeolite for methane-to-methanol conversion from first-principles kinetic modelling: is the [Cu–O–Cu]2+ motif relevant for Ni, Co, Fe, Ag, and Au?†
Adam A.
Arvidsson
 a,
Vladimir P.
Zhdanov
ab,
Per-Anders
Carlsson
a,
Henrik
Grönbeck
a and
Anders
Hellman
*a
a,
Vladimir P.
Zhdanov
ab,
Per-Anders
Carlsson
a,
Henrik
Grönbeck
a and
Anders
Hellman
*a
aCompetence Centre for Catalysis, Department of Physics, Chemistry and Chemical Engineering, Chalmers University of Technology, 412 96 Gothenburg, Sweden. E-mail: anders.hellman@chalmers.se
bBoreskov Institute of Catalysis, Russian Academy of Sciences, Novosibirsk 630090, Russia
First published on 16th February 2017
Abstract
Direct methane-to-methanol conversion is a desired process whereby natural gas is transformed into an energy-rich liquid. It has been realised at ambient pressure and temperature in metal ion-exchanged zeolites, where especially copper-exchanged ZSM-5 has shown promising results. The nature of the active sites in these systems is, however, still under debate. The activity has been assigned to a [Cu–O–Cu]2+ motif. One remaining question is whether this motif is general and also active in other metal-exchanged zeolites. Herein, we use first-principles micro-kinetic modelling to analyse the methane-to-methanol reaction on the [Cu–O–Cu]2+ motif, for Cu and other metals. First, we identify the cluster model size needed to accurately describe the dimer motif. Starting from the [Cu–O–Cu]2+ site, the metal ions are then systematically substituted with Ni, Co, Fe, Ag and Au. The results show that activation of Ag and Au dimer sites with oxygen is endothermic and therefore unlikely, whereas for Cu, Ni, Co and Fe, the activation is possible under realistic conditions. According to the kinetic simulations, however, the dimer motif is a plausible candidate for the active site for Cu only. For Ni, Co and Fe, close-to-infinite reaction times or unreasonably high temperatures are required for sufficient methane conversion. As Ni-, Co- and Fe-exchanged ZSM-5 are known to convert methane to methanol, these results indicate that the Cu-based dimer motif is not an appropriate model system for these metals.
1. Introduction
The known reserves of natural gas at the end of 2014 were 190 trillion cubic meters, which are enough to sustain global consumption for more than half a century.1 Unfortunately, much of the reserves are located in isolated areas, like off-shore oil wells, and as a consequence a large amount of this valuable resource is today flared into carbon dioxide and water. It is estimated that 3.5%, or about 143 billion cubic metres, of the natural gas globally extracted was flared in 2012.2Natural gas consists mainly of methane along with minor amounts of higher alkanes, such as ethane, propane and buthane.3 Owing to the low carbon emission per energy unit of methane, natural gas is often regarded as a transitional energy source while the energy systems of society are transformed to more sustainable solutions.4 Furthermore, methane is an important raw material for other upgraded products.5,6 Hence, methods to convert methane into a liquid, e.g., methanol, are highly desirable, as these would enable easier transportation and thereby allow this valuable resource to become accessible to the chemical industry.
At present, industrial conversion of methane to methanol utilises a two-step process, where methane (natural gas) is transformed into synthesis gas using steam reforming,7 which is later used in methanol synthesis.8 These energy-intensive processes require large centralised plants, which are poorly compatible with the distribution of methane reserves. Direct conversion of methane to methanol would be a considerably more energy-efficient route. However, this reaction is associated with intriguing challenges and has so far not been industrially realised.6,9 One of the main challenges is to activate the highly stable methane for oxidation but still prevent complete oxidation of the reactants, which is thermodynamically favorable.10 Another issue is the activation of molecular oxygen and the formation of a reactive oxygen species that methane can react with. In short, it is a selectivity problem where methane should be oxidised, but not over-oxidised.
Ion-exchanged zeolites with different ions (e.g. Cu, Ni, Co and Fe) have been shown to convert methane to methanol under ambient conditions.11–16 One of the most studied systems for this reaction is copper-exchanged ZSM-5.11,16–26 The first experimental observation of successful methane-to-methanol conversion under ambient conditions, i.e., atmospheric pressure and a temperature of 448 K, for Cu-ZSM-5 was reported by Groothaert et al.11 However, the experiment required O2 activation at a much higher temperature (723 K) and the introduction of a water/acetonitrile mixture for the sequential methanol extraction, which makes the process industrially unattractive. Recent theoretical studies27 have indicated that water reduces the methanol desorption barrier, which could be the underlying reason for adding an extraction solvent. Although a continuous cycle has recently been successfully demonstrated,28,29 an efficient catalyst for methane-to-methanol conversion is still lacking. For progress, it is desirable to scrutinise the active site(s), to understand the oxygen activation and to clarify the influence of extraction-solvation molecules like water in these systems.
There have been many attempts to determine the active sites in metal ion-exchanged zeolites.11,13,17–20,22,24,27,28,30–45 For Cu-exchanged zeolites, a [Cu–O–Cu]2+ motif has been put forth as a possible candidate for the active site. This complex was identified by a UV-vis spectroscopic signature at around 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 cm−1, and the appearance of this signature correlated with the methane-to-methanol conversion.11,23 The [Cu–O–Cu]2+ motif has been extensively characterised, both experimentally and theoretically.23,26,46 The need for a [Cu–O–Cu]2+ site for methane-to-methanol conversion is, however, not conclusive. For instance, recently Narsimhan et al.28 were able to experimentally demonstrate a continuous catalytic methane-to-methanol cycle in Cu-exchanged ZSM-5 without the appearance of the UV-vis signature at around 22700 cm−1. Furthermore, in recent theoretical studies18,47 trinuclear Cu clusters were suggested as active sites for Cu-mordenite and Cu-ZSM-5, while Kulkarni et al.48 suggest a Cu monomer as an active site in addition to the [Cu–O–Cu]2+ and Cu3O3 motifs. Although there exists a controversy as to the exact nature of the active site(s), the use of zeolites seems to be important to prohibit the complete oxidation of methane and steer the selectivity towards methanol. Zeolites are sometimes viewed as inorganic analogues to the naturally occurring enzyme methane monooxygenase (MMO),49,50 which can convert methane to methanol under ambient conditions and is reported to have a porous structure and Fe or Cu double metal ion active sites, similar to zeolites.51
700 cm−1, and the appearance of this signature correlated with the methane-to-methanol conversion.11,23 The [Cu–O–Cu]2+ motif has been extensively characterised, both experimentally and theoretically.23,26,46 The need for a [Cu–O–Cu]2+ site for methane-to-methanol conversion is, however, not conclusive. For instance, recently Narsimhan et al.28 were able to experimentally demonstrate a continuous catalytic methane-to-methanol cycle in Cu-exchanged ZSM-5 without the appearance of the UV-vis signature at around 22700 cm−1. Furthermore, in recent theoretical studies18,47 trinuclear Cu clusters were suggested as active sites for Cu-mordenite and Cu-ZSM-5, while Kulkarni et al.48 suggest a Cu monomer as an active site in addition to the [Cu–O–Cu]2+ and Cu3O3 motifs. Although there exists a controversy as to the exact nature of the active site(s), the use of zeolites seems to be important to prohibit the complete oxidation of methane and steer the selectivity towards methanol. Zeolites are sometimes viewed as inorganic analogues to the naturally occurring enzyme methane monooxygenase (MMO),49,50 which can convert methane to methanol under ambient conditions and is reported to have a porous structure and Fe or Cu double metal ion active sites, similar to zeolites.51
Zeolites are formed by corner-sharing SiO4 and AlO4 tetrahedra or T-sites. Such sites can be joined in many different ways, creating a multitude of structures, including e.g. mordenite, chabasite, and MFI.52,53 ZSM-5 is an example of the latter. By replacing Si4+ with Al3+, different cations, such as H, Cu, Fe, or Co, can be introduced to compensate for the change in charge in the zeolite structure. For any given Si:Al ratio, there exists a distribution of Al T-sites.54 Depending on the number of Al T-sites per cation, their oxidation states can vary. The common oxidation states for Cu are 1+ and 2+,55 indicating that one cation can in principle compensate for the charge of one or two Al T-sites.56 The distribution of Al and the number of Al pairs can be controlled to some degree by tuning the synthesis method.57,58 However, studies have shown that for a Cu dimer site, and especially the [Cu–O–Cu]2+ motif, there is a clear energetically favourable configuration within the ZSM-5 structure at the crossing between the straight and sinusoidal channels.46
An equally clear candidate for the active site in other metal-exchanged zeolites, e.g. Fe-, Ni- and Co-ZSM-5, has yet to be identified. One remaining question to address is whether the [Cu–O–Cu]2+ motif is also an active site in other metal-exchanged zeolites. This question is well suited for first-principles studies and can be addressed by exchanging Cu with known active elements (Ni, Co, Fe), and further scrutinised by also including Ag and Au, which have the same electronic configuration as Cu. Henceforth, we will denote the motif as [M–O–M]2+, which is the structure obtained when replacing Cu with one of the other metals. Furthermore, the level of description of active sites in the ZSM-5 structure needs to be investigated. Small cluster models have been used,23,46 and the question remains whether such small models adequately capture the effect of a pore structure.
Here, we investigate the [M–O–M]2+ motif in the ZSM-5 structure and its implications on the reaction kinetics of methane-to-methanol conversion by combining first-principles methods and micro-kinetic modelling. To determine the model size needed to accurately describe the reaction on the dimer motif, we study cluster models of different sizes for the Cu-ZSM-5 structure, and compare these to the periodic crystal structure. The focus is on the kinetics of the removal of the oxygen linking the two Cu atoms in the [Cu–O–Cu]2+ motif, which is the α-oxygen. The mechanism for adding the α-oxygen is not studied here as it is not fully understood. One possibility is the formation of a peroxo intermediate25 with subsequent removal of the first and second oxygen atoms. The removal of the first oxygen has been proposed20,22 to be assisted by a Cu+ spectator site. If this is the case, our model describes the removal of the second oxygen, which is expected to be rate-limiting. The influence of water is not scrutinised, as experiments are typically carried out without water in the reaction feed.11,21,59 After finding a suitable cluster model system, we address the question of how generic the [M–O–M]2+ motif is, via replacing the Cu by Ni, Co, Fe, Ag or Au, and investigate the transient and steady-state kinetics of methane-to-methanol conversion.
2. Method
The first-principles calculations were performed using density functional theory (DFT), as implemented in the DMol3 software.60–62 The one-electron Kohn–Sham orbitals were expanded in an all-electron double-numeric basis set together with polarization functions (dnp), and the cut-off radius was set to 5 Å. The density was converged until the difference was less than 10−6 a.u. between two subsequent self-consistent steps. The Coulomb potential was obtained by projection of the charge density onto angular dependent weighting functions centered at each atom. The Kohn–Sham equations were solved self-consistently using an integration technique based on weighted overlapping spheres centered at each atom. In the fully periodic model, only the gamma point was considered in the reciprocal space integration over the Brillouin zone. The exchange-correlation was approximated using the Perdew–Burke–Ernzerhof (PBE) spin-polarised formula.63 A sensitivity analysis was performed by separately changing the energy of each intermediate and transition state along the reaction pathway by ±0.25 eV and calculating the response in the kinetics. The analysis shows that the conclusions are not affected, and that the PBE level of accuracy is sufficient.Transition states (TS) were found using the climbing image nudge elastic band (NEB) method,64 and the ionic positions were optimised until the maximum force was less than 0.05 eV Å−1 for the intermediates and the transition states. For the Cu system, the quadratic synchronous transit (QST) method together with a linear synchronous transit (LST) algorithm as implemented in the DMol3 software was also used to find the transition state for methane dissociation.
Entropic contributions to the rate constants of the reaction steps were calculated from vibrational frequencies obtained by displacing the atoms in the [M–O–M]2+ site and the C and H atoms belonging to the methane molecule, i.e. in total, eight atoms were displaced. Central differences were used to obtain the Hessian matrix with a displacement of 0.01 Å. Low frequencies were truncated at 100 cm−1 for numerical stability.
2.1. Structure modelling
Several structure models, based on the experimental structure,65,66 were evaluated in order to accurately describe the [Cu–O–Cu]2+ motif. For all cluster models, the terminal Si atoms were saturated with H atoms in order to remove the dangling bonds and mimic the ZSM-5 zeolite framework. The Si–H distance was set to 1.491 Å, which is the relaxed distance of the SiH4 molecule, in the direction of the replaced Si–O bond.The smallest model, consisting of 20 T-sites (small model), is very close to the one that was earlier suggested by Tsai et al.46 The Al atoms were placed in the crossing between the straight and the sinusoidal pores in the ZSM-5 framework and separated by two T-sites from each other (see the ESI†). This configuration was found to be the most energetically favourable.46 A second model (expanded model) was created by including two T-sites at the bottom of the pore channel, i.e. beneath the active site (in total, 22 T-sites). Finally, T-sites belonging to the top of the pore were included to form a ring model (in total, 40 T-sites). The periodic unit cell (periodic model) with 96 T-sites was also studied with the Al atoms in the same configuration.
The two Cu atoms were placed on top of the Al T-sites, similarly to Tsai et al.46 For the ring and the periodic models, the favourable configuration was found when the two Cu atoms without any oxygen were in adjacent hollow sites on the channel wall. For the small and expanded models, the empty Cu–Cu site was obtained from the [Cu–O–Cu]2+ site by removing the α-O atom located between the Cu atoms and relaxing the system. For further details, see the ESI.† For Ni, Co, Fe, Ag and Au, the same structures as for the Cu-ZSM-5 ring model were used but with the metal atoms exchanged. With the exception of the H atoms in the Si–H bonds, all atoms were allowed to relax during the geometry optimisation.
Relevant zero-point corrected energies were calculated with the [M–O–M]2+ site plus methane in the gas phase as reference, i.e.
| E = EA–Z − (EO–Z + ECH4), | (1) |
2.2. Micro-kinetic modelling
The kinetics of the methane-to-methanol conversion was investigated using a micro-kinetic model with input parameters solely based on the DFT results. The rate constants of the desorption and activated conversion of adsorbed reactants were expressed using the conventional transition state theory (TST),67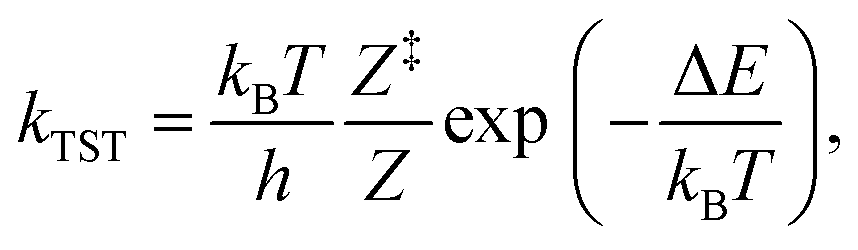 | (2) |
 | (3) |
 | (4) |
 | (5) |
 for the vacant site) and gas-phase methanol (Zrot and Zvib, which is considered as an ideal gas68), and the denominator contains Zviba which is the partition function for the adsorbed state (including the metal ions).
for the vacant site) and gas-phase methanol (Zrot and Zvib, which is considered as an ideal gas68), and the denominator contains Zviba which is the partition function for the adsorbed state (including the metal ions).
Starting from the metal dimer site with an oxygen atom, the [M–O–M]2+ motif, the most direct mechanism for methane-to-methanol conversion is investigated. Specifically, we consider dissociative adsorption of methane with the formation of adsorbed –OH and –CH3 groups, subsequent recombination of these groups resulting in the formation of adsorbed methanol (*CH3OH), and a methanol desorption step. To calculate the time scale characterising the corresponding transient kinetics of this reaction, we first neglect the back-adsorption of methanol from the gas phase. In this case, the kinetic equations describing the reaction steps are:
 | (6) |
 | (7) |
 | (8) |
Eqn (6)–(8) correspond to the situation when methanol is removed from the reactor containing the zeolite catalyst and methanol adsorption is negligible. To clarify the role of the latter process, we have also calculated the steady-state kinetics including methanol adsorption by modifying eqn (8) to include a methanol adsorption term as
 | (9) |
3. Results and discussion
The gas-phase C–H distances in methane were calculated to be 1.10 Å which compares well with the experimental value of 1.09 Å.69 For methanol, the C–H, C–O, and O–H distances were calculated to be 1.10 Å, 1.43 Å, and 0.97 Å, respectively. These values compare well with the experimental distances of 1.10 Å, 1.43 Å and 0.96 Å, respectively.70 The formation energy of methanol from methane and oxygen was calculated to be −1.08 eV, which compares reasonably well with the experimental value of −1.28 eV.713.1. Structure model
The size of the cluster model was converged with respect to the oxygen and methanol adsorption energies and compared to the values for the fully periodic Cu-ZSM-5 zeolite. The oxygen adsorption energies are calculated to be −2.13 eV, −1.73 eV and −1.59 eV with increasing cluster size (see Fig. 1). Compared to the adsorption energy of the periodic model which is −1.62 eV, it is clear that the smaller and expanded models overestimate the adsorption energy of oxygen. The methanol adsorption energy does not have the same monotonic decrease. Instead the calculated values are −1.52 eV, −1.18 eV and −1.50 eV for the small, expanded and ring models, respectively, which should be compared to −1.63 eV for the periodic model. Although absolute adsorption values are important, the relative energy difference between reaction intermediates affects the kinetics even more. Here, we see that the difference between the oxygen and methanol adsorption energies changes from 0.61 eV to 0.55 eV, 0.09 eV, and 0.01 eV for the small, expanded, ring, and periodic models, respectively. The difference between the latter two values is small, and we judge the ring model to be a good compromise between accuracy in energies (left vertical axis) and computational cost (right vertical axis). It is clear that the smallest cluster size, which is closest to the one previously used,46 is not sufficient. | ||
| Fig. 1 Oxygen and methanol adsorption energies of the [Cu–O–Cu]2+ motif (left vertical axis) and computational cost in CPU hours for the last SCF cycle (right vertical axis) as a function of the model size. Atomic colour code: silicon (yellow), oxygen (red), aluminium (green), copper (orange), and hydrogen (white). | ||
It is also clear (see Table 1) that the ring model agrees well with the periodic structure concerning such properties as Cu–O–Cu angle, Cu–Cu distance and Mulliken charge of the α-oxygen. The results obtained for the ring and periodic models differ by less than 0.3%, while for the small and expanded models the difference to the periodic model is around 2% with respect to the Cu–O–Cu angle and Cu–Cu distance, and 7% with respect to the charge of the α-oxygen. The conclusion from this is clear: the small and expanded models are not sufficient to describe the [Cu–O–Cu]2+ motif.
| Model | ∠Cu–O–Cu (°) | Cu–Cu (Å) | α-O charge |
|---|---|---|---|
| Small | 138.7 | 3.32 | −0.61 |
| Expanded | 138.8 | 3.32 | −0.61 |
| Ring | 142.6 | 3.39 | −0.57 |
| Periodic | 142.2 | 3.39 | −0.57 |
3.2. Energy landscapes
Using the ring model for the dimer site, the oxygen adsorption energies for Cu, Ni, Co, Fe, Ag and Au are calculated considering one half of an O2 or N2O molecule as a reference (see Table 2). The formation of α-oxygen in the Ag and Au zeolites is calculated to be energetically unfavourable, while the opposite holds for Cu, Ni, Co and Fe. Based on these findings, we do not consider the Au- and Ag-dimer sites in the micro-kinetic model, as oxygen activation of these sites is unrealistic. and N2O
and N2O
| Metal ions | Ref. O2 (eV) | Ref. N2O (eV) |
|---|---|---|
| Cu | −1.59 | −1.80 |
| Ni | −2.67 | −2.87 |
| Co | −3.81 | −4.01 |
| Fe | −5.14 | −5.35 |
| Ag | 1.29 | 1.08 |
| Au | 1.22 | 1.02 |
Using the ring model, the energy landscapes were calculated with the considered mechanism and different metal ions (see Fig. 2). It is important to note that these energy landscapes start from an already oxygen-activated site.
 | ||
| Fig. 2 Reaction energy landscapes for the direct conversion of methane to methanol over ZSM-5 for Cu, Ni, Co and Fe dimer sites. Two curves for Cu and TS1 are shown, wherein the solid darker line represents the surface-stabilised TS whereas the dashed line is the radical-like TS. The atomic models show selected structures for the Cu system. Atomic colour code: silicon (yellow), oxygen (red), aluminium (green), carbon (gray), hydrogen (white), and copper (orange). | ||
We consider the geometrically shortest reaction route for methane dissociation (TS1), through what resembles a surface-stabilised transition state with a CH3-group bound to one of the copper ions. There exists another path through a CH3 radical-like transition state, similar e.g. to that found by Latimer et al.72 for a copper-dimer site in mordenite. Using the climbing image NEB method, we did not find the radical barrier. However, with the alternative LSTQST method we were able to find the radical-like TS for Cu. This barrier, which was confirmed by vibrational analysis, is 0.39 eV lower than the surface mediated barrier. Although the radical-like barrier is lower than the surface-stabilised barrier for Cu, the sensitivity analysis shows that it is only for the [Cu–O–Cu]2+ motif that the first TS is a rate-controlling step. For Ni, Co, and Fe, the results are not affected by changing the methane dissociation barrier.
The second barrier (TS2) is the recombination of the methyl group (bound to one of the metal ions) and the hydroxyl group (in between the two metal ions) into adsorbed methanol. This adsorbed methanol species can then desorb in the last step of the reaction mechanism.
We see that Cu has smaller barriers than the other metals: 0.85 eV for TS1 (0.45 eV for the radical-like TS1) and 0.78 eV for TS2, compared to 0.61 and 1.98 eV for Ni, 0.83 and 2.36 eV for Co, and 1.37 and 2.30 eV for Fe. For Cu, the energies of the two different TS1 found for the methane dissociation, in particular the surface-stabilised TS, agree reasonably well with previously reported results, e.g. 0.7 eV (ref. 23) and 0.96 eV (ref. 18). The reaction is also the least endothermic for Cu with 0.41 eV, starting from the [Cu–O–Cu]2+ site and gas-phase methane and ending with the [Cu–Cu]2+ site and gas-phase methanol. For Ni, Co and Fe, the energy cost for producing methanol is much larger: 1.48 eV, 2.62 eV and 3.96 eV, respectively. Note that the general trend found in the calculated reaction landscapes will not be affected by the inclusion of dispersion forces owing to the large difference (in the range of several eVs) between the different [M–O–M]2+ sites.
3.3. Micro-kinetic modelling
Starting from oxygen-covered [M–O–M]2+ sites and using realistic experimental conditions such as a methane partial pressure of 1 mbar and a temperature of 523 K, we have calculated the transient reaction kinetics by employing eqn (6)–(8). In particular, Fig. 3a) shows the fraction of empty sites as a function of time. Owing to the simplicity of our reaction mechanism, this fraction is proportional to the amount of desorbed methanol molecules. The time scale of the reaction can in this case be quantified using the time τ1/2, which is the time needed to convert half of the α-oxygen and methane into gas-phase methanol. This is shown for Cu, Ni, Co and Fe as a function of temperature in Fig. 3b). The time scale is reasonable for Cu, but too long for Ni, Co and Fe. There exists, however, experimental evidence for methane-to-methanol conversion on Ni-, Co- and Fe-ZSM-5 samples.12–14 This means that for Ni, Co and Fe, either the proposed reaction mechanism is not correct, or the dimer motif is not the active site. | ||
| Fig. 3 a) Coverage of empty sites (methanol produced) for the Cu, Ni, Co and Fe dimer sites as a function of time at 523 K and b) the reaction time τ1/2 when half of the [M–O–M]2+ sites have reacted with methane as a function of temperature. The dashed line is for the radical-like TS1 for Cu. | ||
The steady-state reaction kinetics have been studied using eqn (6), (7) and (9) for a methanol pressure of 10−6 bar. The corresponding steady-state fractions of empty sites are shown in Fig. 4 as a function of temperature. The [Cu–O–Cu]2+ site converts methane to methanol at reasonable temperatures (around 500 K), while Ni, Co and Fe require unrealistically high temperatures. Note that the zeolite framework breaks at around 1400 K.73 We see that the two different barriers for Cu influence the transient kinetics in Fig. 3, but do not affect the steady-state kinetics in Fig. 4. This confirms our conclusion from the transient kinetics that the [M–O–M]2+ site is not a plausible candidate for the active site(s) for Ni, Co and Fe.
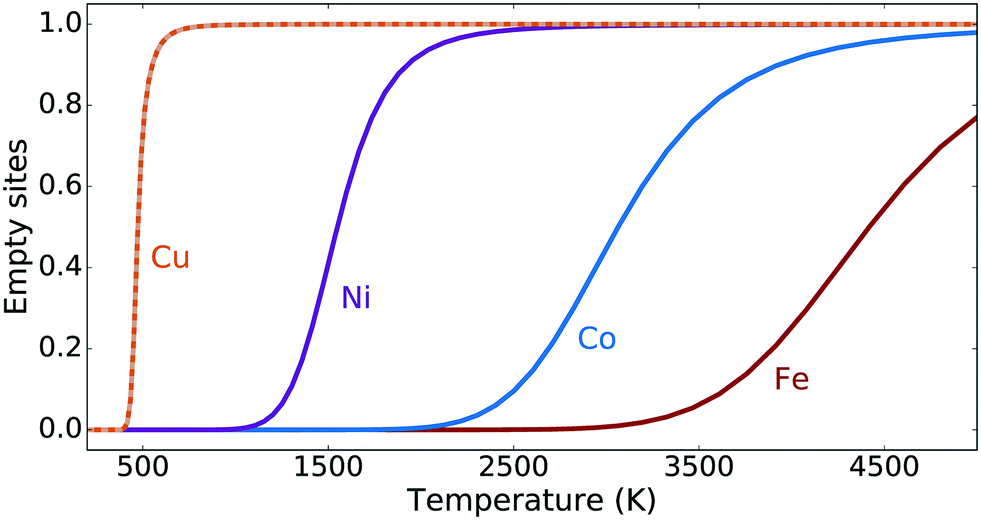 | ||
| Fig. 4 Fractions of empty sites for the Cu, Ni, Co and Fe dimer sites under steady-state conditions with inclusion of methanol adsorption, as a function of temperature. The kinetics for Cu is shown for the surface-supported (solid line) and the radical-like TS1 (dashed line). | ||
3.4. Sensitivity analysis
By changing each energy separately in the energy landscape by ±0.25 eV for Cu, Ni, Co and Fe, we have performed a sensitivity analysis of the micro-kinetic model. The outcome demonstrates that our results are robust, and that a different energy description (e.g. including van der Waals interactions) will not change the conclusion that the dimer site is reasonable for Cu, but not for Ni, Co or Fe. In this context, note that our energy change, ±0.25 eV, is of the same order of magnitude as the shift in adsorption energies attributed by Göltl et al.74 to van der Waals interactions in zeolites. For further details on the results of the sensitivity analysis, see the ESI.†Furthermore, the sensitivity analysis shows which energies are important for the overall reaction in general and the reaction half-time τ1/2 in particular. It demonstrates that the kinetics for all the elements (Cu, Ni, Co and Fe) are dependent on the energy of the oxygen-activated [M–O–M]2+ site. The kinetics for Cu is the most sensitive with respect to the TS1 energy, while for Ni and Co, the TS2 energy is more important. For Fe, the stability of the bare ion site is the most important, as the barriers are small compared to the overall energy cost for producing methanol from methane. We note that for Cu, the radical pathway to TS1 affects τ1/2, but that the order (Cu, Ni, Co and Fe) will not be affected by either barrier. This analysis confirms that Cu has the shortest reaction half-time, followed by Ni and Co, and that the half-time for Fe is unrealistic.
4. Conclusions
We showed that a large cluster model size is needed in order to accurately describe the [Cu–O–Cu]2+ motif in the ZSM-5 structure. In order to investigate if the widely studied [Cu–O–Cu]2+ motif is a relevant candidate for the active site in other ion-exchanged ZSM-5 systems, the energy landscape for methane-to-methanol conversion was calculated for Cu, Ni, Co, Fe, Ag and Au. The oxygen activation of Ag and Au was found to be energetically unfavorable. Thereby the dimer motif is not likely for these elements. The transient and steady-state behaviour of the proposed micro-kinetic model for the Cu, Ni, Co and Fe sites was analysed. The results show that for Cu, the dimer site is a plausible candidate for the active site, with a reasonable reaction time at realistic temperatures. Since no activity under reasonable conditions was found for Ni-, Co- and Fe-dimer sites, which experimentally are known to be active for methane-to-methanol conversion, the dimer site is judged to be not the correct candidate for the active sites in these systems. The strong oxygen binding of, for instance, Fe, indicates that a more highly oxidised Fe dimer motif is a more plausible candidate for the active site.The sensitivity analysis confirms that the results are robust and relatively insensitive to errors in the energies, for instance with respect to the surface-stabilised or radical-like transition state for methane dissociation. It also shows that the reaction kinetics are the most sensitive to errors in the methane-dissociation barrier for Cu, while the sensitivity is higher with respect to the second barrier in the cases of Ni and Co. For Fe, the overall energy difference between methane and methanol is what determines the reaction time.
Acknowledgements
This work is done within the Röntgen-Ångström collaboration “Time-resolved in situ methods for design of catalytic sites within sustainable chemistry” which is financially supported by the Swedish Research Council and the Competence Centre for Catalysis. The Knut and Alice Wallenberg Foundation via the project “Atomistic Design of Catalysts” is also gratefully acknowledged. The Competence Centre for Catalysis is hosted by Chalmers University of Technology and financially supported by the Swedish Energy Agency and the member companies AB Volvo, ECAPS AB, Haldor Topsøe A/S, Scania CV AB, Volvo Car Corporation AB and Wärtsilä Finland Oy. The calculations were performed at C3SE (Göteborg) via a SNIC grant.References
- BP, Statistical Review of World Energy, 2015, http://www.bp.com/statisticalreview Search PubMed.
- C. D. Elvidge, M. Zhizhin, K. Baugh, F.-C. Hsu and T. Ghosh, Energies, 2016, 9, 1–15 Search PubMed.
- Composition of natural gas, 2013, http://naturalgas.org/overview/background/ Search PubMed.
- S. Pacala and R. Socolow, Science, 2004, 305, 968–972 CrossRef CAS PubMed.
- A. Caballero and P. J. Pérez, Chem. Soc. Rev., 2013, 42, 8809–8820 RSC.
- A. I. Olivos-Suarez, À. Szécsényi, E. J. M. Hensen, J. Ruiz-Martinez, E. A. Pidko and J. Gascon, ACS Catal., 2016, 6, 2965–2981 CrossRef CAS.
- J. R. Rostrup-Nielsen, Catal. Today, 1993, 18, 305–324 CrossRef.
- I. Wender, Fuel Process. Technol., 1996, 48, 189–297 CrossRef CAS.
- A. Holmen, Catal. Today, 2009, 142, 2–8 CrossRef CAS.
- P. Tang, Q. Zhu, Z. Wu and D. Ma, Energy Environ. Sci., 2014, 7, 2580–2591 CAS.
- M. H. Groothaert, P. J. Smeets, B. F. Sels, P. A. Jacobs and R. A. Schoonheydt, J. Am. Chem. Soc., 2005, 127, 1394–1395 CrossRef CAS PubMed.
- J. Shan, W. Huang, L. Nguyen, Y. Yu, S. Zhang, Y. Li, A. I. Frenkel and F. F. Tao, Langmuir, 2014, 30, 8558–8569 CrossRef CAS PubMed.
- N. V. Beznis, B. M. Weckhuysen and J. H. Bitter, Catal. Lett., 2010, 136, 52–56 CrossRef CAS.
- E. V. Starokon, M. V. Parfenov, L. V. Pirutko, S. I. Abornev and G. I. Panov, J. Phys. Chem. C, 2011, 115, 2155–2161 CAS.
- S. E. Bozbag, E. M. C. Alayon, J. Pecháček, M. Nachtegaal, M. Ranocchiari and J. A. van Bokhoven, Catal. Sci. Technol., 2016, 6, 5011–5022 CAS.
- M. J. Wulfers, S. Teketel, B. Ipek and R. F. Lobo, Chem. Commun., 2015, 51, 4447–4450 RSC.
- N. V. Beznis, B. M. Weckhuysen and J. H. Bitter, Catal. Lett., 2010, 138, 14–22 CrossRef CAS.
- G. Li, P. Vassilev, M. Sanchez-Sanchez, J. A. Lercher, E. J. M. Hensen and E. A. Pidko, J. Catal., 2016, 338, 305–312 CrossRef CAS.
- P. J. Smeets, M. H. Groothaert and R. A. Schoonheydt, Catal. Today, 2005, 110, 303–309 CrossRef CAS.
- P. J. Smeets, R. G. Hadt, J. S. Woertink, P. Vanelderen, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, J. Am. Chem. Soc., 2010, 132, 14736–14738 CrossRef CAS PubMed.
- P. Tomkins, A. Mansouri, S. E. Bozbag, F. Krumeich, M. B. Park, E. M. C. Alayon, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., 2016, 128, 5557–5561 CrossRef.
- P. Vanelderen, R. G. Hadt, P. J. Smeets, E. I. Solomon, R. A. Schoonheydt and B. F. Sels, J. Catal., 2011, 284, 157–164 CrossRef CAS PubMed.
- J. S. Woertink, P. J. Smeets, M. H. Groothaert, M. A. Vance, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18908–18913 CrossRef CAS PubMed.
- M. H. Mahyuddin, A. Staykov, Y. Shiota and K. Yoshizawa, ACS Catal., 2016, 8321–8331 CrossRef CAS.
- T. Yumura, Y. Hirose, T. Wakasugi, Y. Kuroda and H. Kobayashi, ACS Catal., 2016, 2487–2495 CrossRef CAS.
- P. Haack, A. Kärgel, C. Greco, J. Dokic, B. Braun, F. F. Pfaff, S. Mebs, K. Ray and C. Limberg, J. Am. Chem. Soc., 2013, 135, 16148–16160 CrossRef CAS PubMed.
- Z.-J. Zhao, A. Kulkarni, L. Vilella, J. K. Nørskov and F. Studt, ACS Catal., 2016, 3760–3766 CrossRef CAS.
- K. Narsimhan, K. Iyoki, K. Dinh and Y. Román-Leshkov, ACS Cent. Sci., 2016, 2, 424–429 CrossRef CAS PubMed.
- B. Ipek and R. F. Lobo, Chem. Commun., 2016, 52, 13401–13404 RSC.
- M. H. Groothaert, K. Pierloot, A. Delabie and R. A. Schoonheydt, Phys. Chem. Chem. Phys., 2003, 5, 2135–2144 RSC.
- B. F. Mentzen and G. Bergeret, J. Phys. Chem. C, 2007, 111, 12512–12516 CAS.
- T. Yumura, M. Takeuchi, H. Kobayashi and Y. Kuroda, Inorg. Chem., 2009, 48, 508–517 CrossRef CAS PubMed.
- N. V. Beznis, A. N. C. van Laak, B. M. Weckhuysen and J. H. Bitter, Microporous Mesoporous Mater., 2011, 138, 176–183 CrossRef CAS.
- E. V. Starokon, K. A. Dubkov, L. V. Pirutko and G. I. Panov, Top. Catal., 2003, 23, 137–143 CrossRef CAS.
- C. Hammond, M. M. Forde, M. H. Ab Rahim, A. Thetford, Q. He, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, N. F. Dummer, D. M. Murphy, A. F. Carley, S. H. Taylor, D. J. Willock, E. E. Stangland, J. Kang, H. Hagen, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2012, 51, 5129–5133 CrossRef CAS PubMed.
- D. Nachtigallová, P. Nachtigall, M. Sierka and J. Sauer, Phys. Chem. Chem. Phys., 1999, 1, 2019–2026 RSC.
- A. B. Ene, M. Bauer, T. Archipov and E. Roduner, Phys. Chem. Chem. Phys., 2010, 12, 6520–6531 RSC.
- G. Li, E. A. Pidko, R. A. van Santen, C. Li and E. J. M. Hensen, J. Phys. Chem. C, 2013, 117, 413–426 CAS.
- P. Vanelderen, B. E. R. Snyder, M.-L. Tsai, R. G. Hadt, J. Vancauwenbergh, O. Coussens, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, J. Am. Chem. Soc., 2015, 137, 6383–6392 CrossRef CAS PubMed.
- E. M. C. Alayon, M. Nachtegaal, A. Bodi and J. A. van Bokhoven, ACS Catal., 2014, 4, 16–22 CrossRef CAS.
- E. M. C. Alayon, M. Nachtegaal, A. Bodi, M. Ranocchiari and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2015, 17, 7681–7693 RSC.
- F. Göltl, C. Michel, P. C. Andrikopoulos, A. M. Love, J. Hafner, I. Hermans and P. Sautet, ACS Catal., 2016, 8404–8409 CrossRef.
- M. He, J. Zhang, X. Sun, B.-H. Chen and Y.-G. Wang, J. Phys. Chem. C, 2016, 27422–27429 CAS.
- C. Paolucci, A. A. Parekh, I. Khurana, J. R. Di Iorio, H. Li, J. D. Albarracin Caballero, A. J. Shih, T. Anggara, W. N. Delgass, J. T. Miller, F. H. Ribeiro, R. Gounder and W. F. Schneider, J. Am. Chem. Soc., 2016, 138, 6028–6048 CrossRef CAS PubMed.
- L. Vilella and F. Studt, Eur. J. Inorg. Chem., 2016, 2016, 1514–1520 CrossRef CAS.
- M.-L. Tsai, R. G. Hadt, P. Vanelderen, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, J. Am. Chem. Soc., 2014, 136, 3522–3529 CrossRef CAS PubMed.
- S. Grundner, M. A. C. Markovits, G. Li, M. Tromp, E. A. Pidko, E. J. M. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2015, 6, 7546 CrossRef PubMed.
- A. R. Kulkarni, Z.-J. Zhao, S. Siahrostami, J. K. Nørskov and F. Studt, ACS Catal., 2016, 6531–6536 CrossRef CAS.
- A. C. Rosenzweig, C. A. Frederick, S. J. Lippard and P. Nordlund, Nature, 1993, 366, 537–543 CrossRef CAS PubMed.
- A. E. Shilov and A. A. Shteinman, Russ. Chem. Rev., 2012, 81, 291 CrossRef CAS.
- E. I. Solomon, J. W. Ginsbach, D. E. Heppner, M. T. Kieber-Emmons, C. H. Kjaergaard, P. J. Smeets, L. Tian and J. S. Woertink, Faraday Discuss., 2010, 148, 11–39 RSC.
- M. Beller, A. Renken and R. A. van Santen, Catalysis, Wiley-VCH, Weinheim, 2012 Search PubMed.
- J. J. Pluth, J. V. Smith and W. J. Mortier, Mater. Res. Bull., 1977, 12, 1001–1007 CrossRef CAS.
- B. R. Goodman, K. C. Hass, W. F. Schneider and J. B. Adams, Catal. Lett., 2000, 68, 85–93 CrossRef CAS.
- G. T. Palomino, P. Fisicaro, S. Bordiga, A. Zecchina, E. Giamello and C. Lamberti, J. Phys. Chem. B, 2000, 104, 4064–4073 CrossRef CAS.
- F. Göltl, P. Sautet and I. Hermans, Catal. Today, 2016, 267, 41–46 CrossRef.
- O. H. Han, C.-S. Kim and S. B. Hong, Angew. Chem., 2002, 114, 487–490 CrossRef.
- V. Gábová, J. Dědeček and J. Čejka, Chem. Commun., 2003, 1196–1197 RSC.
- S. Grundner, W. Luo, M. Sanchez-Sanchez and J. A. Lercher, Chem. Commun., 2016, 52, 2553–2556 RSC.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- B. Delley, J. Phys. Chem., 1996, 100, 6107–6110 CrossRef CAS.
- We have used DMol3, within Materials Studio, version 7.0.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- H. van Koningsveld, H. van Bekkum and J. C. Jansen, Acta Crystallogr., Sect. B: Struct. Sci., 1987, 43, 127–132 CrossRef.
- Accelrys Software Inc., Materials Studio Release Notes, Release 7.0, Accelrys Software Inc., San Diego, 2014 Search PubMed.
- I. Chorkendorff and J. W. Niemantsverdriet, Concepts of Modern Catalysis and Kinetics, John Wiley & Sons, 2006 Search PubMed.
- T. L. Hill, An Introduction to Statistical Thermodynamics, Dover Publications Inc, New York, 1960 Search PubMed.
- E. Hirota, J. Mol. Spectrosc., 1979, 77, 213–221 CrossRef CAS.
- P. Venkateswarlu and W. Gordy, J. Chem. Phys., 1955, 23, 1200–1202 CrossRef CAS.
- L. V. Gurvich, Pure Appl. Chem., 2009, 61, 1027–1031 Search PubMed.
- A. A. Latimer, A. R. Kulkarni, H. Aljama, J. H. Montoya, J. S. Yoo, C. Tsai, F. Abild-Pedersen, F. Studt and J. K. Nørskov, Nat. Mater., 2017, 16, 225–229 CrossRef CAS PubMed.
- Y. Hirata, T. Kodama, K. Shimada and Y. Ishihara, Nippon Seramikkusu Kyokai Gakujutsu Ronbunshi, 1991, 99, 1255–1259 CrossRef CAS.
- F. Göltl, A. Grüneis, T. Bučko and J. Hafner, J. Chem. Phys., 2012, 137, 114111 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cy02521h |
| This journal is © The Royal Society of Chemistry 2017 |