
The development of ruthenium(II) polypyridyl complexes and conjugates for in vitro cellular and in vivo applications
Fergus E.
Poynton
 *a,
Sandra A.
Bright
b,
Salvador
Blasco
a,
D. Clive
Williams
b,
John M.
Kelly
c and
Thorfinnur
Gunnlaugsson
*a
*a,
Sandra A.
Bright
b,
Salvador
Blasco
a,
D. Clive
Williams
b,
John M.
Kelly
c and
Thorfinnur
Gunnlaugsson
*a
aSchool of Chemistry and Trinity Biomedical Sciences Institute (TBSI), Trinity College Dublin, The University of Dublin, Dublin 2, Ireland. E-mail: poyntonf@tcd.ie; gunnlaut@tcd.ie
bSchool of Biochemistry and Immunology and Trinity Biomedical Sciences Institute (TBSI), Trinity College Dublin, The University of Dublin, Dublin 2, Ireland
cSchool of Chemistry, Trinity College Dublin, The University of Dublin, Dublin 2, Ireland
First published on 27th November 2017
Abstract
Ruthenium(II) [Ru(II)] polypyridyl complexes have been the focus of intense investigations since work began exploring their supramolecular interactions with DNA. In recent years, there have been considerable efforts to translate this solution-based research into a biological environment with the intention of developing new classes of probes, luminescent imaging agents, therapeutics and theranostics. In only 10 years the field has expanded with diverse applications for these complexes as imaging agents and promising candidates for therapeutics. In light of these efforts this review exclusively focuses on the developments of these complexes in biological systems, both in cells and in vivo, and hopes to communicate to readers the diversity of applications within which these complexes have found use, as well as new insights gained along the way and challenges that researchers in this field still face.
 Fergus E. Poynton | Fergus Poynton obtained his PhD in Chemistry from Trinity College in 2016 under the supervision of Prof. Gunnlaugsson and Prof. Kelly, investigating the excited-state processes and reactivity of ruthenium(II) polypyridyl complexes in solution and in the presence of DNA, with a focus on exploiting these properties to design luminescent probes and light-activated anti-cancer agents. He was subsequently awarded both the Irish RIA and international IUPAC-SOLVAY Award for Young Chemists for the most outstanding PhD thesis in the chemical sciences. He is now a postdoctoral researcher in the translational medicine group of Prof. Seamas Donnelly, developing new therapeutics for respiratory diseases. |
 Sandra A. Bright | Sandra Bright completed her PhD in Biochemistry in 2009 in Trinity College Dublin, researching the effects of novel pyrollo-1,5-benzoxazepines in chronic myeloid leukaemia (CML). Sandra then undertook post-doctoral positions with Prof. Williams and Prof. Gunnlaugsson. Her research interests include oncology, drug development, photodynamic therapy and fluorescent sensors. Sandra currently works in the field of regulation of medicines. |
 Salvador Blasco | Salvador Blasco graduated in Chemistry and Chemical Engineering in the University of Valencia, Spain. He did his PhD research in Prof. Enrique García-España's group and received a PhD in 2011 for a research on antioxidant biomimetics. In 2013 he moved to Ireland to join Prof. Gunnlaugsson's group as a postdoc with a Marie Curie fellowship where he researched on Ruthenium compounds with anti-cancer properties. In 2016 he went back to Spain where he is now a Senior Researcher in the University of Valencia and Associated Professor in UCH-CEU. |
 D. Clive Williams | Clive Williams obtained his BSc and PhD from Cardiff University in 1970 and 1973. After a period in the University Chemical Laboratory, Cambridge with Prof Sir Alan Battersby FRS, he joined Trinity College Dublin in 1977, was Head of Biochemistry from 1997–2004, University Bursar from 2005–2008, and Dean of Faculty of Engineering and Science from 2008–2015. He is currently Fellow Emeritus and Professor Emeritus. He obtained a Personal Chair in Chemical Biology in 2012 and was elected Member of the Royal Irish Academy (MRIA) in 1996. His current research interests are focussed on the interaction of small molecules with proteins. |
 John M. Kelly | John M. Kelly, FRSC, MRIA., obtained his BSc from the University of Manchester, his MSc in organic photochemistry from McMaster University and his PhD in photo-physical chemistry from the University of London (supervisor, George Porter) in 1970. He joined Trinity College Dublin in 1973, was the Head of Department from 1994–2000 and is currently Fellow Emeritus. His recent research interests are focussed on the transient spectroscopy and photochemistry of DNA and metal containing compounds. In 2016, with Prof. Christine Cardin and Dr. Susan Quinn he was awarded the Cornforth medal by the RSC. |
 Thorfinnur Gunnlaugsson | Thorfinnur (Thorri) Gunnlaugsson MRIA, is a Professor of Chemistry in the School of Chemistry, Trinity College Dublin (TCD) and a PI in the Trinity Biomedical Sciences Institute (TBSI). His research interests lie broadly within the fields of medicinal, organic, inorganic supramolecular and materials chemistries. He is a Fellow of TCD, and was elected as a Member of The Royal Irish Academy in 2011. In 2014, he was awarded The Institute of Chemistry of Ireland (ICI) Annual Award for Chemistry (Eva Philbin Lecturer). He is the author of over 220 papers and has an H-index of 70. |
Introduction
The synthesis of Ru(II) polypyridyl complexes and investigation into their salient properties has become a major area of research over the past few decades.1–5 This stems largely from their appealing photophysical and photochemical properties. Such complexes absorb visible light and emit long wavelength light within the red and near-infrared spectral regions. They possess long-lived triplet excited states and can show reversible redox processes.6,7 This has made such complexes highly desirable across numerous research fields, including catalysis, solar energy, sensors, etc.8–15 Moreover, their photophysical and photochemical properties can be tuned by varying the nature and the numbers of the polypyridyl ligands around the Ru(II) metal centre.16,17 Furthermore, due to the octahedral geometry of a number of these complexes, chemists can readily gain access to molecules with complicated 3-dimensional architectures. Hence, they have found increasing applications within the realm of supramolecular chemistry,18 and importantly, in biological systems, where they have been used to gain greater insights into the non-covalent binding of molecules to DNA,19,20 and as luminescent probes.21,22Most drug molecules elicit their biological activity through non-covalent (supramolecular) interactions with biomolecules. As such, the knowledge gained from such solution-based studies into the noncovalent interactions between Ru(II) polypyridyl complexes and biomolecules lends itself well to designing new classes of therapeutic agents.23 Within the last decade, considerable research has focused on the translation of Ru(II) polypyridyl complexes into biological systems for use as cellular imaging and diagnostic agents, and to develop new classes of therapeutic agents. The aim of this review is to highlight these particular developments, rather than discuss the nature of the binding mechanism, or the photophysical properties of Ru(II) based polypyridyl complexes with nucleic acids, as this has been well documented in recent times.19,24–27 Moreover, while investigations into the interactions of Ru(II) polypyridyl complexes with DNA remains the primary focus within this field, the interactions of such complexes with proteins can have a profound influence on their behaviour in vitro and in vivo, as will be illustrated in this review.
This review begins with discussion on the nature of the uptake and localisation of Ru(II) polypyridyl complexes in cells, with particular focus on their use as luminescent cellular probes. This discussion is followed by examining the application of two-photon activation of Ru(II) complexes; a technique that is becoming of increasing importance in biological imaging and light activatable therapeutics. We then examine the influence of the chirality of Ru(II) polypyridyl complexes on their uptake and localisation within cells. Subsequently, we detail the biological applications of Ru(II) polypyridyl complexes as novel therapeutic agents. Finally, we examine the diverse range of molecular structures that Ru(II) polypyridyl complexes have been incorporated into or onto, in the development of new classes of diagnostic and therapeutic agents.
Cellular uptake & localisation
When translating solution-based research to a cellular context, the first major hurdle for Ru(II) complexes is for them to be successfully internalised into cells. There are a number of mechanisms by which molecules can enter cells, such as passive diffusion, facilitated diffusion, active transport and endocytosis, as illustrated in Fig. 1.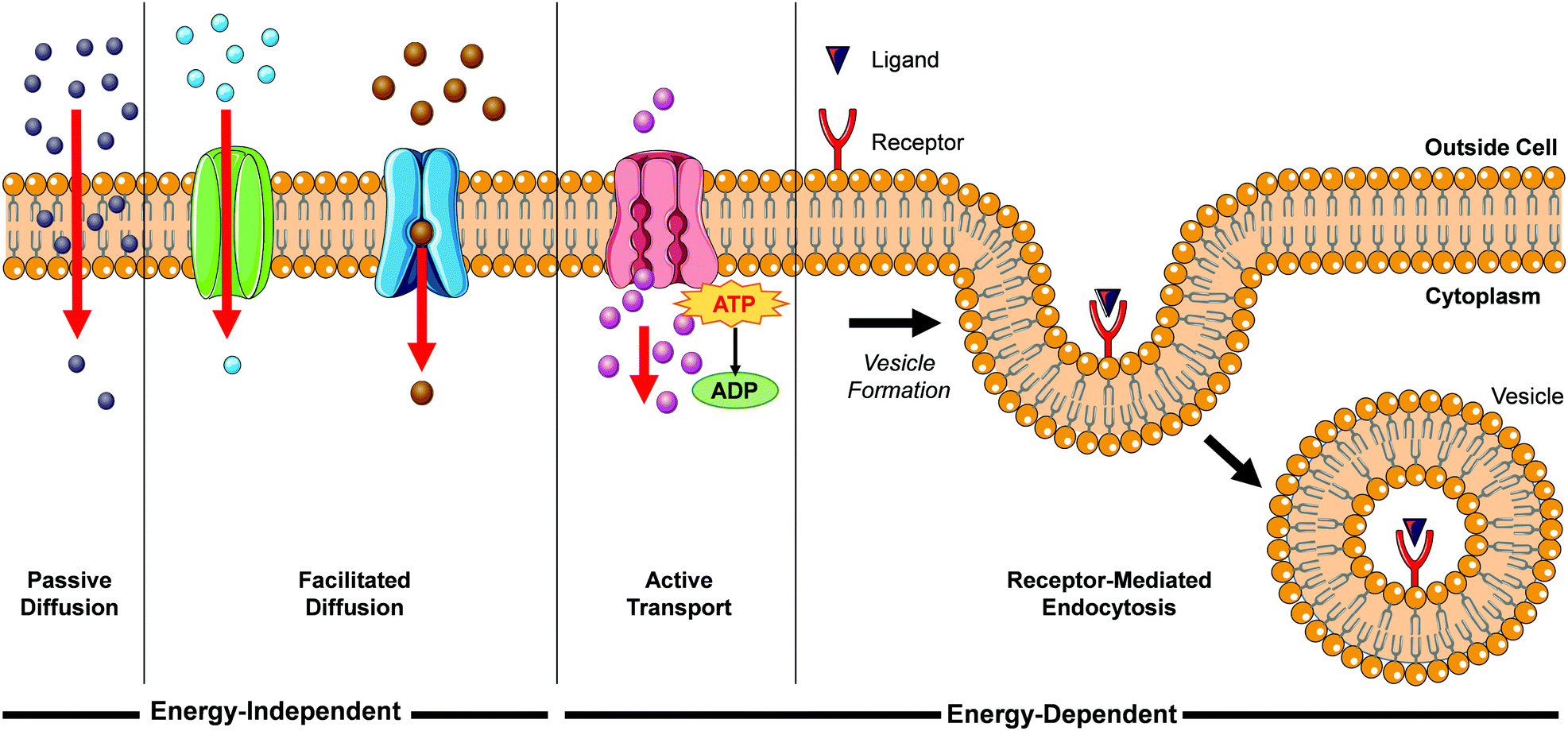 | ||
| Fig. 1 Schematic representation of a number of mechanisms by which molecules can enter cells. | ||
During passive diffusion, a molecule dissolves in the phospholipid bilayer, diffuses through it and then dissolves into the aqueous medium inside the cell. For this mechanism of uptake the lipophilicity and size of a molecule play very important roles.28 In addition, this mode of uptake is not cell type specific, which may be an important consideration for researchers if trying to develop more cell-specific targeted therapeutics. Most biological molecules, however, as well as many drug molecules, are unable to diffuse through the cell membrane. Cells have specific transport proteins for such cases that allow them to selectively control the passage of small molecules across their membrane. During facilitated diffusion of molecules into a cell these proteins can either (i) bind to specific molecules and undergo a conformation change that allows the molecule to pass through the membrane and be released on the other side (carrier proteins) or (ii) form pores in the membrane through which molecules of the appropriate size and charge can pass (channel proteins). Both passive and facilitated diffusion are driven by the concentration gradient of the molecule across the cell membrane and do not require energy to occur. The uptake of cationic molecules (such as Ru(II) complexes) by these mechanisms can also be driven by the negative membrane potential of cells (typically ca. −60 mV).29,30 Proteins may also transport molecules against their concentration gradients in a process called active transport. The energy to do this is provided by the hydrolysis of ATP or by the co-transport of another molecule down its concentration gradient. More than 400 membrane transporters have been annotated in the human genome and numerous drug molecules have been found to “hitch-hike” on such carriers and transporters, whose natural functions are to transport endogenous substances such as glucose, organic cations, organic anions, oligopeptides etc.31,32
While the above uptake mechanisms are typically used by cells to transport small molecules, endocytosis facilitates the uptake of macromolecules and particles, as well as fluids, from the surrounding medium. During this process, the material to be internalised is surrounded by part of the plasma membrane, which then buds off inside the cell to form a vesicle containing the material. This in an energy-dependent process and there are a number of distinct endocytic pathways by which molecules can enter cells, which are discussed in more detail elsewhere.33–35 One endocytic process that is of particular interest for this review is receptor-mediated endocytosis. This involves binding of a specific ligand to a receptor located on the surface of the cell, which initiates its endocytic uptake. Compounds are first contained in membrane-bound vesicles known as ‘early endosomes’ before being transferred to acidic late endosomes. ‘Late endosomes’ then fuse with lysosomes which contain hydrolytic enzymes for the breakdown of macromolecules, or they can fuse with other organelles such as mitochondria, the endoplasmic reticulum, or the Golgi network This mechanism enables cells to selectively internalise specific macromolecules and also provides researchers with a method to target specific cells expressing these receptors.36 One important consequence of uptake by endocytosis is that the molecules may become trapped if they are unable to escape the vesicle, thereby preventing them from reaching their intended subcellular targets.37 In contrast, compounds that are taken up into cells by the mechanisms previously discussed are not restricted to endosomes and are free to localise to any intracellular organelle. In their 2012 review, Gill et al. highlighted the significant influence of lipophilicity in the interactions of Ru(II) polypyridyl complexes with cells.19 Such an importance in lipophilicity is consistent with the well-established importance of this physico-chemical property in determining their pharmacokinetics, potencies and toxicities.38–43 In addition, the cytoplasm of a cell contains numerous membranous structures and organelles, which include the endoplasmic reticulum, Golgi apparatus, lysosomes, mitochondrial membranes, nuclear envelope, peroxisomes and vesicles. As well as influencing the interactions between Ru(II) complexes and lipid membranes, modifications to the lipophilicity of complexes has also been found to significantly influence the interactions of various complexes with proteins, which will be discussed in more detail later in this section. As such, the lipophilicity of Ru(II) complexes can also have a significant influence on their localisation within cells. In recent years researchers have employed various strategies to modulate the lipophilicity of Ru(II) complexes, which include varying the hydrophobicity of their ligands, changing the charge of the complexes and switching their counterion, with a number of interesting results reported, which will be presented in the coming sections.
Due to its significant importance, where possible, the lipophilicity of complexes will also be quoted in the following text. This is most commonly quantified as the logarithm of a molecule's partition coefficient between an organic solvent, such as octanol, and water (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P). However, if the complex possesses acidic/basic functional groups, the logarithm of the distribution coefficient of the complex between octanol and water or buffered solution at a stated pH is reported (logDpH).
P). However, if the complex possesses acidic/basic functional groups, the logarithm of the distribution coefficient of the complex between octanol and water or buffered solution at a stated pH is reported (logDpH).
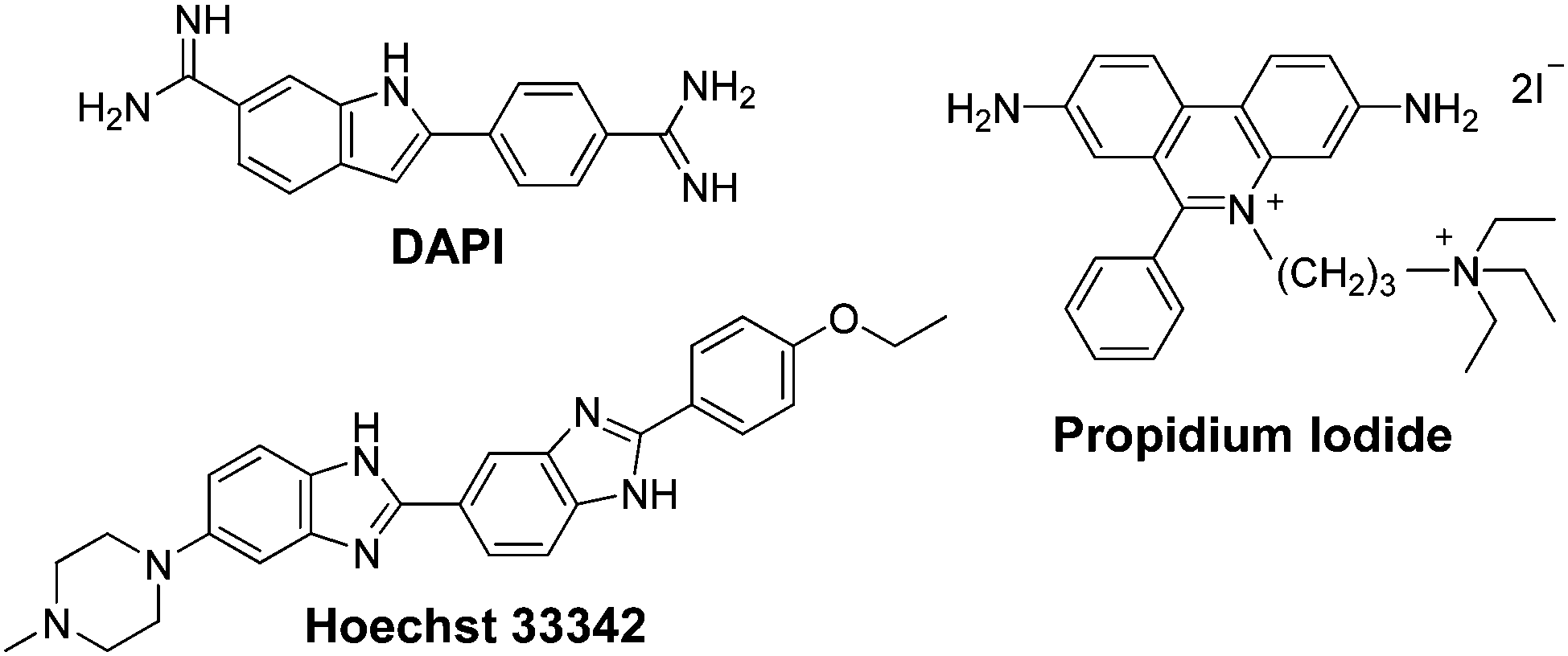
As such, lipophilic molecules have positive logP and logD values, whereas hydrophilic molecules have negative values. To put these values into context, those of commonly used fluorescent nuclear stains will first be presented. The nuclear stains 4′,6-diamidino-2-phenylindole (DAPI) and Hoechst 33342, are in equilibrium between a hydrophilic cationic form (logP = −2.8 and −4.1, respectively) and neutral lipophilic form (logP = +0.8 and +4.7, respectively). This results in the respective molecules being semi-permeable and fully permeable to the cellular membrane and thus can enter live cells and stain nuclear DNA. In contrast, propidium iodide, which is used to stain the nucleic acid of cells whose plasma membranes are damaged, has a logP of −5.1 and therefore cannot permeate intact membranes.44
Investigation into the cellular uptake of Ru(II) polypyridyl complexes and the importance of their lipophilicity was spearheaded in 2007 by the Barton research group investigating complexes incorporating the dipyrido[3,2-a:2′,3′-c]phenazine (dppz) ligand.45 The researchers found that a complex incorporating two lipophilic, 4,7-diphenyl-1,10-phenanthroline (dip) ancillary ligands [Ru(dip)2(dppz)]2+ (1, logP = +1.30 as its chloride salt) was internalised by cells to a greater extent than more hydrophilic derivatives in the study.45 Confocal microscopy studies showed the luminescence from the complex to be localised in the cytoplasm of live HeLa cells (as shown in Fig. 2a) and mechanistic studies indicated that the complex entered cells by passive diffusion.30,45 Soon after, Wölfl and co-workers used graphite furnace atomic absorption spectroscopy to measure the uptake of Ru(II) complexes in cells and found that increasing the aromatic surface area of the polypyridyl ligands resulted in increased cellular uptake.50 However, one important consequence of highly lipophilic complexes is their poor water solubility, which can present challenges for administration of such complexes if they are to be developed for in vivo applications.30,45,51–53 Gunnlaugsson and co-workers later demonstrated that incorporation of two ancillary TAP ligands (TAP = 1,4,5,8-traazaphenanthrene), yielded the water soluble hydrophilic [Ru(TAP)2(dppz)]2+ complex (2), which was also found to localise in the cytoplasm of live HeLa cells, as shown in Fig. 2b.46 In recent years, the influence of chemical modification of the dppz ligand on cellular uptake and localisation has also been investigated. Of the chemical modifications reported, fluorination of the 11-position of the dppz ligand (11-F-dppz) had a dramatic influence on both the uptake and localisation of the resulting complexes. In contrast to the weak luminescence of [Ru(bpy)2(dppz)]2+ within live cells and undetectable nuclear localisation in HeLa cells,45 the 11-F-dppz analogue [Ru(bpy)2(11-F-dppz)]2+ (3) was internalised by HeLa cells within 1 h and localised in the nucleus, as shown in Fig. 2c.47 Aromatic fluorination is commonly used by medicinal chemists to increase the lipophilicity of a drug molecule,54,55 however no logP values were reported for the complex.
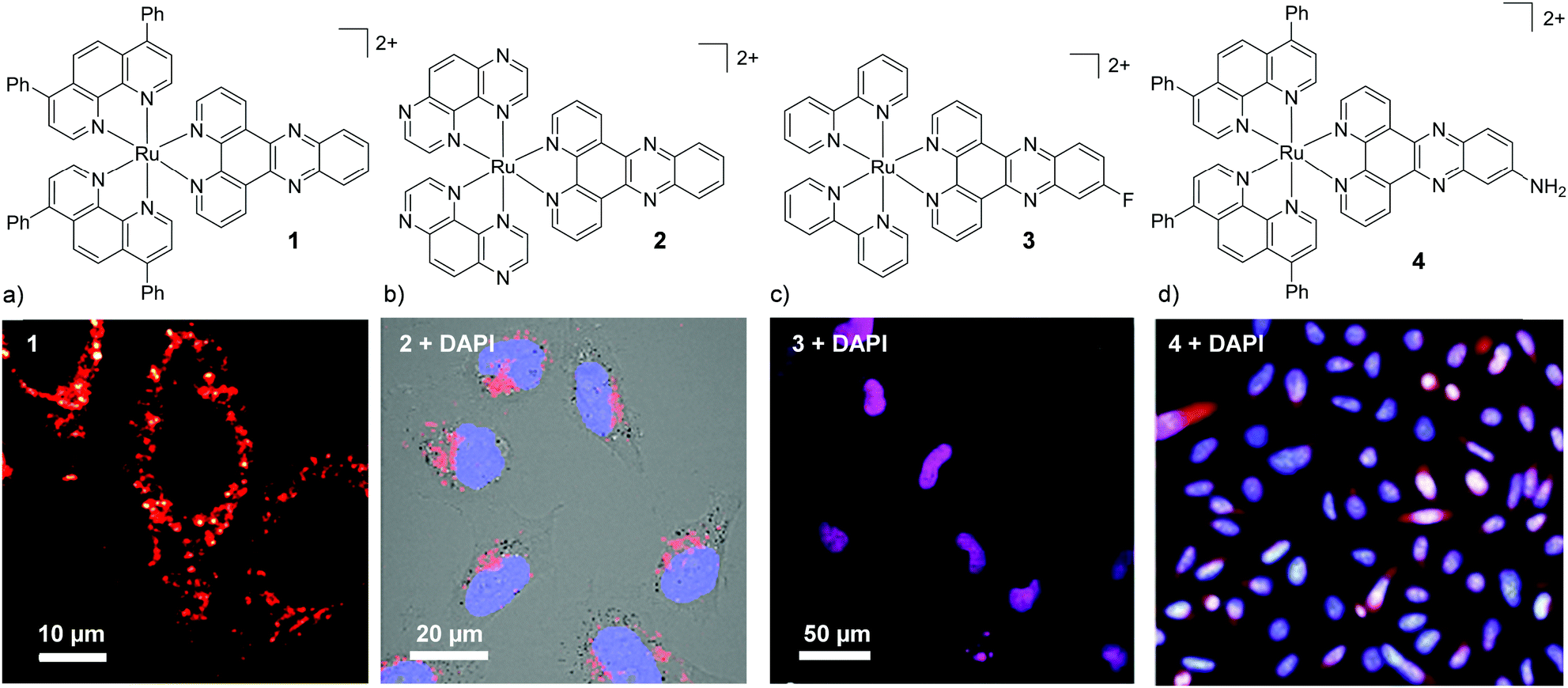 | ||
| Fig. 2 Chemical structures and confocal microscopy images of HeLa cells incubated with (a) complex 1 (5 μM) for 2 h, (b) complex 2 (100 μM) for 24 h, (c) complex 3 (10 μM) for 2 h. (d) Fluorescence microscope images of BEL-7402 cells incubated with 4 (25 μM) for 24 h. The ruthenium emission is shown in red, the nuclear stain DAPI is shown in blue and the overlay of the ruthenium emission with DAPI is shown in purple. Images reproduced with permission from ref. 45–48. | ||
Amino-functionalisation at the 11-position of the dppz ligand, resulted in the complexes distributing throughout the cytoplasm and nuclei of live BEL-7402 cells, as shown in Fig. 2d for [Ru(dip)2(11-NH2-dppz)]2+ (4).48 While the authors did not report the influence of the amino functionality on the lipophilicity of the complex, Gasser and co-workers examined the effect of a variety of functional groups at the 11-position of dppz, as shown in Fig. 3a.49 Interestingly, the amino-functionalised dppz complex was found to be the most lipophilic complex of the series, which was attributed to the lone pair being strongly delocalised into the dppz ligand. The logD7 values of the PF6− salts of the complexes were found to be in the order of NH2 (−0.27) > CH2Cl (−0.36) > OMe ∼ OH (−0.42) > CH2OH (−0.62) > OAc (−0.74). High-resolution continuum source atomic absorption spectrometry revealed that only the NH2- and OMe-dppz derivatives 5 and 6, respectively, entered cells to an appreciable extent. Confocal microscopy studies in fixed HeLa cells showed complex 6 to predominantly localise in the nuclei of treated cells (shown in Fig. 3b), while complex 5 was found to distribute throughout treated cells, consistent with the findings of Jiang et al. for its dip-analogue, complex 4, in live cells.48,49 The researchers also highlighted, however, that amino functionalisation of the dppz ligand resulted in a significant decrease in the luminescence quantum yield of the complex and, as such, very weak luminescence was detected from treated cells.49
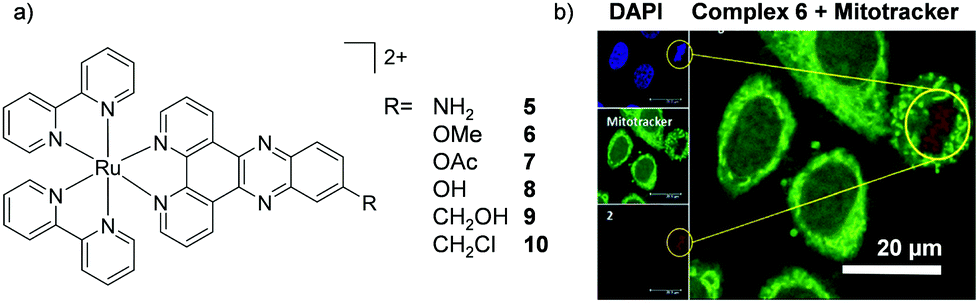 | ||
| Fig. 3 (a) Chemical structure of the Ru(II) complexes 5–10 and (b) confocal microscopy images of HeLa cells treated with 6 (100 μM) for 2 h and fixed in formaldehyde (shown in red) and stained with the nuclear stain DAPI (blue colour) and mitochondrial stain Mitotracker green (green colour). Image reproduced with permission from ref. 49. | ||
Di-substitution of the dppz ligand has also been show to impact the uptake and localisation of the complexes, whereby complexes incorporating either an 11,12-diamino- or 11,12-dichloro-dppz successfully enter cells and disperse throughout the cells.56,57 In contrast, incorporation of C4H9 and C6H13 alkyl chains at the 11- and 12-positions of the dppz ligand resulted in the complexes binding to the plasma membrane of live cells, without being internalised.58 Overall, the nature of the ancillary ligands as well as functionalisation of the dppz-ligand have been found to play important roles in the uptake and localisation of these complexes, which have been shown to localise in the:
– cytoplasm45,56,59,60
– nucleus47,49,61–67
– mitochondria46,59
– cell membrane58,68 and
– dispersed throughout the cell.48,49,57,69
In addition to the widely investigated dppz-based complexes, numerous Ru(II) complexes incorporating the imidazo-phenanthroline motif have also been studied in cells. The synthetic route of this ligand has allowed the creation of libraries of related compounds, incorporating various functionalities. For a number of these complexes, their lipophilicities were important to their uptake by cells70–72 and the uptake of this class of complex have been reported to occur by a number of different mechanisms: both energy-independent mechanisms70 and energy-dependent mechanisms, such as endocytosis,73 energy-dependent non-endocytic mechanisms,74,75 and receptor-mediated endocytosis.70 These complexes have been found to localise in various regions of the cell, such as the nucleus73,74,76–79 mitochondria,70,71,80 cytoplasm,81–83 and lysosomes.84 An interesting result has been shown for two indole-functionalised imidazo-phenanthroline-based complexes. When the complex incorporated two ancillary phen ligands (complex 11, shown in Fig. 4) it entered HeLa cells by an energy-dependent non-endocytic pathway, initially localising in the lysosomes and subsequently escaping and localising throughout other parts of the cytoplasm, as shown in Fig. 4a.74 In contrast, another indole-functionalised imidazo-phenanthroline complex (12), which incorporated two 2,2′-dipyridylamine (hdpa) ancillary ligands, was found to localise in the nuclei and cytoplasm of the cells within 2 h, as shown in Fig. 4b.72 Interestingly, Barton and co-workers found that incorporation of ancillary hdpa ligands into their rhodium complexes significantly enhanced and accelerated the cellular uptake of the resulting complexes, and also influenced their subcellular localisation.85 This demonstrated that the hdpa ligand motif could have a profound influence on the uptake and localisation of complexes.
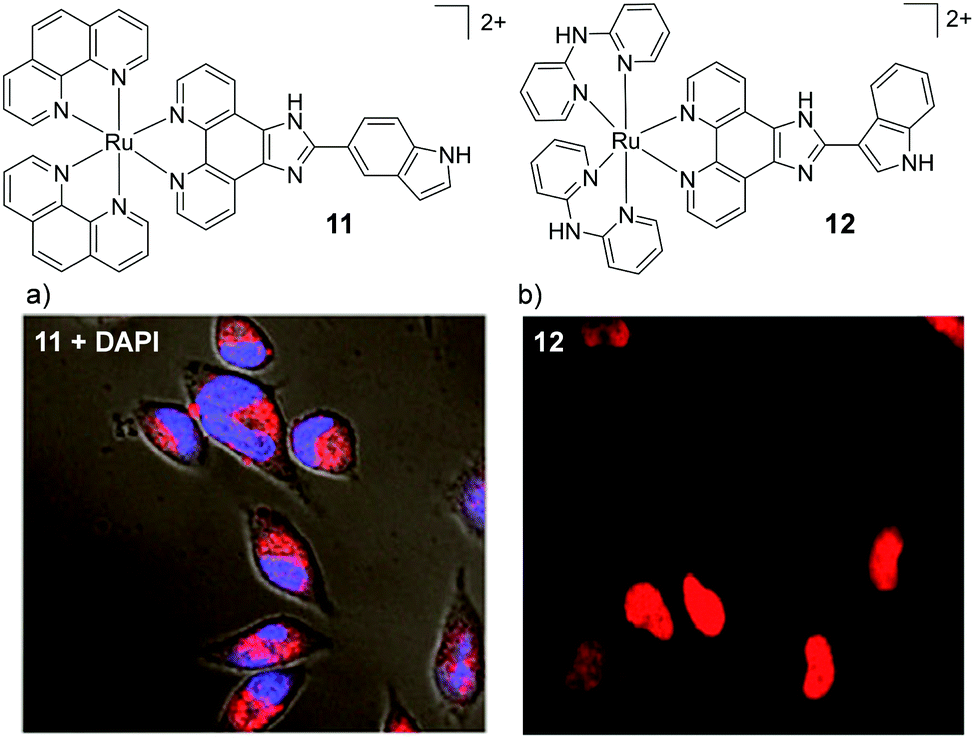 | ||
| Fig. 4 Chemical structures of complexes 11 and 12 and the confocal microscopy images of HeLa cells incubated with (a) complex 11 (10 μM) for 12 h and stained with DAPI (blue) and (b) complex 12 (50 μM) for 2 h. Images reproduced with permission from ref. 74 and 72. | ||
While increasing the lipophilicity of molecules can increase their uptake by cells, it has also been found to increase the likelihood of binding to multiple targets such as the hydrophobic pockets within proteins and cell membranes.38–40,58 Both of these outcomes have been reported by the Thomas research group in recent years. In 2009, they reported a binuclear Ru(II) complex [(phen)2Ru(tpphz)Ru(phen)2]4+ (13), that selectively stained the nuclei of live MCF-7 human breast cancer cells.86 Substitution of the four phen ligands with four bpy ligands, resulted in a significant reduction in the lipophilicity of the complex (logP = −0.96 and −1.61, respectively, as their chloride salts) and the inability of the complex to enter live cells.86 Conversely, substitution of the four phen ligands with dip ligands significantly increased the lipophilicity of the resulting complex 14 (logP = +1.52 as its nitrate salt),87 but dramatically altered its localisation. Instead of localising in the nucleus of cells, complex 14 localised in the membrane-rich endoplasmic reticulum and perinuclear region of treated cells, as shown in Fig. 5a.87 The complex was shown to be taken into cells via an energy-dependent manner, similar to complex 13, while, in addition, a small amount of uptake was found to occur by an energy-independent mechanism.86,87
Another method of increasing the lipophilicity of Ru(II) polypyridyl complexes, which results in minimal structural changes to the molecules, has been to decrease the positive charge on the complex via cyclometalation. This involves the exchange of one or more of the coordinating nitrogen atoms of the polypyridyl ligands for an isoelectronic carbon anion, with each such exchange reducing the cationic charge on the metal centre.88 Thomas and co-workers utilised this method in the design of tricationic dinuclear Ir–Ru complexes, which were based on a tetracationic complex 13.86,89 Incorporation of two cyclometalating 2-phenylpyridine (phpy) ligands around the Ir centre resulted in the complex [(phpy)2Ir(tpphz)Ru(bpy)2]3+ (15) which stained the nuclei of live MCF-7 cells.89 In addition, the authors found that by fluorinating the cyclometalating phpy ligand, the resulting complex 16 showed a higher rate of nuclear uptake than its non-fluorinated analogue 15.89 Moreover, complex 16 effectively stained the nuclei of live cells (as shown in Fig. 5b) at a 50-fold lower incubation concentration of 10 μM, compared to 500 μM required for the tetracationic complex 13.86,89 Interestingly, a subsequent study by the authors revealed that incorporation of two benzo[h]quinolone cyclometalating ligands around the Ir centre resulted in complex 17 displaying an even higher rate of nuclear accumulation, relative to complex 16.90 However, when cells were incubated with complex 17 in serum-containing medium, both the uptake and localisation of the complex dramatically changed, as shown in Fig. 5c. In serum-containing medium the complex exhibited significantly weaker luminescence intensity within cells and the luminescence was found to be localised in the cytoplasm, with no nuclear staining observed. Solution studies revealed that the complex displayed a high binding affinity for serum albumin and, as such, the change in cellular uptake and localisation of the complex was attributed to binding of the complex to serum proteins in the medium.90 Thus, when increasing the lipophilicity of Ru(II) complexes, the possible impact on their binding to proteins should be kept in mind. This also raises the question as to whether other complexes studied in the literature may in fact be bound to proteins in the cell culture medium and subsequently taken up by cells and whether this influences the observed intracellular localisation.
 | ||
| Fig. 5 Chemical structures of complexes 13–17 and confocal microscopy images of (a) MCF-7 cells incubated 14 (5 μM) for 24 h, (b) HeLa cells incubated with complex 16 (10 μM) in serum-free medium for 80 min, (c) MCF-7 cells incubated with 17 (10 μM) in serum-containing and serum-free media for the stated times. The Ru emission is shown in red and the nuclear stain DAPI emission is shown in blue. Images reproduced with permission from ref. 87, 89 and 90. | ||
While protein binding had undesirable consequences on the uptake and localisation of complex 17, which was designed as a nuclear stain, it should also be noted that binding of drug molecules to proteins in the blood, such as transferrin, can increase their accumulation in malignant cells in vivo, which tend to overexpress the transferrin receptor.35,91 For example, the clinically investigated anti-cancer Ru(III) complexes KP1019 and NAMI-A, have been shown to bind to the blood plasma proteins transferrin and albumin upon administration in vivo.92,93 This protein binding is believed to be a contributing factor to the low toxicity of these drug candidates and, in the case of transferrin binding, to result in drug enrichment in tumours for reasons mentioned above.94 Indeed other Ru(II) polypyridyl complexes have been found to bind to transferrin and serum albumin, and in one particular instance this was shown to result in an enhancement in the photophysical properties and tumour targeting ability of the complex, which will be discussed later in the review.95–100
Cyclometalation of the Ru metal centre has also been demonstrated to have a significant impact on the cellular uptake and localisation of complexes. In the case of [Ru(bpy)2(dppz)]2+, for example, exchange of one of the ancillary bpy ligands for the cyclometalating phpy ligand resulted in a significant increase in the lipophilicity of the resulting [Ru(bpy)(phpy)(dppz)]1+ complex 18 (logP = +1.0 as its perchlorate salt).62,63 Inductively coupled plasma mass spectrometry (ICP-MS) studies of cells incubated with 18 showed that 90% of the internalised complexes localised in the nuclei of HeLa cells within 2 h, which contrasts sharply with the poor cellular uptake of [Ru(bpy)2(dppz)]2+. Mechanistic studies revealed that complex 18 entered cells by an energy-independent mechanism, consistent with its high lipophilicity.62 Unlike cyclometalated Ir complexes, which can be strongly luminescent,101,102 Ru cyclometalates tend to exhibit very weak luminescence and therefore are not always suited for imaging applications.103 However, they are showing promising biological activity with regard to the development of new classes of chemotherapeutic agents, as will be discussed later in this review.
In 2015, by incorporating the 6-chloro-5-hydroxylpyrido[3,2-a]-phenazine (CQM) ligand into their complexes, which was designed to mimic the extended dppz ligand,104 Ding et al. were able to decrease the positive charge of their Ru complexes, while avoiding such loss of luminescence. While the complex [Ru(bpy)2(CQM)]1+ (19) was emissive, it did not show the “luminescence light-switching” activity seen for [Ru(bpy)2(dppz)]2+.104,105 As with the cyclometalated dppz analogues, this monocationic complex was significantly more lipophilic (logP = +0.10 as its perchlorate salt) than [Ru(bpy)2(dppz)]2+. Complex 19 was found to be taken up by human liver carcinoma HepG2 cells and localise in both the nucleus and mitochondria of cells, as shown in Fig. 6a. The ancillary ligands of this complex were also demonstrated to be of importance, whereby upon substitution of the two ancillary bpy ligands by phen ligands, the [Ru(phen)2(CQM)]1+ complex (20, logP = +0.18) localised in the nuclei of HepG2 cells, as shown in Fig. 6b.104 In addition to decreasing the charge of Ru complexes, the CQM ligand-type is now being used to move the absorbance of Ru complexes into the 600–700 nm region to achieve long wavelength activation.106 The modulation in the lipophilicities of these complexes is summarised in Table 1 to highlight the influence of the ligand and charge on the lipophilicities of these Ru complexes.
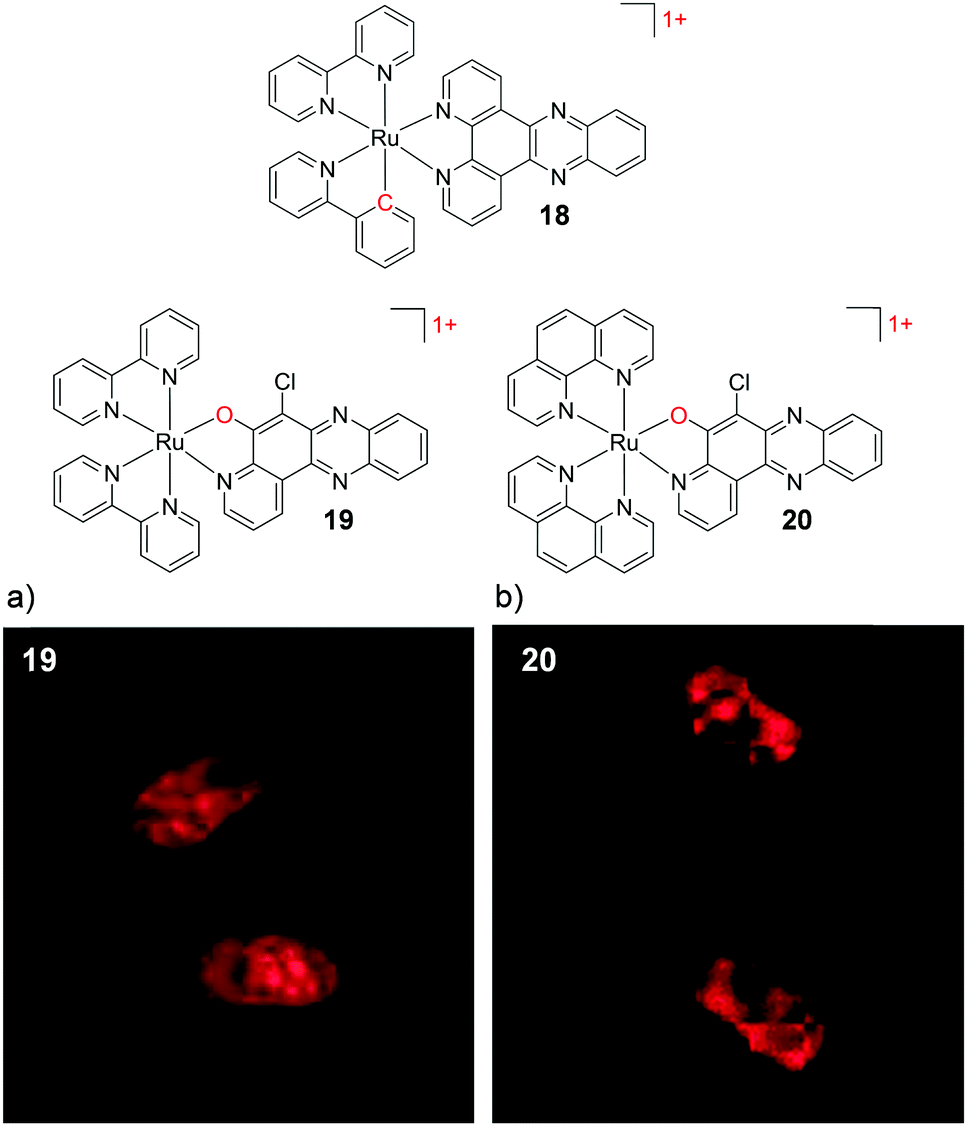 | ||
| Fig. 6 Chemical structures of complexes 18–20, where the charge of the complex and atom changes are highlighted in red. The confocal microscopy images of human liver carcinoma HepG2 cells incubated with (a) 19 (200 μM) and (b) 20 (200 μM) for 2 h. Images reproduced with permission from ref. 104. | ||
P values of Ru(II) polypyridyl complexes showing the influence of the ligand structure and charge of the complexes on their lipophilicities45,104
Recently, Zhu et al. demonstrated that the uptake and localisation of Ru(II) complexes could be dramatically altered by choosing an appropriate counterion for the complex. They found that the conjugate bases of three weak acids with bulky hydrophobic aromatic moieties (pentachlorophenol (PCP), carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone (FCCP) and tolfenamic acid (TA) shown in Fig. 7) formed stable ion-pairs with a number of Ru(II) polypyridyl complexes. For example, when live cells were treated with [Ru(bpy)2(dppz)]2+, and PCP, FCCP or TA, the complex was readily internalised by cells and preferentially localised in the nucleus, as shown in Fig. 7.107 This was in direct contrast to the incubation of cells with the complex in the absence of these reagents, where the complex showed extremely poor uptake by cells and no nuclear localisation.45,107 The uptake of the ion-paired complex was found to occur by an energy-independent mechanism and the authors proposed that this was a result of ion-pairing of the hydrophilic Ru(II) complex and the deprotonated hydrophobic weak acids, to form overall neutral lipophilic adducts, which were capable of crossing cellular membranes. This enhancement in cellular uptake and nuclear localisation was also demonstrated for the complexes, [Ru(phen)2(dppz)]2+, [Ru(phen)3]2+ and [Ru(bpy)3]2+, which demonstrates that this technique could be successfully applied to other Ru(II) complexes.107
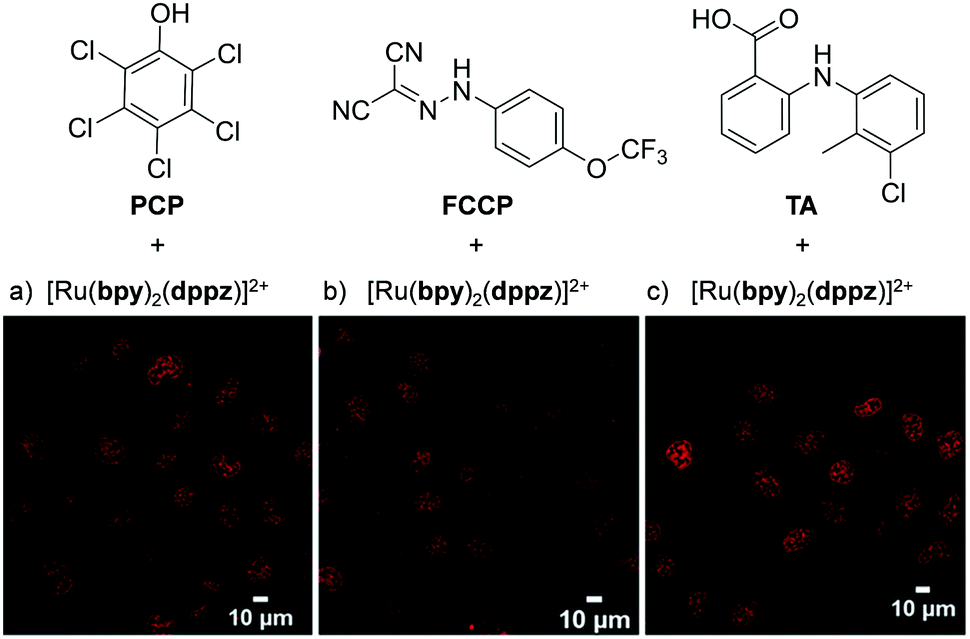 | ||
| Fig. 7 Chemical structures of the weak acids PCP, FCCP and TA and the confocal microscopy images of live QSG-7701 cells treated with (a) [Ru(bpy)2(dppz)]2+ (100 μM) and PCP (300 μM) for 3 h in complete medium, (b) [Ru(bpy)2(dppz)]2+ (200 μM) and FCCP (50 μM) for 1 h in serum-free medium and (c) [Ru(bpy)2(dppz)]2+ (300 μM) and TA (300 μM) for 3 h in complete medium. The chloride salts of [Ru(bpy)2(dppz)]2+ was used in these experiments. Images reproduced with permission from ref. 107. | ||
While lipophilicity can be important in the uptake of Ru complexes it is important to note that highly charged and hydrophilic complexes will not necessarily be precluded from entering cells. For example, Glazer et al. demonstrated that a tetra-anionic complex incorporating two sulfonic acid groups on each of its three dip ligands (complex 21) was successfully internalised by cells, despite its low lipophilicity.108 This hydrophilic complex (logP value of −2.2) was found to localise in the cytosol of A549 lung cancer cells, as shown in Fig. 8a. It should however be noted that this complex did show significantly lower levels of uptake by cells than its unsulfonated [Ru(dip)3]2+ analogue (22), which was highly lipophilic (logP = +1.8) and showed significantly different subcellular localisation, primarily localising in the lysosomes and the mitochondria, as shown in Fig. 8b. Interestingly, this difference in subcellular localisation between the two complexes was also suggested to be responsible for the low toxicity of complex 21, in contrast to complex 22 which was inherently quite toxic to cells.108
 | ||
| Fig. 8 Chemical structures of the tetra-anionic complex 21 and the di-cationic complex 22 and the confocal microscopy images of live A549 cells treated with the fluorescent lysosome stain LysoTracker® Green DND-26 and (a) 21 (20 μM) and (b) 22 (5 μM) for 8 h. Ru emission is shown in red and LysoTracker emission is shown in green. Yellow coloured regions indicate colocalisation of the Ru and lysosome stain. Images reproduced with permission from ref. 108. | ||
Thus far this section has examined some of the intrinsic factors that influence the uptake and localisation of Ru(II) polypyridyl complexes (their structures, functional groups, charges, lipophilicities etc.). However, extrinsic factors such as the concentration at which cells are treated with a complex, the choice of cell line, the environmental conditions (e.g. serum containing/serum-free medium, O2 partial pressure), treatment time point or cell manipulation, such as fixation, can also have a profound effect, which will now be briefly discussed.
The concentration at which cells are incubated with compounds, particularly with respect to the concentrations at which they show toxicity to cells, was shown to have a considerable impact on the uptake of a Ru(II) polypyridyl complex (23) by Zava et al.109 The researchers found that when A2780 human ovarian cancer cells were incubated with low concentrations of the complex (1 μM), luminescence was observed in discrete packets within the cytoplasm. However, when cells were incubated at higher concentrations (10 μM), the luminescence from the complex originated from the cell membrane, as shown in Fig. 9.109 This extreme difference in localisation was attributed to inhibition of the cellular machinery responsible for the uptake of the complexes at higher concentrations, thus preventing their internalisation.109
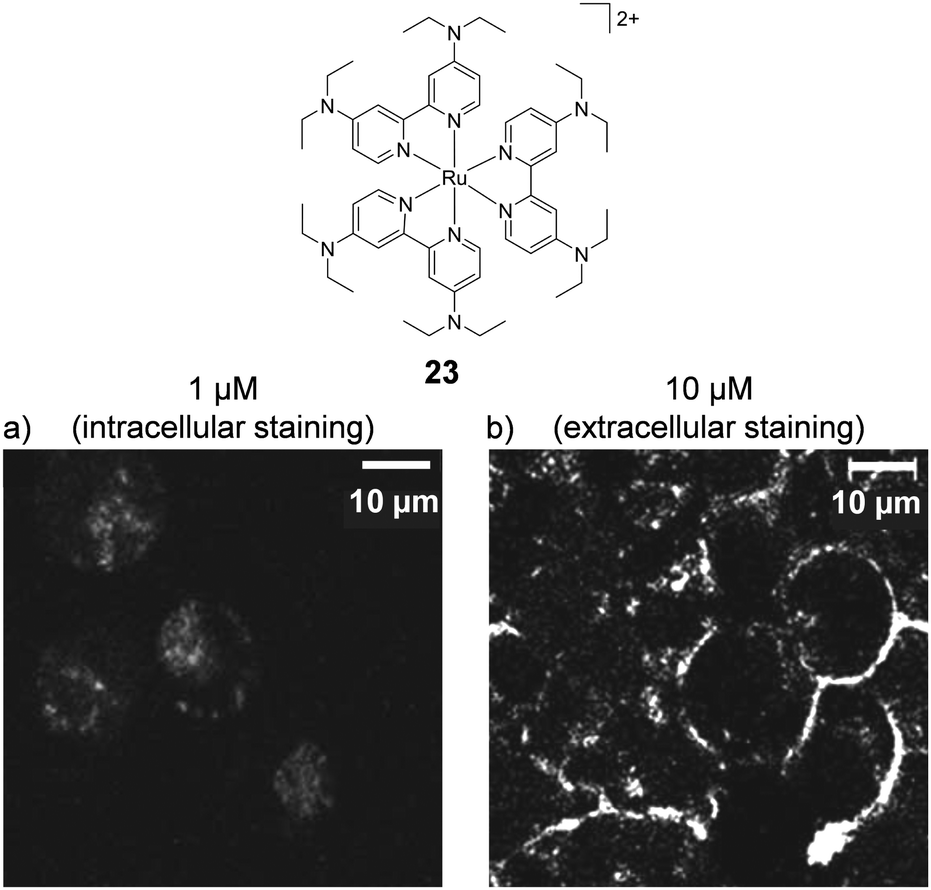 | ||
| Fig. 9 Chemical structure of complex 23 and the confocal microscopy images of live A2780 ovarian cancer cells treated with complex 23 (a) at 1 μM and (b) at 10 μM for 24 h. Images reproduced with permission from ref. 109. | ||
A number of studies by Keene and co-workers highlighted the potential importance of cell line and treatment time point to the localisation of Ru(II) complexes. Initial studies conducted in murine L1210 leukaemia cells showed that a series of dinuclear complexes, shown in Fig. 10, localised predominantly in the mitochondria of cells after incubation with the complexes for 4 h.110,111 These complexes incorporated flexible linkers of various lengths between the metal centres (n = 2, 5, 7, 10, 12 and 16) and by varying the number of methylene groups in the linker the researchers were able to modulate the lipophilicity of the complexes. Indeed flow cytometry indicated that as the chain length of the complexes increased so too did their levels of cellular uptake. The most lipophilic complex 24 (n = 16, logP = −1.9 as its chloride salt)112 showed the highest uptake in L1210 cells and mechanistic studies showed that it entered cells predominantly via an energy-independent pathway, with a small contribution from protein-mediated active transport.111 However, dramatically different subcellular localisation was reported for complex 25 (n = 12, logP = −2.9)112 in baby hamster kidney (BHK), human embryonic kidney (HEK-293) and human liver carcinoma (Hep-G2) cell lines.113 After treatment of these cells for 4 h, complex 25 was found to localise predominantly in the cytoplasm, as shown in Fig. 10b, with the endoplasmic reticulum suggested to be the target organelle. However, at longer incubation times (20 h), the complex localised in the nucleoli and nuclear envelopes of cells, as shown in Fig. 10b.113 At present, the origin of this change in selectivity between the mitochondria of one cell line and the nucleoli of other cell lines remains unclear but suggests that uptake studies of Ru(II) complexes may show variability between cell lines.
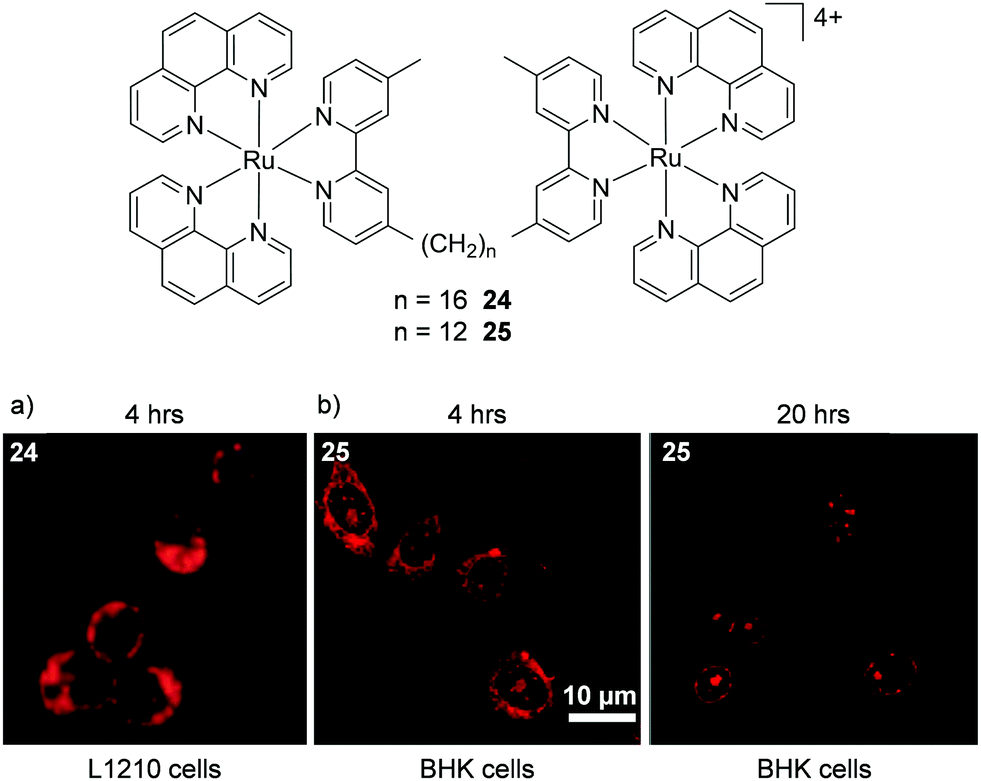 | ||
| Fig. 10 Chemical structures of complexes 24 and 25 and the confocal microscopy images of (a) L1210 cells incubated with complex 24 (50 μM) and (b) BHK cells incubated with complex 25 (55 μM) for the states times. Images reproduced with permission from ref. 113 and 111. | ||
Another study by Chao and colleagues demonstrated that the subcellular distribution of a Ru cyclometalate was influenced by oxygen concentration in HeLa cells.114 Studies by ICP-MS showed that when cells were incubated with the complex under normoxic conditions (20% O2) 60% of the cellular Ru was found in the nucleus and 28% in the mitochondria. Conversely, when cells were treated with the complex under hypoxic conditions (1% O2), 50% of the cellular Ru was found in the mitochondria, with only 32% in the nucleus.114 As solid tumours tend to possess regions of low oxygen concentrations,115 this study raised the possibility that the microenvironment of a tumour could also influence the subcellular distribution of complexes, which could be of particular importance to researchers developing Ru complexes for in vivo cancer applications.
Finally, luminescence based techniques have been the most widely used methods to quantify the uptake of complexes and report on their cellular localisation. However, since the luminescence quantum yields of these complexes can be extremely sensitive to their environment, for example the presence/concentration of quenching species such as oxygen,116 water105 or certain biomolecules,117 it is important that techniques such as transmission electron microscopy (TEM), ICP-MS and atomic absorption spectroscopy be used in addition to, or in place of, luminescence based techniques, as noted in the examples above.
The importance of chirality
Tris(bidentate) octahedral complexes are chiral by virtue of the configurations of the chelating ligands around the metal centre, as shown in Fig. 11. This results in the formation of either a left-handed Λ-enantiomer or a right-handed Δ-enantiomer. Solution studies have shown that the two enantiomers of Ru(II) polypyridyl complexes can interact differently with chiral biomolecules such as DNA119–124 and proteins.100,125 In 2013 the crystal structure of both the Λ- and Δ-enantiomers of [Ru(phen)2(dppz)]2+ bound at a TG/CA step of a DNA duplex hexamer was reported, which provided direct observation of the difference between the binding geometries of the two enantiomers and DNA, as shown in Fig. 11.119 In recent years the differences in the interaction of the enantiomers of Ru(II) complexes in a cellular environment have been investigated and, in a number of cases, significant differences have been observed.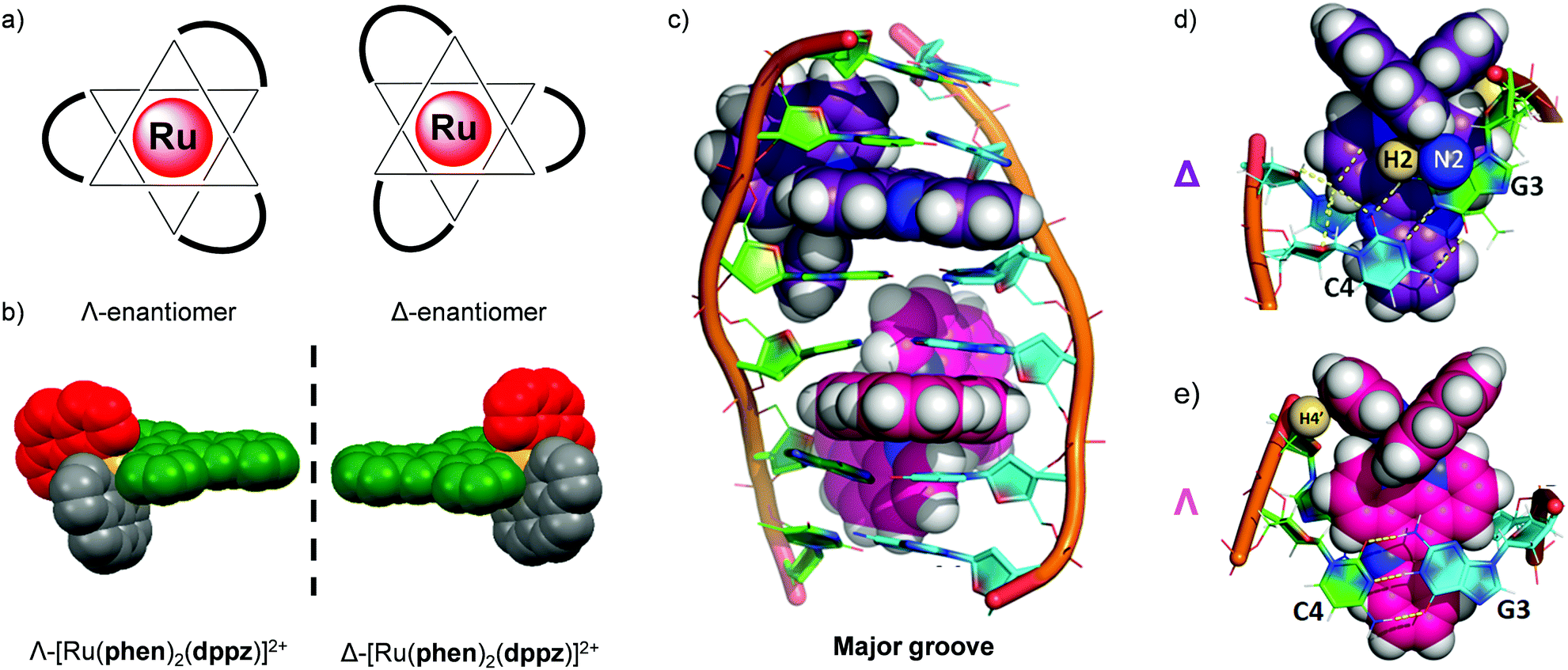 | ||
| Fig. 11 (a) The octahedral coordination geometry of three bidentate ligands around a Ru(II) centre, giving rise to two optical isomers- the left-handed Λ and the right-handed Δ enantiomers and (b) the spacefill diagram of [Ru(phen)2(dppz)]2+, obtained from the crystal structure in ref. 118 (CCDC 1038710), (c) structure of the Δ- and Λ-enantiomers of [Ru(phen)2(dppz)]2+ (coloured purple and pink, respectively) bound to the DNA duplex d(ATGCAT)2 when viewed into the major groove. The different binding geometries of (d) Δ- and (e) Λ-[Ru(phen)2(dppz)]2+ when bound to this DNA duplex by intercalation of the dppz ligand. Images reproduced with permission from ref. 119. | ||
In 2012 Svensson et al. demonstrated that two dinuclear Ru(II) complexes showed enantiomer-specific staining of methanol-fixed Chinese hamster ovary (CHO)-K1 cells.126 For one of the dinuclear complexes (26), the researchers found that the ΔΔ-complex showed more prominent nuclear staining than the ΛΛ-complex, as shown in Fig. 12a and b.126 In contrast, when live cells were incubated with these complexes, the luminescence from the internalised ΛΛ- and ΔΔ-complexes was found to originate from discrete packets within the cytoplasm, with no differences observed between the staining patterns of the two enantiomers, as shown in Fig. 12c and d. This was attributed to entrapment of the ΛΛ- and ΔΔ-complexes in endosomes of live cells when the complexes were taken up by endocytosis.126 This demonstrated that, although the enantiomers of Ru(II) complexes may be capable of exhibiting enantiomer-specific interactions within cells, the mechanism of uptake may prevent such differential interactions from being observed for live-cell imaging.
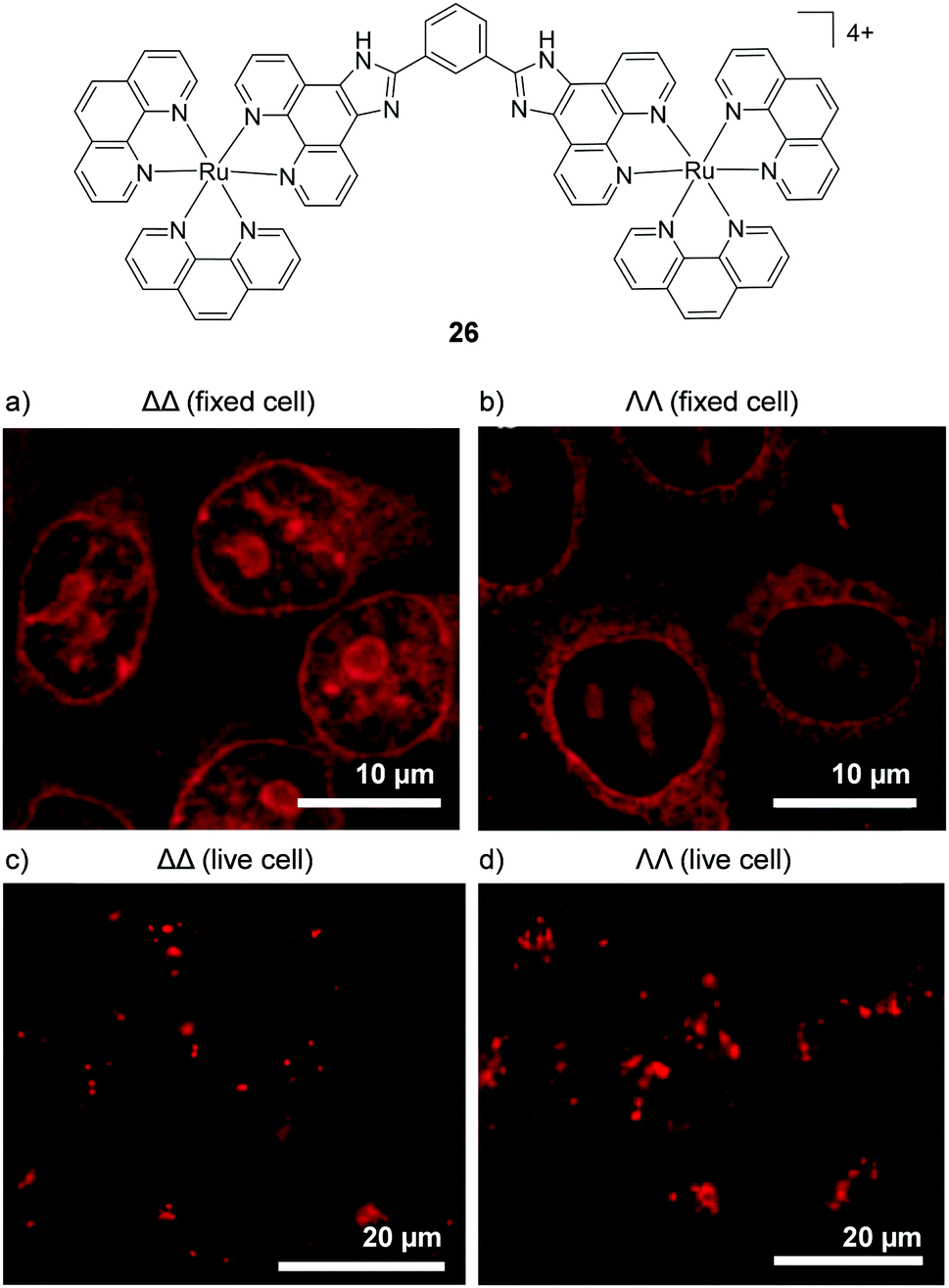 | ||
| Fig. 12 Confocal microscopy images of CHO-K1 cells (a) fixed and treated with ΔΔ-26 (5 μM) for 4 h, (b) fixed and treated with ΛΛ-26 (5 μM) for 4 h and live cells treated with (c) ΔΔ-26 (5 μM) and (d) ΛΛ-26 (5 μM) for 1 h. Images reproduced with permission from ref. 126. | ||
More recently, enantiomer-specific interactions have also been observed for the dppz-based complexes [Ru(bpy)2(dppz)]2+ and [Ru(phen)2(dppz)]2+ in live cells. Using the ion-pair methodology discussed previously (shown in Fig. 7), the cellular uptake and nuclear localisation of the Δ- and Λ-enantiomers of both complexes was achieved, facilitated by PCP counterions.107 For both complexes, a significant difference was observed between the luminescence intensities of the Δ- and Λ-enantiomers. In both cases, the Δ-enantiomers displayed significantly higher luminescence within the nuclei of live cells than their Λ-counterparts, as demonstrated in Fig. 13.107 This stronger luminescence intensity of the Δ-enantiomers was consistent with previous solution studies which showed the Δ-enantiomers to be more emissive than their Λ-counterparts when bound to isolated B-DNA.121,127
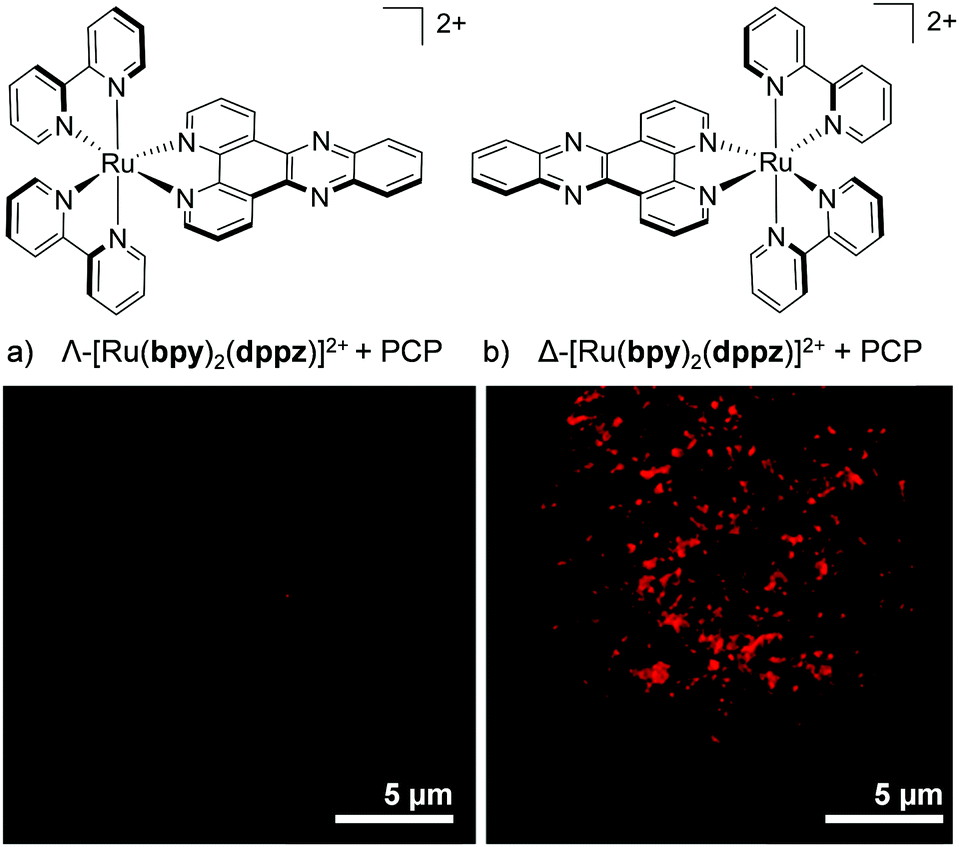 | ||
| Fig. 13 Confocal microscopy images showing the difference in luminescence intensity between the nucleus of a live QSG-7701 human hepatocyte cell treated with (a) the Λ- and (b) the Δ-enantiomer of [Ru(bpy)2(dppz)]2+ (100 μM) + PCP (300 μM) for 4 h. Images reproduced with permission from ref. 107. | ||
The most dramatic difference, so far, in the cellular staining between enantiomers was recently demonstrated by Mei and co-workers, who showed that the Λ-enantiomer of complex 27 predominantly localised in the nucleus of live MDA-MB-231 breast cancer cells, whereas the Δ-enantiomer localised mainly in the cytoplasm, as shown in Fig. 14.73 Mechanistic studies were conducted with Λ-27 and indicated that the complex was taken up by cells via endocytosis and that the nuclear localisation of Λ-27 occurred by an ATP-dependent active transport pathway.73 An earlier study by Yu et al. found a similar result to these authors, albeit with a less distinct staining pattern between their enantiomers.128 The researchers showed the Λ-enantiomer of their Ru(II) complex to localise in the cytoplasm of HepG2 cell, with a partial staining of the cell nucleus, whereas the Δ-enantiomer showed exclusively cytoplasmic staining.128
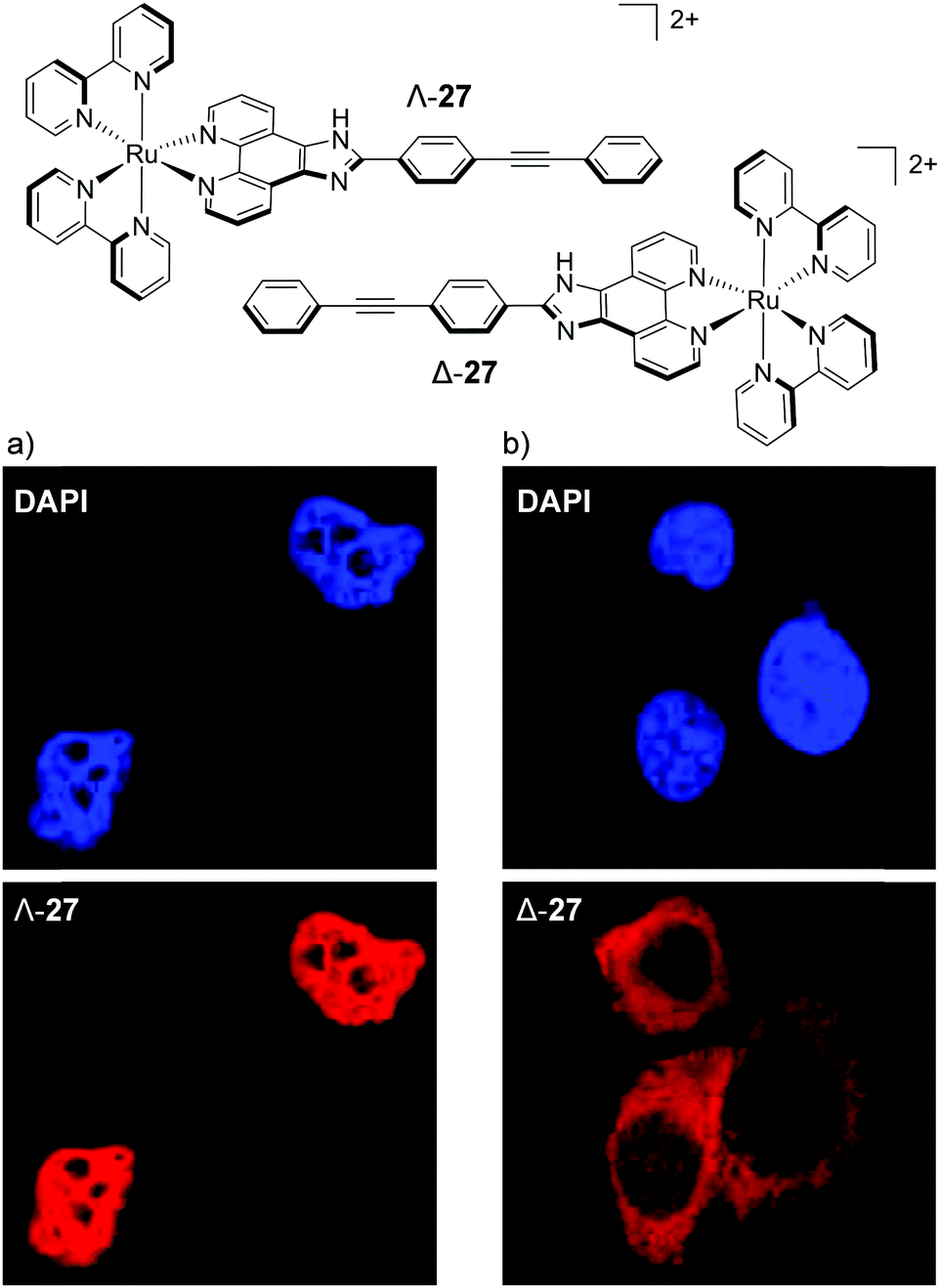 | ||
| Fig. 14 Chemical structures of the Λ- and Δ-enantiomers of complex 27 and the colocalisation studies of live MDA-MB-231 cells incubated with the nuclear stain DAPI and (a) Λ-27 (5 μM) and (b) Δ-27 (5 μM) for 6 h. DAPI emission is blue Ru emission is red. Images reproduced with permission from ref. 73. | ||
It should be noted that studies with resolved Λ- and Δ-enantiomers have also been reported where no differences were observed between the cellular staining of the different enantiomers.113 However, the examples above illustrate that in the chiral environment within a cell the Δ- and Λ-enantiomers of certain complexes are capable of acting as distinct chemical entities. Later in the review, differences between the biological activities of the enantiomers of Ru(II) polypyridyl complexes in vitro and in vivo will be discussed.
Multiphoton cellular imaging
A significant challenge towards the development of Ru(II) polypyridyl complexes as luminescence probes and photo-reactive agents for in vivo applications is the relatively high-energy wavelength of excitation required. These complexes typically absorb light between 400–500 nm, and while such wavelengths are suitable for histology and in vitro cellular studies, wavelengths below 650 nm only penetrate a small distance into biological tissues due to absorption and scattering of the light.129 For deeper tissue penetration (in the >500 μm to cm range), near infrared (NIR) light between 650–900 nm is required.130 Two-photon excitation of Ru(II) complexes is one way by which researchers have succeeded to excite Ru(II) complexes at these longer wavelengths. This technique involves the simultaneous absorption of two photons, which each possess half of the energy required to excite the Ru(II) complex from the ground to the excited state, as depicted in Fig. 15a.132 The efficiency of two-photon absorption by a molecule is quantified by its “two-photon absorption cross-section” (σ2), in units of GM (1 GM = 10−50 cm4 s per photon) and typically femtosecond lasers or diode lasers have been used for such experiment with Ru(II) polypyridyl complexes. Organic fluorophores typically have cross-sections between 0.1–100 GM. For example, the nuclear stain DAPI has a two-photon absorption cross-section of 0.16 GM at 700 nm.133 With regards to Ru(II) polypyridyl complexes, cross-sections ranging from ca. 10–650 GM in the 800 nm region have been reported from investigations into their two-photon applications in biological systems.81,82,113,134–142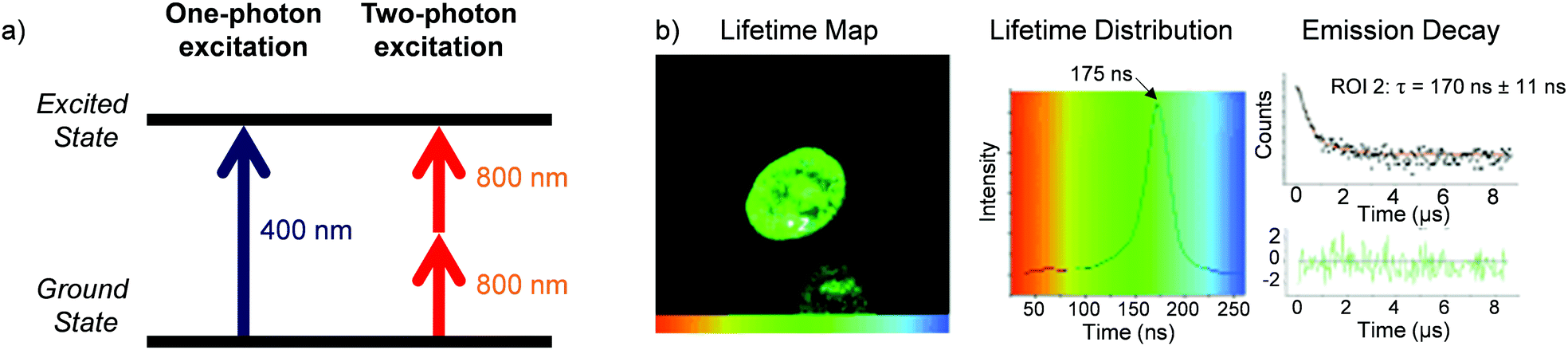 | ||
| Fig. 15 (a) Simplified energy diagram showing electronic transitions following one-photon and two-photon excitation of a chromophore, (b) two-photon phosphorescence lifetime image (λex = 800 nm) of live MCF-7 cells incubated with complex 13 [(phen)2Ru(tpphz)Ru(phen)2]4+, 500 μM for 1 h in serum-free medium. Images reproduced with permission from ref. 131. | ||
Thomas and co-workers were the first to investigate Ru(II) polypyridyl complexes as two-photon luminescence lifetime probes for cellular DNA.131 The researchers were able to image and record the luminescence lifetimes of their dinuclear complex 13 when bound to DNA in live cells, upon two-photon excitation at 800 nm (as shown in Fig. 15b). Moreover, the two-photon absorption cross-section of complex 13 was 142 GM at 800 nm, which is more than two orders of magnitude greater than that of DAPI.131,133 A series of dinuclear complexes based on the imidazo-phenanthroline motif (shown in Fig. 16) were investigated as two-photon cellular probes by Chao and co-workers and possessed cross-sections of between 322–386 GM (at 830 nm).81 The complexes showed low toxicity towards cells at the concentrations required for cellular imaging (10 μM) and interestingly showed only small differences between their subcellular localisation. The complexes predominantly localised in the cytoplasm of cells, where they could be visualised using two-photon excitation (λex = 830 nm), as shown in Fig. 16 for the fluoro derivative (31).81
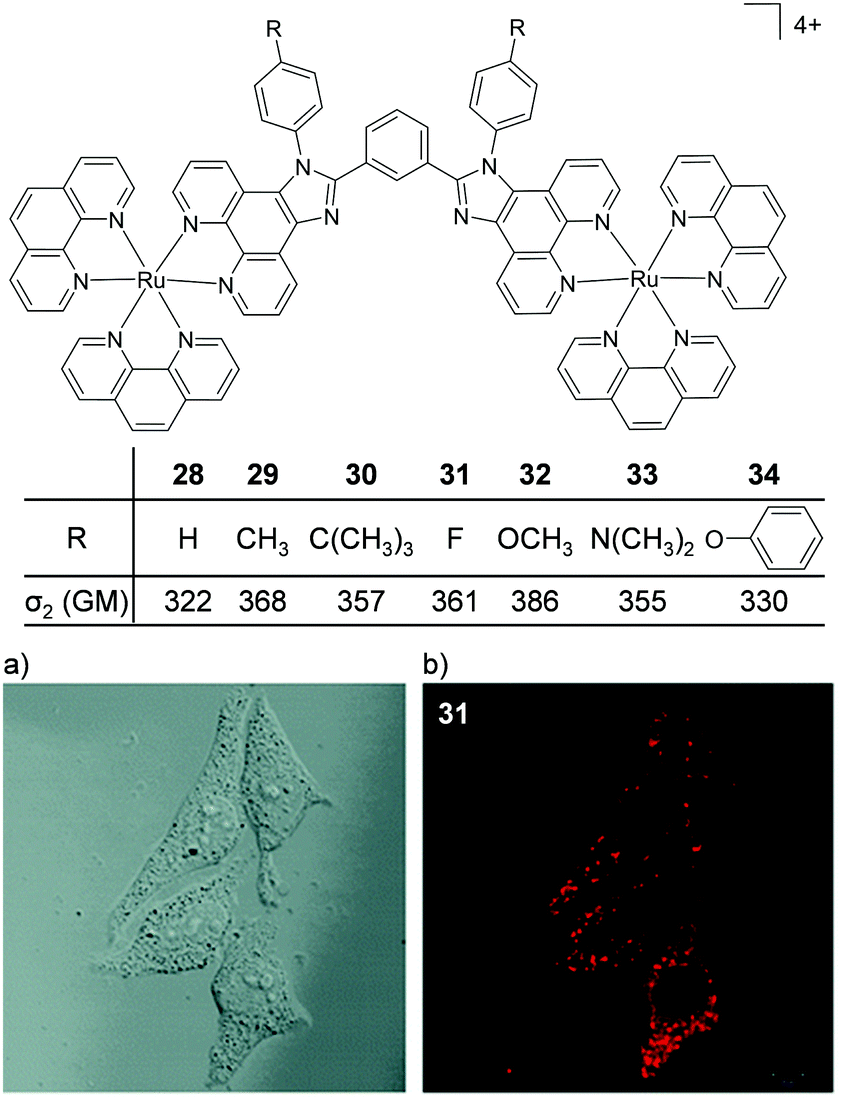 | ||
| Fig. 16 Chemical structures and two-photon absorption cross-sections at 830 nm of complexes 28–34. (a) the bright-field image and (b) the two-photon confocal microscopy image (λex = 830 nm) of live HeLa cells incubated with complex 31 (10 μM) for 4 h. Images reproduced with permission from ref. 81. | ||
By substituting the central phenyl moiety of the imidazo-phenanthroline-based ligand for a pyridine, these researchers were able to incorporate a metal binding pocket into their dinuclear complex 35, shown in Fig. 17. The complex was found to selectively bind Cu2+ ions in aqueous medium, even in the presence of other cations, such as Na+, K+, Mg2+ and Ca2+, which are abundant in biological systems. The complex possessed a cross-section of 400 GM (at 850 nm) and upon binding to Cu2+ its luminescence was quenched, allowing it to function as a two-photon luminescent probe for Cu2+.82 Within 1 h of incubation with 35 (10 μM), the complex was found to localise in the cytoplasm of HeLa cells, where it could be visualised by two-photon excitation at 850 nm. However, within 5 min of addition of CuCl2 (20 μM) to the cell culture medium, the intracellular luminescence of the complex was effectively quenched, as shown in Fig. 17.
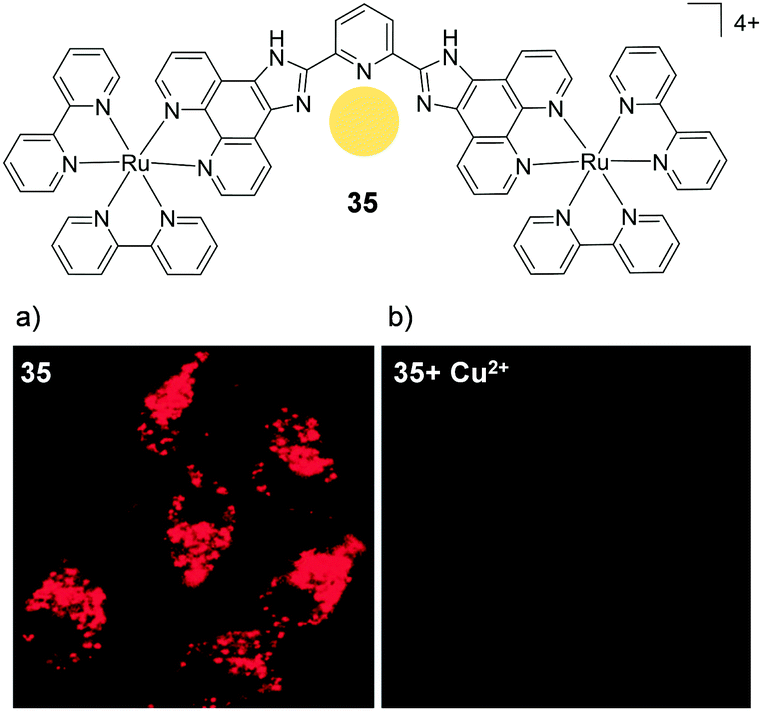 | ||
| Fig. 17 Chemical structures of complex 35 showing the binding pocket for Cu2+ in yellow and the two-photon confocal microscopy image (λex = 850 nm) of live HeLa cells incubated with complex 35 (10 μM) for 1 h (a) in the absence and (b) in the presence of CuCl2 (20 μM) and pyrrolidine dithiocarbamate (100 μM) added to the medium. Images reproduced with permission from ref. 82. | ||
A mononuclear complex incorporating an N-hexyl phenothiazine moiety (36), which possessed a cross-section of 219 GM (at 760 nm), was found to be impermeable to the cell membrane of HepG2 cells and was taken into cells via an endocytic pathway.139 Within one hour of treatment with 36 (10 μM), the complex could be visualised within cells by two-photon microscopy (λex = 760 nm) and was found to localise predominantly in the mitochondria of treated cells, as shown in Fig. 18, where it was suggested to bind specifically to the mitochondrial DNA.139 In addition, the researchers reported that the complex could be used as a stain for two-photon microscopy studies of fixed brain tissue samples.139
 | ||
| Fig. 18 Chemical structure of complex 36 and the two-photon confocal microscopy image of HepG2 incubated with complex 36 (10 μM, λex = 760 nm shown in red) for 30 min followed by incubation for 15 min with the mitochondrial stain Mitotracker (shown in green). Areas of co-localisation of complex 36 and Mitotracker are coloured yellow. Images reproduced with permission from ref. 139. | ||
Two interesting applications have recently been reported by Chao and co-workers. The authors utilised a series of tetranuclear complexes (37–41, shown in Fig. 19) as DNA delivery agents and exploited their high two-photon absorption cross-sections to monitor their uptake and localisation in living cells by two-photon luminescence microscopy.137 These complexes were found to condense plasmid DNA into particles of approximately 100 nm in diameter and the DNA within these particles was found to resist degradation by the DNase-I endonuclease. The Ru(II)–DNA particles were internalised by cells via endocytosis and flow cytometry revealed that increasing the length of the alkyl chains resulted in greater luminescence intensity from the complexes within cells, which indicated that the hydrophobic effect played an important role in their cellular uptake. Upon internalisation, the Ru(II)–DNA particles were subsequently found to localise within endosomes or in the cytoplasm of treated cells, as shown for complex 39 in Fig. 19.137 In another study a series of mononuclear complexes incorporating a redox-active anthraquinone moiety (complexes 42–44, shown in Fig. 20a) were developed as in situ probes to monitor cycling (intermittent) hypoxia in cells and tissues;143 a process that can result in resistance to anti-cancer therapies and promote tumour metastasis.144 Under normoxic conditions (20% O2) the anthraquinone moiety quenched the luminescence of the complexes, whereas under hypoxic conditions (1% O2) it was reduced to its hydroquinone form, which did not quench the luminescence of the Ru centres, as shown schematically in Fig. 20a. After uptake by A549 cancer cells the complexes localised predominantly in the mitochondria of cells and under normoxic conditions negligible intracellular luminescence was detected. In contrast, under hypoxic conditions, strong two-photon luminescence was observed from the treated cells. The researchers went further to show that these probes could report on hypoxia within 3D multicellular tumour spheroids. These are heterogeneous cellular aggregates possessing a number of properties which make them valuable in vitro models for in vivo tumour microenvironments.145 Due to the limited diffusion of oxygen into tissue beyond approximately 130 μm in thickness, spheroids can possess hypoxic regions.145 Indeed, stronger luminescence was detected from cells deeper within the tumour spheroid. Moreover, using two-photon excitation (λex = 800 nm), the researchers were able to probe deeper into the tumour spheroids than when using one-photon excitation (λex = 460 nm). The researchers also went on to demonstrate that the probes could be used in vivo to monitor cerebral anoxia (no oxygen present) within the brains of live zebrafish, following treatment with 10 μM of complex 44, as shown in Fig. 20b.143
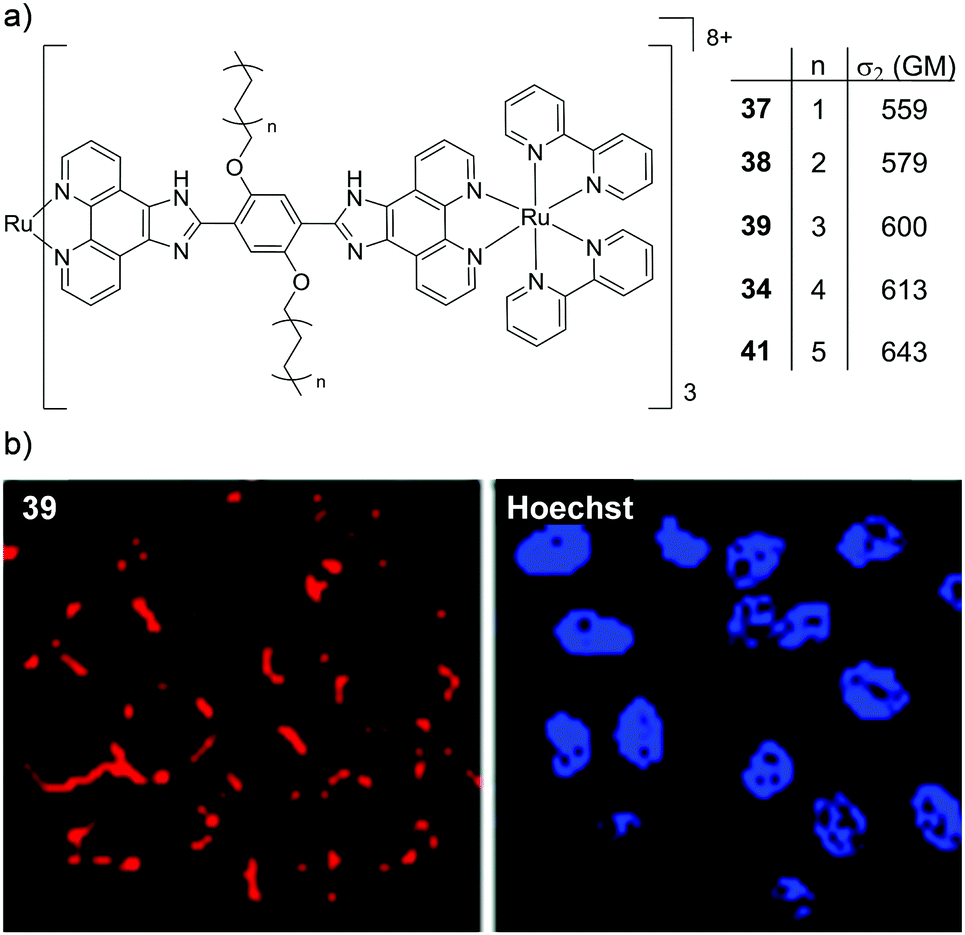 | ||
| Fig. 19 (a) Chemical structures of complexes 37–41, their two-photon absorption cross-sections at 830 nm and (b) the two-photon confocal microscopy images (λex = 830 nm) of HeLa cells incubated with the DNA–Ru particles formed from complex 39 for 4 h (shown in red) and stained with the nuclear stain Hoechst 33258 (shown in blue). Images reproduced with permission from ref. 137. | ||
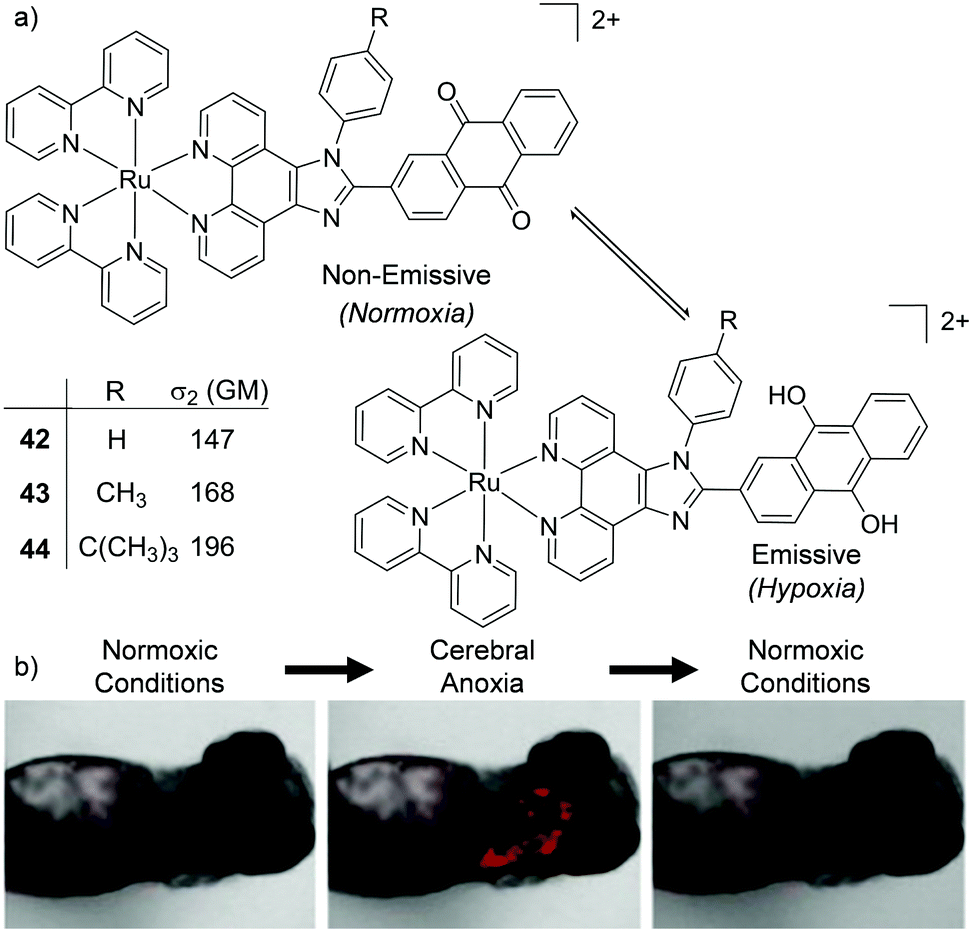 | ||
| Fig. 20 (a) Schematic illustration of the reversible conversion of the anthraquinone moieties of complexes 42–44 to their hydroquinone forms under normoxic and hypoxic conditions, and their two-photon absorption cross-sections at 800 nm. (b) the two-photon confocal microscopy images (λex = 830 nm) of live zebrafish incubated with complex 44 (10 μM) for 1 h and imaged under normoxic or anoxic conditions, as indicated. Images reproduced with permission from ref. 143. | ||
Two-photon studies have also been carried out for Ru(II) polypyridyl complexes conjugated to other molecules, such as porphyrins and nanoparticles, which will be discussed the final section of this review. Moreover, in addition to luminescence imaging, two-photon excitation has also been used in the development of biologically active Ru(II) polypyridyl complexes for light-activatable therapies, which will be discussed in the next section.
In vitro and in vivo biological activity
The previous sections have focused on the application of Ru(II) polypyridyl complexes as diagnostic and imaging agents, predominantly for use in cell culture. However, if these applications are to be translated to therapeutics for clinical use, the biodistribution, activity and the toxicology of these complexes needs to be investigated to assess their efficacy and safe use. Researchers have long demonstrated that these complexes interact strongly with biomolecules through non-covalent interactions, with numerous detailed investigations published, particularly in the case of DNA. Indeed, such non-covalent interactions with biomolecules are the underlying mechanism through which countless pharmaceutical drugs elicit their activity in vivo. As such, this has motivated researchers to exploit these strong interactions with biomolecules in the development of new Ru(II)-based therapeutic agents. Moreover, by exploiting the salient photophysical and photochemical properties of this class of complex, new classes of Ru(II)-based anti-cancer agents, including light-activatable drug candidates have been designed. This section will illustrate the various approaches that researchers are taking in the development of therapeutic Ru(II) polypyridyl complexes and their activity both in vitro and in vivo.Anti-cancer activity
In vitro (no light activation)
There is continuing demand for new classes of anti-cancer therapeutics owing to the development of tumours with resistance to chemotherapeutics over the course of their treatments.146 Furthermore, many clinically used anti-cancer drugs cause considerable side effects and collateral toxicity to non-cancerous cells, which is fuelling the search for more selective and targeted anti-cancer agents. Ru complexes have attracted increasing interest as promising candidates and, of the numerous reported Ru complexes showing potential as anti-cancer therapeutics, one Ru(II) polypyridyl complex (TLD1433) and two Ru(III) complexes (NAMI-A and KP1019) have progressed to clinical trials.147–153 These results have motivated researchers to translate new Ru complexes into a clinical setting.The aim of anti-cancer therapeutics is to either arrest the proliferation of cancer cells (cytostatic agents) or elicit cell death by damaging cells (cytotoxic agents). One of the primary methods that has been used to assess and quantify the anti-cancer activity of the numerous Ru(II) polypyridyl complexes reported to date, has been to conduct cell viability assays. The potencies of the compounds are often quoted as the half-maximal inhibitory concentration (IC50), i.e. the concentration that elicits 50% inhibition of cell viability. Such studies have revealed that Ru(II) polypyridyl complexes show a range of activities against cancer cells, with IC50 values from the nanomolar to millimolar range.62,109,154 In the case of cytotoxic compounds, the majority of complexes have been reported to induce cell death via programmed cell death pathways, such as apoptosis. This is the programmed and controlled pathway by which cells die and are subsequently engulfed by the surrounding cells. In contrast, there are relatively few examples of complexes that induce non-apoptotic cell death, such as necrosis.64,109,155 Unlike apoptosis, necrosis elicits a proinflammatory response in vivo, which can result in the recruitment of cytotoxic immune cells to the tumour site, thereby aiding tumour destruction. However, it can also result in damage to normal tissues.156 A number of different mechanisms have been reported through which these complexes exert their activities, which is not surprising given the variety of molecular structures that have been investigated, as well as the differences reported in their subcellular localisation.62,109,154 These mechanisms include:
– disrupting the binding of transcription factors to DNA,62
– stalling DNA replication forks,64
– inducing endoplasmic reticulum stress157 and
– causing mitochondrial dysfunction;59,65,66,80,97,155,158–171
the latter being the most commonly reported. While a considerable number of cytotoxic pathways have been identified, in many cases the specific molecular target(s) with which the Ru(II) polypyridyl complexes interact to elicit their anti-cancer activities remain unknown. As such, more detailed characterisation of the molecular pharmacology of such Ru(II) complexes is still required.172 Indeed target identification is noted as one of the most important steps in the development of new drugs, as this can provide valuable insights as to whether a particular cancer type is likely to respond to the drug, what dosages will be required and what potential adverse side effects may be encountered in vivo.174–177 Cell type-specific activity has in fact been reported for numerous Ru(II) complexes. For example, Wölfl and co-workers found a striking difference between the activity of [Ru(bpy)3]2+ in the MCF-7 human breast cancer and HT-29 human colorectal adenocarcinoma cell lines. The complex showed low activity against the MCF-7 cell line (IC50 = 778.3 μM after 96 h incubation) whereas it was quite active against the HT-29 cell line (IC50 = 32.3 μM after a shorter incubation time period of 72 h).50 Moreover, using atomic absorption spectroscopy the authors demonstrated that similar levels of the complex were found in both cell lines, with 9 ± 3 and 5 ± 3 ng of Ru found per mg cell protein in the MCF-7 and HT-29 cell lines, respectively (after treatment with 500 μM of the complex).50 While the origin of this 24-fold difference in activity between cell lines was not commented upon, Luo et al. reported an interesting hypothesis for such cell line-specific activity in their study into the biological activities of [Ru(bpy)3]2+ and a series of Ru(II) complexes where the planarity of the ligands and consequently the lipophilicity of the complexes were incrementally increased, as shown in Fig. 21a.173 The authors found that all of the complexes inhibited the activity of the enzyme thioredoxin reductase. This is a selenoenzyme found in the mitochondria and cytoplasm of cells, which plays an important role in protecting cells from oxidative stress and maintaining a redox balance within cells.178 Moreover, it was found that as the ligand surface area increased, the complexes became more potent inhibitors of the enzyme, both within A375 melanoma cells and cell lysates.173 When the biological activity of the complexes against five cancer cell lines was investigated it was found that as the ligand surface area increased, so too did the activity of the complex but also, that there was a considerable difference between the activity of each complex, depending on the cancer cell line tested. Based on their hypothesis regarding the intracellular molecular target of these complexes, the authors concluded that the inhibition of the thioredoxin reductase resulted in increased intracellular ROS levels and subsequent induction of apoptosis in treated cells. Moreover, they proposed that the observed differences in sensitivity to the Ru(II) complexes may be due to differences in the cellular protein expression profiles between the cancer cell lines.173
 | ||
| Fig. 21 (a) Chemical structures and logP values of the perchlorate salts of complexes 45, 46, [Ru(phen)3]2+ and [Ru(bpy)3]2+ their IC50 values (μM) against A375 melanoma cells after 48 h treatment. Values taken from the study of Luo et al.173 | ||
A key strategy used in the development of new therapeutics, both in academia and industry, has been investigating structure–activity relationships of new drug candidates, i.e. trying to determine the relationship between chemical structure and biological activity for a series of compounds, thereby facilitating the rational design and optimisation of new drug candidates.179–182 A number of such studies for Ru(II) polypyridyl complexes have been conducted. For example, Gasser and co-workers constructed a structure–activity relationship around the 2-(pyridin-2-yl)pyrimidine moiety of their bis-dppz complex, shown in Fig. 22. When functionalised with a carboxylic acid, complex 47 was found to localise in the mitochondria of HeLa cells and showed strong activity against cells (IC50 = 10 μM after 48 h).59 However, chemical modifications to the 2-(pyridin-2-yl)pyrimidine moiety, such as those seen in Fig. 22 for complexes 48–51, resulted in a loss of the preferential targeting of the mitochondria and the derivatives showed reduced activity in cells. As such, the researchers concluded that accumulation of the complex in the mitochondria was crucial to its anti-cancer activity and illustrated the importance of intracellular localisation on the activity of complex 47.59,69,183
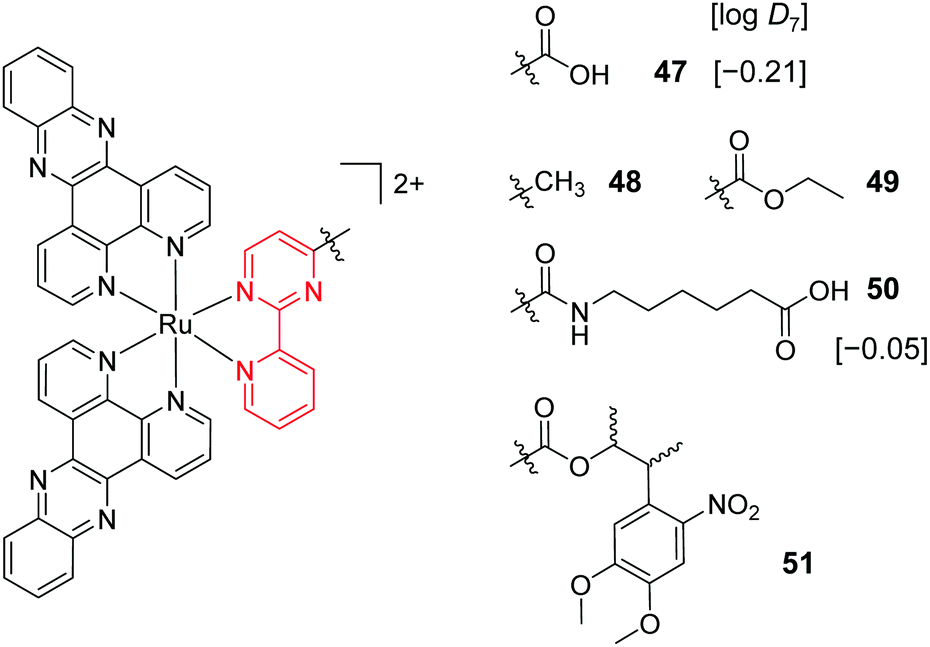 | ||
| Fig. 22 Chemical structures of the complexes investigated as part of the structure–activity relationship around the 2-(pyridin-2-yl)pyrimidine moiety of the bis-dppz complex studies by Gasser et al. where the highlighted in red. logD7 values were obtained from ref. 59 and correspond to their hexafluorophosphate salts. | ||
In a number of studies the size of the polypyridyl ligand coordinated to the Ru(II) centre was found to be of considerable importance to its biological activity, whereby complexes incorporating more extended aromatic ligands tended to show greater activity.59,61,63,86,99,173,184,185 Exceptions to this have been found, however; for example, in the study of Wölfl and co-workers when one of the bpy ligands of [Ru(bpy)3]2+ was substituted for a phen ligand, the biological activity of the resulting [Ru(bpy)2(phen)]2+ complex against MCF-7 cells decreased 6-fold.50
Of course, modulating the aromatic surface area considerably influences the shape and lipophilicity of Ru(II) complexes. Numerous studies have shown a strong correlation between the increased lipophilicity of Ru(II) polypyridyl complexes and their biological activity,50,98,109,110,113,155,158,168,173,186 and in a number of these studies it was remarked that this enhancement in biological activity was likely due to increased intracellular concentrations of the complexes.61,98,110,113,168 This has been clearly demonstrated for cyclometalated Ru complexes where quantitative methods such as ICP-MS showed a significant increase in the intracellular concentrations of the more lipophilic mono-cationic cyclometalates, relative to their di-cationic non-cyclometalated counterparts. This increased cellular uptake was accompanied by a concomitant enhancement in their activities against various cancer cell lines and 3D tumour spheroids, typically in the nanomolar to low micromolar range.62,63,114,158,187–190 This enhancement in lipophilicity and biological activity upon cyclometalation is highlighted in Table 2 for the dppz- and dppn-based complexes (dppn = benzo[i]dipyrido[3,2-a:2′,3′-c]phenazine). In addition, researchers have found Ru cyclometalates to be less sensitive to a number of the mechanisms by which cancer cells become resistant to cisplatin.191 However, while modulating the lipophilicity of drugs can result in increased cellular uptake, it can also change the nature of the interactions with/identity of the molecular targets of a drug, as well as alter their distribution throughout the cell, as has already been seen for a number of Ru(II) polypyridyl complexes in this review. For example, Zava et al. demonstrated that, for a series of Ru(II) complexes, the most lipophilic and biologically active complex (23, logD7 = +0.55 as the chloride salt), which had an IC50 of <1 μM, appeared to induce cell death by targeting the plasma membrane of the cell, as opposed to via an intracellular target.109 The considerable importance of lipophilicity on the biological activity of Ru(II) polypyridyl complexes that has been reported is unsurprising given that the lipophilicity of drug molecules has long been established as one of the most important physico-chemical properties in determining their potency and toxicity.38–43
| Complex | Charge | logP |
IC50 (μM) | Cell Line | Ref. |
|---|---|---|---|---|---|
| [Ru(bpy)2(dppz)]2+ | 2+ | −2.5 | 159.9 | HeLa | 62 |
| [Ru(bpy)(phpy)(dppz)]1+ (18) | 1+ | +1.0 | 0.6 | HeLa | 62 |
| [Ru(bpy)2(dppn)]2+ (52) | 2+ | ∼−0.6 | 95.2 | HeLa | 63 |
| [Ru(bpy)(phpy)(dppn)]1+ (53) | 1+ | ∼+1.0 | 2.0 | HeLa | 63 |
| Cisplatin | 2+ | −2.21 | 16.4 | HeLa | 63 and 196 |
Another structural attribute that can have a considerable influence on the activity of drug molecules is their chirality.192 Of the studies investigating differences between the activities of the Δ- and Λ-enantiomers of Ru(II) complexes, most often only modest differences between the enantiomers was observed, if at all.110,111,113,128,159,167,193–195 However, more studies are needed to gain a better understanding of the enantioselective interactions of Ru(II) polypyridyl complexes in cells.

Another property which a number of researchers have correlated with biological activity is the binding affinity of complexes for DNA, based on solution measurements.61,155,186,197–200 The results of such studies have been mixed, with some authors reporting a correlation,155,199,200 while others found no correlation.61,186,197,198 However, in a number of studies where correlation was claimed between DNA binding and anti-cancer activity, little or no evidence was presented to support the hypothesis that the complexes did in fact reach the DNA of cells or that DNA was the molecular target responsible for their activity.
One limitation of the structure–activity studies to date is that individual studies have generally tested relatively small libraries of compounds, whereas large libraries would be needed to construct more comprehensive and definitive structure–activity relationships.181,201 One approach to overcome this is to conduct a meta-analysis of the various studies to date,202,203 however such a study is beyond the scope of this review. In such meta-analyses, quantitative structure–activity relationships (QSAR) models, where drug molecules are described in terms of quantifiable properties when comparing their biological activities, are of considerable importance. The most important properties to describe a drug in such studies are the 3D shape, volume, charge, electrostatic potential and lipophilicity (e.g. logP value) of a drug molecule.181,204–206 Given the importance that lipophilicity has on the uptake, localisation and biological activity of Ru(II) polypyridyl complexes, it would undoubtedly be of great benefit to future reviewers intending to conduct such a QSAR study, if researchers were to report the experimentally derived lipophilicities (e.g. logP values via the “shake-flask” method)207 of new complexes alongside their biological studies in future publications.
It should be noted, however, that comparison of the activity of Ru(II) complexes between studies presents a number of challenges, as will now be highlighted. As seen in this section, the biological activity of Ru(II) polypyridyl complexes can show considerable variation with cell line. Within the various studies to date, a variety of different cell lines have been used by researchers to evaluate the biological activity of Ru(II) complexes and quite often there are no cell lines in common between studies, thereby hindering direct comparison of the activities of complexes. A useful facility that is available to researchers is the NCI-60 Human Tumor Cell Lines Screen,208 which offers a free screening of potential anti-cancer compounds against 60 different human tumour cell lines and could help researchers to establish whether their complexes show selectivity for particular cancer cell types.
An additional factor that changes between reported studies is the time period over which cells are incubated with the complexes under investigation, which has a considerable effect on the biological activity. Finally, a number of different assays have been used between studies to quantify the anti-cancer activity of Ru(II) polypyridyl complexes. It has been established that the degree of activity of cytotoxic compounds against cultured cells changes considerably depending on the type of assay used to assess the activity.209,210 This is also apparent for a number of Ru(II) complexes. The activity of [Ru(bpy)2(dppz)]2+ against MCF-7 cell lines showed a dramatic difference between studies employing different assays. When its activity was assessed by crystal violet staining, an IC50 value of 90.2 μM was determined after 24 h incubation,50 whereas researchers using an MTT assay reported an IC50 value of 780 μM after twice the incubation time (48 h).154 Another example is seen for the dppn-containing complex 52, where a 10-fold difference in its activity against MCF-7 cell lines after 48 h treatment when crystal violet staining50 and a Calcein AM cell viability assay63 were used, with IC50 values of 3.3 μM and 35.1 μM, reported respectively. For [Ru(bpy)3]2+, its activity against HeLa cells after 48 h showed a considerable difference between studies when determined by the LIVE/DEAD® viability/cytotoxicity assay211 and MTT cell viability assay,212 with IC50 values of >500 μM and 152 μM determined using the respective assays.
In addition, even if comparing different studies using the same cell line and same Ru(II) complex, large variations may still be observed due to a lack of standardisation, for example, using different equipment, different protocols, different cell culture medium, and performing cytotoxicity experiments at different cell line passage numbers etc.
In vivo (no light activation)
While in vitro cell studies have been a cornerstone in the development and assessment of new anti-cancer therapeutics, oftentimes the results derived from such studies show little applicability to studies in vivo.213,214 As such, characterisation of the anti-cancer activity and toxicity of complexes in vivo is essential to their development. Such in vivo investigations began as far back as 1952 by Dwyer et al. with the toxicology studies of Λ- and Δ-[Ru(phen)3](ClO4)2 in mice.215 The complexes had little effect on the health of mice when administered orally, due to poor uptake, however, when administered via intraperitoneal injection, the complexes caused paralysis and death by respiratory failure.215 Furthermore, a ca. two-fold difference was observed between the toxicity of the two enantiomers, with the Λ-(+)-enantiomer showing greater toxicity than the Δ-(−)-enantiomer (9.2 mg kg−1 and >18.4 mg kg−1, respectively).215–217 Studies with radiolabelled 106Ru revealed that 15 min after intraperitoneal injection at sub-lethal doses, the complex predominantly localised in the liver and kidney and did not appear to accumulate in other tissue types. In addition, the rate at which the Λ-(+)-enantiomer entered the blood stream from the peritoneal cavity was approximately twice that of the Δ-(−)-enantiomer.218 Within 24 h, 97–99% of the complex was excreted intact from the mice, with 78–91% present in the urine. This demonstrated that the complex was not metabolised within the body and therefore the observed toxicity was caused by the intact complex as opposed to one of its metabolites.218 Solution studies revealed that [Ru(phen)3]2+ acted as a competitive inhibitor of the enzyme acetylcholinesterase, with 80% enzymatic inhibition at 10−5 M concentrations of the complex, with the Λ-(+)-enantiomer being 1.5–2 times more potent than its Δ-(−)-counterpart.215,219 Further experiments demonstrated that the complex blocked activity at the neuromuscular junctions of the diaphragm-phrenic nerve in rats, where it was suggested to compete with acetylcholine, thereby causing diaphragmatic paralysis and respiratory failure.219More recently, Wang et al. studied the pharmacokinetics and biodistribution of a complex incorporating an imidazo-phenanthroline-based ligand (complex 54) in mice after intravenous injection of the complex as its PF6− salt (3 mg kg−1);220 where the concentration of Ru in biological samples was determined by inductively coupled plasma atomic emission spectroscopy. The complex had an elimination half-life (t1/2β) of 55.1 h and after 72 h the mice were sacrificed and the biodistribution of the complex was assessed. Of the organs examined, the kidneys showed the highest concentrations of Ru, with lower quantities found in the heart, liver, lungs and spleen. Furthermore, the researchers also found evidence of possible damage to the liver, heart and kidneys 72 h post-administration of the complex.220
In addition to these in vivo toxicological studies, recently the anti-cancer activity of Ru(II) polypyridyl complexes have been assessed in mouse allograft or xenograft models, which involve the growth of tumours after implanting mice with cultured cancer cells originating from the same species or a different species, respectively, as illustrated in Fig. 23. A number of these studies have revealed that Ru(II) polypyridyl complexes can show similar anti-cancer activity to cisplatin, but with considerably fewer side effects and collateral toxicity.157,221
 | ||
| Fig. 23 Schematic diagram showing the procedure for conducting in vivo xenograft and allograft studies. | ||
MacDonnell and co-workers evaluated the in vivo anti-cancer activity of the enantiomers of a mono-nuclear and di-nuclear Ru(II) polypyridyl complex, 55 and 56 respectively, which incorporated the (tatpp) ligand, as shown in Fig. 24. In vitro cell culture studies had shown the complexes to be active under hypoxic conditions and hence, could exert preferential activity against tumour cells, as these tend to have hypoxic environments. The complexes were administered via intraperitoneal injection and found to repress tumour growth in models of xenografted H358 human non-small cell lung cancer cells in mice.157,222 The maximum tolerated dose (MTD, the highest dose that does not cause unacceptable side effects) observed for each complex was similar to that of cisplatin (67 mg kg−1 for rac-55 and 64, 43 and 43 mg kg−1 for ΛΛ-, ΔΔ- and the meso isomer ΔΛ-56, respectively).222 In control mice, a rapid and near linear tumour growth was observed, however, after six days of treatment with either Δ-55 or ΔΔ-56, tumour growth slowed with only a gradual increase in dimensions over the course of the experiment, followed by arrest of tumour growth between days 30 and 60 of treatment, as seen in Fig. 24a and b.
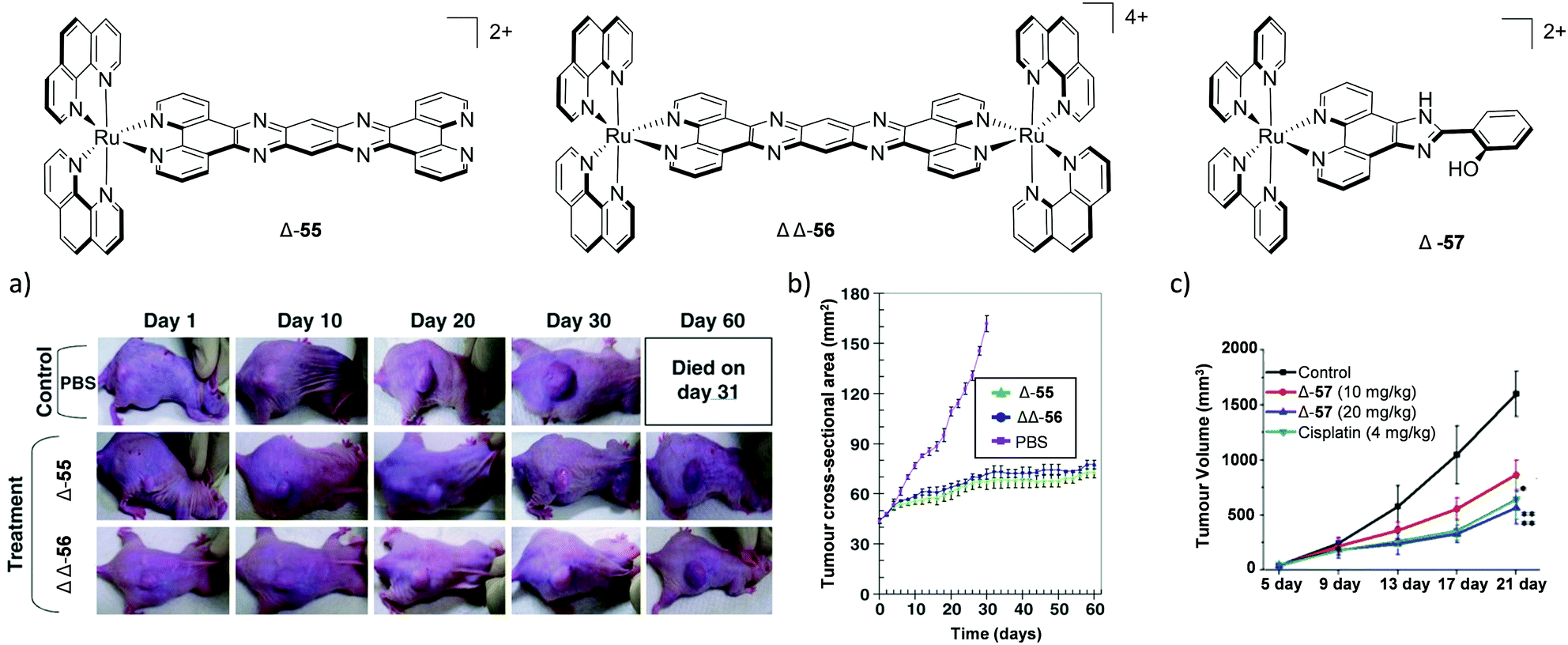 | ||
| Fig. 24 The chemical structures of complexes 55, 56 and 57 and their in vivo anti-cancer activity. (a) photographs and (b) graph of the effect of complexes Δ-55 and ΔΔ-56 (as their chloride salts at 1 mg) on the tumour area (mm2) of H358 human lung cancer cells in a nude mouse xenograft model over the course of 60 days. The mice were treated with the complexes on days 0, 15 and 30 by intraperitoneal injection, each injection contained 1 mg of complex (ca. 50% of MTD) dissolved in phosphate buffered saline (PBS). (c) Graph of the effect of cisplatin and complex Δ-57 (as its perchlorate salt) on the tumour volumes (mm3) of HeLa xenografts in nude mice over the course of 21 days. These drugs were administered by intraperitoneal injection every four days with each injection containing 4 mg kg−1 for cisplatin and either 10 mg kg−1 or 20 mg kg−1 for Δ-57, as indicated, dissolved in saline. Images and graphs were reproduced with permission from ref. 222 and 221. | ||
Another in vivo study by Chao and colleagues examined the anti-cancer activity of the Δ-enantiomer of their Ru(II) polypyridyl complexes 57 in models of xenografted HeLa cells in nude mice.221In vitro cell culture studies had previously shown the complex to localise in the mitochondria of HeLa cells and induce apoptosis, with IC50 values of 9.3 μM after 48 h of treatment.159 The complex was administered to mice by intraperitoneal injection and was found to significantly reduce the rate of tumour growth, with similar efficacy to cisplatin, as shown in Fig. 24c. Furthermore, in contrast to cisplatin, the researchers found no evidence of damage to the kidneys, livers or sensory nerves of the mice at the end of the treatment.221In vivo studies of a Ru cyclometalate, complex 58, have also been undertaken, where intraperitoneal injection of the complex was found to reduce the volume and weight of the tumours by 40% in allograft models of B16F10 mouse melanoma cells in nude mice, relative to untreated mice, as shown in Fig. 25. This anti-cancer activity was similar to that of cisplatin but in contrast to cisplatin it did not cause adverse side effects to the liver, kidneys, or neuronal sensory system.157
 | ||
| Fig. 25 Chemical structure of complex 58 and a graph of the tumour volumes (mm3) over the course of 22 days in allograft models of B16F10 mouse melanoma cells in nude mice treated with cisplatin or complex 58 (as its PF6− salt). The drugs were administered by intraperitoneal injection (13.3 μmol kg−1) twice a week after 10 days of implanting tumour cells into mice. Graph reproduced with permission from ref. 157. | ||
Photo-activation of drug molecules
While the above examples demonstrate preferential cytostatic and cytotoxic activity towards tumour cells, chemotherapeutics will nearly always exhibit some degree of toxicity towards healthy non-cancerous cells (unwanted side effects). With a view to increasing the therapeutic index of cancer drugs, researchers have been exploiting the rich photochemistry of transition metal complexes to develop new classes of light-activatable therapeutics, which will now be highlighted.The therapeutic use of light in modern medicine has its roots in the Nobel prize-winning work of Finsen on the UV light treatment of lupus vulgaris in 1896.223,224 However, it was fifty years before the systematic use of UV light with an administered drug became clinically established when a psoralen derivative was used to treat vitiligo and psoriasis, thus opening the door to ‘photochemotherapy’ or ‘photoactivated chemotherapy’.201,225–227 The considerable appeal of this therapeutic strategy is that it allows clinicians to administer a drug which can disperse throughout the body, and then control both its spatial and temporal activation by selectively illuminating areas of the body requiring treatment. This can significantly reduce collateral damage to healthy tissues and adverse effects to patients, particularly in the case of cancer therapy. In addition, as a result of considerable technological developments over the last number of decades in light sources and light delivery technologies (such as fiber optic light guides),229 light can now be delivered deep into tissues. This has transformed the field from the treatment of diseases that affect the surface of the body to those affecting many different sites within the body, including the brain, bladder, lungs, liver and prostate.230 However, despite its significant advantages, this mode of therapy remains clinically underexploited and as such this is an active area of research across numerous fields.
Ru(II) polypyridyl complexes have shown great promise as photo-activatable therapeutic agents, with a range of different mechanisms identified by which their biological activity can be elicited, as depicted in Fig. 26. There have been a number of excellent reviews of this subject,227,231–233 and therefore this section will provide key examples to illustrate these mechanisms and, where possible, present more recent examples that have emerged since the publication of these listed review articles. In this review we will consider separately reports where a role for oxygen is established and those where Ru(II) complexes have been designed so as not to rely on the presence of oxygen. However, as will become apparent over the course of this section, Ru(II) polypyridyl complexes can undergo a number of different mechanisms simultaneously (both oxygen-dependent and -independent), the relative amounts of which depend on various factors such as the nature of the Ru(II) complex, the environmental conditions (such as O2 concentration) and its subcellular localisation/the presence of target biomolecules. Moreover, solid tumours may possess regions of higher and lower oxygen concentration in vivo and therefore the mechanism of action of such Ru(II)-based drugs may be foreseen to vary across these regions.
 | ||
| Fig. 26 Flow chart illustrating some of the pertinent photophysical and photochemical mechanisms by which the therapeutic responses of photo-activated Ru(II) polypyridyl complexes can be elicited. Please note that there are numerous applications of light in photomedicine and this figure only highlights topics of interest to this review. *The term photoactivated chemotherapy only applies to therapies in which the photo-activatable drug molecules incorporate a d-block element, as opposed to purely organic based molecules such as porphyrins and psoralens. The relationship between the terms phototherapy, photochemotherapy and PDT are based on those recommended in ref. 228. | ||
Oxygen-dependent mechanisms
Both Type I and Type II reactions can occur simultaneously and the ratio between these processes depends on factors, such as the properties of photosensitiser used, the concentration of biomolecules and dioxygen in the vicinity of the excited photosensitiser, and the binding affinity of the photosensitiser for a particular biomolecule.238 In general PDT treatment leads to cell death by either apoptosis or necrosis. A number of factors influence the extent of photo-damage and cytotoxicity, as well as the mechanism by which cell death occurs, following PDT. These include the type of photosensitiser, the intensity of administered light, the cell type investigated and the extracellular/intracellular localisation of the photosensitiser.230,238–241 In addition, in vivo, the time between administration of the photosensitiser and application of light can have a significant impact on the efficacy of the treatment.
Traditionally, porphyrin-based photosensitisers have dominated clinical PDT. These include the first generation photosensitisers hematoporphyrin derivative (HpD) and Photofrin®; as well as the chlorin-based second generation photosensitisers Foscan® and NPe6.242 However, issues such as poor light absorption and prolonged photosensitivity encountered with many of these photosensitisers has motivated research into new classes of photosensitisers. With regards to Ru(II) polypyridyl complexes, their excited-state properties can be tuned to allow optimal 1O2 generation. In fact, a number of Ru(II) polypyridyl photosensitisers have been developed that are capable of producing 1O2 with quantum yields of close to unity.243,244 However, while 1O2 can elicit significant damage to biomolecules, resulting in cellular damage and cell death,245 this highly reactive molecule only diffuses a short distance within cells (ca. 100 nm);246,247 and as such, the subcellular localisation of a photosensitiser determines what molecular components of the cell are damaged during treatment and the subsequent fate of the cell.
Gunnlaugsson and co-workers demonstrated that a bis-TAP complex incorporating an extended dppz ligand (complex 59) showed little activity against HeLa cells in the dark (IC50 (dark) > 100 μM), however upon irradiation of the treated cells with visible light (λex > 400 nm, 18 J cm−2) the complex became highly active against cells, with an IC50 (light) = 8.8 μM.46 Similar light-induced activity was also observed for the dppz analogue 2. Detailed mechanistic studies showed complex 59 to predominantly localise in the mitochondria of cells where, upon irradiation with light, it induced apoptosis through ROS generation. Furthermore, in the presence of inhibitors of ROS, the light-induced activity displayed by the complex was significantly reduced.46 In contrast, when both ancillary TAP ligands of 59 were substituted by phen ligands, the resulting complex (60), showed little activity against treated cells with or without light activation.46
While Ru cyclometalates tend to exhibit strong biological activity, with reported IC50 values typically below 5 μM, Sainuddin et al. recently reported that a Ru complex incorporating a cyclometalating dppn-analogue 4,9,16-triazadibenzo[a,c]napthacene (pbpn) (complex 61), showed little activity against SK-MEL-28 human malignant melanoma cells in the dark (IC50 (dark) > 300 μM, after 80 h).248 This low activity is particularly striking given that its cyclometalated analogue 53 showed very high intrinsic activity against cells, after 48 h (IC50 = 2 μM, see Table 2), albeit, different cell viability assays and cell lines were used in these studies.63 However, the authors demonstrated that when the cyclometalating pbpn-ligand was substituted for a cyclometalating dppz-ligand (complex 62), the intrinsic activity of the complex was restored as complex 62 was highly active against SK-MEL-28 cells in the dark (IC50 (dark) = 1.9 μM, after 80 h).248 Upon activation with visible light (100 J cm−2), complex 61 showed a substantial increase in activity (IC50 (light) = 200 nM), which corresponded to an extremely large phototherapeutic index (i.e. the ratio of its IC50 in the light and dark) of >1400. Similar light-induced activity was also seen for complex 62. By monitoring the green ligand-centred emission from complex 61 within cells by confocal microscopy, the researchers found that the complex localised in the nuclei of treated cells. Mechanistic studies showed that photo-activation of the complex resulted in the reduction of dioxygen to produce superoxide (O2˙−), which was hypothesised to be at least partly responsible for the photo-toxicity of the complex towards cells.248
A particularly exciting development within the field has emerged from the McFarland research group, who licenced a number of Ru- and Os-based complexes to the Canadian-based company Theralase Technologies Inc. One of these Ru(II) complexes, designated TLD1433 (shown in Fig. 27), has recently commenced phase Ib clinical trials in Canada as the photosensitiser in the treatment of non-muscle invasive bladder cancer by PDT.148,249 In addition, Theralase has also announced plans to expand the application of their compounds to include lung, melanoma and brain cancers.250 Studies into TLD1433 revealed the complex to generate 1O2 with a quantum yield of near unity.251 The complex showed little dark toxicity towards various cell lines in vitro, at tested concentrations ≤10 μM after 6 h incubation. However, a strong photodynamic effect was observed in U87 human glioblastoma and murine CT26.WT colon carcinoma cells when incubated with the complex (1 μM) for 6 h. After cells were irradiated with green light (λex ≈ 525 nm, 45 J cm−2), the complex induced 100% cell death, with corresponding lethal dose 50 (LD50) values of 51 nM and 21 nM in the U87 and CT26.WT cell lines, respectively. Furthermore, under hypoxic conditions (<0.5% O2) the PDT effect was completely lost, which demonstrated its dependence on the presence of oxygen.
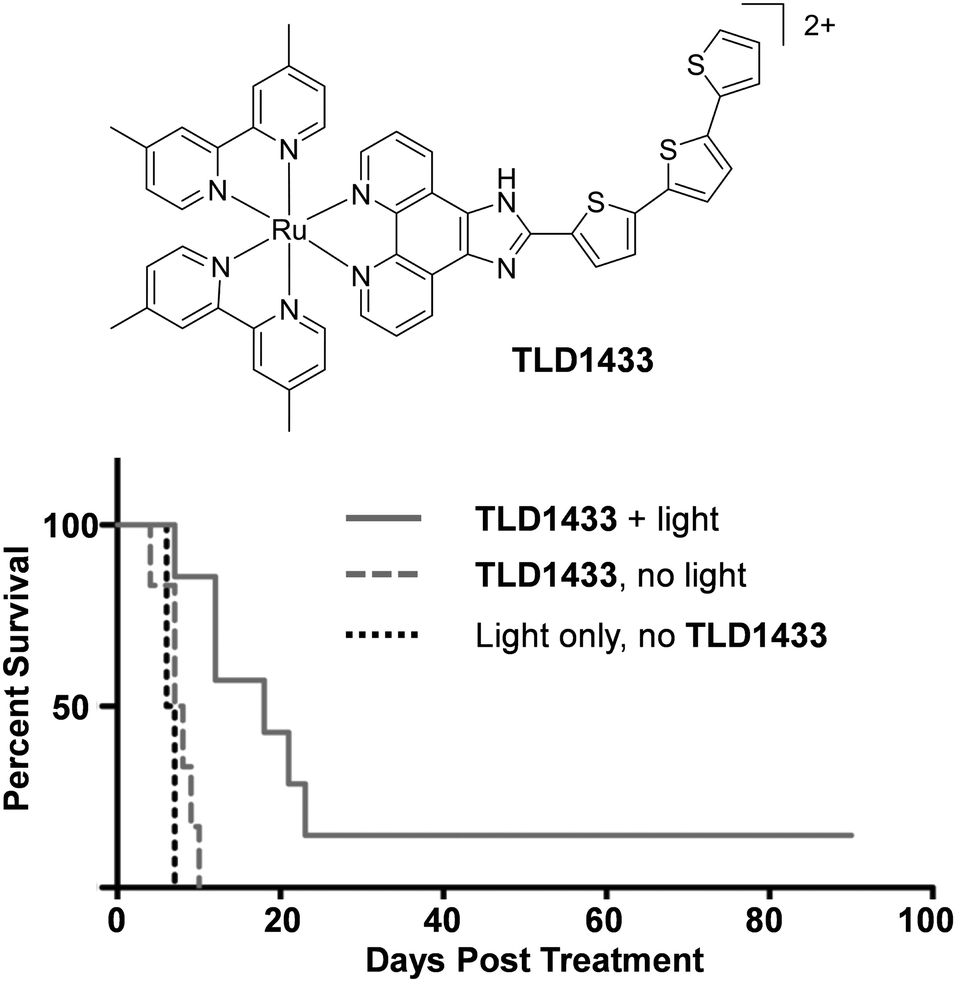 | ||
| Fig. 27 Chemical structure of TLD1433 and Kaplan–Meier survival curves of mice bearing CT26.WT colon adenocarcinoma tumours post TLD1433-mediated PDT. Mice were administered with TLD1433 (5 mg kg−1) via intratumoral injection and after 4 h the tumours were irradiated with continuous wave light (λex = 525–530 nm, 192 J cm−2). Graph reproduced with permission from ref. 252. | ||
In vivo studies showed TLD1433 to have a MTD50 of 103 mg kg−1 in mice. The biodistribution of the complex was assessed after either intraperitoneal or intratumoral injection of mice with TLD1433 at 50% of the MTD50 and recording the luminescence from homogenised tissue samples. These studies showed rapid clearance of the complex from most tissues, with retention of the complex in the liver (>100 μM), tumour (16.1 μM) and the brain (11.5 μM) after 24 h. The in vivo PDT activity of the complex was tested in mice with subcutaneous allografts of CT26.WT colon adenocarcinoma cells. The complex was administered by injection into the tumour and after four hours the tumours were irradiated with light (λex = 525–530 nm, 192 J cm−2). Upon photo-activation, the complex induced significant tumour damage and slowed tumour growth in a dose-dependent manner, which resulted in an increase in animal survival, as shown in Fig. 27. Furthermore, the nature of the light source was found to be of considerable importance to the efficacy of the treatment, where greater tumour destruction was achieved using continuous wave, rather than pulsed light.252
More recently, the authors demonstrated that when TLD1433 was incubated with the iron-binding glycoprotein transferrin, a remarkable improvement in the properties of the Ru(II)–transferrin complex (designated Rutherrin) were observed. This included an enhancement in its molar absorption coefficient, extension of its absorbance into the 660 nm to NIR region, enhancement in luminescence quantum yield and greater photo-stability.95 As discussed previously, malignant cells tend to overexpress the transferrin receptor, which can therefore allow enhanced accumulation of drug molecules bound to transferrin in these cell types.35,91 Indeed, the Rutherrin complex showed increased PDT efficacy in AY27 rat bladder carcinoma cells, relative to TLD1433 alone, but curiously no such enhancement was seen in HT1376 human bladder carcinoma cells. Furthermore, in vivo studies in mice showed the Rutherrin complex to be less toxic and exhibit improved PDT activity, relative to the complex alone. In light of this enhanced activity, the authors have subsequently patented the use of transferrin, as well as other glycoproteins, with metal-based coordination complexes for use as in vivo diagnostic agents and cancer therapeutics.253
Two Ru(II) complexes (63 and 64, shown in Fig. 28) have been recently reported for two-photon PDT.211 The two-photon absorption cross-section (σ2) of the complexes were 250 GM (at 800 nm)211 and 198 GM (between 810–830 nm),254 respectively, which are significantly higher than those reported for readily available photosensitisers such as Photofrin®, which has a cross-section of 10 GM at 800 nm.256 However it must be noted that Photofrin® was developed for clinical use using single-photon (ca. 630 nm), as opposed to two-photon activation.257 Both complexes displayed little dark toxicity towards HeLa multicellular tumour spheroids, however, upon two-photon activation in the 800 nm wavelength region, both complexes exhibited activities in the low micromolar range, as illustrated in Table 3.
 | ||
| Fig. 28 Chemical structures of complexes 63, 64 and 65, with the azo moiety highlighted in yellow. Also shown is the reduction of the azo moiety by intracellular glutathione. | ||
| IC50 (dark) (μM) | IC50 (light) (μM) | λ ex (nm) | Light dose (J cm−2) | σ 2 (GM) | |
|---|---|---|---|---|---|
| a 100 mW, 80 MHz, 100 fs for 30 min. | |||||
| 63 | >500 | 1.9 | 800 | 10 | 250 |
| 64 | >100 | 1.9 | 830 | 800 | 198 |
| 65 | >100 | 5.7 | 810 | 210 | |
As mentioned previously, the subcellular localisation of photosensitisers has a significant influence on their effectiveness in PDT.258,259 The high charge of complex 63 (8+) was designed with the intention of avoiding localisation in the mitochondria or nuclei of cells in an attempt to minimise dark toxicity.211 The complex was found to enter cells by endocytosis and localise in the lysosomes of HeLa cells, whereupon two-photon activation of the complex (λex = 800 nm) resulted in cell death by necrosis.211 This is consistent with studies of other classes of photosensitisers which found that photosensitisers that localise in the lysosomes tend to induce either apoptosis or necrosis.259 In contrast, complex 64 was designed to target the mitochondria because the light activation of photosensitisers localised in this organelle has been generally shown to induce apoptosis, as opposed to necrosis.259 Mitochondrial targeting was achieved by the incorporation of a lipophilic triphenylphosphonium moiety into the complex.260 Such lipophilic cations are well documented to accumulate in the mitochondria of cells due to the large negative membrane potential across the inner membrane of mitochondria (typically between 140–180 mV).261 While complex 64 was indeed found to localise in the mitochondria of cells, the mechanism of cell death was not reported.254
Recently, Zeng et al. designed a glutathione-activatable Ru(II)-azo photosensitizer (complex 65), in which the strongly electron-withdrawing azo-functionalised bpy ligands quenched the luminescence of the complex and its ability to generate 1O2. However, reduction of the azo group by intracellular glutathione, illustrated in Fig. 28, resulted in a restoration of the luminescence of the complex and its ability to generate 1O2.255 The complex localised in the mitochondria of HeLa cells and two-photon excitation of its reduced form (σ2 = 210 GM at 810 nm) resulted in the generation of intracellular ROS and apoptosis of both cultured cell monolayers and 3D multicellular tumour spheroids, with low micromolar light activity, as shown in Table 3.255
For clinical applications, new two-photon photosensitisers would ideally possess high two-photon cross-sections, so as to avoid the need for potentially damaging high light intensities to achieve a therapeutic effect. As mentioned, conventional PDT drugs have been found to possess relatively small cross-sections (e.g. σ2 = 10 GM for Photofrin® at ca. 800 nm).262 In contrast, conjugated porphyrin dimers have been developed with large cross-sections (σ2 = 17000 GM) in the 900 nm region and have shown extremely promising use in vivo.263 In addition, nanomaterials such as quantum dots and gold nanostars have been shown to possess extremely high cross-sections (σ2 = 103–106 GM) in the 800 nm region which has made such materials of particular interest to researchers towards the development of two-photon activated drug therapies.264 Indeed a number of Ru(II) complexes have been conjugated to porphyrins and nanomaterials and their two-photon properties have been studied, as will be detailed in the final section of this review. To the best of our knowledge, no Ru(II) polypyridyl complex with two-photon cross-sections in this range have been reported in cells. However, photosensitisers may not actually require large two-photon cross-sections for use in two-photon therapies. In 2014, Kachynski et al. upconverted NIR beams to generate visible light deep within tissues to activate intracellular chlorin e6 photosensitisers by single-photon activation to elicit PDT responses in cells.265 While this technique has yet to be applied to Ru(II) polypyridyl complexes, it circumvents the need for two-photon absorption by a photosensitiser and therefore could be of considerable use to researchers in the future.
Oxygen-independent mechanisms
One significant limitation to the clinical application of conventional PDT agents is their reliance on the presence of oxygen within tumours. However, in vivo, solid tumours often possess regions of low oxygen concentration, which has been shown to limit the efficacy of such agents.115,146,266 Moreover, the use of PDT agents in vivo may also induce hypoxia in tissues as a result of damage to the local vasculature and photochemical consumption of oxygen.146,266,267 As such, the development of light-activatable drugs that do not rely on the generation of ROS to elicit their activities is highly desirable and will now be discussed.There is increasing interest in the use of external stimuli, particularly light, to provide spatial and temporal control over the release of biologically active molecules in the body. One such concept involves the inactivation of drug molecules using a protecting group (a process termed “caging”),268 which can be removed upon light irradiation (“uncaging”). This concept is often referred to as “photo-triggered” release.269 With regards to cancer therapy, using this treatment strategy, drug molecules with selective activity for specific cellular targets can be released in target cells and tissues. This could represent a considerable advantage over PDT, where ROS production results in oxidative damage to numerous types of biomolecules, thereby potentially activating/altering numerous biochemical pathways within cells and tissues during treatment.
Within this area, two design strategies can be seen in the development of Ru(II) polypyridyl complexes, where the Ru(II) core is either used as the biologically active molecule or as the photo-cleavable protecting group, whose main purpose is to inactivate drug molecules. For ease of discussion these design strategies have been designated ‘case I’ and ’case II’, respectively.
Case I: two distinct methods have been employed to inactivate the biological activity of Ru(II) polypyridyl complexes: through the coordination of a ligand to the Ru(II) metal centre, whereby the bond between the Ru and the ligand is cleaved upon light activation (case Ia); or by attachment of a photo-labile protecting group to one of the ligands, which is removed upon light activation, but the coordination sphere of the Ru(II) metal centre remains unaltered (case Ib).
Case Ia: while some Ru(II) polypyridyl complexes are quite photostable, those possessing distorted octahedral geometries can undergo photo-induced ligand loss due to the population of low-lying metal-centred excited states, which possess strong antibonding character. One way that complexes have been designed to undergo efficient photo-induced ligand loss is to incorporate sterically demanding ligands into the coordination sphere of the Ru(II) metal centre. Such complexes are capable of inducing light-triggered toxicity with IC50 (light) values in the low micromolar range in cultured cell monolayers and 3D tumour spheroids, whilst displaying low toxicity in the dark.270–274 For example, when complex 66, which incorporates two bulky 6,6′-dimethyl-2,2′-bipyridine (dmb) ancillary ligands, was irradiated with visible light (7 mW cm−2) in water, the complex underwent rapid ejection of one of the dmb ligands, as shown in Scheme 1, with a t1/2 value of 275 seconds.273 Such photochemically generated bis-aqua species have been demonstrated to covalently bind to guanosine monophosphate (GMP) and plasmid DNA via substitution of the thermally labile Ru–OH2 bonds.273,275,276 This binding mode is similar to that of the anti-cancer drug cisplatin and offers the notable advantage of spatial and temporal selectivity in its activation.232 In addition to undergoing photo-induced ligand loss, complex 66 was also shown to produce 1O2 with a quantum yield of 0.42, and therefore could undergo both oxygen-dependent and oxygen-independent photo-reactivity. Interestingly, when only one thiophene unit was present in the extended ligand, only 3% 1O2 was generated by the complex upon light activation, although the rate of ligand loss was significantly higher. The complex showed little activity against HL-60 human promyelocytic leukaemia cells in the absence of light activation (IC50 (dark) = 150 μM after 80 h). However, upon irradiation with visible light (100 J cm−2) the complex became highly active, with an IC50 (light) value of 0.2 μM, corresponding to a phototoxicity index of 750.273
 | ||
| Scheme 1 Photo-triggered ligand loss in complex 66 due to the sterically crowded coordination sphere of the Ru(II) mental centre. | ||
In another study, Hufziger et al. incorporated a 6,6′-OH-bpy ligand in complex 67 (pKa1 = 5.26 and pKa2 = 7.27) so that the ligand would undergo photo-triggered release at low pH, but not at higher pH values, where both hydroxyl groups were deprotonated. It was envisioned that this pH sensitivity would enhance its selectivity for cancer cells due to the lower pH commonly found in tumour masses. Despite this, however, the complex showed low photo-induced activity against HeLa cells, with IC50 (light) values of 88 μM.212
In addition to bidentate ligands, complexes with one or more monodentate ligands can undergo photo-triggered release. Turro and co-workers showed that photo-excitation of the cyclometalated complex 58 (λex = 690 nm, 5 J cm−2) resulted in the ejection of the monodentate acetonitrile ligands. The complex was highly active against OVCAR-5 human ovary carcinoma cells in the dark, with IC50 (dark) values of 1 μM after 39 h, which was attributed to thermal ligand exchange in the presence of intracellular glutathione.277 Indeed other researchers also demonstrated similarly strong activity of complex 58 against U87 glioblastoma cells, where the complex was found to induce endoplasmic reticulum stress and apoptosis in treated cells.157 Photo-activation of the complex resulted in an enhancement in activity by a factor of 14, with IC50 (light) values of 70 nM.277 By incorporating a dppn ligand into the coordination sphere of the Ru(II) metal centre of complex 68, the same authors designed a dual-action complex, capable of 1O2 production and photo-induced ligand loss, although the quantum efficiency of ligand loss was very low (<1%). The complex showed low activity against HeLa cells in the dark (IC50 (dark) = 330 μM after >72 h); however upon activation with 466 ± 20 nm light (6.5 mW cm−2 for 20 min), the complex became highly active against cells, with an IC50 (light) value of 470 nM.278
While aquated forms of Ru(II) complexes ([Ru(L)n(OH2)n]2+) have been shown to covalently bind to and cross-link DNA in solution,233,273,275 evidence to confirm that DNA is in fact the cellular target of such complexes has yet to be reported and therefore further such studies are still required.
Case Ib: the incorporation of organic photo-labile protecting groups into biologically active molecules has seen intensive study across various fields.279–281 However, there has been relatively little investigation into controlling the biological activity of Ru(II) polypyridyl complexes with photo-labile protecting groups, which do not involve the breaking of the Ru ligand bond. One example has been reported by Gasser and co-workers, who incorporated a photo-labile 3-(4,5-dimethoxy-2-nitrophenyl)-2-butyl ester to inactivate their biologically active complex 47, as shown in Scheme 2.69 Solution studies demonstrated that upon UV-A light activation, the protecting group underwent photolytic cleavage with a quantum yield of 0.038. When HeLa cells were incubated with the inactivated complex 51, little dark activity was observed after 4 h of treatment (IC50 (dark) = >100 μM). However, upon irradiation with UV-A light (2.6 J cm−2), the activity of the complex was enhanced considerably with IC50 (light) values of 17 μM, which were similar to the IC50 (dark) values seen in cells treated with the biologically active complex 47 (16 μM).69
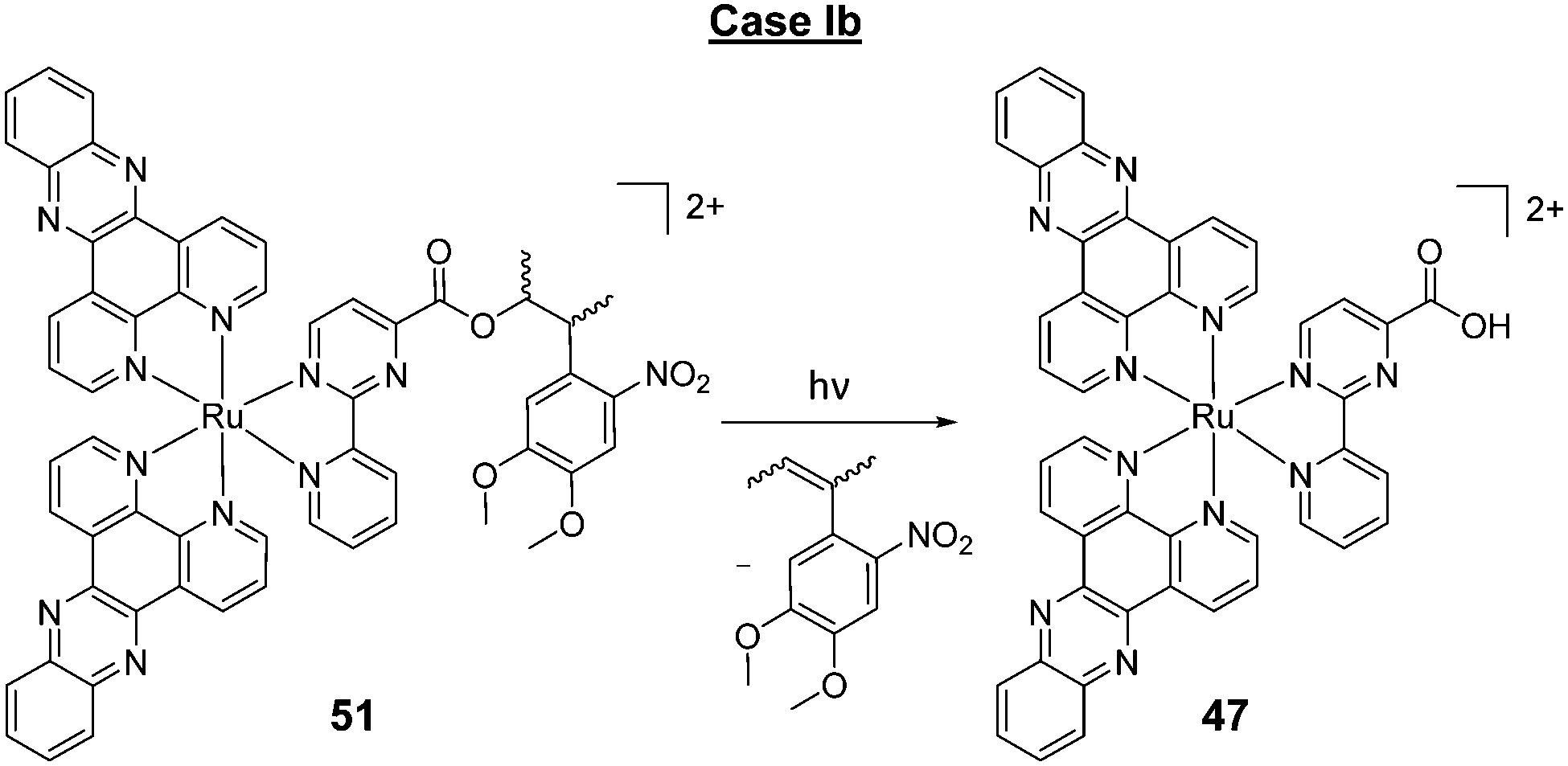 | ||
| Scheme 2 Photo-deprotection of complex 51 to yield the biologically active complex 47. | ||
Case II: a variety of biologically active molecules have been inactivated by their coordination to a Ru(II) metal centre, which may be removed upon photo-activation. These include enzyme inhibitors,282,283 neuroactive 4-aminopurine,284 nicotine;285 butylamine, tyramine, tryptamine and serotonin,286 the neurotransmitters γ-amino butyric acid (GABA)287,288 and glutamate;289,290 and the fluorescent probe Rhodamine.291 Many of these have been investigated in cells.282,285,287,288,290,291 In one such study, Fino et al. exploited two-photon photo-triggered release of glutamate from complex 69 to activate neurons in tissue samples to enable researchers to probe complicated neural circuit dynamics.287 Upon light activation the complex underwent rapid ejection of the caged glutamate molecule, as shown in Fig. 29a, with a quantum yield of ca. 0.13 in water at pH 7 and a corresponding two-photon uncaging efficiency (“functional cross-section”) of 0.14 GM at 800 nm.287,290 By using two-photon activation (λex = 800 nm) the researchers achieved significantly higher spatial resolution in the activation of 69 relative to one-photon activation, and could induce glutamate release in dendritic spines of single neurons.287
 | ||
| Fig. 29 (a) The photo-triggered release of the neurotransmitter glutamate from complex 69 upon one-photon (λex = 532 nm) and two-photon (λex = 800 nm) excitation and (b) a pyramidal neuron in which the photo-triggered release of glutamate from complex 69 was achieved by laser excitation in the regions highlighted in green. Image reproduced with permission from ref. 287. | ||
In addition to releasing inactivated drug molecules from the coordination sphere of the Ru(II) centre, this process also generates the potentially biologically active aquated Ru(II) complex, and as such, this photo-activation of the caged complex can generate two biologically active molecules: a [Ru(OH2)]2+ species and the free drug cargo, as illustrated in Fig. 30 for complex 70. Upon photo-activation of complex 70 with 400 nm light in deoxygenated water, two of the biologically active 5-cyanouracil ligands were ejected, with the concomitant generation of the bis-aqua Ru(II) species, with a quantum yield of 0.022.292,293 Complex 70 showed no dark toxicity towards HeLa cells after 2 h incubation; however, upon visible light activation for 1 h, an LC50 value of 160 μM was observed. This relatively low activity was similar to that seen for cells treated with 5-cyanouracil alone. Furthermore it was not determined whether complex 70 was internalised by cells before photo-activation, or whether the observed photo-induced activity in fact originated from extracellular complexes.292
 | ||
| Fig. 30 The photo-triggered release of the biologically active bis-aqua Ru(II) complex and two 5-cyanouracil molecules from complex 70. | ||
Another way by which the photo-reactivity of Ru(II) complexes has been exploited for light-activatable therapies is the design of complexes which possess strongly oxidising excited states, capable of photo-oxidising target biomolecules in their vicinity. As shown in Fig. 26, the radical species generated from such electron transfer reactions can interact with oxygen to generate ROS (Type I PDT mechanism) but can also undergo additional oxygen-independent radical reactions. Such reactions can result in damage to biomolecules, as well as the formation of new covalent bonds.294–297 Through the incorporation of π-deficient ligands (such as TAP) into the coordination sphere of the Ru(II) metal centre, the Kirsch-De Mesmaeker and Moucheron research groups have developed various Ru(II) polypyridyl complexes, capable of photo-oxidising and forming covalent bonds to target biomolecules for applications such as anti-cancer and anti-sense gene therapies,298,299 which will be highlighted in the next section.
This section has discussed how various properties of the excited states of Ru(II) polypyridyl complexes can be harnessed to develop new classes of photo-activatable therapeutic agents. However, the characterisation of the ultrafast processes occurring within these complexes upon absorption of light, which ultimately lead to their sought-after reactivity, has predominantly been carried out in solution.300,301 In an effort to enhance our understanding of the processes occurring within the excited states of Ru(II) complexes when internalised by cells, Dietzek and colleagues recently investigated the ultrafast dynamics of [Ru(bpy)2(dppz)]2+ within fixed HepG2 cells by transient absorption microscopy.302 Confocal microscopy studies showed the emission from the complex to originate from the nuclei of the fixed cells, as shown in Fig. 31a. Similarly, transient absorption signals (λpump = 430 nm and λprobe = 580 nm) from the complex were only detected from the nuclei of treated cells, with no signals originating from the cytoplasm, as shown in Fig. 31b–d.302 This work demonstrated for the first time that ultrafast photo-induced processes occurring in Ru(II) complexes can be investigated within cells. Moreover, such studies may enable researchers to gain a more detailed understanding of the interplay between the ultrafast dynamics within these complexes and their biological activities.
 | ||
| Fig. 31 (a) Confocal microscopy image of fixed HepG2 cells, which had been incubated with of [Ru(bpy)2(dppz)]2+ (50 μM) for 24 h prior to fixation, (b) transient absorption intensity map at a 10 ps delay of the same cells (λpump = 430 nm and λprobe = 580 nm), (c) overlay of images (a) and (b); and (d) transient absorption kinetics traces observed in the indicated positions of the cell displayed on the inset (black square indicates point 1 while the yellow dashed line denotes the nucleus). The data for the kinetics traces were recorded with λpump = 430 nm and λprobe = 580 nm. Images reproduced with permission from ref. 302. | ||
X-Ray sensitisers (radiosensitisers)
While photochemotherapy typically refers to the use of non-ionising radiation to activate a drug molecule (light in the UV to NIR regions),303 light in this spectral region has a finite penetration depth in vivo. In contrast, high-energy radiation, such as X-rays and γ-rays, has no such limit and can therefore irradiate any part of the body, regardless of tissue depth.304,305 The use of drug molecules to enhance the susceptibility of tumours to ionising radiation is clinically well established. Such drugs, termed “radiosensitisers” include platinum-containing drugs and taxanes, and this combination of chemotherapy and radiation therapy is termed “chemoradiotherapy”.306,307 In recent years, researchers have begun to evaluate the potential of Ru(II) polypyridyl complexes in such treatment regimes and have demonstrated that upon exposure to ionising radiation, a synergistic decrease in survival can be achieved in cancer cells pre-treated with Ru(II) polypyridyl complexes.64,220,308,309 For example, Gill et al. demonstrated that when HeLa cells were pre-treated with complex 71 (40 μM) for 20 h and exposed to 137Cs-γ-rays with a dose of 6 Gy, a 7-fold decrease in cancer cell survival was achieved relative to radiation alone, as shown in Fig. 32b.64 Furthermore, the complex showed low toxicity towards cells in the absence of radiation.64 In addition to acting as a radiosensitiser, the complex also displayed intrinsic anti-cancer activity. ICP-MS studies revealed that, of the total Ru content in cells, 58% was found in the cytosol and 20% in the nucleus, where it stalled DNA replication fork progression, resulting in the activation of DNA damage checkpoints and impeding cell-cycle progression. This DNA-based mechanism of action therefore imparted complex 71 with preferential activity against rapidly proliferating cancer cells over non-malignant cells, as demonstrated by its IC50 values of 38 μM and >100 μM after 24 h, in HeLa breast cancer cells and non-malignant human foreskin fibroblasts (HFFs), respectively.64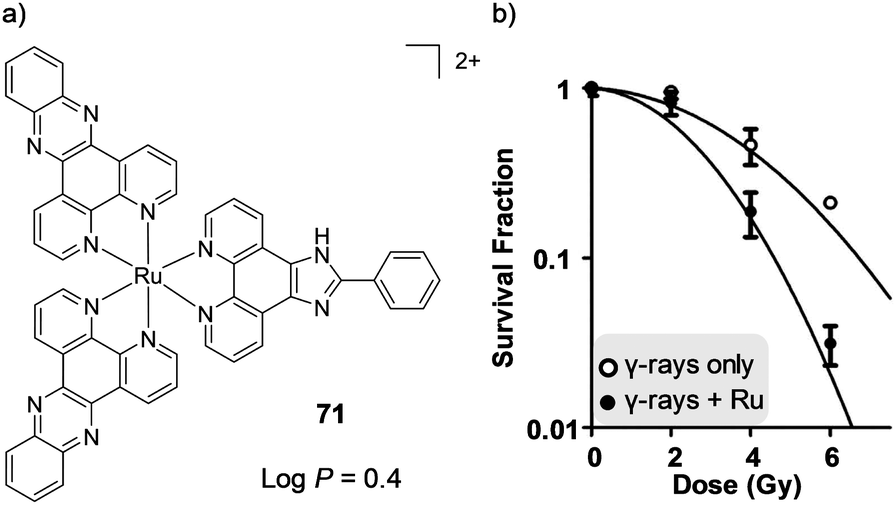 | ||
| Fig. 32 (a) Chemical structure of complex 71 and the logP value of its PF6− salt and (b) plot of the survival fraction of HeLa cells irradiated with varying doses of 137Cs-γ-rays, in the absence of complex 71 and after incubation with complex 71 (40 μM) for 20 h. Graph reproduce with permission from ref. 64. | ||
Nomenclature and terminology
This section has illustrated a diverse range of mechanisms by which Ru(II) polypyridyl complexes may elicit their biological activity upon light activation. Indeed, across numerous fields new classes of photo-activatable drug molecules with diverse mechanisms of action are being developed in tandem with new terminology. However, the majority of these terms have yet to be assigned official definitions. As such, the use of a number of these terms can be seen to vary slightly between researchers, both between different fields and within the Ru field. With this in mind, a glossary is provided below, which presents the definition of the most widely adopted terms, based, where possible, on the publications in which they were first coined.Photochemotherapy – “the interaction of light and drug that results in a beneficial effect on disease”. Parrish et al. 1974.201
Photodynamic therapy (PDT) – the therapeutic application of the photodynamic effect, i.e. “photoinduced damage requiring the simultaneous presence of light, photosensitizer and molecular oxygen”. (IUPAC).237
The term arose from mechanistic investigations by researchers between 1900 and 1907, who noted that topically applied eosin could treat skin tumours upon light activation via an oxygen-dependent mechanism, which they termed “photodynamic action”.226 Furthermore, in 1941 it was recommended that the term “photodynamic” only be applied to photosensitised reaction in biological systems in which oxygen is consumed.310,311
Type I PDT: radical-mediated photo-oxidative damage involving hydrogen or electron transfer reactions.
Type II PDT: 1O2-mediated photo-oxidative damage involves energy transfer.
Type III PDT: this term is not universally adopted and has multiple reported definitions making it an ambiguous term. These definitions include:
(i) The interaction between excited triplet photosensitisers and native free radicals in tumours.312–314
(ii) The ejection of a hydroxyl radical directly from a photo-excited bacteriochlorophyll. The term was introduced by Scherz et al. as the authors noted that this process did not fall under either the Type I or Type II mechanisms.315
(iii) The direct reaction of a photo-excited molecule with a biomolecule, resulting in the destruction of the photo-excited molecule and damage to the biomolecule. This mechanism is highlighted as likely to be oxygen-independent in nature.316
(iv) To denote that the photo-reaction(s) does not involve oxygen.317,318
However, based on the established definition of PDT, wherein the presence of oxygen is a critical component, one might ask whether oxygen-independent mechanisms can in fact be classified as PDT, under its current definition.
Photoactivated chemotherapy (PACT) – exploiting the deactivation pathways of photo-excited transition metal complexes (both photophysical and photochemical), for therapeutic application. These mechanisms of action can be oxygen-dependent and/or oxygen-independent in nature. Sadler and co-workers, 2009.227
It should be noted that the acronym PACT is also used for the term ‘photodynamic antimicrobial chemotherapy’.
Photo-triggered release – the release of a chemical species (such as a drug molecule) by light-induced cleavage of a photo-labile protecting group.
Caged – a chemical species (such as a drug molecule) whose activity has been “switched-off” through the attachment of a photo-labile protecting group. Kaplan, 1978.268
Ru(II)-Conjugates
The ability to selectively target specific cell types and molecular targets, and control the subcellular localisation of a compound is of considerable interest to researchers when designing new imaging and therapeutic agents. One strategy that has shown great promise in enhancing these capabilities has been the conjugation of Ru(II) polypyridyl complexes to targeting molecules. In addition, the conjugation of functional molecules to Ru(II) complexes, can impart new advantageous properties to the resulting systems. This section highlights various types of conjugates that have been developed and assessed in cells and in vivo to date.Ru–Nucleotide conjugates
There has been considerable research into the development of oligonucleotides and their analogues as therapeutic agents for the treatment of various diseases.319–321 The most famous mechanism of action through which they exert their therapeutic effect is antisense therapy. This involves the binding of complementary engineered oligonucleotides to a specific target messenger RNA (mRNA) sequence, through base pairing, to prevent translation of the mRNA into a protein; thereby silencing the gene from which the mRNA molecule was transcribed.322Reschner et al. conjugated a Ru(II)–TAP complex to a 21mer antisense oligonucleotide, targeting an E6 oncogene which is associated with the Human Papillomaviruses (HPV)-linked cancers.299 The researchers had previously demonstrated that Ru(II) complexes incorporating at least two π-deficient TAP ligands, could photo-oxidise guanine and form covalent photo-adducts with the guanine nucleobase of DNA, as shown in Fig. 33a.323–328 Indeed, solution studies demonstrated that conjugate 72 (illustrated in Fig. 33b) formed covalent photo-adducts with target sequences upon light activation and was therefore anticipated to exhibit photo-induced gene silencing in cells. Furthermore, in the absence of a target sequence, the [Ru(TAP)2]2+ core formed intramolecular photo-adducts, as illustrated in Fig. 33c, thereby neutralising the photo-reactivity of the conjugate and preventing off-target reactions. The biological activity of 72 was assessed in HPV16-infected SiHa human cervical cancer cells. Cells were incubated overnight with conjugate 72 (200 nM) in the presence of a transfection regent, to promote internalisation of the conjugate, washed and illuminated with 380–480 nm light for 2.5 h. After 24 h, cells were shown to undergo a selective repression of E6 at the protein level, reactivation of p53 tumour suppressor protein expression, and a reduced cell proliferation of 52%.299
 | ||
| Fig. 33 (a) Chemical structure of the photo-adduct of [Ru(TAP)2(phen)]2+ with GMP, (b) chemical structure of the [Ru(TAP)2(phen)]2+-oligonucleotide conjugate 72 and (c) schematic representation of the behaviour of conjugate 72 (coloured purple) in the absence and presence of a complementary target strand (coloured green). Figure adapted from ref. 299. | ||
There are, however, a number of challenges faced when using nucleotide conjugates, which include poor cellular uptake and their degradation by nucleases within cells. To overcome such issues, researchers have developed nucleotide analogues such as pyrrole-imidazole polyamides and peptide nucleic acids (PNAs) for antisense applications.329–338 However, while the synthesis of a number of Ru(II)–PNA conjugates has been reported, evaluation of their application in cells has yet to be investigated.339–341
Peptide conjugates
Peptide conjugation has found various applications in chemical biology and drug development. Conjugation can impart water solubility to hydrophobic molecules, enhance cellular uptake, act as targeting motifs and improve biocompatibility to reduce toxicity.342,343 With regards to Ru(II) polypyridyl complexes, conjugation of peptides to complexes has been used effectively to:(1) Target receptors expressed on the cell membrane of particular cell types.183,344,345
These peptides include bombesin,183 arginylglycylaspartic acid (RGD)344 and somatostatin peptides.346 These peptide conjugates showed high affinities for their molecular targets and in a number of cases, the conjugates were shown to be predominantly internalised by the target cell types, relative to other cell types.
Weil and co-workers reported the first receptor-targeting Ru(II)–peptide conjugate for use as a PDT agent.346 The peptide hormone somatostatin was conjugated to a [Ru(bpy)3]2+ core (shown in Fig. 34) with the aim of increasing the selectivity of the resulting Ru(II)-conjugate 73 for cancer cells that overexpress the somatostatin receptors; thereby minimising collateral damage to non-cancerous cells in the vicinity during PDT.346 Peptide conjugation resulted in a significant enhancement in uptake by A549 cells, which expressed the receptor, and uptake was found to occur by receptor-mediated endocytosis. The conjugate showed low dark toxicity towards cells (IC50 (dark) = 300 μM), however upon activation with 470 nm light (7 J cm−2), the complexes were highly active against cells, with IC50 (light) values of 13 μM.346
 | ||
| Fig. 34 The structures of complex 73 and 74. The sequence of the somatostatin peptide denoted in blue from the N- to C-terminus is Ala-Gly-Cys-Lys-Asn-Phe-Phe-Trp-Lys-Thr-Phe-Thr-Ser-Cys. The TAT peptide sequence is Gly-Arg-Lys-Lys-Arg-Arg-Gln-Arg-Arg-Arg-NH2. | ||
(2) Enhance cellular uptake of Ru(II) complexes.
This has been achieved either by triggering receptor-mediated endocytosis346 or using cell penetrating peptides347–350 These are short amphiphilic and cationic peptides (typically below 30 amino acid residues in length) that are rapidly internalised by cells.349,351 Examples of these include oligoarginine and the trans-activating transcriptional (TAT) protein of HIV-1. The mechanisms of uptake of cell penetrating peptides are still a subject of investigation, with both energy-independent and energy-dependent processes having been reported.347–349 These peptides have been exploited for the delivery of various types of molecular cargo into cells, such as imaging agents, nucleic acids, drugs, nanoparticles,352 and are now being evaluated for use with Ru(II) complexes.298,353–356 For example, Kirsch-De Mesmaeker and co-workers conjugated the cell penetrating TAT peptide (sequence GRKKRRQRRR) to a bis(TAP) Ru(II) complex (conjugate 74 in Fig. 34) for application as a photo-activated therapeutic agent.298 In the absence of the peptide, the complex was unable to enter cells; however upon incorporation of the peptide, the conjugate was readily internalised by HeLa cells. Solution studies showed that upon light activation of conjugate 74 in the presence of guanine-containing DNA, the complex formed covalent bonds with the guanine residue and was therefore anticipated to exhibit photo-toxicity in cells. However, the conjugate showed no photo-induced toxicity towards cells,298 in contrast to the bis(TAP) complexes 2 and 59, which have been shown to be highly active against HeLa cells when irradiated with visible light.46 The authors attributed the lack of activity of conjugate 74 to its subcellular localisation. The conjugate was found predominantly in endosomal and endoplasmic reticulum vesicles, and therefore had no access to its intended DNA or RNA molecular targets, which highlighted the importance of the subcellular localisation of this class of photo-activated therapeutic.258,259,298
While conjugation to the TAT cell penetrating peptide did not afford nuclear localisation, it is interesting to note that in addition to facilitating cell uptake, cell penetrating peptides that are rich in positively charged amino acid residues, such as arginine or lysine, can promote nuclear localisation via active transport through the nuclear pore complex.359
(3) Control the intracellular localisation of the Ru(II) complexes.
Within cells, specific signalling molecules such as peptides of a particular sequence are utilised to ‘tag’ molecules and mediate their delivery to a target organelle.360,361 By conjugating peptides of the appropriate sequence to Ru(II) complexes, researchers have succeeded in controlling the localisation of the resulting conjugates within cells. The Keyes research group have been particularly active in this area and showed that conjugation of a nuclear localisation signal (VQRKRQKLMP-NH2) derived from the transcription factor NF-κB,362 facilitated the nuclear localisation of Ru(II) complexes, which in the absence of the peptide had been either unable to enter live cells or had otherwise localised in the cytoplasm.363
In another study, two cell-impermeable complexes were conjugated to a mitochondrial penetrating peptide (FrFKFrFK-CONH2, where r = D-arginine) and the resulting dinuclear conjugate 75 was readily internalised by HeLa cells by an energy-dependent mechanism and selectively localised in the mitochondria, as shown in Fig. 35a. Furthermore, the conjugates could be used to monitor dynamic changes in oxygen and ROS levels within the mitochondria of live HeLa cells using fluorescence lifetime imaging.357
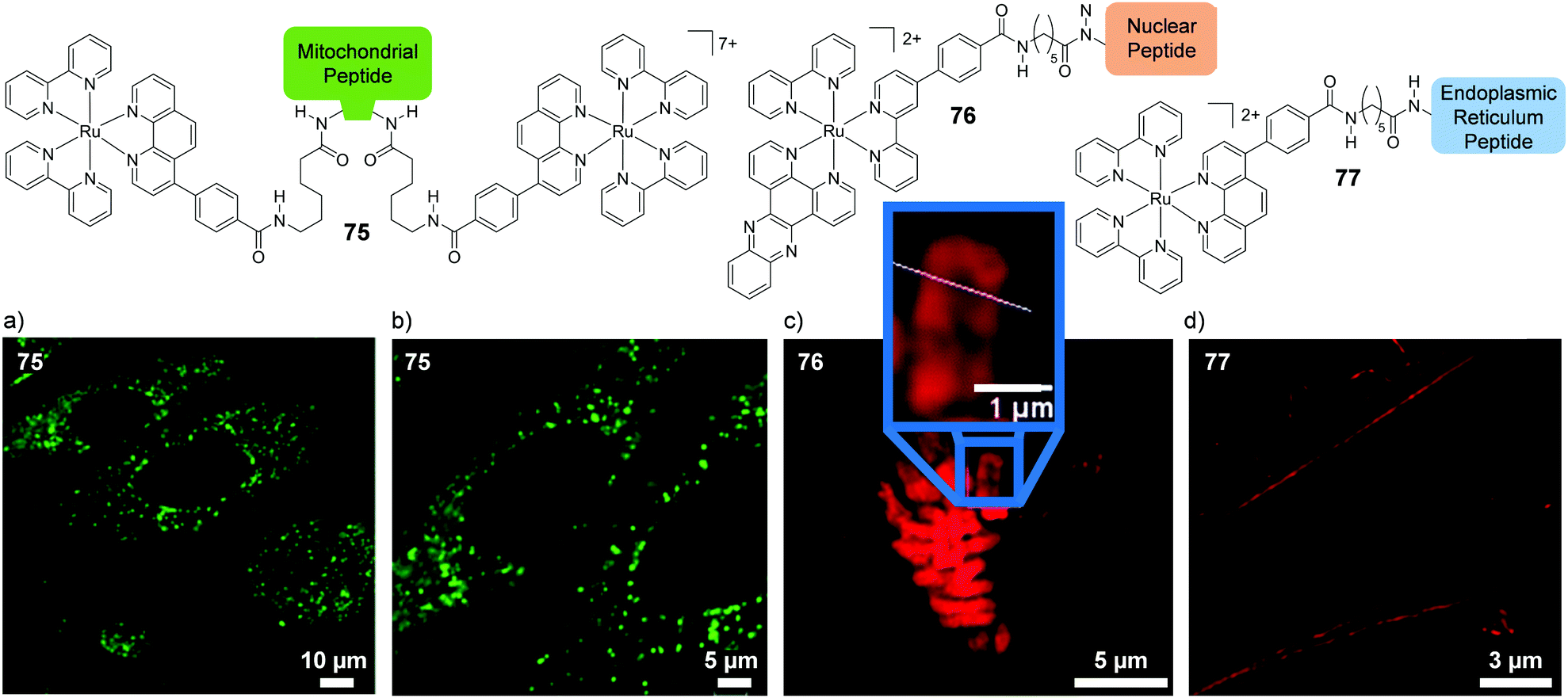 | ||
| Fig. 35 Structures of the Ru(II)-conjugates 75, 76 and 77 and (a and b) the confocal microscopy images of HeLa cells incubated with conjugate 75 (75 μM, for 2 h, Ru emission coloured green). The time-gated STED microscopy images of (c) 76 bound to chromosomal DNA in the nucleus of a HeLa cell during metaphase (cells were incubated with 76 (40 μM) for 24 h) and (d) HeLa cells fixed after incubation with 77 (70 μM) for 4 h. The mitochondrial targeting sequence is Phe-(D-Arg)-Phe-Lys-Phe-(D-Arg)-Phe-Lys-CONH2, the nuclear targeting sequence is Val-Gln-Arg-Lys-Arg-Gln-Lys-Leu-Met-Pro and the ER targeting sequence is Arg-Gln-Ile-Lys-Ile-Trp-Phe-Gln-Asn-Arg-Arg-Met-Lys-Trp-Lys-Lys. Images reproduced with permission from ref. 357 and 358. | ||
In a more recent study, Ru(II)–peptide conjugates were utilised as luminescent stains for the imaging of DNA and the endoplasmic reticulum by stimulated emission depletion (STED) microscopy.358 STED is an ultra-resolution microscopic technique in which resolution below the diffraction limit can be achieved. This technique has allowed researchers to probe the microstructures within cells in unprecedented detail, with resolution below 50 nm reported.364–366 The nuclear stain (conjugate 76) consisted of a DNA “light-switch” complex, incorporating the dppz ligand, conjugated to the aforementioned nuclear localisation signal (VQRKRQKLMP-NH2), whereas the endoplasmic reticulum stain (conjugate 77), incorporated a [Ru(bpy)2]2+ core conjugated to an endoplasmic reticulum targeting peptide (RQIKIWFQNRRMKWKK), as shown in Fig. 35. Confocal microscopy studies demonstrated that both 76 and 77 selectively stained their target organelles in live HeLa cells. Subsequent STED microscopic studies of conjugate 76 in live HeLa cells revealed high resolution and high contrast images of the chromosomes within the nucleus and in the case of conjugate 77, detailed features of the tubular endoplasmic reticulum structure within fixed HeLa cells were visualised, as shown in Fig. 35c and d, respectively.
Both complexes exhibited excellent photostability under the high intensity of the STED lasers and due to their long luminescence lifetimes, the stains could be used for time-gated STED spectroscopy, which reduced background signals from the samples thereby achieving higher image resolution.358 This represented the first example of luminescent metal complexes for use in STED microscopy and highlighted the excellent suitability of Ru(II) polypyridyl complexes as imaging agents for this emerging microscopic technique.
While peptide conjugation has been demonstrated to result in the successful targeting of specific cell types, enhance the uptake of Ru(II) polypyridyl complexes and control their intracellular distribution within cells, a study conducted by the Gasser research group highlighted some of the challenges that can be encountered with such systems. They investigated the influence of peptide conjugation on the intracellular localisation of their bis-dppz complex 47, which had been shown to localise predominantly in the mitochondria of HeLa cells, as shown in Fig. 36a.59 The researchers found that conjugating the complex to either a nuclear or mitochondrial targeting sequence (-RRRK and -RFK, respectively) resulted in both conjugates localising in the nucleoli and cytoplasm of HeLa cell.183 In addition, the authors found that the nature of the spacer between the Ru(II) complex and the peptide had a dramatic effect on the uptake and localisation of the conjugates. When a peptide of random sequence (GPGVK) was directly attached to complex 47, the resulting conjugate was not internalised into cells. In contrast, when a flexible hexyl spacer was inserted between the peptide and complex, the resulting conjugate (78) was internalised into cells and predominantly localised in the nucleus, as shown in Fig. 36b.183 This suggests that the nature of the linker between the Ru(II) complexes and targeting peptides can have a significant impact on their successful use in cellular studies. It should be noted however that this study used fixed cells, which has been reported to possibly alter peptide uptake and localisation,349,351,367 thereby potentially comprising the intracellular location of the conjugates.
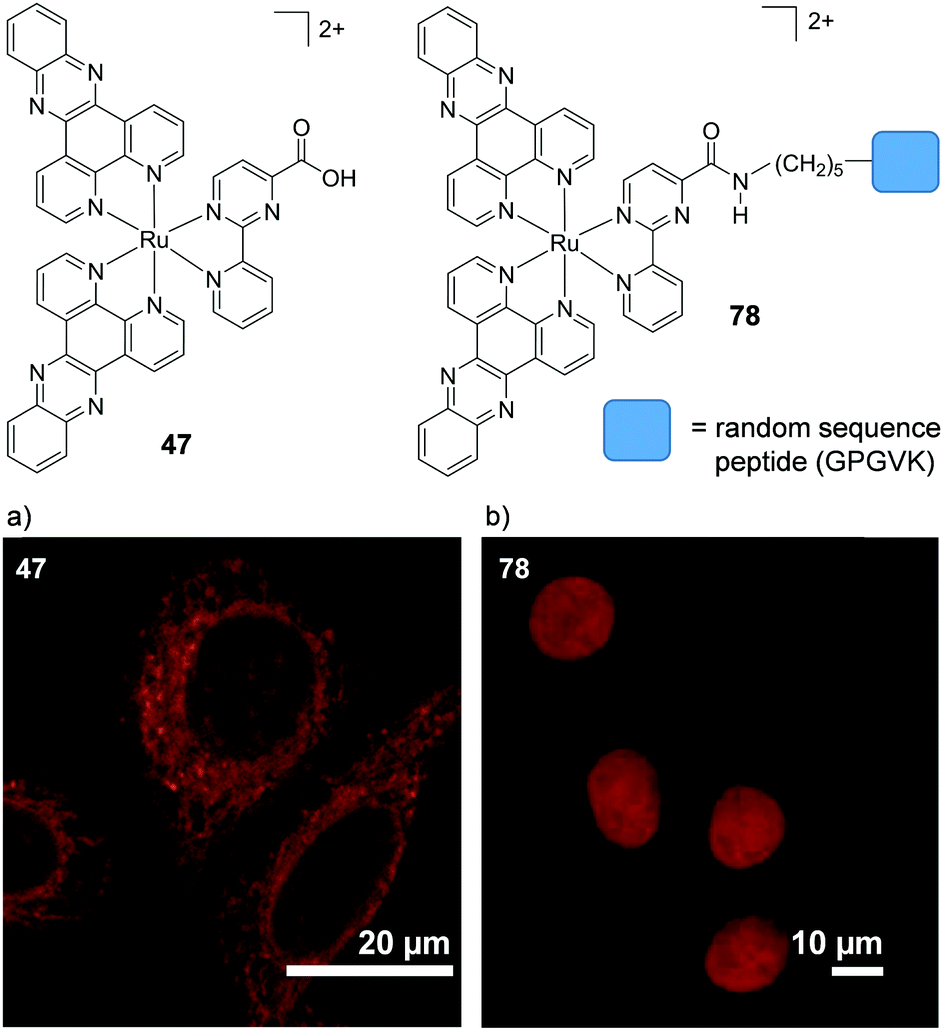 | ||
| Fig. 36 Structures of complexes 47 and 78 and confocal microscopy images of HeLa cells fixed after incubation with (a) complex 47 (20 μM) for 2 h and (b) complex 78 (100 μM) for 2 h. Images reproduced with permission from ref. 59 and 183. | ||
It is interesting to note that the targeting Ru(II)-conjugates reported by the Keyes group, such as those shown in Fig. 35, also contained flexible spacers between the peptides and Ru(II) complexes.357,358,363 Indeed the importance of the nature of the linker between peptides and their molecular cargo has been highlighted in various other systems.368–370
While the conjugates presented thus far have incorporated short oligopeptides, Weil and co-workers recently developed a ca. 67 kDa polypeptide conjugate for photodynamic applications.371 The macromolecular conjugate (79), illustrated in Fig. 37a, consisted of a modified polycationic human serum albumin (HSA) protein, in which ca. 10 of its exposed tyrosine residues were conjugated to a Ru(II) polypyridyl complex, as depicted in Fig. 37b. In addition, lipophilic triphenylphosphonium cations were conjugated to the protein to achieve mitochondrial targeting and poly(ethylene oxide) side groups were incorporated into the protein to impart water solubility and reduce any non-specific interactions. The researchers found that protein conjugation significantly improved the photophysical properties of the Ru(II) complex and enhance its 1O2 quantum yields, compared to the complex alone;371 similar to the observations previously discussed for the Ru(II) complex TLD1433 when bound non-covalently to the transferrin glycoprotein.253 As anticipated, conjugate 79 was found to localise predominantly in the mitochondria of HeLa cells. The conjugate showed little activity against cells in the dark, however upon light activation, the conjugate was highly active, with IC50 (light) values of 35 nM in HeLa cells which had been incubated with 79 for 4 h and irradiated with 470 nm light (5 min, 20 mW cm−2). In addition, conjugate 79 had a two-photon absorption cross-section of 50 GM at ca. 800 nm, which was noted to permit the use of light at longer wavelengths to excited the conjugate for future possible in vivo applications.371
 | ||
| Fig. 37 (a) Schematic representation of the macromolecular conjugate 79 consisting of a modified polycationic HSA protein covalently functionalised with Ru(II) complexes (red), triphenylphosphonium cations (green) and poly(ethylene oxide) side groups (grey). (b) Illustration of the chemical structure of part of the HSA polypeptide sequence (purple). HSA structure obtained from the PDB (code 1BKE)372 and the figure was modified with permission from ref. 371. | ||
Ru–Carbohydrate conjugates (glycoconjugates)
Similarly to peptides, carbohydrates can enhance water solubility and increase the biocompatibility of drug molecules. In addition, they can also target specific carbohydrate-binding proteins (lectins) on the surface of cells.375 However, to date, there have been relatively few investigations into the biological application of Ru(II) glycoconjugates.In 2010, researchers examined the cellular uptake of a [Ru(bpy)3]2+ complex incorporating either D-glucose, D-galactose or D-mannose thioglycosides (conjugates 80–82, respectively), in HepG2 cells, as shown in Fig. 38.373 The glycoconjugates showed slow uptake by cells, requiring incubation times of 24 h and, of the sugars investigated, the glucose-substituted conjugate showed the greatest uptake, with the galactose- and mannose-conjugates showing lower uptake and a different cellular staining pattern in formalin-fixed cells, as seen in Fig. 38a and b. The authors postulated that uptake of the conjugates occurred by an endocytic mechanism as opposed to via a transporter, however, they were unable to conduct experiments to prove this.
 | ||
| Fig. 38 Chemical structure of glycoconjugates 80–84 and confocal microscopy images of (a and b) formalin-fixed HepG2 cells after incubation with each conjugate 80 and 81, respectively, (500 μM) for 24 h and (c and d) paraformaldehyde-fixed A2780 cells after incubation with conjugate 83 and 84, respectively, (100 μM) for 3 h in serum-free medium. Ru emission is shown in red and nuclear stain emission is shown in blue. Images reproduced with permission from ref. 373 and 374. | ||
More recently, researchers examined the anti-cancer activity of two Ru(II)–Au(I) complexes conjugated to acetylated thioglucose (conjugates 83 and 84, shown in Fig. 38).374 The biological activities of both complexes increased upon incorporation of the sugar moiety and showed similar activities to cisplatin against the A2780 ovarian carcinoma cell line with IC50 (dark) values of 1 and 3 μM, for 83 and 84, respectively. Mechanistic studies revealed that the conjugates were internalised into cells via an active or facilitated transport mechanism and that the glucose transporter GluT-1 was not involved in their uptake. The conjugates showed quite different localisation in paraformaldehyde-fixed A2780 cells, with 83 distributing throughout the cytoplasm, while 84 accumulated in the nuclei of cells, as shown in Fig. 38c and d.
In another study, researchers examined the influence of the chirality of a glucose moiety (L- and D-) on the uptake and localisation of the Ru(dppn) complex (conjugate 85 shown in Scheme 3).376 The conjugate showed very weak emission and no difference was seen in the localisation of the L- and D-conjugates, with both found to localise in the mitochondria of A549 and MCF-7 cancer cells. In contrast, a difference was observed between the activities of the L- and D-conjugates, whereby the D-conjugate was twice as active against cells in the dark, with effective concentrations 50 (EC50) values of 19 and 50 μM, respectively, in A549 cells after 24 h. The conjugates were designed for dual-action photoactivated chemotherapy, whereby in the presence of oxygen the complex generated 1O2 with a quantum yield of 0.7, whereas in the absence of oxygen in water, the complex was shown to undergo photo-cleavage of the thioether linkage with a quantum yield of 0.001 to yield a Ru-aqua species. Indeed, the activity of both conjugates significantly increased upon activation with 454 nm light (3.1 J cm−2), with EC50 (light) values below 1 μM in both cell lines.376
 | ||
| Scheme 3 Photo-triggered loss of the D-glucose thioether ligand from conjugate 85 upon light activation. | ||
Ru–Lipid conjugates
As shown in this review, modulation of the lipophilicity of Ru(II) polypyridyl complexes has had a considerable impact on the uptake and localisation of the resulting complexes. One way to increase this is through the conjugation of lipid molecules to complexes. The biological evaluation of three such conjugates have been reported. In the first study, a [Ru(bpy)2]2+ core was conjugated to a derivative of the natural lipid squalene (conjugate 86), which was formulated as nanoassemblies of ca. 300 nm diameter in water. Biological assessment showed the conjugate to be non-toxic (IC50 > 100 μM) and it displayed intense nuclear staining in acetone:methanol-fixed HT-29 cells, as shown in Fig. 39a. This was in contrast to its squalene-free derivative, which showed only weak luminescence inside fixed cells, suggesting poor uptake of the un-conjugated complex. Interestingly, in fixed MCF-7 cells, conjugate 86 was found to localise mainly in the cytoplasm (Fig. 39b). However, no live cell imaging was conducted in the study to confirm whether the conjugate was internalised by live cells or entered cells upon chemical fixation. Co-nanoassembly with squalene derivatives of the anti-cancer drugs paclitaxel and gemcitabine was also successfully demonstrated with Ru:drug ratios of 1:20 and 1:10 and diameters of 162 nm and 226 nm, respectively. However, the biological evaluation of these Ru:drug nanoassemblies remains to be investigated.377
 | ||
| Fig. 39 Chemical structure of conjugate 86 and the confocal microscopy images of acetone:methanol-fixed (a) HT-29 and (b) MCF-7 cells after incubation with 86 (50 μM) for 2 h. Images reproduced with permission from ref. 377. | ||
Two studies examined the activity of two steroid-based Ru(II)-conjugates. In one study, researchers linked estradiol moieties to various Ru(II) polypyridyl complexes (such as conjugate 87, shown in Fig. 40) to target the oestrogen receptor-α of cells, for which estradiol is known to have a high binding affinity.378,379 The conjugate showed low activity against HeLa cells (IC50 (dark) = 166 μM after 48 h) and was found to localise in the cytoplasm of treated cells, as shown in Fig. 40. In addition, temperature-dependent uptake studies revealed that the complex was internalised by an energy-dependent mechanism.378
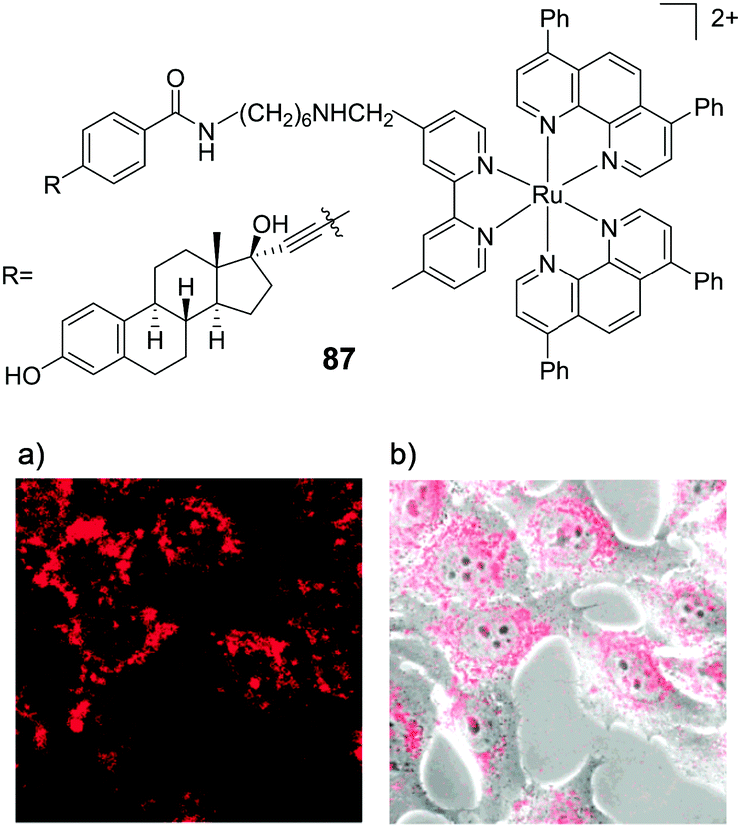 | ||
| Fig. 40 Chemical structure of conjugate 87 and the confocal microscopy images of HeLa cells incubated with conjugate 87 (5 μM) for 24 h, showing (a) the Ru emission and (b) overlay of Ru emission and bright field image. Images reproduced with permission from ref. 378. | ||
In another study, researchers conjugated cholesterol to a Ru(II) complex via a photo-cleavable Ru–S bond, as shown in Scheme 4, to develop a new photoactivated chemotherapeutic, akin to their glucose-based system (conjugate 85) shown previously.380 Interestingly, however, the Ru(II)–cholesterol conjugate (88) was highly active against cells in the dark, with EC50 values of between 5–6 μM against various cancer cell lines. The researchers found that the conjugate formed aggregates at concentrations above 3 μM, and when cells were incubated above this concentration the aggregates extracted lipids and proteins from the membranes of cells, in the same fashion as detergents, resulting in the permeabilisation of cells and subsequently necrosis. At concentrations below 3 μM, the conjugates existed as monomers and were found to insert into the membrane of cells.
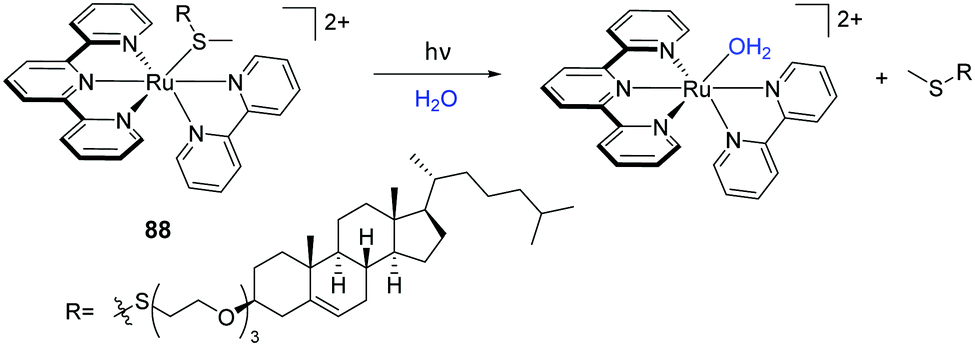 | ||
| Scheme 4 Photo-triggered loss of the thioether–cholesterol ligand from conjugate 88 upon light activation. | ||
This highlighted the care that must be taken when working with such amphiphilic conjugates in cells.380 The authors also found that the time after which cells were irradiated with light was critical to the observation of any light-induced enhancement in biological activity. Upon light activation of conjugate 88 with 455 nm light (6.3 J cm−2) after 6 h incubation with A549 cells, no enhancement in activity was observed; whereas after 24 h incubation, light activation did result in an enhancement in the activity of the conjugates. This time-dependence was attributed to the orientation of the conjugate in the membrane of cells. After 6 h, the cholesterol moiety of conjugate 88 was proposed to insert into the outer membrane of cells with the Ru(II)-centre facing into the extracellular medium. As such, the photo-generated Ru-aqua species was released outside of the cell, which was shown by ICP-MS to be unable to enter cells and therefore exerted no toxicity. However, at longer times (24 h), conjugate 88 was proposed to flip into the inner leaflet of the cell membrane. As such, subsequent light activation released the Ru-aqua species inside the cells, where it could interact with its intracellular molecular targets.380
Ru–Drug conjugates
Ru(II) polypyridyl compounds have also been complexed with suberanilohydroxamic acid (SAHA), a histone deacetylase inhibitor (FDA approved for the treatment of patients with persistent or recurrent cutaneous T-cell lymphoma).382,383 In 2013, Ye et al. complexed SAHA to a number of Ru(II) polypyridyl conjugated to SAHA, in which the surface area of the ancillary ligands was incrementally increased, as shown in Fig. 41.381 Conjugates 89 and 90 were inactive against cells (IC50 values >100 μM) most likely due to their low cellular uptake. In contrast, conjugate 91, which incorporated more lipophilic dip ancillary ligands, was rapidly taken into cells and showed high activity across a range of cell lines, with IC50 values in the 1–2 μM range. Moreover, it showed preferential activity for cancer over normal cells. Interestingly 91 was found to localise in the cytoplasm of live HeLa cells, as shown in Fig. 41a. However, when cells were fixed with formaldehyde, the conjugates were found to relocate to the nuclei of fixed cells, as shown in Fig. 41b. When compared to SAHA alone, conjugate 91 induced apoptosis in cells through the induction of mitochondrial dysfunction and the generation of ROS.381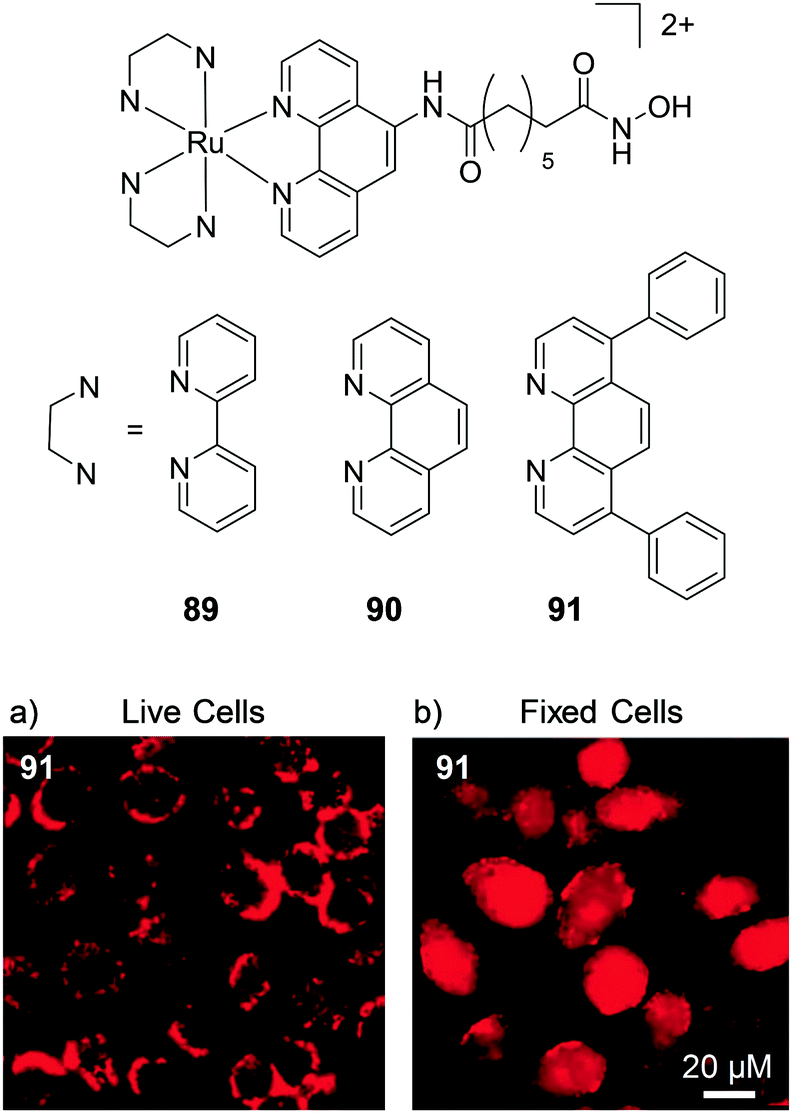 | ||
| Fig. 41 Chemical structures of conjugates 89–91 and the confocal microscopy images of (a) live and (b) formaldehyde-fixed HeLa cells after incubation with conjugate 91 (12 μM) for 3 h. Images reproduced with permission from ref. 381. | ||
In another study, Ru(II) polypyridyl complexes were conjugated to 4-anilinoquinazolines, molecules known to inhibit the epidermal growth factor receptor, which is overexpressed in a number of cancer cell types and therefore represents a therapeutic target in cancer therapies.384–386 The resulting conjugates were found to be active against a series of cancer cell lines overexpressing the epidermal growth factor receptor, with IC50 values of between 9–50 μM after 48 h incubation. However, of the series of conjugates synthesised, only one conjugate (92) showed greater anti-cancer activity than the 4-anilinoquinazoline ligand alone.384
While the previous studies used drug conjugation to enhance the biological activity of the Ru(II) complex, Lee et al. conjugated a [Ru(bpy)3]2+ core to a peptoid drug to impart photo-activity to their drug molecule, to develop a new class of reagent for the targeted, light-mediated inactivation of proteins.387 Peptoids are peptidomimetic oligomers and are composed of N-substituted glycine units.388 The peptoid drug used in this study, GU40C, targeted the extracellular domain of the vascular endothelial growth factor receptor 2 (VEGFR2). This receptor is important in the formation of new blood vessels (angiogenesis) and is often deregulated in tumour cells, making it an important target for anti-angiogenic therapy in cancer treatments.389,390 Upon visible light irradiation, inactivation of the VEGFR2 in cultured endothelial cells by the Ru(II)–peptoid conjugate 93 increased 800-fold, with an inhibitory IC50 (dark) value of 49 μM and IC50 (light) value of 59 nM. This light-enhanced inhibitory activity was largely attributed to the generation of 1O2 by the photo-excited [Ru(bpy)3]2+ core.387
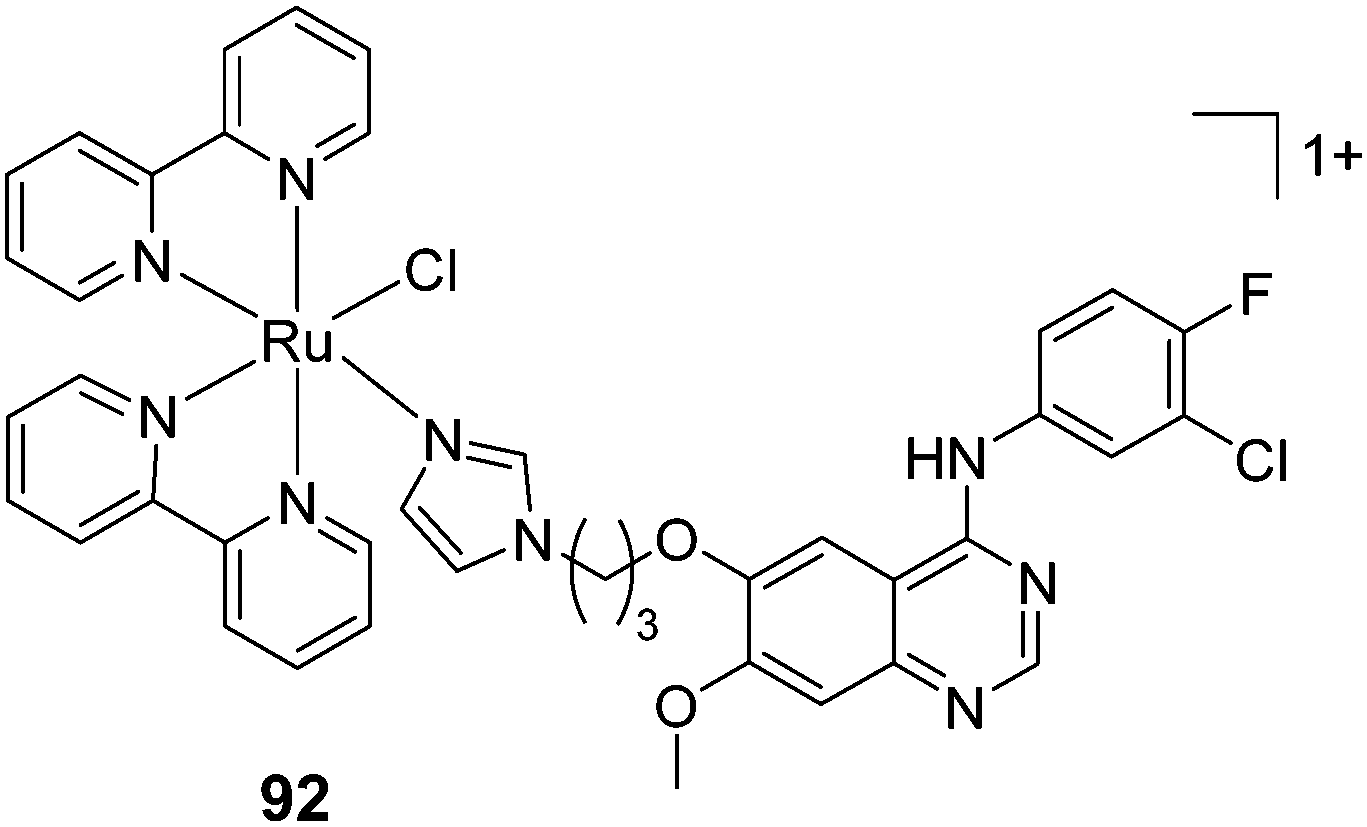
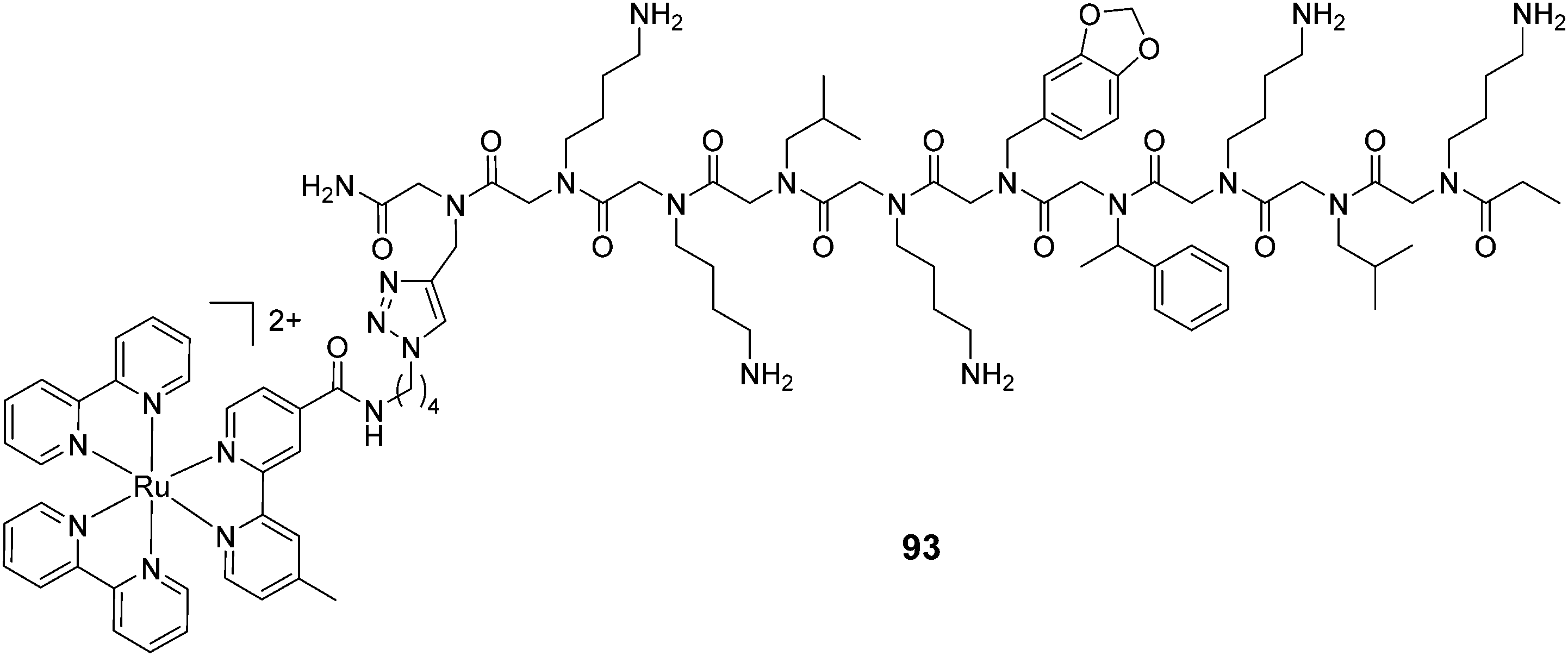
Another way by which Ru(II) complexes can impart photo-activity to clinically used drug molecules is to inactivate the biological activity of the drug molecules by coordinating them to the Ru metal centre. As illustrated in Fig. 29 in the previous section, upon irradiation with light, the drug molecule is released and its biological activity is restored in a spatially and temporally controlled manner.233,269
Ru–Porphyrin conjugates
Porphyrins have long been the established frontline drug candidates for PDT. Porphyrin and Ru–arene compounds have previously been the focus of an in-depth review.231 In contrast, this section centres on porphyrin and Ru(II) polypyridyl compounds. These compounds are amphiphilic with hydrophobic porphyrin and hydrophilic Ru(II) cores, which can improve their water solubility. In recent years, Wong and colleagues have published a number of papers on a series of such Ru(II)–porphyrin conjugates, using free bases and metalated porphyrins containing Zn(II).391–396 The conjugates analysed include Zn(II)-94 where the Ru(II) and porphyrin moieties were joined via an acetylide linker;39395–97 as the free base and Zn(II)-metalates joined by a variety of linkers,395 where the linker originates at the meso-position of the porphyrin ring, as shown in Fig. 42. In addition, conjugates with the linker at the at the β-position of the porphyrin ring have been integrated, such as conjugates 98 (as the free base and Zn(II) metalate) linked via an acetylide linker394 and 99, in which the Ru(II) moiety is linked via a Schiff base linkage, the structures of which are shown in Fig. 42.396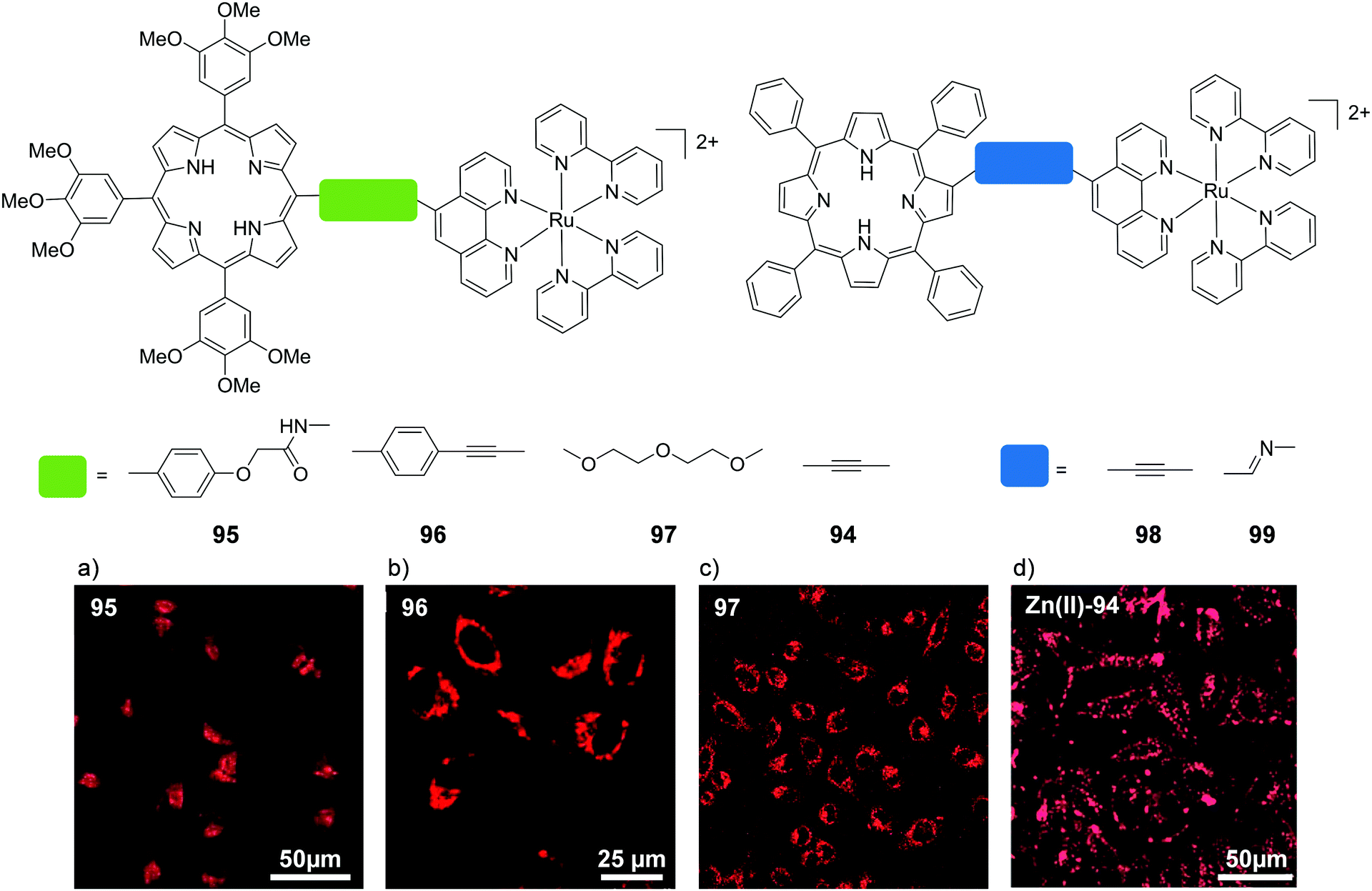 | ||
| Fig. 42 Two-photon confocal microscopy images (λex = 850 or 860 nm) of HeLa cells after incubation with (a) 95, (b) 96, (c) 97 (2 μM) for 6 h and (d) Zn(II)-94 (50 μM) for 1 h. Images reproduced with permission from ref. 395 and 393. | ||
All of the Ru(II)–porphyrin conjugates listed above showed high singlet oxygen quantum yields and were successfully imaged by two-photon microscopy. The conjugates were rapidly internalised into cells and localised in the cytoplasm. Whilst some compounds were not analysed further for subcellular localisation, 95 was found to localise predominantly in the lysosomes of human nasopharyngeal carcinoma HK-1 and HeLa cells.395 Zn(II)–94 and 96 localised in the mitochondria and 97 did not localise in any tested organelle in HeLa cells and appeared to simply localise in the cytoplasm.393,395 Similarly, as expected for porphyrin complexes, all compounds displayed potent light-activated toxicity with IC50 values of between 1–4 μM, depending on the intensity of light used to activate the conjugates. Furthermore, a number of the conjugates exhibited low dark toxicity, with IC50 values of >8 to >250 μM reported. Two-photon induced toxicity was also confirmed by microscopy. The authors found no direct link between the toxicities of the conjugates and either their rates of cellular uptake or their 1O2 quantum yields and instead attributed the differences in their toxicities to differences in their subcellular localisation.395 They speculated that photoexcitation of 96 produces mitochondrial 1O2-mediated oxidative stress which can readily elicit apoptotic cell death. Photo-excitation of 95 produces 1O2 that can damage the lysosomal membrane, causing the release of the lysosomal enzymes which leads to necrosis; while photo-excited 97, which is more hydrophilic, produces 1O2 in the cytosol where a considerable proportion of the 1O2 produced could become deactivated before inducing cell death.395
Ru–Naphthalimide conjugates
Within the Gunnlaugsson research group the potential of Ru(II) complexes conjugated to 1,8-napthalimedes for use in cells has been investigated. 1,8-Napthalimedes possess rich photophysical properties, which has spurred on investigation into their use as imaging agents.399,400 Moreover, a number of these compounds have shown high antitumor activity and have entered clinical trials.401Mono- and bis-1,8-naphthalimide-conjugated Ru(II)–polypyridyl complexes 100 and 101 show strong binding to DNA through the naphthalimide arms and a cytoplasmic localisation was observed in HeLa cells, as shown in Fig. 43a and b.397 Linear and circular dichroism (LD and CD) spectroscopy revealed close association of the [Ru(bpy)3]2+ core with DNA in the case of the mono-100 complex whilst intriguingly the second naphthalimide arm of complex 101 was found to displace the [Ru(bpy)3]2+ centre from the DNA backbone. In solution studies this ‘negative allosteric effect’ was found to reduce photo-induced damage of DNA and in cellular studies, photo-activation of complex 101 did not significantly affect cell viability, in contrast to the mono-naphthalimide containing complex 100.397
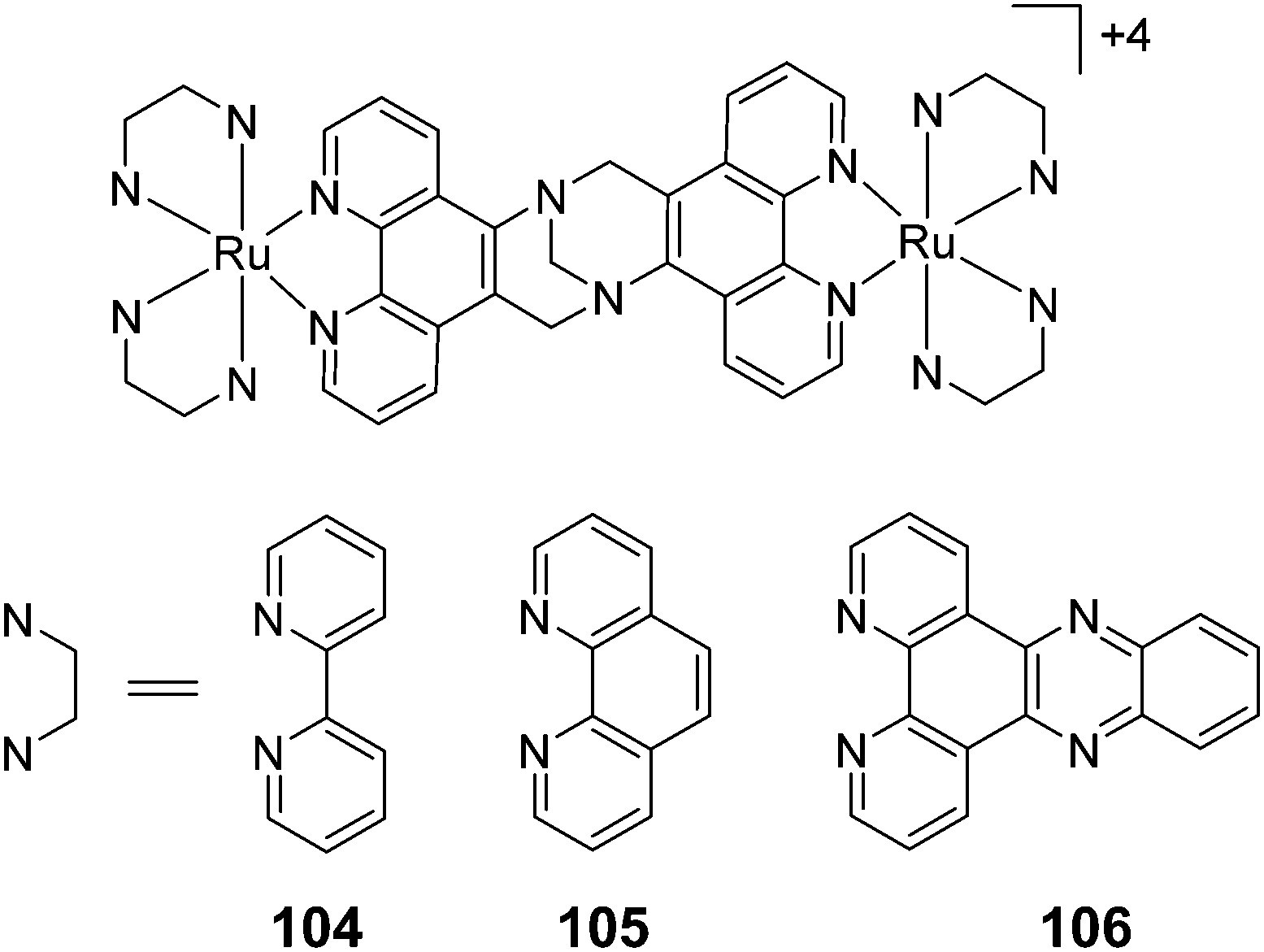
 | ||
| Fig. 43 Chemical structures of complexes 100–103 and the confocal microscopy images of HeLa cells incubated with the nuclear stain DAPI (blue) and (a) 100 (30 μM) for 24 h and (b) 101 (30 μM) for 4 h, (c) 102 (10 μM) for 4 h and (d) 103 (10 μM) for 4 h (red). Images reproduced with permission from ref. 397 and 398. | ||
Gunnlaugsson and co-workers have also incorporated the Tröger's base linkage to synthesise rigid dinuclear Ru(II) complexes based on the naphthalimide motif.398,402,403 Tröger's base was first discovered in 1887 and is a highly strained, chiral structure where the two aryl groups are close to being orthogonal to each other causing a ‘cleft-like’ structure, which has found use in supramolecular chemistry as a structural scaffold. Tröger's bases have also been used in the development of novel luminescent materials, and for probing DNA structure, Kirsch-De Mesmaeker and co-workers formed the first example of Ru(II) polypyridyl complexes of Tröger's bases as potential DNA binders in 1997.404 Since then, two papers have been published on Ru(II)-linked Tröger's base compounds tested in human cells. In both cases, these compounds were dinuclear Ru(II) polypyridyl complexes. Within our group, we found that two rigid dinuclear Ru(II)-complexes 102 and 103, derived from 4-amino-1,8-naphthalimides linked via a Tröger's base, localised within the cytoplasm of HeLa cells and exhibited strong luminescence in vitro at low μM concentrations and did not induce cytotoxicity in cells when kept in the dark, as shown in Fig. 43c and d.398 We are currently investigating the photo-active potential of these compounds.
Ezadyar et al. used bis-phenanthroline Tröger's base as a bridging ligand and demonstrated a good reduction in HeLa cell viability with IC50 values ranging between 8 and 30 μM. Treatment with complexes 104 (bpy) and 105 (phen) induced apoptosis, mitochondrial fragmentation and partial photo-cleavage of DNA. In contrast, complex 106 (dppz) induced necrosis with intact mitochondria and full photo-cleavage of DNA. However the authors did not demonstrate the intracellular location of complexes or the effect of photo-activation on cellular viability.405
Ru-Nanoconjugates
Nanoparticle research is an area of intense growth where particles can range from ultrafine (1–100 nm) to fine (100–2500 nm) and coarse (2500–10000 nm).406,407 Ultrafine nanoparticles have long been considered for anti-cancer therapies as their size and the leaky vasculature of tumours leads to an enhanced permeation and retention rate (EPR) within tumours, which results in the accumulation of nanomaterials at these sites, thereby allowing for more direct targeting of tumours. In addition, nanomaterials can be decorated with a variety of molecules, such as drug molecules and targeting moieties, making these systems extremely versatile. Indeed, there has been considerable interest among researchers to conjugate Ru(II) polypyridyl complexes to nanomaterials of various compositions to design new classes of probes and drug molecules. In such nanoconjugates, the Ru(II) complexes have been most commonly incorporated onto the surface of the nanomaterials, but examples of encapsulated Ru(II) complexes have also been reported. In many cases, nanoparticles were also coated with biological ligands to encourage enhanced uptake by target cell types.
Functionalised gold nanoparticles (AuNPs) have been the subject of intensive studies for various biomedical applications owing to their biocompatibility, unique size- and shape-dependence, and optoelectronic properties.411 While the surface plasmon of AuNPs can quench the emission of nearby luminophores, this effect is dependent on both the distance and method of attachment of molecules to the gold surface.141 In fact, conjugation of Ru(II) complexes to AuNPs via alkane thiols has been shown to result in highly luminescent nanoparticles in which the tethered Ru(II) complexes possess longer luminescence lifetimes than their free counterparts.410,412
Within the Gunnlaugsson research group such Ru(II)-nanoconjugates of 4 nm and 15 nm diameter (conjugates 107–111 illustrated in Fig. 44) have been used for luminescence imaging within live cells. The nanoparticles were internalised into cells and localised in the cytoplasm (as shown in Fig. 44a and b for conjugates 107 and 111) with little or no toxicity towards cells after 24 h in the dark.408,409 In the case of the larger diameter conjugates (15 nm), the individual particles could be visualised within cells using TEM, where they were observed within single membrane vesicles in the cytoplasm, which suggested the route of uptake to be endocytosis. In addition, these nanoparticles were shown to remain intact within these vesicles for up to 24 h, as demonstrated in Fig. 44c.409
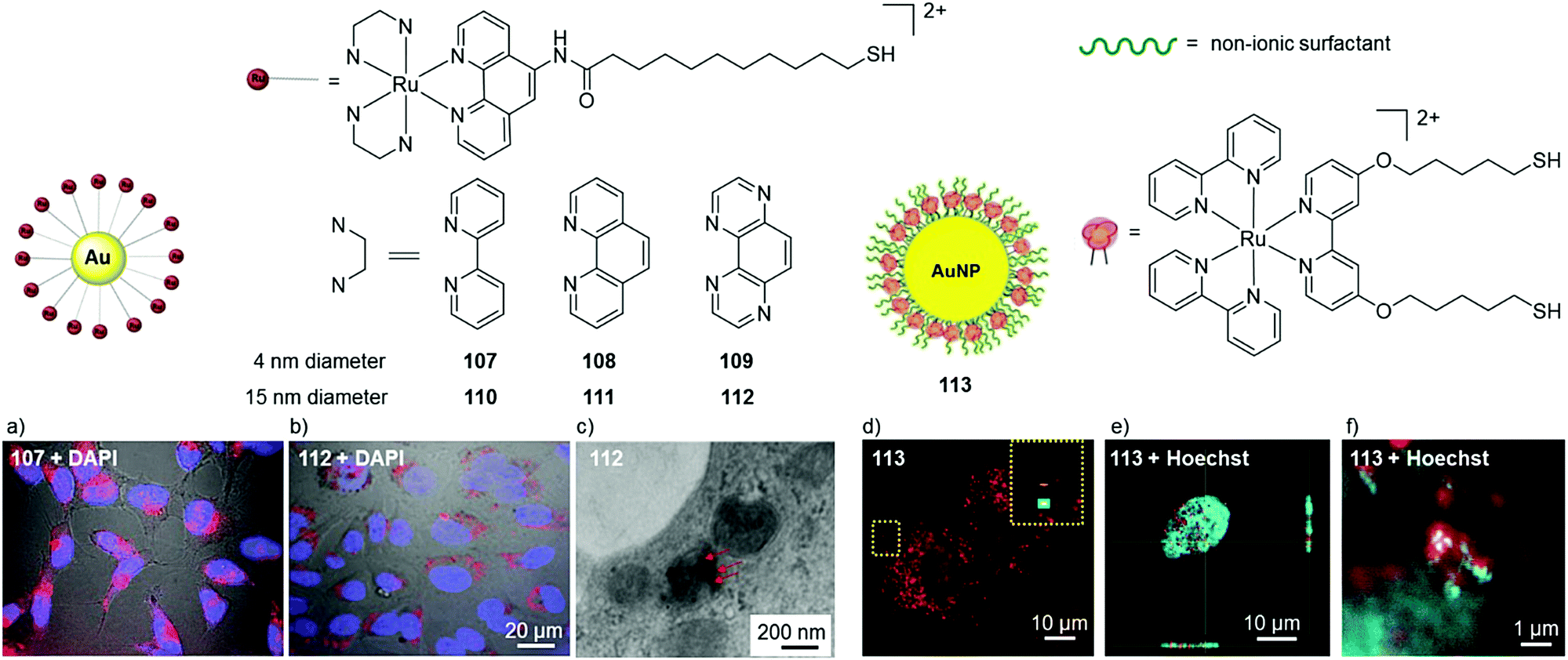 | ||
| Fig. 44 The structures of AuNPs 107–111 and 113 and the bright field confocal microscopy images of live HeLa cells incubated with DAPI (blue) and (a) the 4 nm nanoparticle 107 (ca. 16 nM gold concentration) for 4 h and (b) the 15 nm nanoparticle 111 (20 μM Ru concentration) for 24 h. (c) TEM images of HeLa cells incubated with 111 (20 μM Ru concentration) for 24 h, showing vesicle-entrapped conjugates denoted by red arrows. Confocal microscopy images of (d) methanol-fixed A549 cells after treatment with the 100 nm nanoparticle 113 (40 pM) for 24 h, (e) a single methanol-fixed A549 cell, co-stained with the Hoechst nuclear stain and (f) magnification of the nucleus of a treated cell showing association of 113 with chromatin. Images reproduced with permission from ref. 408, 409 and 410. | ||
Rogers et al. have also investigated the cellular imaging of 100 nm diameter AuNPs coated with ca. 105 Ru(II) complexes and non-ionic surfactant molecules (113 in Fig. 44).410 The nanoparticles showed no toxicity towards A549 cells under the experimental conditions employed and were stable after 24 h incubation with cells. Furthermore, due to their large size, individual nanoparticles could be visualised within cells by luminescence imaging Fig. 44d, where they were shown to distribute throughout methanol-fixed cells, as shown in Fig. 44d–f.410
In addition to cellular imaging, Ru(II)–AuNP conjugates have been utilised for photothermal cancer therapy, whereby they generate heat upon absorption of light, which results in the thermal ablation of cancer cells.413 Zhang et al. grafted Ru(II) complexes, which possessed strong two-photon absorption at 808 nm (σ2 = 176–394 GM), onto the surface of AuNPs of various sizes. One of the Ru(II)–AuNP conjugates, 114 (of 45 nm diameter), depicted in Fig. 45, was shown to localise predominantly in the cytoplasm of HeLa cells and exhibited little toxicity in the dark at concentrations below 200 μg mL−1 after 24 h. However, irradiation of treated HeLa cells with 808 nm light (0.8 W cm−2 for 5 min) resulted in a dramatic reduction in cell viability. Furthermore, the authors demonstrated that 114 could be used for photothermal therapy in vivo, as shown in Fig. 45. After intratumoral injection of 114, the subcutaneous tumours in mice were irradiated with 808 nm light (0.8 W cm−2 for 5 min). This resulted in the formation of black scars, as shown in Fig. 45b, as well as tumour shrinkage or even disappearance of the tumour after 10 days. In contrast, no tumour shrinkage was observed upon laser irradiation of tumours in mice injected with either the ungrafted AuNPs or the Ru(II) complex alone.141
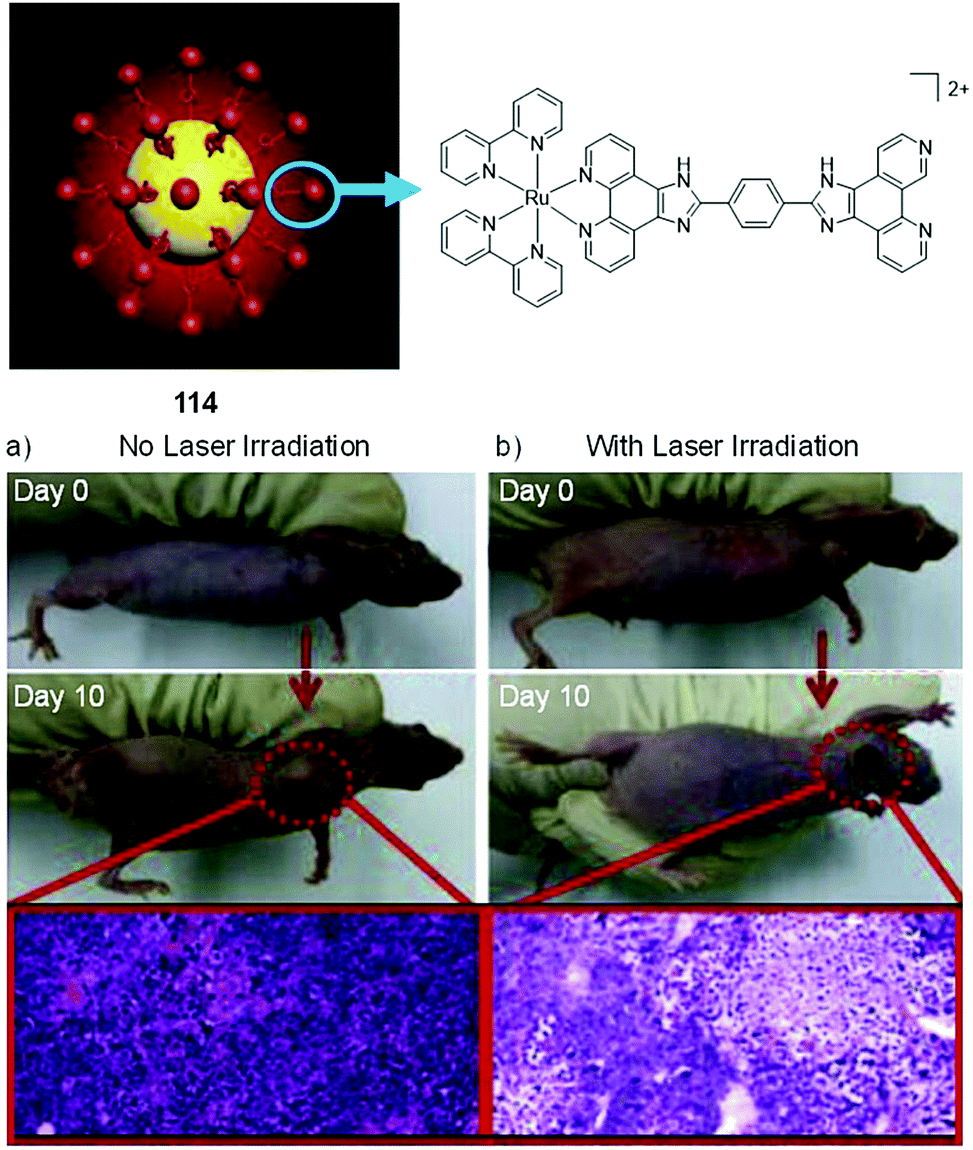 | ||
| Fig. 45 Illustration of the structure of Ru(II)–AuNP conjugate 114 and photographs of tumours derived from xenografted HeLa cells in nude mice after administration of 114via intratumoral injection (200 mg/20 g body weight), followed by (a) no irradiation and (b) irradiation of the tumour with 808 nm light (0.8 W cm−2, 5 min). Histological examination of the tumours without/with laser treatments on day 10 is shown in purple. Images reproduced with permission from ref. 141. | ||
Xiang et al. developed a tumour-targeting system based on carbon-doped TiO2 nanoparticles for use in photoactivated chemotherapy.414 The surfaces of the 4 nm diameter nanoparticles were functionalised with receptor-targeting folate molecules and Ru(II) complexes, which possessed a lysosome-targeting morpholine moiety. The conjugates (115) showed low dark toxicity towards HeLa cells at the tested concentrations (10–200 μg mL−1) and were found to localise in their target organelle, as shown in Fig. 46. Subsequent light-activation of the conjugates (λex = 808 nm, 600 mW cm−2, 10 min), resulted in the generation of ROS and photo-induced loss of nitric oxide, the quantum yields of which were 0.23 and 0.017 mol per Einstein, respectively (in solution), which resulted in a significant decrease in the viability of treated cells, with a corresponding IC50 (light) value of 20 μg mL−1.414
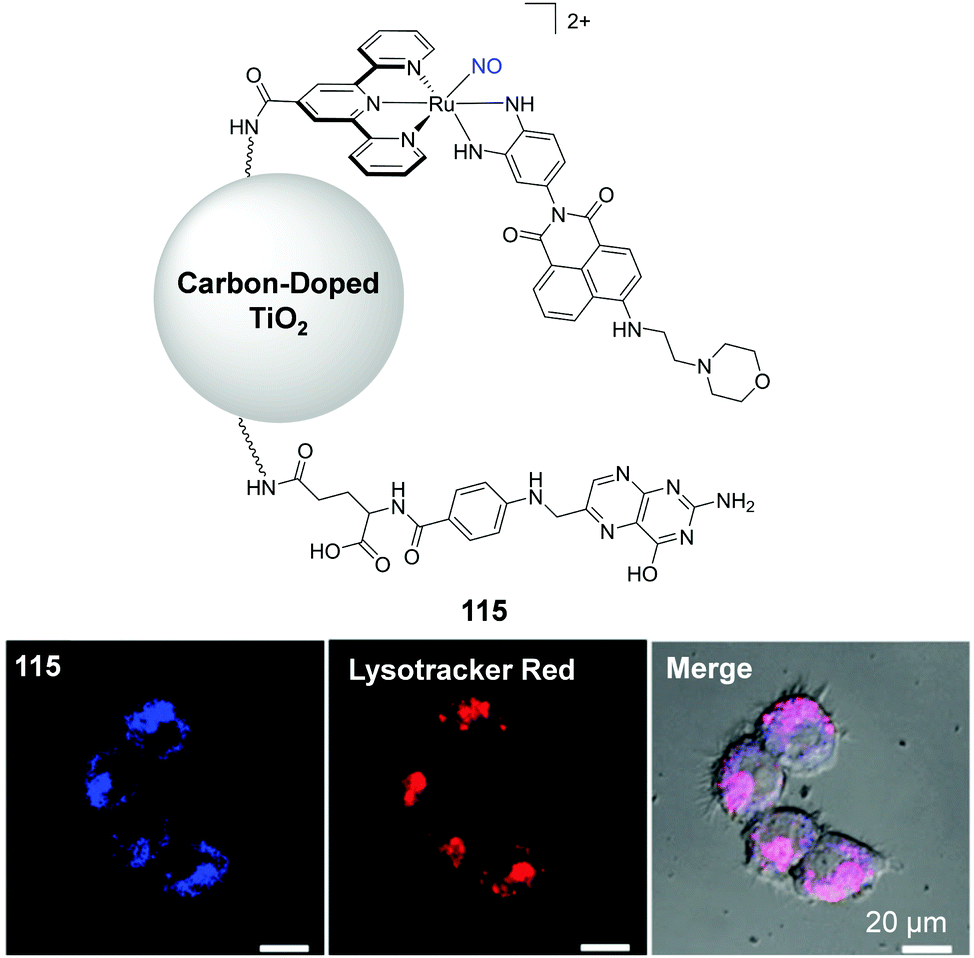 | ||
| Fig. 46 Illustration of the chemical structure of conjugate 115, in which the photo-cleavable nitric oxide ligand is highlighted in blue. The confocal microscopy images of HeLa cells incubated with conjugate 115 for 8 h (shown in blue) and co-stained with Lysotracker Red. Images reproduced with permission from ref. 414. | ||
A number of silica-based nanoparticles have been investigated in which Ru(II) complexes have decorated the surface of the particles or have been entrapped within them. In addition, other molecules such as MRI contrast agents,415 anti-cancer agents,416 and targeting molecules have also been incorporated into such nanoparticles for use as theranostic agents and light-activatable drug compounds.417,418 For example, Frasconi et al. functionalised the surface of mesoporous silica nanoparticles (MSNPs) of ca. 120 nm diameter with a Ru(II)–dppz complex for development of theranostic agents.416 The resulting conjugate 116 localised in the cytoplasm of MDA-MB-468 breast cancer cells, as shown in Fig. 47. Subsequent light activation of the Ru(II) complexes (λex > 450 nm) resulted in photo-induced ligand loss, thereby releasing Ru-aqua species from the MSNP, in a similar manner to that seen previously in Fig. 30. However, no significant light-induced increase in cytotoxicity was observed for 116 against treated cells. In contrast, when the researchers encapsulated the anti-cancer drug paclitaxel into the MSNPs, light-activation of the 116-Ptx system resulted in a significant enhancement in its cytotoxicity due to the release of paclitaxel.416 On the other hand, Chen and co-workers encapsulated their Ru(II) complex (54) within MSNPs of ca. 118 nm diameter, the surface of which was coated with RGD tripeptides (Arg–Gly–Asp) to yield conjugate 117, the structure of which is illustrated in Fig. 48. Incorporation of the RGD peptides led to a significant enhancement in uptake of the conjugates into cancer cells overexpressing the integrin receptor via receptor-mediated endocytosis.417 Once inside the target cells, endosomes lysed with lysosomes resulting in the pH-mediated release of complex 54 and the subsequent induction of apoptosis. Impressive IC50 values in the nanomolar range were displayed across a selection of cancer cell lines, and in the micromolar range for non-cancer cells.417 A similar study from the same group used complex 54 to form folate-conjugated selenium nanoparticles (SeNPs) (with average diameters of 180 nm).419 These conjugates could effectively antagonise against multidrug resistance in liver cancer through inhibition of ABC family protein expression. Conjugation of folate molecules to the surface of the SeNPs significantly enhanced their cellular uptake by overexpressed folate receptors in cancer cells, with a lower uptake in normal cells. Within cells, the conjugates caused ROS over-production and induced apoptosis by activating p53 and MAPKs pathways. Moreover, the Ru(II)–SeNPs exhibited low in vivo acute toxicity, which supported the safety of the system in vivo.419 Other Ru(II)–SeNP conjugates have also been investigated and shown to inhibit angiogenesis and suppress tumour growth in vivo.420,421
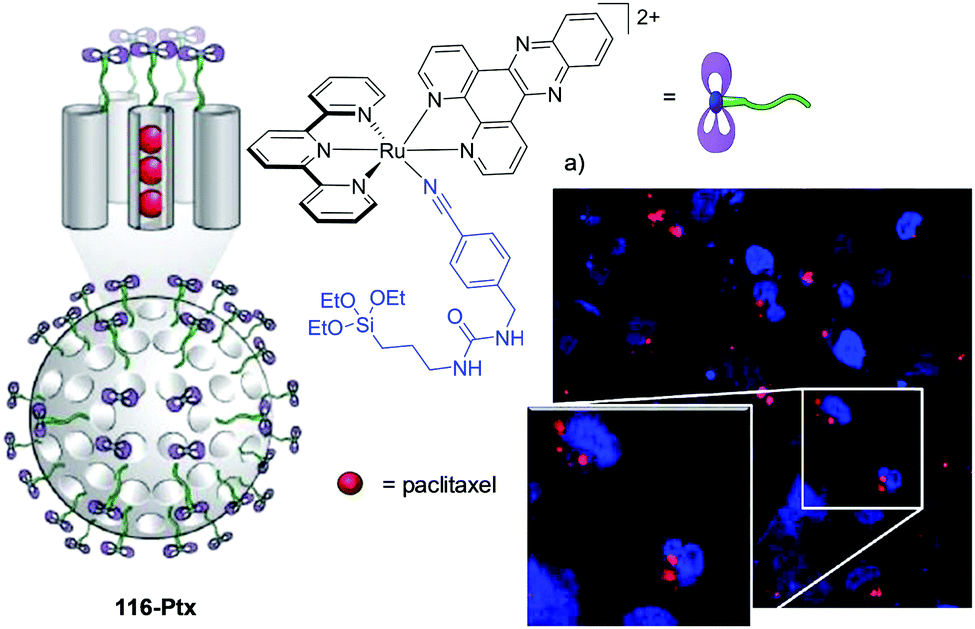 | ||
| Fig. 47 Illustration of the chemical structure of the silica-based nanoparticle 116-Ptx, where the photolabile ligand of the Ru(II)-dppz complex is highlighted in blue and the confocal microscopy images of MDA-MB-468 cells after incubation with 116 (red) for 6 h and the nuclear stain DAPI (blue). Images reproduced with permission from ref. 416. | ||
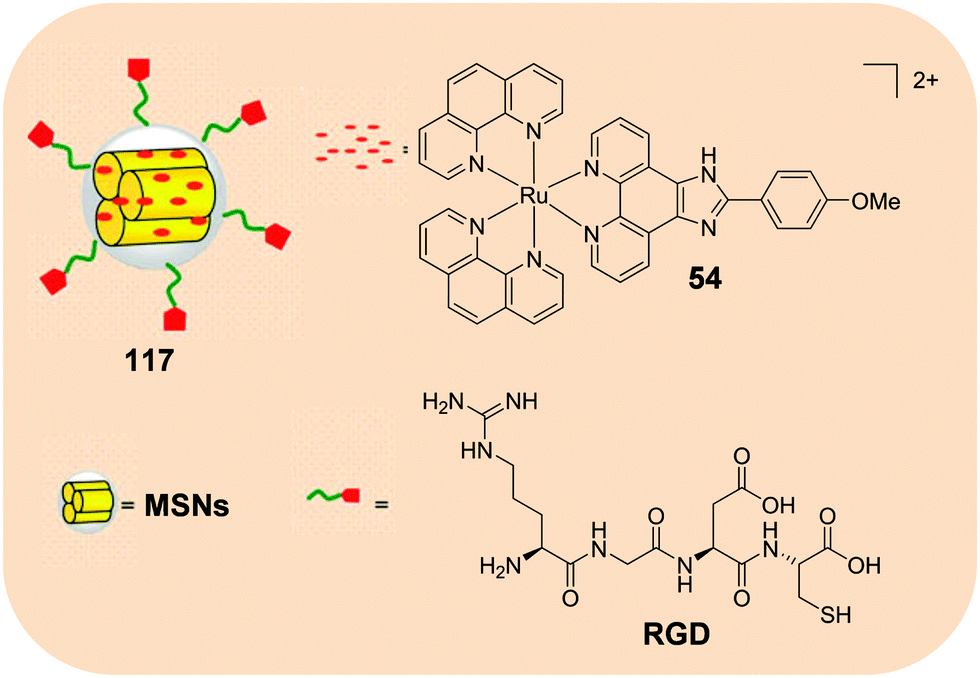 | ||
| Fig. 48 Illustration of the structure of the silica-based nanoparticle 117. Figure modified from ref. 417. | ||
Ru(II) complexes have also been encapsulated into polymer-based nanoparticles. Choi et al. developed nanoparticles of 40 nm diameter, in which the Ru(II) polypyridyl complexes were entrapped in a cross-linked polymer composed of urethane acrylate nonionomer.422 The resulting conjugates (118) were used as luminescent probes for the quantitative imaging of the distribution of oxygen in vitro and in vivo. In vitro, 3D scaffolds were seeded with tumour cells and assessed for toxicity. While the free Ru(II) complex was quite toxic towards cells (>75% toxicity at 112.5 nM), the encapsulated complex in 118 was non-toxic at a similar concentration, both in the dark and when illuminated. In vivo, mice injected with these nanoparticles showed no acute toxicity or adverse effects for up to a month after the injection. Furthermore, using 118, the researchers were able to monitor the O2 concentration in the arteries of sedated mice after intracardiac injection of the conjugate, as shown in Fig. 49.422
Researchers have also encapsulated Ru(II) polypyridyl complexes in other polymer-based nanoparticles, derived from poly(ethylene)glycol–poly(aspartate) (PEG–ASP)423 and poly(lactic-co-glycolic acid) (PLGA),424 as well as in phospholipids-based liposomes.425
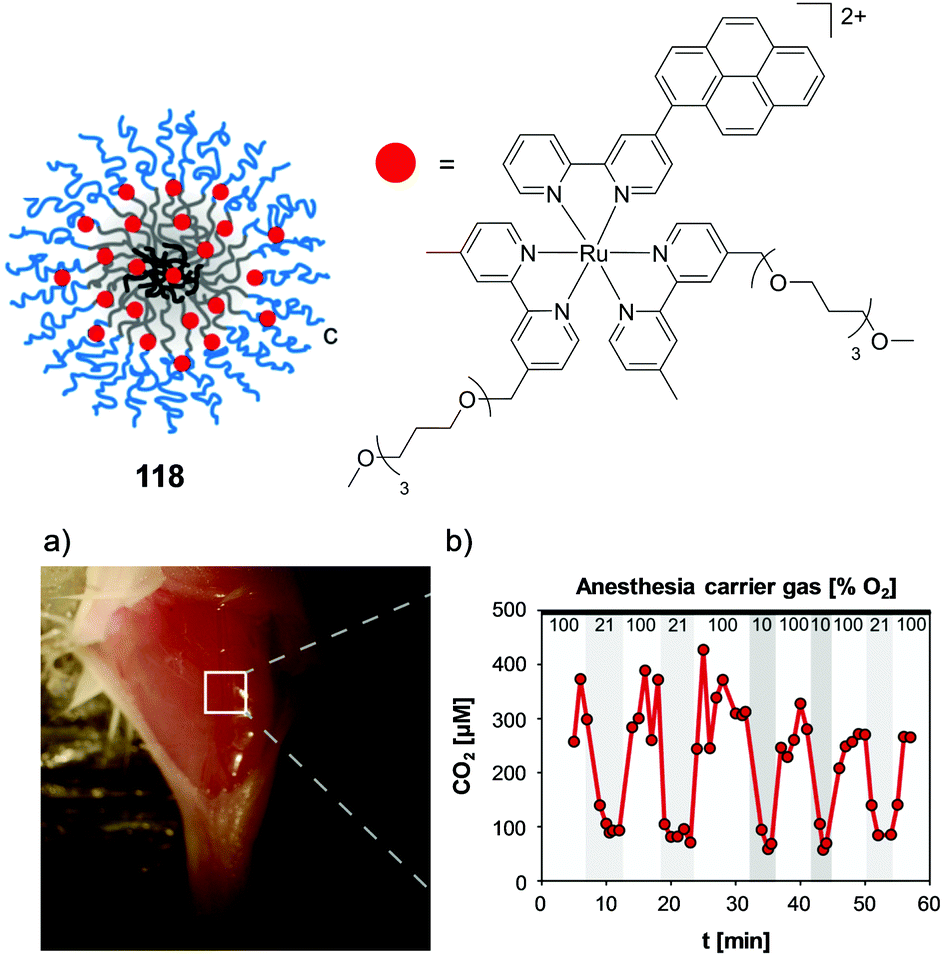 | ||
| Fig. 49 Illustration of the structure of the polymer-based nanoparticle 118 and (a) image of the blood vessels of an anesthetised mouse, exposed by an incision through the skin and (b) the measured concentration of O2 using 118 in the artery (white square in (a)) of a mouse while the partial pressure of O2 in the anaesthesia carrier gas was varied. The nanoparticles were administered by intracardiac injection and the O2 concentration was determined based on the luminescence lifetime of 118. Images reproduced with permission from ref. 422 and adapted. | ||
In fact, polymer-based nano/microparticles and liposomes have seen considerable development across numerous academic and industrial areas, for the encapsulation of various classes of drug compounds. Indeed, various drug formulations utilising these materials have already received FDA approval and have been in clinical use since the 1990's.426,427 Recently, encapsulation of [Ru(phen)2(dppz)]2+ into PEGylated liposomes of ca. 80 nm in diameter (illustrated in Fig. 50) was shown to increase the intracellular concentration of the complex 15-fold in MDA-MB-231 breast cancer cells, after 6 h incubation with 119.425 Interestingly, Ru emission was found to arise from the nuclei of treated cells, when incubated with either 119 or the free complex for 6 h at 5 μM Ru, as shown in Fig. 50a and b. In contrast, however, Tan et al. found that in HeLa cervical cancer cells treated with [Ru(phen)2(dppz)]2+ at a higher concentration of 40 μM for 24 h, the complex was largely excluded from the nuclei of cells.155 While the free complex showed little activity against breast cancer cell lines (IC50 >200 μM), there was a significant decrease in the viability of cells incubated with 119, with IC50 values of 1.4 μM after 72 h incubation.425 Mechanistic studies revealed that cells treated with 119 exhibited double-strand DNA breaks, which was proposed to induce cell cycle arrest and apoptosis. The biodistribution and activity of 119in vivo was assessed in orthotopic tumour models of xenografted MDA-MB-231 cells in nude mice. Two hours after intravenous injection, 119 was found to localise primarily in the liver (34%) and tumour (30%). Moreover, cancer cell proliferation within the implanted tumours was dramatically suppressed after treatment with 119 (5 mg kg−1) once a week for four weeks.425
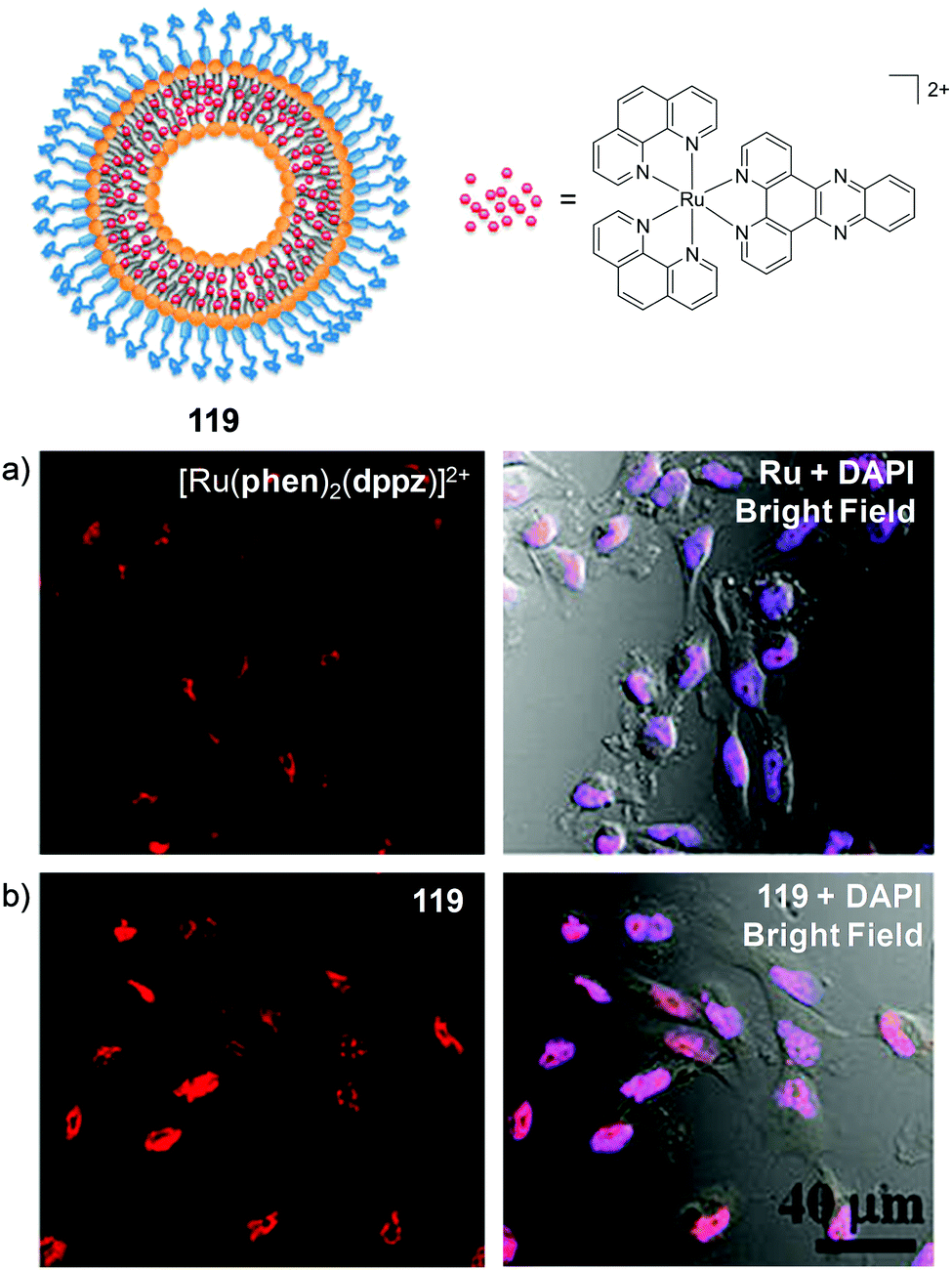 | ||
| Fig. 50 Illustration of the chemical structure of Ru–liposome 119 and the confocal microscopy images of MDA-MB-231 breast cancer cells incubated with (a) [Ru(phen)2(dppz)]2+ and (b) 119, both at a Ru concentration of 5 μM for 6 h (shown in red) and the nuclear stain DAPI (shown in blue). Co-localisation of Ru and DAPI is shown in purple. Images reproduced with permission from ref. 425. | ||
In another study into polymer-based nanoparticles, the Ru(II) complexes were covalently linked to polymer chains, which self-assembled into nanoparticles with average diameters of 180 nm, as illustrated in Fig. 51.428 Solution studies of the resulting nanoparticles (120) showed that photo-activation of the Ru(II) complexes resulted in the cleavage of the monodentate ligands, in a similar manner to that seen previously in Fig. 30, to liberate bis-aqua Ru(II) species, which was shown to exert similar activity to cisplatin against various cancer cell lines. Indeed, when internalised in the cytoplasm of cells, photo-activation of 120 at 656 nm (50 mW cm−2, 30 min) resulted in a significant decrease in the viability of cells, which was attributed to the generation of 1O2 and the release of the Ru-aqua species. Furthermore, nanoparticles of 120, loaded with the fluorescent dye Cy®5, were shown to localise in xenografted tumours in mice, within 12 h of intravenous injection via the tail vein, as shown in Fig. 51. Subsequent irradiation of the tumour sites, 12 h post-administration of 120, with 655 nm light (0.2 W cm−2, 10 min) then resulted in inhibition of tumour growth.428
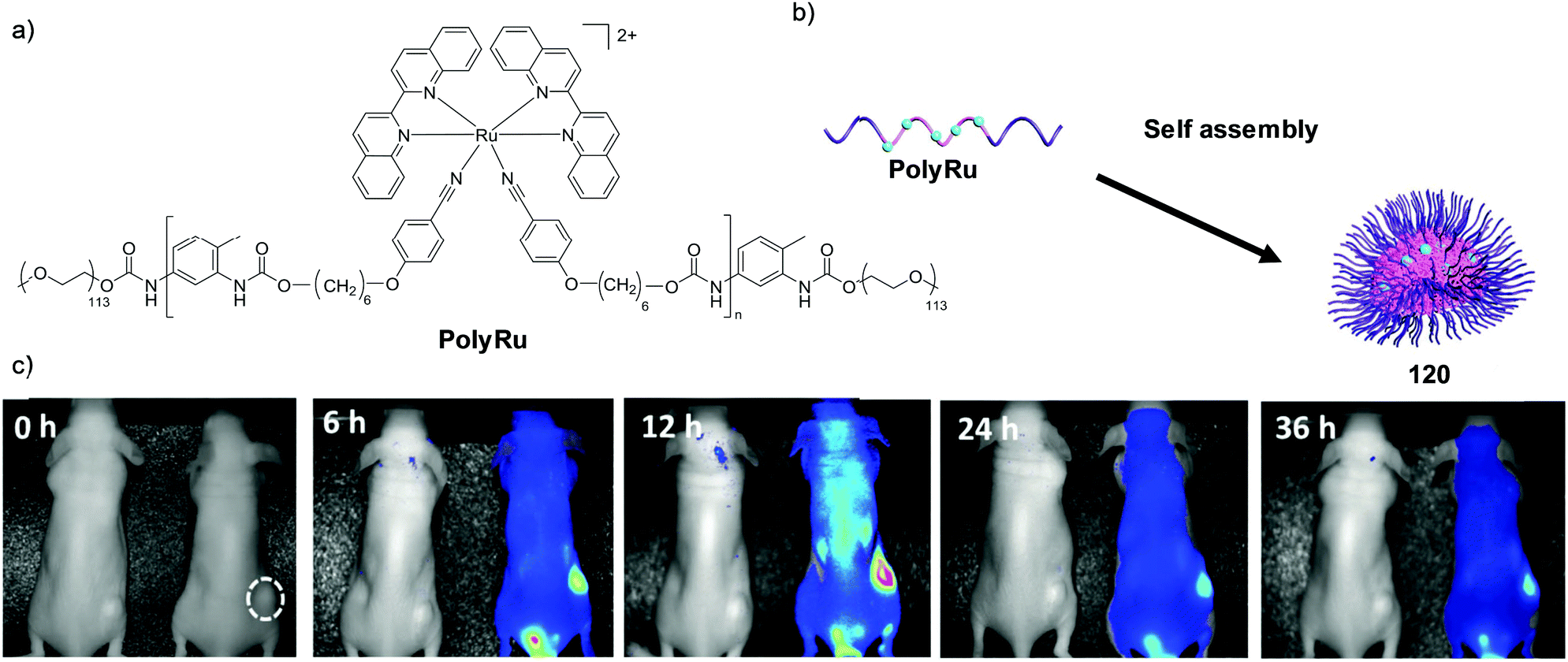 | ||
| Fig. 51 (a) Chemical structure of the amphiphilic polymer PolyRu and (b) an illustration of the self-assembly of PolyRu into nanoparticles (conjugate 120). (c) In vivo luminescence images of tumour-bearing mice after intravenous injection of saline (left, control) and Cy®5 dye-loaded nanoparticles (right). Images were taken at 0, 6, 12, 24, and 36 h after injection. The dashed circle indicates the tumour. Figure adapted and were images reproduced with permission from ref. 428. | ||
The same researchers who developed the Ru(II)–AuNP conjugate 114, reported the use of single-walled carbon nanotubes loaded with Ru(II) polypyridyl complexes via noncovalent π–π interactions to develop dual photothermal and two-photon PDT agents.429 Irradiation of nanotubes loaded with either complex 121 or 122, shown in Fig. 52, with 808 nm light (0.25 W cm−2) resulted in a photothermal effect, which released the loaded Ru(II) complexes, which in turn could generate 1O2via two-photon activation under these conditions. These conjugates showed greater photo-induced anti-cancer activity in both cultured cell monolayers and 3D multicellular tumour spheroids, than either the nanotubes or Ru(II) complexes alone. Furthermore, the conjugates were successfully used for tumour ablation in vivo. The conjugates were administered to mice bearing xenografted HeLa cell-derived tumours via intratumoral injection and following irradiation of the tumours with 808 nm light (0.25 W cm−2, 5 min), the photothermal effect of the nanotubes could be observed in thermographs, as shown in Fig. 52a, where a rapid increase in the temperature of the tumour region from 37 °C to ca. 60 °C was observed for both conjugates. This treatment resulted in the shrinkage or even disappearance of the tumours several days post-treatment, as shown in Fig. 52b. In addition, no evidence of toxicity towards mice was detected following administration of the conjugates or following photothermal therapy.429 Another study by Wang et al. demonstrated that loading the Ru(II) complex 54 into carbon nanotubes reduced its toxicity in vivo and prolonged its blood circulation.220 Moreover, the biodistribution of the complex was also altered upon loading into nanotubes: there was a reduction in the levels of Ru in the spleen and lung, and an increased accumulation of Ru in the liver, when incorporated into the nanotubes, relative to the free complex.220
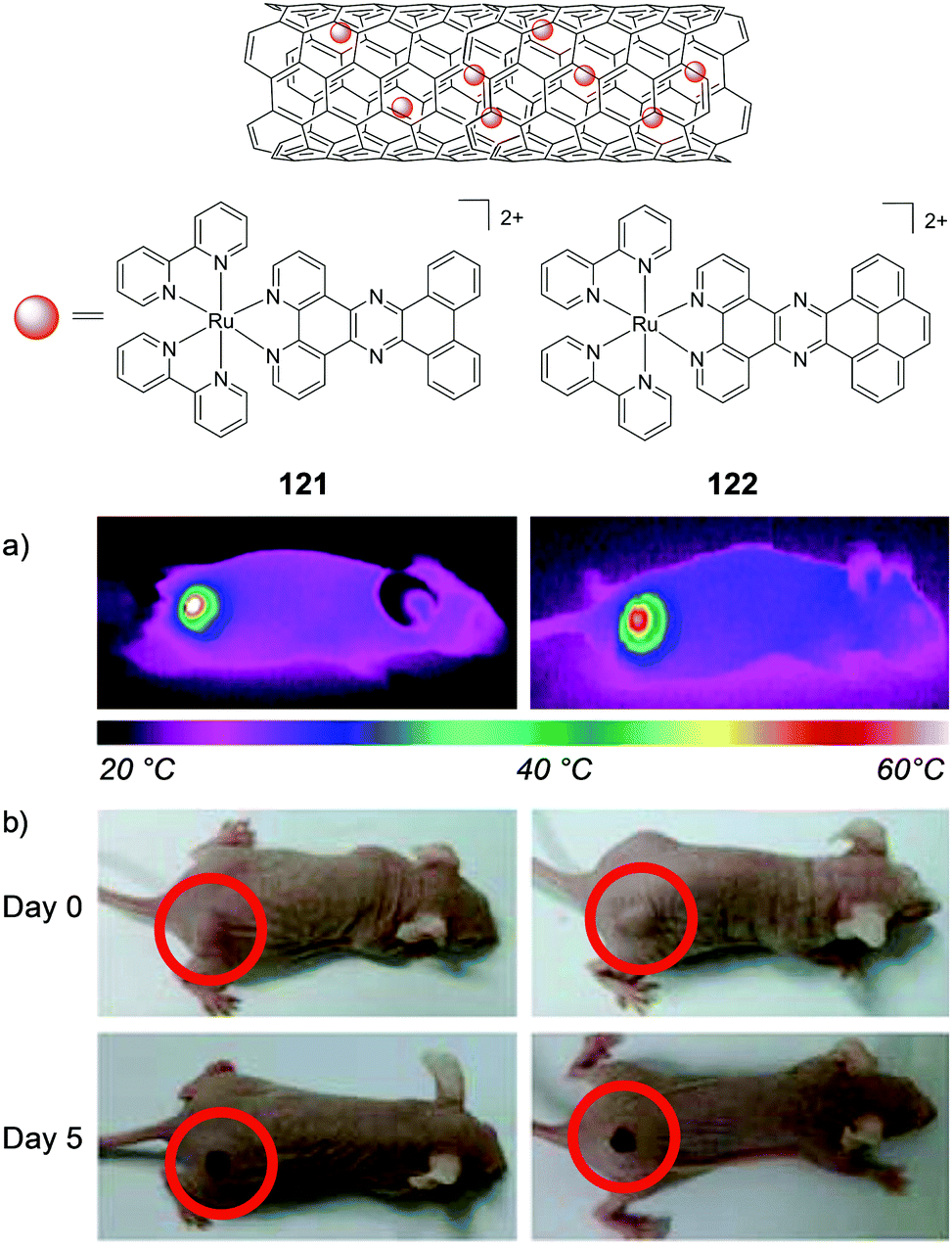 | ||
| Fig. 52 Illustration of the chemical structure of the Ru(II) encapsulated carbon nanotubes 120z and 121z and (a) thermographs of mice bearing xenografted HeLa cell-derived tumours after intratumoral injection of an aqueous dispersion (1 mg mL−1) of conjugate 120z (left) or 121z (right) and receiving photothermal treatment by laser irradiation of the tumour (λex = 808 nm, 0.25 W cm−2 for 5 min). (b) Photographs of mice administered with either conjugate 0 and 5 days after photothermal treatment (808 nm) laser light (λex = 808 nm, 0.8 W cm−2, 5 min). Images modified and reproduced with permission from ref. 429. | ||
Finally, Ru(II) complexes have even been incorporated into zirconium-based metal organic framework (MOF) nanoparticles, with average diameters of 92 nm, for two-photon imaging and PDT applications.135 Incorporation of [Ru(bpy)3]2+ into the pores of the MOFs resulted in an enhancement in the luminescence quantum yield, two-photon absorption cross-section (σ2 = 21.9 GM at 880 nm), 1O2 generation and photostability relative to the free complex. Two-photon confocal microscopy (λex = 880 nm) revealed that the Ru(II)–MOFs localised in the cytoplasm of A549 cells after 20 h incubation and showed low toxicity towards cells in the dark at concentrations of 200 μg mL−1. However exposure of treated cells to white light (200 mW cm−2 for 10 min), resulted in a significant reduction in the viability of cells.135
Conclusions and future perspectives
Considerable progress has been made within the last decade translating Ru(II) polypyridyl complexes from use as purely solution-based probes to a biological setting as diagnostic and therapeutic agents.With regards to cellular uptake and localisation, the lipophilicity of complexes has been further shown to play a profound role in both processes.47,63,86,89,90,104 For example, the formation of lipophilic ion-pairs has recently been demonstrated to promote the cellular uptake and nuclear localisation of complexes which had previously been unable to enter live cells;107 a strategy which could prove beneficial for other systems in the future. However, while increasing the lipophilicities of complexes can improve their uptake, it can also have a considerable impact on the nature of their interactions with biomolecules, such as proteins and membranes, which may in fact hinder their uptake, alter their subcellular localisation and/or influence the nature of their interactions with (or even the identity of) their molecular targets within cells.87,90 Researchers have developed complexes capable of selectively targeting and visualising specific organelles within cells, including the nucleus, mitochondria, lysosomes and endoplasmic reticulum, and have shown Ru(II) polypyridyl complexes to be excellent candidates for use as luminescent probes for ultra-resolution STED microscopy and two-photon imaging.87,131,143,211,357,358 Furthermore, by conjugating functional molecules, such as targeting peptides, drug molecules and nanomaterials to Ru(II) complexes, the resulting conjugates can be targeted for specific cell types or subcellular organelles and imparted with additional properties such as enhanced solubility, multimodal drug action, altered biodistribution and enhanced circulation times in vivo, etc. As such, the scope for the use of Ru(II) polypyridyl complexes in biological applications is ever increasing.
In addition to their use as luminescent probes, complexes possessing high intrinsic anti-cancer activities against various cancer cell lines have been developed. However, in many cases, the identity of the molecular target(s), with which they interact to elicit their anti-cancer activity, remains unclear. In fact, while many of these complexes have been designed to bind to DNA in solution, in a cellular environment the complexes show no evidence of reaching their intended target. As the foundations of this field were laid by researchers focused on the binding of small molecules to DNA, it is unsurprising that DNA remains the primary focus within this field. However, while publications in this area typically include detailed DNA binding studies, their binding interactions with proteins have seen significantly less attention, despite the vast quantities of these biomolecules in cellular media, cell membranes and throughout cells. Indeed, as shown in this review, publications demonstrating the importance of Ru(II)–protein interaction, both in vitro and in vivo, highlight that this field could benefit greatly from increasing our understanding of the factors influencing the interaction of Ru(II) polypyridyl complexes with proteins.90,95,173,215,219 Furthermore, such studies could providing key insights into, not only the cellular uptake of complexes, but also explain why complex localise in specific regions of the cell, as well as the origin of their biological activities. As such, more detailed biological studies will be required before a comprehensive picture can be drawn for the mechanism(s) underlying the observed anti-cancer activity of Ru(II) polypyridyl complexes.
While there has now been a considerable number of studies examining the interactions of Ru(II) polypyridyl complexes within cells, and growing numbers of in vivo studies, relatively few studies have examined difference between the behaviour of the Λ- and Δ-enantiomers of tris(bidentate) complexes, with most studies using the racemic mixture of the two enantiomers. Indeed, as shown in this review, enantiomer-specific interactions have been observed both in cultured cells and in vivo and therefore more studies of this kind will be required to gain a deeper understanding of the enantiomer-specific interactions of these complexes in a biological setting.73,107,215
In addition to the intrinsic anti-cancer activity of numerous complexes, the photophysical and photochemical properties of this class of complex have been exploited towards the development of light-activated therapeutics with various mechanisms of action, which have been highlighted in this review, and will undoubtedly continue to be an area of intensive research in the coming years. While Ru(II) complexes tend to absorb light outside of the therapeutic window (650–900 nm),130 two-photon activation of complexes is now permitting the use of deeper tissue penetrating wavelengths of light (ca. 800 nm) to activate complexes for both imaging and therapeutic applications in vivo.
With the increasing number of in vivo studies, a more detailed picture is emerging regarding the bioavailability, toxicology and pharmacology of Ru(II) polypyridyl complexes, which will be invaluable in the future development of both diagnostic and therapeutic agents. For example, [Ru(phen)3]2+, which typically shows relatively low activity against cancer cells in vitro,222 was found to be a quite potent neurotoxin in vivo, acting on the neuromuscular junction of mice.215,219 On the other hand, other Ru(II) complexes have been much better tolerated by mice.222,252 Furthermore, a number of complexes show similar or greater intrinsic anti-cancer activity to cisplatin in subcutaneous tumour models, while exhibiting fewer side effects.157,221,222 Further still, by exploiting the photophysical and photochemical properties of complexes, new light-activatable therapeutics with diverse mechanisms of action have been developed. One such Ru(II) polypyridyl complex, TLD1433, has now advanced to clinical trials as a photosensitiser in the treatment of cancer, which will undoubtedly fuel efforts to translate more Ru(II) complexes to clinical applications.148 In light of these developments, and the increasing numbers of chemists investigating the therapeutic potential of Ru(II) polypyridyl complexes, now is an opportune time to survey the field of preclinical drug discovery in academia to take note of what lies ahead as this field continues to move and grow in this direction.
Across numerous fields within academia, there are increasing numbers of academic researchers and universities now engaging in preclinical drug discovery.430,431 This has led to the establishment of organisations such as the Academic Drug Discovery Consortium (ADDC, http://addconsortium.org),432 which aims to build a collaborative network between this growing number of academic drug discovery scientists and centres. Indeed numerous marketed drugs have their origins in academic research centres.433,434 However, amid this innovation a serious issue currently faces researchers engaged in preclinical drug discovery, as well as basic and translational oncology. That is, that many researchers are reporting serious difficulties reproducing results from the literature.435 Although the issue of irreproducibility is a widespread problem across the life sciences and has been discussed in the literature for decades,436,437 it has become so prolific and gained such a degree of public attention that it has been dubbed the “reproducibility crisis”.438–441 For example, in the US alone, an estimated $28 billion each year is spent on biomedical research that cannot be repeated successfully.436 A considerable proportion of this negative attention has been focused on preclinical research, in light of the high rates of attrition of new drug compounds during clinical trials and the increasing costs involved in drug development.442–444 Furthermore, the practices within academic research groups engaged in preclinical research has seen considerable scepticism and critiquing.435,440,445,446 The factors that have been highlighted in contributing to this irreproducibility are complex and multifactorial and are discussed further elsewhere.447–456 As already seen in this review, the biological activity of a Ru(II) polypyridyl complexes can be found to show significant differences between research groups, even when the same assays and cell lines are used for their evaluation. Indeed, reproducibility and quality issues have now been cited as significant barriers to drug development,435,446 and could pose a significant challenge to researchers in this field looking to licence their compound or engage in collaboration with industry or secure funding from private sources such as venture capitalists,457,458 as both sources are now openly voicing their concerns as to the reliability of results from academic preclinical studies.459,460 There are, however, a number of recent initiatives aimed at tackling this reproducibility issue,461 such as Science Exchange's Reproducibility Initiative,462 the Reproducibility2020 Initiative of the Global Biological Standards Institute,463 more stringent checklists for publications,464 post-publication peer review (e.g. PubMed Commons and PubPeer) and platforms promoting the publication of negative results, to combat publication bias (e.g. Figshare and PLOS ONE).465
As this field is still in the early stages of expanding into the field of preclinical drug discovery, now may be the best chance to take heed of such concerns and open discussion among researchers in this field to ensure the continued realisation of the potential of Ru(II) polypyridyl complexes in the years to come.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the Irish Research Council (PhD studentship to FEP, grant RS/2011/580) and R. E. A. for the IEF Marie Curie Fellowship of SB. We thank Science Foundation Ireland for Financial support (SFI PI Awards 10/IN.1/B2999 and 13/IA/1865, to TG).Notes and references
- J. G. Vos and J. M. Kelly, Dalton Trans., 2006, 4869 RSC.
- V. Marin, E. Holder, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 618 RSC.
- C. Daniel, Coord. Chem. Rev., 2015, 282–283, 19 CrossRef CAS.
- S. Fantacci and F. De Angelis, Coord. Chem. Rev., 2011, 255, 2704 CrossRef CAS.
- J. P. Paris and W. W. Brandt, J. Am. Chem. Soc., 1959, 81, 5001 CrossRef CAS.
- S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, Top. Curr. Chem., 2007, 280, 117 CrossRef CAS.
- D. W. Thompson, A. Ito and T. J. Meyer, Pure Appl. Chem., 2013, 85, 1257 CrossRef CAS.
- D. Chatterjee, Catal. Surv. Asia, 2009, 13, 132 CrossRef CAS.
- A. Vlček, in Electron Transfer in Chemistry, ed. V. Balzani, Wiley-VCH, Germany, 2008, ch. 5, vol. 2, pp. 804–877 Search PubMed.
- A. Adeloye and P. Ajibade, Molecules, 2014, 19, 12421 CrossRef PubMed.
- A. Islam, H. Sugihara and H. Arakawa, J. Photochem. Photobiol., A, 2003, 158, 131 CrossRef CAS.
- Y. Amao and I. Okura, Sens. Actuators, B, 2003, 88, 162 CrossRef CAS.
- J. A. Kitchen, E. M. Boyle and T. Gunnlaugsson, Inorg. Chim. Acta, 2012, 381, 236 CrossRef CAS.
- R. B. P. Elmes and T. Gunnlaugsson, Tetrahedron Lett., 2010, 51, 4082 CrossRef CAS.
- K. Ocakoglu and S. Okur, Sens. Actuators, B, 2010, 151, 223 CrossRef CAS.
- M. Abrahamsson, H. Wolpher, O. Johansson, J. Larsson, M. Kritikos, L. Eriksson, P.-O. Norrby, J. Bergquist, L. Sun, B. Åkermark and L. Hammarström, Inorg. Chem., 2005, 44, 3215 CrossRef CAS PubMed.
- L. Zayat, O. Filevich, L. M. Baraldo and R. Etchenique, Philos. Trans. R. Soc., A, 2013, 371, 20120330 CrossRef PubMed.
- J. Yang, M. Bhadbhade, W. A. Donald, H. Iranmanesh, E. G. Moore, H. Yan and J. E. Beves, Chem. Commun., 2015, 51, 4465 RSC.
- M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179 RSC.
- B. J. Pages, D. L. Ang, E. P. Wright and J. R. Aldrich-Wright, Dalton Trans., 2015, 44, 3505 RSC.
- C. S. Burke, A. Byrne and T. E. Keyes, in Advances in Imaging and Sensing, ed. S. Tang and D. Saeedkia, CRC Press, Florida, USA, 2016, ch. 11, pp. 227–254 Search PubMed.
- N. P. Cook, V. Torres, D. Jain and A. A. Martí, J. Am. Chem. Soc., 2011, 133, 11121 CrossRef CAS PubMed.
- P. Imming, C. Sinning and A. Meyer, Nat. Rev. Drug Discovery, 2006, 5, 821 CrossRef CAS PubMed.
- G. Li, L. Sun, L. Ji and H. Chao, Dalton Trans., 2016, 45, 13261 RSC.
- L. Marcélis, W. Vanderlinden and A. K.-D. Mesmaeker, in Inorganic Chemical Biology, ed. G. Gasser, John Wiley & Sons, Ltd, West Sussex, England, 2014, ch. 6, pp. 183–213 Search PubMed.
- J. P. Hall, P. M. Keane, H. Beer, K. Buchner, G. Winter, T. L. Sorensen, D. J. Cardin, J. A. Brazier and C. J. Cardin, Nucleic Acids Res., 2016, 44, 9472 CrossRef CAS PubMed.
- P. M. Keane, F. E. Poynton, J. P. Hall, I. V. Sazanovich, M. Towrie, T. Gunnlaugsson, S. J. Quinn, C. J. Cardin and J. M. Kelly, Angew. Chem., Int. Ed., 2015, 54, 8364 CrossRef CAS PubMed.
- R. A. Conradi, P. S. Burton and R. T. Borchardt, in Lipophilicity in Drug Action and Toxicology, ed. V. Pliška, B. Testa and H. van de Waterbeemd, Wiley-VCH, Germany, 2008, ch. 14, vol. 4, pp. 233–252 Search PubMed.
- B. Ehrenberg, V. Montana, M. D. Wei, J. P. Wuskell and L. M. Loew, Biophys. J., 1988, 53, 785 CrossRef CAS PubMed.
- C. A. Puckett and J. K. Barton, Biochemistry, 2008, 47, 11711 CrossRef CAS PubMed.
- P. D. Dobson and D. B. Kell, Nat. Rev. Drug Discovery, 2008, 7, 205 CrossRef CAS PubMed.
- K. M. Giacomini, S. M. Huang, D. J. Tweedie, L. Z. Benet, K. L. Brouwer, X. Chu, A. Dahlin, R. Evers, V. Fischer, K. M. Hillgren, K. A. Hoffmaster, T. Ishikawa, D. Keppler, R. B. Kim, C. A. Lee, M. Niemi, J. W. Polli, Y. Sugiyama, P. W. Swaan, J. A. Ware, S. H. Wright, S. W. Yee, M. J. Zamek-Gliszczynski and L. Zhang, Nat. Rev. Drug Discovery, 2010, 9, 215 CrossRef CAS PubMed.
- G. J. Doherty and H. T. McMahon, Annu. Rev. Biochem., 2009, 78, 857 CrossRef CAS PubMed.
- A. C. Komor and J. K. Barton, Chem. Commun., 2013, 49, 3617 RSC.
- L. Rajendran, H.-J. Knolker and K. Simons, Nat. Rev. Drug Discovery, 2010, 9, 29 CrossRef CAS PubMed.
- L. M. Bareford and P. W. Swaan, Adv. Drug Delivery Rev., 2007, 59, 748 CrossRef CAS PubMed.
- S. Ahn, E. Seo, K. Kim and S. J. Lee, Sci. Rep., 2013, 3, 1997 CrossRef PubMed.
- P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881 CrossRef CAS PubMed.
- C. Hansch, Acc. Chem. Res., 1969, 2, 232 CrossRef CAS.
- H. van de Waterbeemd, D. A. Smith, K. Beaumont and D. K. Walker, J. Med. Chem., 2001, 44, 1313 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3 CrossRef CAS.
- Lipophilicity in Drug Action and Toxicology, ed. V. Pliska, B. Testa and H. van de Waterbeemd, Series Editors: R. Mannhold, H. Kubinyi and H. Timmerman, Wiley-VCH, Germany, 2008 Search PubMed.
- C. Hansch and W. J. Dunn, J. Pharm. Sci., 1972, 61, 1 CrossRef CAS PubMed.
- R. W. Horobin, J. C. Stockert and F. Rashid-Doubell, Histochem. Cell Biol., 2006, 126, 165 CrossRef CAS PubMed.
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2007, 129, 46 CrossRef CAS PubMed.
- S. M. Cloonan, R. B. P. Elmes, M. Erby, S. A. Bright, F. E. Poynton, D. E. Nolan, S. J. Quinn, T. Gunnlaugsson and D. C. Williams, J. Med. Chem., 2015, 58, 4494 CrossRef CAS PubMed.
- N. Deepika, Y. Kumar, C. Shobha Devi, P. Reddy, A. Srishailam and S. Satyanarayana, J. Biol. Inorg. Chem., 2013, 18, 751 CrossRef CAS PubMed.
- G.-B. Jiang, Y.-Y. Xie, G.-J. Lin, H.-L. Huang, Z.-H. Liang and Y.-J. Liu, J. Photochem. Photobiol., B, 2013, 129, 48 CrossRef CAS PubMed.
- C. Mari, V. Pierroz, R. Rubbiani, M. Patra, J. Hess, B. Spingler, L. Oehninger, J. Schur, I. Ott, L. Salassa, S. Ferrari and G. Gasser, Chem. – Eur. J., 2014, 20, 14421 CrossRef CAS PubMed.
- U. Schatzschneider, J. Niesel, I. Ott, R. Gust, H. Alborzinia and S. Wolfl, ChemMedChem, 2008, 3, 1104 CrossRef CAS PubMed.
- M. A. Walker, Expert Opin. Drug Discovery, 2014, 9, 1421 CrossRef CAS PubMed.
- A. Fahr and X. Liu, Expert Opin. Drug Delivery, 2007, 4, 403 CrossRef CAS PubMed.
- P. van Hoogevest, X. Liu and A. Fahr, Expert Opin. Drug Delivery, 2011, 8, 1481 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- B. E. Smart, J. Fluorine Chem., 2001, 109, 3 CrossRef CAS.
- Y.-Y. Xie, G.-J. Lin, G.-B. Jiang, Z.-H. Liang, H.-L. Huang and Y.-J. Liu, Transition Met. Chem., 2013, 38, 563 CrossRef CAS.
- B.-J. Han, G.-B. Jiang, J.-H. Yao, W. Li, J. Wang, H.-L. Huang and Y.-J. Liu, Spectrochim. Acta, Part A, 2015, 135, 840 CrossRef CAS PubMed.
- F. R. Svensson, M. Matson, M. Li and P. Lincoln, Biophys. Chem., 2010, 149, 102 CrossRef CAS PubMed.
- V. Pierroz, T. Joshi, A. Leonidova, C. Mari, J. Schur, I. Ott, L. Spiccia, S. Ferrari and G. Gasser, J. Am. Chem. Soc., 2012, 134, 20376 CrossRef CAS PubMed.
- B. C. Poulsen, S. Estalayo-Adrian, S. Blasco, S. A. Bright, J. M. Kelly, D. C. Williams and T. Gunnlaugsson, Dalton Trans., 2016, 45, 18208 RSC.
- M. R. Gill, H. Derrat, C. G. W. Smythe, G. Battaglia and J. A. Thomas, ChemBioChem, 2011, 12, 877 CrossRef CAS PubMed.
- H. Huang, P. Zhang, B. Yu, Y. Chen, J. Wang, L. Ji and H. Chao, J. Med. Chem., 2014, 57, 8971 CrossRef CAS PubMed.
- H. Huang, P. Zhang, H. Chen, L. Ji and H. Chao, Chem. – Eur. J., 2015, 21, 715 CrossRef CAS PubMed.
- M. R. Gill, S. N. Harun, S. Halder, R. A. Boghozian, K. Ramadan, H. Ahmad and K. A. Vallis, Sci. Rep., 2016, 6, 31973 CrossRef CAS PubMed.
- S.-H. Lai, G.-B. Jiang, J.-H. Yao, W. Li, B.-J. Han, C. Zhang, C.-C. Zeng and Y.-J. Liu, J. Inorg. Biochem., 2015, 152, 1 CrossRef CAS PubMed.
- G.-J. Lin, G.-B. Jiang, Y.-Y. Xie, H.-L. Huang, Z.-H. Liang and Y.-J. Liu, J. Biol. Inorg. Chem., 2013, 18, 873 CrossRef CAS PubMed.
- C. Griffith, A. S. Dayoub, T. Jaranatne, N. Alatrash, A. Mohamedi, K. Abayan, Z. S. Breitbach, D. W. Armstrong and F. M. MacDonnell, Chem. Sci., 2017, 8, 3726 RSC.
- F. R. Svensson, M. Abrahamsson, N. Strömberg, A. G. Ewing and P. Lincoln, J. Phys. Chem. Lett., 2011, 2, 397 CrossRef CAS PubMed.
- T. Joshi, V. Pierroz, C. Mari, L. Gemperle, S. Ferrari and G. Gasser, Angew. Chem., Int. Ed., 2014, 53, 2960 CrossRef CAS PubMed.
- Z. Zhao, Z. Luo, Q. Wu, W. Zheng, Y. Feng and T. Chen, Dalton Trans., 2014, 43, 17017 RSC.
- L. Zeng, Y. Chen, J. Liu, H. Huang, R. Guan, L. Ji and H. Chao, Sci. Rep., 2016, 6, 19449 CrossRef CAS PubMed.
- C. Shobha Devi, P. Nagababu, S. Natarajan, N. Deepika, P. Venkat Reddy, N. Veerababu, S. S. Singh and S. Satyanarayana, Eur. J. Med. Chem., 2014, 72, 160 CrossRef CAS PubMed.
- Z.-P. Zeng, Q. Wu, F.-Y. Sun, K.-D. Zheng and W.-J. Mei, Inorg. Chem., 2016, 55, 5710 CrossRef CAS PubMed.
- Q. Yu, Y. Liu, L. Xu, C. Zheng, F. Le, X. Qin, Y. Liu and J. Liu, Eur. J. Med. Chem., 2014, 82, 82 CrossRef CAS PubMed.
- L. Xu, Y.-Y. Liu, L.-M. Chen, Y.-Y. Xie, J.-X. Liang and H. Chao, J. Inorg. Biochem., 2016, 159, 82 CrossRef CAS PubMed.
- H.-L. Huang, Z.-Z. Li, Z.-H. Liang, J.-H. Yao and Y.-J. Liu, Eur. J. Med. Chem., 2011, 46, 3282 CrossRef CAS PubMed.
- J.-Q. Wang, Z.-Z. Zhao, H.-B. Bo and Q.-Z. Chen, J. Coord. Chem., 2016, 69, 177 CrossRef CAS.
- H.-j. Yu, Y. Chen, L. Yu, Z.-f. Hao and L.-h. Zhou, Eur. J. Med. Chem., 2012, 55, 146 CrossRef CAS PubMed.
- Y.-J. Liu, Z.-H. Liang, Z.-Z. Li, J.-H. Yao and H.-L. Huang, DNA Cell Biol., 2011, 30, 829 CrossRef CAS PubMed.
- Y. Du, X. Fu, H. Li, B. Chen, Y. Guo, G. Su, H. Zhang, F. Ning, Y. Lin, W. Mei and T. Chen, ChemMedChem, 2014, 9, 714 CrossRef CAS PubMed.
- W. Xu, J. Zuo, L. Wang, L. Ji and H. Chao, Chem. Commun., 2014, 50, 2123 RSC.
- P. Zhang, L. Pei, Y. Chen, W. Xu, Q. Lin, J. Wang, J. Wu, Y. Shen, L. Ji and H. Chao, Chem. – Eur. J., 2013, 19, 15494 CrossRef CAS PubMed.
- P. Liu, J. Liu, Y.-Q. Zhang, B.-Y. Wu and K.-Z. Wang, J. Photochem. Photobiol., B, 2015, 143, 89 CrossRef CAS PubMed.
- P. Liu, B.-Y. Wu, J. Liu, Y.-C. Dai, Y.-J. Wang and K.-Z. Wang, Inorg. Chem., 2016, 55, 1412 CrossRef CAS PubMed.
- A. C. Komor, C. J. Schneider, A. G. Weidmann and J. K. Barton, J. Am. Chem. Soc., 2012, 134, 19223 CrossRef CAS PubMed.
- M. R. Gill, J. Garcia-Lara, S. J. Foster, C. Smythe, G. Battaglia and J. A. Thomas, Nat. Chem., 2009, 1, 662 CrossRef CAS PubMed.
- M. R. Gill, D. Cecchin, M. G. Walker, R. S. Mulla, G. Battaglia, C. Smythe and J. A. Thomas, Chem. Sci., 2013, 4, 4512 RSC.
- J.-P. Djukic, J.-B. Sortais, L. Barloy and M. Pfeffer, Eur. J. Inorg. Chem., 2009, 817 CrossRef CAS.
- A. Wragg, M. R. Gill, D. Turton, H. Adams, T. M. Roseveare, C. Smythe, X. Su and J. A. Thomas, Chem. – Eur. J., 2014, 20, 14004 CrossRef CAS PubMed.
- A. Wragg, M. R. Gill, L. McKenzie, C. Glover, R. Mowll, J. A. Weinstein, X. Su, C. Smythe and J. A. Thomas, Chem. – Eur. J., 2015, 21, 11865 CrossRef CAS PubMed.
- T. R. Daniels, T. Delgado, G. Helguera and M. L. Penichet, Clin. Immunol., 2006, 121, 159 CrossRef CAS PubMed.
- O. Dömötör, C. G. Hartinger, A. K. Bytzek, T. Kiss, B. K. Keppler and E. A. Enyedy, J. Biol. Inorg. Chem., 2013, 18, 9 CrossRef PubMed.
- K. Śpiewak and M. Brindell, J. Biol. Inorg. Chem., 2015, 20, 695 CrossRef PubMed.
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925 RSC.
- P. Kaspler, S. Lazic, S. Forward, Y. Arenas, A. Mandel and L. Lilge, Photochem. Photobiol. Sci., 2016, 15, 481 CAS.
- F. Li, M. Feterl, J. M. Warner, A. I. Day, F. R. Keene and J. G. Collins, Dalton Trans., 2013, 42, 8868 RSC.
- S.-H. Lai, W. Li, J.-H. Yao, B.-J. Han, G.-B. Jiang, C. Zhang, C.-C. Zeng and Y.-J. Liu, J. Photochem. Photobiol., B, 2016, 158, 39 CrossRef CAS PubMed.
- O. Mazuryk, K. Magiera, B. Rys, F. Suzenet, C. Kieda and M. Brindell, J. Biol. Inorg. Chem., 2014, 19, 1305 CrossRef CAS PubMed.
- V. Rajendiran, M. Palaniandavar, V. S. Periasamy and M. A. Akbarsha, J. Inorg. Biochem., 2012, 116, 151 CrossRef CAS PubMed.
- E. Meggers, Chem. Commun., 2009, 1001 RSC.
- Ł. Skórka, M. Filapek, L. Zur, J. G. Małecki, W. Pisarski, M. Olejnik, W. Danikiewicz and S. Krompiec, J. Phys. Chem. C, 2016, 120, 7284 Search PubMed.
- K. K.-W. Lo and K. Y. Zhang, RSC Adv., 2012, 2, 12069 RSC.
- C. Kreitner and K. Heinze, Dalton Trans., 2016, 45, 13631 RSC.
- Y. Ding, Q. Wu, K. Zheng, L. An, X. Hu and W. Mei, RSC Adv., 2015, 5, 63330 RSC.
- A. E. Friedman, J. C. Chambron, J. P. Sauvage, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1990, 112, 4960 CrossRef CAS.
- Y. Zhang, Q. Zhou, N. Tian, C. Li and X. Wang, Inorg. Chem., 2017, 56, 1865 CrossRef CAS PubMed.
- B.-Z. Zhu, X.-J. Chao, C.-H. Huang and Y. Li, Chem. Sci., 2016, 7, 4016 RSC.
- M. Dickerson, Y. Sun, B. Howerton and E. C. Glazer, Inorg. Chem., 2014, 53, 10370 CrossRef CAS PubMed.
- O. Zava, S. M. Zakeeruddin, C. Danelon, H. Vogel, M. Grätzel and P. J. Dyson, ChemBioChem, 2009, 10, 1796 CrossRef CAS PubMed.
- M. J. Pisani, D. K. Weber, K. Heimann, J. G. Collins and F. R. Keene, Metallomics, 2010, 2, 393 RSC.
- M. J. Pisani, P. D. Fromm, Y. Mulyana, R. J. Clarke, H. Körner, K. Heimann, J. G. Collins and F. R. Keene, ChemMedChem, 2011, 6, 848 CrossRef CAS PubMed.
- A. K. Gorle, M. Feterl, J. M. Warner, L. Wallace, F. R. Keene and J. G. Collins, Dalton Trans., 2014, 43, 16713 RSC.
- X. Li, A. K. Gorle, T. D. Ainsworth, K. Heimann, C. E. Woodward, J. Grant Collins and F. Richard Keene, Dalton Trans., 2015, 44, 3594 RSC.
- L. Zeng, Y. Chen, H. Huang, J. Wang, D. Zhao, L. Ji and H. Chao, Chem. – Eur. J., 2015, 21, 15308 CrossRef CAS PubMed.
- J. M. Brown, Methods Enzymol., 2007, 435, 295 Search PubMed.
- S. Ji, W. Wu, W. Wu, P. Song, K. Han, Z. Wang, S. Liu, H. Guo and J. Zhao, J. Mater. Chem., 2010, 20, 1953 RSC.
- L. Marcelis, J. Ghesquiere, K. Garnir, A. Kirsch-De Mesmaeker and C. Moucheron, Coord. Chem. Rev., 2012, 256, 1569 CrossRef CAS.
- F. E. Poynton, J. P. Hall, P. M. Keane, C. Schwarz, I. V. Sazanovich, M. Towrie, T. Gunnlaugsson, C. J. Cardin, D. J. Cardin, S. J. Quinn, C. Long and J. M. Kelly, Chem. Sci., 2016, 7, 3075 RSC.
- J. P. Hall, D. Cook, S. R. Morte, P. McIntyre, K. Buchner, H. Beer, D. J. Cardin, J. A. Brazier, G. Winter, J. M. Kelly and C. J. Cardin, J. Am. Chem. Soc., 2013, 135, 12652 CrossRef CAS PubMed.
- M. Eriksson, M. Leijon, C. Hiort, B. Norden and A. Graeslund, Biochemistry, 1994, 33, 5031 CrossRef CAS PubMed.
- C. Hiort, P. Lincoln and B. Norden, J. Am. Chem. Soc., 1993, 115, 3448 CrossRef CAS.
- J. K. Barton, L. A. Basile, A. Danishefsky and A. Alexandrescu, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 1961 CrossRef CAS.
- J. K. Barton, J. M. Goldberg, C. V. Kumar and N. J. Turro, J. Am. Chem. Soc., 1986, 108, 2081 CrossRef CAS.
- P. M. Keane, F. E. Poynton, J. P. Hall, I. P. Clark, I. V. Sazanovich, M. Towrie, T. Gunnlaugsson, S. J. Quinn, C. J. Cardin and J. M. Kelly, J. Phys. Chem. Lett., 2015, 6, 734 CAS.
- I. J. Dmochowski, J. R. Winkler and H. B. Gray, J. Inorg. Biochem., 2000, 81, 221 CrossRef CAS PubMed.
- F. R. Svensson, J. Andersson, H. L. Åmand and P. Lincoln, J. Biol. Inorg. Chem., 2012, 17, 565 CrossRef CAS PubMed.
- J.-G. Liu, B.-H. Ye, Q.-L. Zhang, X.-H. Zou, Q.-X. Zhen, X. Tian and L.-N. Ji, JBIC, J. Biol. Inorg. Chem., 2000, 5, 119 CrossRef CAS PubMed.
- Q. Yu, Y. Liu, C. Wang, D. Sun, X. Yang, Y. Liu and J. Liu, PLoS One, 2012, 7, e50902 CAS.
- A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710 CrossRef CAS PubMed.
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316 CrossRef CAS PubMed.
- E. Baggaley, M. R. Gill, N. H. Green, D. Turton, I. V. Sazanovich, S. W. Botchway, C. Smythe, J. W. Haycock, J. A. Weinstein and J. A. Thomas, Angew. Chem., Int. Ed., 2014, 53, 3367 CrossRef CAS PubMed.
- F. Helmchen and W. Denk, Nat. Methods, 2005, 2, 932 CrossRef CAS PubMed.
- C. Xu and W. W. Webb, J. Opt. Soc. Am. B, 1996, 13, 481 CrossRef CAS.
- S. C. Boca, M. Four, A. Bonne, B. van der Sanden, S. Astilean, P. L. Baldeck and G. Lemercier, Chem. Commun., 2009, 4590 RSC.
- R. Chen, J. Zhang, J. Chelora, Y. Xiong, S. V. Kershaw, K. F. Li, P. K. Lo, K. W. Cheah, A. L. Rogach, J. A. Zapien and C.-S. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 5699 CAS.
- K. Qiu, B. Yu, H. Huang, P. Zhang, L. Ji and H. Chao, Dalton Trans., 2015, 44, 7058 RSC.
- B. Yu, C. Ouyang, K. Qiu, J. Zhao, L. Ji and H. Chao, Chem. – Eur. J., 2015, 21, 3691 CrossRef CAS PubMed.
- P. Shi, B. J. Coe, S. Sánchez, D. Wang, Y. Tian, M. Nyk and M. Samoc, Inorg. Chem., 2015, 54, 11450 CrossRef CAS PubMed.
- H. Wang, X. Tian, L. Guan, Q. Zhang, S. Zhang, H. Zhou, J. Wu and Y. Tian, J. Mater. Chem. B, 2016, 4, 2895 RSC.
- J. Hess, H. Huang, A. Kaiser, V. Pierroz, O. Blacque, H. Chao and G. Gasser, Chem. – Eur. J., 2017, 23, 9888 CrossRef CAS PubMed.
- P. Zhang, J. Wang, H. Huang, B. Yu, K. Qiu, J. Huang, S. Wang, L. Jiang, G. Gasser, L. Ji and H. Chao, Biomaterials, 2015, 63, 102 CrossRef CAS PubMed.
- K. Qiu, J. Wang, C. Song, L. Wang, H. Zhu, H. Huang, J. Huang, H. Wang, L. Ji and H. Chao, ACS Appl. Mater. Interfaces, 2017, 9, 18482 CAS.
- P. Zhang, H. Huang, Y. Chen, J. Wang, L. Ji and H. Chao, Biomaterials, 2015, 53, 522 CrossRef CAS PubMed.
- C. Michiels, C. Tellier and O. Feron, Biochim. Biophys. Acta, 2016, 1866, 76 CAS.
- C. R. Thoma, M. Zimmermann, I. Agarkova, J. M. Kelm and W. Krek, Adv. Drug Delivery Rev., 2014, 69–70, 29 CrossRef CAS PubMed.
- C. A. Rabik and M. E. Dolan, Cancer Treat. Rev., 2007, 33, 9 CrossRef CAS PubMed.
- W. Zheng, Y. Zhao, Q. Luo, Y. Zhang, K. Wu and F. Wang, Sci. China: Chem., 2016, 59, 1240 CrossRef CAS.
- Theralase Technologies Inc., Theralase Successfully Achieves Primary and Secondary Objectives for First Three Patients Treated Using Anti-Cancer Technology for Bladder Cancer, http://theralase.com/pressrelease/theralase-successfully-achieves-objectives-for-patients-treating-using-anti-cancer-technology-for-bladder-cancer/, (accessed June 2017).
- S. Leijen, S. A. Burgers, P. Baas, D. Pluim, M. Tibben, E. van Werkhoven, E. Alessio, G. Sava, J. H. Beijnen and J. H. M. Schellens, Invest. New Drugs, 2015, 33, 201 CrossRef CAS PubMed.
- F. Lentz, A. Drescher, A. Lindauer, M. Henke, R. A. Hilger, C. G. Hartinger, M. E. Scheulen, C. Dittrich, B. K. Keppler and U. Jaehde, Anti-Cancer Drugs, 2009, 20, 97 CrossRef CAS PubMed.
- E. Alessio, Eur. J. Inorg. Chem., 2017, 1549 CrossRef CAS.
- A. Bergamo and G. Sava, Dalton Trans., 2007, 1267 RSC.
- B. M. Blunden and M. H. Stenzel, J. Chem. Technol. Biotechnol., 2015, 90, 1177 CrossRef CAS.
- F. Gao, X. Chen, J.-Q. Wang, Y. Chen, H. Chao and L.-N. Ji, Inorg. Chem., 2009, 48, 5599 CrossRef CAS PubMed.
- C. Tan, S. Lai, S. Wu, S. Hu, L. Zhou, Y. Chen, M. Wang, Y. Zhu, W. Lian, W. Peng, L. Ji and A. Xu, J. Med. Chem., 2010, 53, 7613 CrossRef CAS PubMed.
- M. S. Ricci and W.-X. Zong, Oncologist, 2006, 11, 342 CrossRef CAS PubMed.
- X. Meng, M. L. Leyva, M. Jenny, I. Gross, S. Benosman, B. Fricker, S. Harlepp, P. Hébraud, A. Boos, P. Wlosik, P. Bischoff, C. Sirlin, M. Pfeffer, J.-P. Loeffler and C. Gaiddon, Cancer Res., 2009, 69, 5458 CrossRef CAS PubMed.
- B. Peña, A. David, C. Pavani, M. S. Baptista, J.-P. Pellois, C. Turro and K. R. Dunbar, Organometallics, 2014, 33, 1100 CrossRef.
- J.-Q. Wang, P.-Y. Zhang, C. Qian, X.-J. Hou, L.-N. Ji and H. Chao, J. Biol. Inorg. Chem., 2014, 19, 335 CrossRef CAS PubMed.
- W. Li, B.-J. Han, J.-H. Yao, G.-B. Jiang and Y.-J. Liu, RSC Adv., 2015, 5, 24534 RSC.
- G.-B. Jiang, X. Zheng, J.-H. Yao, B.-J. Han, W. Li, J. Wang, H.-L. Huang and Y.-J. Liu, J. Inorg. Biochem., 2014, 141, 170 CrossRef CAS PubMed.
- W. Li, B.-J. Han, J. Wang, G.-B. Jiang, Y.-Y. Xie, G.-J. Lin, H.-L. Huang and Y.-J. Liu, Inorg. Chim. Acta, 2014, 423, 229 CrossRef CAS.
- C. Zhang, C.-C. Zeng, S.-H. Lai, D.-G. Xing, W. Li, B.-J. Han and Y.-J. Liu, Polyhedron, 2016, 106, 115 CrossRef CAS.
- G.-B. Jiang, J.-H. Yao, J. Wang, W. Li, B.-J. Han, Y.-Y. Xie, G.-J. Lin, H.-L. Huang and Y.-J. Liu, New J. Chem., 2014, 38, 2554 RSC.
- Y.-Y. Xie, Z.-Z. Li, G.-J. Lin, H.-L. Huang, X.-Z. Wang, Z.-H. Liang, G.-B. Jiang and Y.-J. Liu, Inorg. Chim. Acta, 2013, 405, 228 CrossRef CAS.
- T. Chen, Y. Liu, W.-J. Zheng, J. Liu and Y.-S. Wong, Inorg. Chem., 2010, 49, 6366 CrossRef CAS PubMed.
- T. Chen, W.-J. Mei, Y.-S. Wong, J. Liu, Y. Liu, H.-S. Xie and W.-J. Zheng, MedChemComm, 2010, 1, 73 RSC.
- C. Tan, S. Wu, S. Lai, M. Wang, Y. Chen, L. Zhou, Y. Zhu, W. Lian, W. Peng, L. Ji and A. Xu, Dalton Trans., 2011, 40, 8611 RSC.
- W. Li, G.-B. Jiang, J.-H. Yao, X.-Z. Wang, J. Wang, B.-J. Han, Y.-Y. Xie, G.-J. Lin, H.-L. Huang and Y.-J. Liu, J. Photochem. Photobiol., B, 2014, 140, 94 CrossRef CAS PubMed.
- J. Chen, G. Li, F. Peng, X. Jie, G. Dongye, Y. Zhong, R. Feng, B. Li, J. Qu, Y. Ding and L. Chen, Inorg. Chem. Commun., 2016, 69, 35 CrossRef CAS.
- B. Tang, D. Wan, S.-H. Lai, H.-H. Yang, C. Zhang, X.-Z. Wang, C.-C. Zeng and Y.-J. Liu, J. Inorg. Biochem., 2017, 173, 93 CrossRef CAS PubMed.
- M. E. Bunnage, A. M. Gilbert, L. H. Jones and E. C. Hett, Nat. Chem. Biol., 2015, 11, 368 CrossRef CAS PubMed.
- Z. Luo, L. Yu, F. Yang, Z. Zhao, B. Yu, H. Lai, K.-H. Wong, S.-M. Ngai, W. Zheng and T. Chen, Metallomics, 2014, 6, 1480 RSC.
- J. P. Hughes, S. Rees, S. B. Kalindjian and K. L. Philpott, Br. J. Pharmacol., 2011, 162, 1239 CrossRef CAS PubMed.
- F. T. Unger, I. Witte and K. A. David, Cell. Mol. Life Sci., 2015, 72, 729 CrossRef CAS PubMed.
- Editorial, Nat. Med., 2010, 16, 347 CrossRef PubMed.
- E. Lounkine, M. J. Keiser, S. Whitebread, D. Mikhailov, J. Hamon, J. L. Jenkins, P. Lavan, E. Weber, A. K. Doak, S. Cote, B. K. Shoichet and L. Urban, Nature, 2012, 486, 361 CAS.
- W. Li, J. Bandyopadhyay, H. S. Hwaang, B.-J. Park, J. H. Cho, J. I. Lee, J. Ahnn and S.-K. Lee, Mol. Cells, 2012, 34, 209 CrossRef CAS PubMed.
- I. D. Kuntz, Science, 1992, 257, 1078 CAS.
- R. Perkins, H. Fang, W. Tong and W. J. Welsh, Environ. Toxicol. Chem., 2003, 22, 1666 CrossRef CAS PubMed.
- J. D. McKinney, A. Richard, C. Waller, M. C. Newman and F. Gerberick, Toxicol. Sci., 2000, 56, 8 CrossRef CAS PubMed.
- S. Page and R. Wheeler, Educ. Chem., 2012, 49, 26 CAS.
- T. Joshi, V. Pierroz, S. Ferrari and G. Gasser, ChemMedChem, 2014, 9, 1419 CrossRef CAS PubMed.
- J. Liu, X.-H. Zou, Q.-L. Zhang, W.-J. Mei, J.-Z. Liu and L.-N. Ji, Met.-Based Drugs, 2000, 7, 343 CrossRef CAS PubMed.
- L. Jin, L. Tan, X. Zou, J. Liu and F. Luan, Inorg. Chim. Acta, 2012, 387, 253 CrossRef CAS.
- J. Liu, W. Zheng, S. Shi, C. Tan, J. Chen, K. Zheng and L. Ji, J. Inorg. Biochem., 2008, 102, 193 CrossRef CAS PubMed.
- I. Omae, Coord. Chem. Rev., 2014, 280, 84 CrossRef CAS.
- M. Klajner, C. Licona, L. Fetzer, P. Hebraud, G. Mellitzer, M. Pfeffer, S. Harlepp and C. Gaiddon, Inorg. Chem., 2014, 53, 5150 CrossRef CAS PubMed.
- L. Fetzer, B. Boff, M. Ali, M. Xiangjun, J.-P. Collin, C. Sirlin, C. Gaiddon and M. Pfeffer, Dalton Trans., 2011, 40, 8869 RSC.
- T. Matsui, H. Sugiyama, M. Nakai and Y. Nakabayashi, Chem. Pharm. Bull., 2016, 64, 282 CrossRef CAS PubMed.
- C. Gaiddon, P. Jeannequin, P. Bischoff, M. Pfeffer, C. Sirlin and J. P. Loeffler, J. Pharmacol. Exp. Ther., 2005, 315, 1403 CrossRef CAS PubMed.
- L. A. Nguyen, H. He and C. Pham-Huy, Int. J. Biomed. Sci., 2006, 2, 85 CAS.
- Z. Zhang, Y.-J. Wang, Q. Wu, X.-H. Wu, F.-Q. Sun, B.-G. Wang, W.-J. Mei and S.-D. Chen, Aust. J. Chem., 2015, 68, 137 CrossRef CAS.
- P. Zhang, J. Wang, H. Huang, L. Qiao, L. Ji and H. Chao, Dalton Trans., 2013, 42, 8907 RSC.
- F. Gao, H. Chao, J.-Q. Wang, Y.-X. Yuan, B. Sun, Y.-F. Wei, B. Peng and L.-N. Ji, J. Biol. Inorg. Chem., 2007, 12, 1015 CrossRef CAS PubMed.
- J. J. Wilson and S. J. Lippard, J. Med. Chem., 2012, 55, 5326 CrossRef CAS PubMed.
- O. Novakova, J. Kasparkova, O. Vrana, P. M. van Vliet, J. Reedijk and V. Brabec, Biochemistry, 1995, 34, 12369 CrossRef CAS PubMed.
- E. Corral, A. C. G. Hotze, H. den Dulk, A. Leczkowska, A. Rodger, M. J. Hannon and J. Reedijk, J. Biol. Inorg. Chem., 2009, 14, 439 CrossRef CAS PubMed.
- P. Zhang, J. Chen and Y. Liang, Acta Biochim. Biophys. Sin., 2010, 42, 440 CrossRef CAS PubMed.
- A. Srishailam, Y. P. Kumar, P. Venkat Reddy, N. Nambigari, U. Vuruputuri, S. S. Singh and S. Satyanarayana, J. Photochem. Photobiol., B, 2014, 132, 111 CrossRef CAS PubMed.
- J. A. Parrish, T. B. Fitzpatrick, L. Tanenbaum and M. A. Pathak, N. Engl. J. Med., 1974, 291, 1207 CrossRef CAS PubMed.
- E. Oh, R. Liu, A. Nel, K. B. Gemill, M. Bilal, Y. Cohen and I. L. Medintz, Nat. Nanotechnol., 2016, 11, 479 CrossRef CAS PubMed.
- M. W. Russo, Gastroenterol. Hepatol., 2007, 3, 637 Search PubMed.
- D. A. Winkler, Briefings Bioinf., 2002, 3, 73 CrossRef CAS.
- N. Nikolova and J. Jaworska, QSAR Comb. Sci., 2003, 22, 1006 Search PubMed.
- J. C. Dearden, Environ. Health Perspect., 1985, 61, 203 CrossRef CAS PubMed.
- OECD, Test No. 107: Partition Coefficient (n-octanol/water): Shake Flask Method, OECD Guidelines for the Testing of Chemicals, Section 1, OECD Publishing, 1995.
- NCI-60 Human Tumor Cell Lines Screen, National Institutes of Health, http://https://dtp.cancer.gov/discovery_development/nci-60/, (accessed November 2016).
- M. D. Fellows and M. R. O'Donovan, Mutagenesis, 2007, 22, 275 CrossRef CAS PubMed.
- J. Weyermann, D. Lochmann and A. Zimmer, Int. J. Pharm., 2005, 288, 369 CrossRef CAS PubMed.
- H. Huang, B. Yu, P. Zhang, J. Huang, Y. Chen, G. Gasser, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2015, 54, 14049 CrossRef CAS PubMed.
- K. T. Hufziger, F. S. Thowfeik, D. J. Charboneau, I. Nieto, W. G. Dougherty, W. S. Kassel, T. J. Dudley, E. J. Merino, E. T. Papish and J. J. Paul, J. Inorg. Biochem., 2014, 130, 103 CrossRef CAS PubMed.
- J.-P. Gillet, A. M. Calcagno, S. Varma, M. Marino, L. J. Green, M. I. Vora, C. Patel, J. N. Orina, T. A. Eliseeva, V. Singal, R. Padmanabhan, B. Davidson, R. Ganapathi, A. K. Sood, B. R. Rueda, S. V. Ambudkar and M. M. Gottesman, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 18708 CrossRef CAS PubMed.
- J.-P. Gillet, S. Varma and M. M. Gottesman, J. Natl. Cancer Inst., 2013, 105, 452 CrossRef CAS PubMed.
- F. P. Dwyer, E. C. Gyarfas, W. P. Rogers and J. H. Koch, Nature, 1952, 170, 190 CrossRef CAS PubMed.
- N. L. Kilah and E. Meggers, Aust. J. Chem., 2012, 65, 1325 CrossRef CAS.
- T. J. Rutherford, P. A. Pellegrini, J. Aldrich-Wright, P. C. Junk and F. R. Keene, Eur. J. Inorg. Chem., 1998, 1677 CrossRef CAS.
- J. H. Koch, W. Rogers, F. Dwyer and E. C. Gyarfas, Aust. J. Biol. Sci., 1957, 10, 342 CrossRef CAS.
- F. P. Dwyer, E. C. Gyarfas, R. D. Wright and A. Shulman, Nature, 1957, 179, 425 CrossRef CAS PubMed.
- N. Wang, Y. Feng, L. Zeng, Z. Zhao and T. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 14933 CAS.
- J.-Q. Wang, P.-Y. Zhang, L.-N. Ji and H. Chao, J. Inorg. Biochem., 2015, 146, 89 CrossRef CAS PubMed.
- A. Yadav, T. Janaratne, A. Krishnan, S. S. Singhal, S. Yadav, A. S. Dayoub, D. L. Hawkins, S. Awasthi and F. M. MacDonnell, Mol. Cancer Ther., 2013, 12, 643 CrossRef CAS PubMed.
- H. Hönigsmann, J. Invest. Dermatol., 2013, 133, E18 CrossRef PubMed.
- The Nobel Prize in Physiology or Medicine 1903, http://www.nobelprize.org/nobel_prizes/medicine/laureates/1903/, (accessed October 2016).
- A. Monem El Mofty, J. Egypt. Med. Assoc., 1948, 31, 651 CAS.
- R. Ackroyd, C. Kelty, N. Brown and M. Reed, Photochem. Photobiol., 2001, 74, 656 CrossRef CAS PubMed.
- N. J. Farrer, L. Salassa and P. J. Sadler, Dalton Trans., 2009, 10690 RSC.
- R. Bonnett, Chemical Aspects of Photodynamic Therapy, Gordon and Breach Science Publishers, Amsterdam, 2000 Search PubMed.
- C. W. Brian and S. P. Michael, Phys. Med. Biol., 2008, 53, R61 CrossRef PubMed.
- Z. Huang, H. Xu, A. D. Meyers, A. I. Musani, L. Wang, R. Tagg, A. B. Barqawi and Y. K. Chen, Technol. Cancer Res. Treat., 2008, 7, 309 CrossRef CAS PubMed.
- C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660 RSC.
- J. D. Knoll and C. Turro, Coord. Chem. Rev., 2015, 282–283, 110 CrossRef CAS PubMed.
- J. K. White, R. H. Schmehl and C. Turro, Inorg. Chim. Acta, 2016, 454, 7 CrossRef PubMed.
- R. Bonnett and G. Martínez, Tetrahedron, 2001, 57, 9513 CrossRef CAS.
- M. Triesscheijn, P. Baas, J. H. Schellens and F. A. Stewart, Oncologist, 2006, 11, 1034 CrossRef CAS PubMed.
- C. Moylan, E. M. Scanlan and M. O. Senge, Curr. Med. Chem., 2015, 22, 2238 CrossRef CAS PubMed.
- IUPAC Compendium of Chemical Terminology, (the “Gold Book”), Compiled by A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, 2nd edn, 1997 Search PubMed.
- D. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380 CrossRef CAS PubMed.
- L. Wyld, M. W. R. Reed and N. J. Brown, Br. J. Cancer, 2001, 84, 1384 CrossRef CAS PubMed.
- B. B. Noodt, K. Berg, T. Stokke, Q. Peng and J. M. Nesland, Br. J. Cancer, 1999, 79, 72 CrossRef CAS PubMed.
- P. Mroz, A. Yaroslavsky, G. B. Kharkwal and M. R. Hamblin, Cancers, 2011, 3, 2516 CrossRef CAS PubMed.
- A. E. O'Connor, W. M. Gallagher and A. T. Byrne, Photochem. Photobiol., 2009, 85, 1053 CrossRef PubMed.
- Y. Liu, R. Hammitt, D. A. Lutterman, L. E. Joyce, R. P. Thummel and C. Turro, Inorg. Chem., 2009, 48, 375 CrossRef CAS PubMed.
- R. Lincoln, L. Kohler, S. Monro, H. Yin, M. Stephenson, R. Zong, A. Chouai, C. Dorsey, R. Hennigar, R. P. Thummel and S. A. McFarland, J. Am. Chem. Soc., 2013, 135, 17161 CrossRef CAS PubMed.
- M. J. Davies, Biochem. Biophys. Res. Commun., 2003, 305, 761 CrossRef CAS PubMed.
- M. K. Kuimova, G. Yahioglu and P. R. Ogilby, J. Am. Chem. Soc., 2009, 131, 332 CrossRef CAS PubMed.
- E. Boix-Garriga, B. Rodríguez-Amigo, O. Planas and S. Nonell, in Singlet Oxygen: Applications in Biosciences and Nanosciences, ed. S. Nonell and C. Flors, Royal Society of Chemistry, Cambridge, England, 2016, ch. 2, vol. 13, pp. 23–46 Search PubMed.
- T. Sainuddin, J. McCain, M. Pinto, H. Yin, J. Gibson, M. Hetu and S. A. McFarland, Inorg. Chem., 2015, 55, 83 CrossRef PubMed.
- Intravesical Photodynamic Therapy (PDT) in BCG Refractory High-Risk Non-muscle Invasive Bladder Cancer (NMIBC) Patients, http://https://clinicaltrials.gov/ct2/show/study/NCT03053635, (accessed April 2017).
- Theralase Technologies Inc., Theralase Extends Research Agreement with University Health Network, http://theralase.com/pressrelease/theralase-extends-research-agreement-university-health-network/, (accessed October 2016).
- Y. Arenas, S. Monro, G. Shi, A. Mandel, S. McFarland and L. Lilge, Photodiagn. Photodyn. Ther., 2013, 10, 615 CrossRef CAS PubMed.
- J. Fong, K. Kasimova, Y. Arenas, P. Kaspler, S. Lazic, A. Mandel and L. Lilge, Photochem. Photobiol. Sci., 2015, 14, 2014 CAS.
- A. Mandel, US Pat., US20160206653A1, 2016 Search PubMed.
- J. Liu, Y. Chen, G. Li, P. Zhang, C. Jin, L. Zeng, L. Ji and H. Chao, Biomaterials, 2015, 56, 140 CrossRef CAS PubMed.
- L. Zeng, S. Kuang, G. Li, C. Jin, L. Ji and H. Chao, Chem. Commun., 2017, 53, 1977 RSC.
- A. Karotki, M. Khurana, J. R. Lepock and B. C. Wilson, Photochem. Photobiol., 2006, 82, 443 CrossRef CAS PubMed.
- Highlights of Prescribing Information, U.S. Food and Drug Administration, http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/020451s020lbl.pdf, (accessed March 2017).
- A. P. Castano, T. N. Demidova and M. R. Hamblin, Photodiagn. Photodyn. Ther., 2004, 1, 279 CrossRef CAS PubMed.
- A. C. E. Moor, J. Photochem. Photobiol., B, 2000, 57, 1 CrossRef CAS.
- N. M. Sakhrani and H. Padh, Drug Des., Dev. Ther., 2013, 7, 585 CAS.
- M. P. Murphy, Biochim. Biophys. Acta, 2008, 1777, 1028 CrossRef CAS PubMed.
- M. Khurana, H. A. Collins, A. Karotki, H. L. Anderson, D. T. Cramb and B. C. Wilson, Photochem. Photobiol., 2007, 83, 1441 CrossRef CAS PubMed.
- H. A. Collins, M. Khurana, E. H. Moriyama, A. Mariampillai, E. Dahlstedt, M. Balaz, M. K. Kuimova, M. Drobizhev, X. D. YangVictor, D. Phillips, A. Rebane, B. C. Wilson and H. L. Anderson, Nat. Photonics, 2008, 2, 420 CrossRef CAS.
- Y. Shen, A. J. Shuhendler, D. Ye, J.-J. Xu and H.-Y. Chen, Chem. Soc. Rev., 2016, 45, 6725 RSC.
- A. V. Kachynski, A. Pliss, A. N. Kuzmin, T. Y. Ohulchanskyy, A. Baev, J. Qu and P. N. Prasad, Nat. Photonics, 2014, 8, 455 CrossRef CAS.
- P. Vaupel and L. Harrison, Oncologist, 2004, 9, 4 CrossRef PubMed.
- H. Schouwink, H. Oppelaar, M. Ruevekamp, M. van der Valk, G. Hart, P. Rijken, P. Baas and F. A. Stewart, Radiat. Res., 2003, 159, 190 CrossRef CAS PubMed.
- J. H. Kaplan, B. Forbush and J. F. Hoffman, Biochemistry, 1978, 17, 1929 CrossRef CAS PubMed.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119 CrossRef PubMed.
- B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324 CrossRef CAS PubMed.
- A. N. Hidayatullah, E. Wachter, D. K. Heidary, S. Parkin and E. C. Glazer, Inorg. Chem., 2014, 53, 10030 CrossRef CAS PubMed.
- E. Wachter, D. K. Heidary, B. S. Howerton, S. Parkin and E. C. Glazer, Chem. Commun., 2012, 48, 9649 RSC.
- T. Sainuddin, M. Pinto, H. Yin, M. Hetu, J. Colpitts and S. A. McFarland, J. Inorg. Biochem., 2016, 158, 45 CrossRef CAS PubMed.
- J.-A. Cuello-Garibo, M. S. Meijer and S. Bonnet, Chem. Commun., 2017, 53, 6768 RSC.
- R. N. Garner, L. E. Joyce and C. Turro, Inorg. Chem., 2011, 50, 4384 CrossRef CAS PubMed.
- L. Salassa, T. Ruiu, C. Garino, A. M. Pizarro, F. Bardelli, D. Gianolio, A. Westendorf, P. J. Bednarski, C. Lamberti, R. Gobetto and P. J. Sadler, Organometallics, 2010, 29, 6703 CrossRef CAS.
- A. M. Palmer, B. Peña, R. B. Sears, O. Chen, M. El Ojaimi, R. P. Thummel, K. R. Dunbar and C. Turro, Philos. Trans. R. Soc., A, 2013, 371, 20120135 CrossRef PubMed.
- B. A. Albani, B. Peña, N. A. Leed, N. A. B. G. de Paula, C. Pavani, M. S. Baptista, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2014, 136, 17095 CrossRef CAS PubMed.
- J. Li and P. R. Chen, Nat. Chem. Biol., 2016, 12, 129 CrossRef CAS PubMed.
- G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2006, 45, 4900 CrossRef CAS PubMed.
- G. C. R. Ellis-Davies, Nat. Methods, 2007, 4, 619 CrossRef CAS PubMed.
- T. Respondek, R. Sharma, M. K. Herroon, R. N. Garner, J. D. Knoll, E. Cueny, C. Turro, I. Podgorski and J. J. Kodanko, ChemMedChem, 2014, 9, 1306 CrossRef CAS PubMed.
- T. Respondek, R. N. Garner, M. K. Herroon, I. Podgorski, C. Turro and J. J. Kodanko, J. Am. Chem. Soc., 2011, 133, 17164 CrossRef CAS PubMed.
- L. Zayat, C. Calero, P. Alborés, L. Baraldo and R. Etchenique, J. Am. Chem. Soc., 2003, 125, 882 CrossRef CAS PubMed.
- O. Filevich, M. Salierno and R. Etchenique, J. Inorg. Biochem., 2010, 104, 1248 CrossRef CAS PubMed.
- L. Zayat, M. Salierno and R. Etchenique, Inorg. Chem., 2006, 45, 1728 CrossRef CAS PubMed.
- E. Fino, R. Araya, D. Peterka, M. Salierno, R. Etchenique and R. Yuste, Front. Neural Circuits, 2009, 3, 2 Search PubMed.
- L. Zayat, M. G. Noval, J. Campi, C. I. Calero, D. J. Calvo and R. Etchenique, ChemBioChem, 2007, 8, 2035 CrossRef CAS PubMed.
- M. Salierno, C. Fameli and R. Etchenique, Eur. J. Inorg. Chem., 2008, 1125 CrossRef CAS.
- M. Salierno, E. Marceca, D. S. Peterka, R. Yuste and R. Etchenique, J. Inorg. Biochem., 2010, 104, 418 CrossRef CAS PubMed.
- J. del Mármol, O. Filevich and R. Etchenique, Anal. Chem., 2010, 82, 6259 CrossRef PubMed.
- M. A. Sgambellone, A. David, R. N. Garner, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2013, 135, 11274 CrossRef CAS PubMed.
- G. A. Gentry, P. A. Morse and M. T. Dorsett, Cancer Res., 1971, 31, 909 CAS.
- L. M. Lewis and G. L. Indig, J. Photochem. Photobiol., B, 2002, 67, 139 CrossRef CAS.
- I. S. Hong, H. Ding and M. M. Greenberg, J. Am. Chem. Soc., 2006, 128, 485 CrossRef CAS PubMed.
- W. R. Wilson and M. P. Hay, Nat. Rev. Cancer, 2011, 11, 393 CrossRef CAS PubMed.
- L. Herman, S. Ghosh, E. Defrancq and A. K.-D. Mesmaekera, J. Phys. Org. Chem., 2008, 21, 670 CrossRef CAS.
- L. Marcélis, S. Kajouj, J. Ghesquière, G. Fettweis, I. Coupienne, R. Lartia, M. Surin, E. Defrancq, J. Piette, C. Moucheron and A. Kirsch-De Mesmaeker, Eur. J. Inorg. Chem., 2016, 2902 CrossRef.
- A. Reschner, S. Bontems, S. Le Gac, J. Lambermont, L. Marcelis, E. Defrancq, P. Hubert, C. Moucheron, A. Kirsch-De Mesmaeker, M. Raes, J. Piette and P. Delvenne, Gene Ther., 2013, 20, 435 CrossRef CAS PubMed.
- M. Towrie, G. W. Doorley, M. W. George, A. W. Parker, S. J. Quinn and J. M. Kelly, Analyst, 2009, 134, 1265 RSC.
- C. J. Cardin, J. M. Kelly and S. J. Quinn, Chem. Sci., 2017, 8, 4705 RSC.
- A. De la Cadena, D. y. Davydova, T. Tolstik, C. Reichardt, S. Shukla, D. Akimov, R. Heintzmann, J. Popp and B. Dietzek, Sci. Rep., 2016, 6, 33547 CrossRef CAS PubMed.
- H. Hönigsmann, in Trends in Photobiology, ed. C. Hélène, M. Charlier, T. Montenay-Garestier and G. Laustriat, Springer US, Massachusetts, USA, 1982, pp. 309–320 Search PubMed.
- W. Fan, P. Huang and X. Chen, Chem. Soc. Rev., 2016, 45, 6488 RSC.
- H. Chen, G. D. Wang, Y.-J. Chuang, Z. Zhen, X. Chen, P. Biddinger, Z. Hao, F. Liu, B. Shen, Z. Pan and J. Xie, Nano Lett., 2015, 15, 2249 CrossRef CAS PubMed.
- T. S. Lawrence, Oncology, 2003, 17, 23 Search PubMed.
- T. Y. Seiwert, J. K. Salama and E. E. Vokes, Nat. Clin. Pract. Oncol., 2007, 4, 86 CrossRef CAS PubMed.
- Z. Deng, L. Yu, W. Cao, W. Zheng and T. Chen, ChemMedChem, 2015, 10, 991 CrossRef CAS PubMed.
- S.-H. Liu, J.-H. Zhao, K.-K. Deng, Y. Wu, J.-W. Zhu, Q.-H. Liu, H.-H. Xu, H.-F. Wu, X.-Y. Li, J.-W. Wang and Q.-F. Guo, Spectrochim. Acta, Part A, 2015, 140, 202 CrossRef CAS PubMed.
- H. F. Blum, Photodynamic Action and Diseases Caused by Light, Hafner Publishing Company, New York, USA, 1964 Search PubMed.
- J. D. Spikes, J. Photochem. Photobiol., B, 1991, 9, 369 CrossRef CAS.
- D. Gal, Proc. SPIE, 1999, 3863, 481 CrossRef CAS.
- D. Gal, T. Kriska, T. G. Shutova and A. Nemeth, Proc. SPIE, 2001, 4248, 169 CrossRef CAS.
- T. Kriska, J. Jakus, A. Keszler, R. Vanyur, A. Nemeth and D. Gal, in Photochemotherapy of Cancer and Other Diseases, ed. B. Ehrenberg and K. Berg, SPIE Proceedings, 1999, vol. 3563. pp. 11–17 Search PubMed.
- A. Scherz, S. Katz, Y. Valcrat, V. Brumfeld, E. Gabelmann, J. Zilberstein, D. Leupold, J. R. Norris, H. Scheer and Y. Salomon, in Photosynthesis: Mechanisms and Effects: Volume I–V: Proceedings of the XIth International Congress on Photosynthesis, Budapest, Hungary, August 17–22, 1998, ed. G. Garab, Springer, Netherlands, Dordrecht, 1998, pp. 4207–4212 Search PubMed.
- P. Mroz, G. P. Tegos, H. Gali, T. Wharton, T. Sarna and M. R. Hamblin, in Medicinal Chemistry and Pharmacological Potential of Fullerenes and Carbon Nanotubes, ed. F. Cataldo and T. da Ros, Springer, Netherlands, 2008, vol. 1, pp. 79–106 Search PubMed.
- G. Laustriat, Biochimie, 1986, 68, 771 CrossRef CAS PubMed.
- C. H. Sibata, V. C. Colussi, N. L. Oleinick and T. J. Kinsella, Braz. J. Med. Biol. Res., 2000, 33, 869 CrossRef CAS PubMed.
- K. E. Lundin, O. Gissberg and C. I. E. Smith, Hum. Gene Ther., 2015, 26, 475 CrossRef CAS PubMed.
- Biotech comes to its ‘antisenses’ after hard-won drug approval, K. Jiang, http://blogs.nature.com/spoonful/2013/02/biotech-comes-to-its-antisenses-after-hard-won-drug-approval.html, (accessed October 2016).
- C. W. Crean, Y. T. Kavanagh, C. M. O'Keeffe, M. P. Lawler, C. Stevenson, R. J. H. Davies, P. H. Boyle and J. M. Kelly, Photochem. Photobiol. Sci., 2002, 1, 1024 CAS.
- R. Kole, A. R. Krainer and S. Altman, Nat. Rev. Drug Discovery, 2012, 11, 125 CAS.
- J. P. Lecomte, A. Kirsch-De Mesmaeker, M. M. Feeney and J. M. Kelly, Inorg. Chem., 1995, 34, 6481 CrossRef CAS.
- J. P. Lecomte, A. Kirsch-De Mesmaeker, J. M. Kelly, A. B. Tossi and H. Gorner, Photochem. Photobiol., 1992, 55, 681 CrossRef CAS PubMed.
- I. Ortmans, B. Elias, J. M. Kelly, C. Moucheron and A. Kirsch-DeMesmaeker, Dalton Trans., 2004, 668 RSC.
- L. Marcélis, M. Rebarz, V. Lemaur, E. Fron, J. De Winter, C. Moucheron, P. Gerbaux, D. Beljonne, M. Sliwa and A. Kirsch-De Mesmaeker, J. Phys. Chem. B, 2015, 119, 4488 CrossRef PubMed.
- J. P. Hall, F. E. Poynton, P. M. Keane, S. P. Gurung, J. A. Brazier, D. J. Cardin, G. Winter, T. Gunnlaugsson, I. V. Sazanovich, M. Towrie, C. J. Cardin, J. M. Kelly and S. J. Quinn, Nat. Chem., 2015, 7, 961 CrossRef CAS PubMed.
- R. Blasius, C. Moucheron and A. Kirsch-De Mesmaeker, Eur. J. Inorg. Chem., 2004, 3971 CrossRef CAS.
- S. I. Kirin, I. Ott, R. Gust, W. Mier, T. Weyhermüller and N. Metzler-Nolte, Angew. Chem., Int. Ed., 2008, 47, 955 CrossRef CAS PubMed.
- A. Gross, N. Hüsken, J. Schur, Ł. Raszeja, I. Ott and N. Metzler-Nolte, Bioconjugate Chem., 2012, 23, 1764 CrossRef CAS PubMed.
- E. Ferri, D. Donghi, M. Panigati, G. Prencipe, L. D'Alfonso, I. Zanoni, C. Baldoli, S. Maiorana, G. D'Alfonso and E. Licandro, Chem. Commun., 2010, 46, 6255 RSC.
- G. Gasser, A. M. Sosniak and N. Metzler-Nolte, Dalton Trans., 2011, 40, 7061 RSC.
- A. E. Hargrove, T. F. Martinez, A. A. Hare, A. A. Kurmis, J. W. Phillips, S. Sud, K. J. Pienta and P. B. Dervan, PLoS One, 2015, 10, e0143161 Search PubMed.
- J. A. Raskatov, J. O. Szablowski and P. B. Dervan, J. Med. Chem., 2014, 57, 8471 CrossRef CAS PubMed.
- T. F. Martinez, J. W. Phillips, K. K. Karanja, P. Polaczek, C. M. Wang, B. C. Li, J. L. Campbell and P. B. Dervan, Nucleic Acids Res., 2014, 42, 11546 CrossRef CAS PubMed.
- F. Yang, N. G. Nickols, B. C. Li, G. K. Marinov, J. W. Said and P. B. Dervan, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1863 CrossRef CAS PubMed.
- T. W. Synold, B. Xi, J. Wu, Y. Yen, B. C. Li, F. Yang, J. W. Phillips, N. G. Nickols and P. B. Dervan, Cancer Chemother. Pharmacol., 2012, 70, 617 CrossRef CAS PubMed.
- C. J. Chou, M. E. Farkas, S. M. Tsai, D. Alvarez, P. B. Dervan and J. M. Gottesfeld, Mol. Cancer Ther., 2008, 7, 769 CrossRef CAS PubMed.
- A. Hess and N. Metzler-Nolte, Chem. Commun., 1999, 885 RSC.
- J. C. Verheijen, G. A. van der Marel, J. H. van Boom and N. Metzler-Nolte, Bioconjugate Chem., 2000, 11, 741 CrossRef CAS PubMed.
- T. Joshi, M. Patra, L. Spiccia and G. Gasser, Artif. DNA PNA XNA, 2013, 4, 11 CrossRef PubMed.
- F. Meyer-Losic, C. Nicolazzi, J. Quinonero, F. Ribes, M. Michel, V. Dubois, C. de Coupade, M. Boukaissi, A.-S. Chéné, I. Tranchant, V. Arranz, I. Zoubaa, J.-S. Fruchart, D. Ravel and J. Kearsey, Clin. Cancer Res., 2008, 14, 2145 CrossRef CAS PubMed.
- K. M. Stewart, K. L. Horton and S. O. Kelley, Org. Biomol. Chem., 2008, 6, 2242 CAS.
- K. Adamson, C. Dolan, N. Moran, R. J. Forster and T. E. Keyes, Bioconjugate Chem., 2014, 25, 928 CrossRef CAS PubMed.
- F. Barragán, P. López-Senín, L. Salassa, S. Betanzos-Lara, A. Habtemariam, V. Moreno, P. J. Sadler and V. Marchán, J. Am. Chem. Soc., 2011, 133, 14098 CrossRef PubMed.
- T. Wang, N. Zabarska, Y. Wu, M. Lamla, S. Fischer, K. Monczak, D. Y. W. Ng, S. Rau and T. Weil, Chem. Commun., 2015, 51, 12552 RSC.
- Z. Guo, H. Peng, J. Kang and D. Sun, Biomed. Rep., 2016, 4, 528 CrossRef PubMed.
- M. Di Pisa, G. Chassaing and J.-M. Swiecicki, Biochemistry, 2015, 54, 194 CrossRef CAS PubMed.
- C. Bechara and S. Sagan, FEBS Lett., 2013, 587, 1693 CrossRef CAS PubMed.
- M. Rizzuti, M. Nizzardo, C. Zanetta, A. Ramirez and S. Corti, Drug Discovery Today, 2015, 20, 76 CrossRef CAS PubMed.
- P. Lundberg and Ü. Langel, J. Mol. Recognit., 2003, 16, 227 CrossRef CAS PubMed.
- D. M. Copolovici, K. Langel, E. Eriste and Ü. Langel, ACS Nano, 2014, 8, 1972 CrossRef CAS PubMed.
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2009, 131, 8738 CrossRef CAS PubMed.
- U. Neugebauer, Y. Pellegrin, M. Devocelle, R. J. Forster, W. Signac, N. Moran and T. E. Keyes, Chem. Commun., 2008, 5307 RSC.
- L. Cosgrave, M. Devocelle, R. J. Forster and T. E. Keyes, Chem. Commun., 2010, 46, 103 RSC.
- C. A. Puckett and J. K. Barton, Bioorg. Med. Chem., 2010, 18, 3564 CrossRef CAS PubMed.
- A. Martin, A. Byrne, C. S. Burke, R. J. Forster and T. E. Keyes, J. Am. Chem. Soc., 2014, 136, 15300 CrossRef CAS PubMed.
- A. Byrne, C. S. Burke and T. E. Keyes, Chem. Sci., 2016, 7, 6551 RSC.
- C. W. Pouton, Adv. Drug Delivery Rev., 1998, 34, 51 CrossRef CAS PubMed.
- C. Castro-Fernández, G. Maya-Núñez and P. M. Conn, Endocr. Rev., 2005, 26, 479 CrossRef PubMed.
- M. Mossalam, A. S. Dixon and C. S. Lim, Ther. Delivery, 2010, 1, 169 CrossRef CAS.
- A. D. Ragin, R. A. Morgan and J. Chmielewski, Chem. Biol., 2002, 9, 943 CrossRef CAS PubMed.
- L. Blackmore, R. Moriarty, C. Dolan, K. Adamson, R. J. Forster, M. Devocelle and T. E. Keyes, Chem. Commun., 2013, 49, 2658 RSC.
- S. W. Hell and J. Wichmann, Opt. Lett., 1994, 19, 780 CrossRef CAS PubMed.
- T. A. Klar, S. Jakobs, M. Dyba, A. Egner and S. W. Hell, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8206 CrossRef CAS.
- R. Schmidt, C. A. Wurm, A. Punge, A. Egner, S. Jakobs and S. W. Hell, Nano Lett., 2009, 9, 2508 CrossRef CAS PubMed.
- J. P. Richard, K. Melikov, E. Vives, C. Ramos, B. Verbeure, M. J. Gait, L. V. Chernomordik and B. Lebleu, J. Biol. Chem., 2003, 278, 585 CrossRef CAS PubMed.
- A. Bolhassani, Biochim. Biophys. Acta, 2011, 1816, 232 CAS.
- E. J. Jeong, M. Choi, J. Lee, T. Rhim and K. Y. Lee, Nanoscale, 2015, 7, 20095 RSC.
- P. Zhang, A. G. Cheetham, L. L. Lock and H. Cui, Bioconjugate Chem., 2013, 24, 604 CrossRef CAS PubMed.
- S. Chakrabortty, B. K. Agrawalla, A. Stumper, N. M. Vegi, S. Fischer, C. Reichardt, M. Kögler, B. Dietzek, M. Feuring-Buske, C. Buske, S. Rau and T. Weil, J. Am. Chem. Soc., 2017, 139, 2512 CrossRef CAS PubMed.
- S. Curry, H. Mandelkow, P. Brick and N. Franks, Nat. Struct. Biol., 1998, 5, 827 CrossRef CAS PubMed.
- M. Gottschaldt, U. S. Schubert, S. Rau, S. Yano, J. G. Vos, T. Kroll, J. Clement and I. Hilger, ChemBioChem, 2010, 11, 649 CrossRef CAS PubMed.
- M. Wenzel, A. de Almeida, E. Bigaeva, P. Kavanagh, M. Picquet, P. Le Gendre, E. Bodio and A. Casini, Inorg. Chem., 2016, 55, 2544 CrossRef CAS PubMed.
- K. Jain, P. Kesharwani, U. Gupta and N. K. Jain, Biomaterials, 2012, 33, 4166 CrossRef CAS PubMed.
- L. N. Lameijer, S. L. Hopkins, T. G. Brevé, S. H. C. Askes and S. Bonnet, Chem. – Eur. J., 2016, 22, 18484 CrossRef CAS PubMed.
- F. Dosio, B. Stella, A. Ferrero, C. Garino, D. Zonari, S. Arpicco, L. Cattel, S. Giordano and R. Gobetto, Int. J. Pharm., 2013, 440, 221 CrossRef CAS PubMed.
- K. K.-W. Lo, T. K.-M. Lee, J. S.-Y. Lau, W.-L. Poon and S.-H. Cheng, Inorg. Chem., 2008, 47, 200 CrossRef CAS PubMed.
- G. G. J. M. Kuiper, B. Carlsson, K. Grandien, E. Enmark, J. Häggblad, S. Nilsson and J.-A. k. Gustafsson, Endocrinology, 1997, 138, 863 CrossRef CAS PubMed.
- B. Siewert, V. H. S. van Rixel, E. J. van Rooden, S. L. Hopkins, M. J. B. Moester, F. Ariese, M. A. Siegler and S. Bonnet, Chem. – Eur. J., 2016, 22, 10960 CrossRef CAS PubMed.
- R.-R. Ye, Z.-F. Ke, C.-P. Tan, L. He, L.-N. Ji and Z.-W. Mao, Chem. – Eur. J., 2013, 19, 10160 CrossRef CAS PubMed.
- P. A. Marks, Oncogene, 2007, 26, 1351 CrossRef CAS PubMed.
- D. Siegel, M. Hussein, C. Belani, F. Robert, E. Galanis, V. M. Richon, J. Garcia-Vargas, C. Sanz-Rodriguez and S. Rizvi, J. Hematol. Oncol., 2009, 2, 31 CrossRef PubMed.
- J. Du, Y. Kang, Y. Zhao, W. Zheng, Y. Zhang, Y. Lin, Z. Wang, Y. Wang, Q. Luo, K. Wu and F. Wang, Inorg. Chem., 2016, 55, 4595 CrossRef CAS PubMed.
- A. E. Wakeling, A. J. Barker, D. H. Davies, D. S. Brown, L. R. Green, S. A. Cartlidge and J. R. Woodburn, Breast Cancer Res. Treat., 1996, 38, 67 CrossRef CAS PubMed.
- P. Cohen, Nat. Rev. Drug Discovery, 2002, 1, 309 CrossRef CAS PubMed.
- J. Lee, D. G. Udugamasooriya, H.-S. Lim and T. Kodadek, Nat. Chem. Biol., 2010, 6, 258 CrossRef CAS PubMed.
- R. N. Zuckermann, Pept. Sci., 2011, 96, 545 CrossRef CAS PubMed.
- M. Shibuya, Endothelium, 2006, 13, 63 CrossRef CAS PubMed.
- E. Zwick, J. Bange and A. Ullrich, Endocr.-Relat. Cancer, 2001, 8, 161 CrossRef CAS PubMed.
- J. Pan, L. Jiang, C.-F. Chan, T.-H. Tsoi, K.-K. Shiu, D. W. J. Kwong, W.-T. Wong, W.-K. Wong and K.-L. Wong, J. Lumin., 2017, 184, 89 CrossRef CAS.
- C.-T. Poon, P.-S. Chan, C. Man, F.-L. Jiang, R. N. S. Wong, N.-K. Mak, D. W. J. Kwong, S.-W. Tsao and W.-K. Wong, J. Inorg. Biochem., 2010, 104, 62 CrossRef CAS PubMed.
- H. Ke, H. Wang, W.-K. Wong, N.-K. Mak, D. W. J. Kwong, K.-L. Wong and H.-L. Tam, Chem. Commun., 2010, 46, 6678 RSC.
- J. Zhang, K.-L. Wong, W.-K. Wong, N.-K. Mak, D. W. J. Kwong and H.-L. Tam, Org. Biomol. Chem., 2011, 9, 6004 CAS.
- J. X. Zhang, J. W. Zhou, C. F. Chan, T. C. K. Lau, D. W. J. Kwong, H. L. Tam, N. K. Mak, K. L. Wong and W. K. Wong, Bioconjugate Chem., 2012, 23, 1623 CrossRef PubMed.
- H. Ke, W. Ma, H. Wang, G. Cheng, H. Yuan, W.-K. Wong, D. W. J. Kwong, H.-L. Tam, K.-W. Cheah, C.-F. Chan and K.-L. Wong, J. Lumin., 2014, 154, 356 CrossRef CAS.
- G. J. Ryan, F. E. Poynton, R. B. P. Elmes, M. Erby, D. C. Williams, S. J. Quinn and T. Gunnlaugsson, Dalton Trans., 2015, 44, 16332 RSC.
- R. B. P. Elmes, M. Erby, S. A. Bright, D. C. Williams and T. Gunnlaugsson, Chem. Commun., 2012, 48, 2588 RSC.
- G. J. Ryan, S. Quinn and T. Gunnlaugsson, Inorg. Chem., 2008, 47, 401 CrossRef CAS PubMed.
- S. Banerjee, J. A. Kitchen, S. A. Bright, J. E. O'Brien, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Commun., 2013, 49, 8522 RSC.
- S. Banerjee, E. B. Veale, C. M. Phelan, S. A. Murphy, G. M. Tocci, L. J. Gillespie, D. O. Frimannsson, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2013, 42, 1601 RSC.
- S. Murphy, S. A. Bright, F. E. Poynton, T. McCabe, J. A. Kitchen, E. B. Veale, D. C. Williams and T. Gunnlaugsson, Org. Biomol. Chem., 2014, 12, 6610 CAS.
- E. B. Veale, D. O. Frimannsson, M. Lawler and T. Gunnlaugsson, Org. Lett., 2009, 11, 4040 CrossRef CAS PubMed.
- O. Van Gijte, A. Tatibouët, M. Demeunynck, J. Lhomme and A. Kirsch-De Mesmaeker, Tetrahedron Lett., 1997, 38, 1567 CrossRef CAS.
- S. Ali Ezadyar, A. S. Kumbhar, A. A. Kumbhar and A. Khan, Polyhedron, 2012, 36, 45 CrossRef CAS.
- K. Cho, X. Wang, S. Nie, Z. Chen and D. M. Shin, Clin. Cancer Res., 2008, 14, 1310 CrossRef CAS PubMed.
- G. Chen, I. Roy, C. Yang and P. N. Prasad, Chem. Rev., 2016, 116, 2826 CrossRef CAS PubMed.
- R. B. P. Elmes, K. N. Orange, S. M. Cloonan, D. C. Williams and T. Gunnlaugsson, J. Am. Chem. Soc., 2011, 133, 15862 CrossRef CAS PubMed.
- M. Martinez-Calvo, K. N. Orange, R. B. P. Elmes, B. la Cour Poulsen, D. C. Williams and T. Gunnlaugsson, Nanoscale, 2016, 8, 563 RSC.
- N. J. Rogers, S. Claire, R. M. Harris, S. Farabi, G. Zikeli, I. B. Styles, N. J. Hodges and Z. Pikramenou, Chem. Commun., 2014, 50, 617 RSC.
- L. A. Dykman and N. G. Khlebtsov, Acta Naturae, 2011, 3, 34 CAS.
- S. A. M. Osborne and Z. Pikramenou, Faraday Discuss., 2015, 185, 219 RSC.
- L. Cheng, C. Wang, L. Feng, K. Yang and Z. Liu, Chem. Rev., 2014, 114, 10869 CrossRef CAS PubMed.
- H.-J. Xiang, Q. Deng, L. An, M. Guo, S.-P. Yang and J.-G. Liu, Chem. Commun., 2016, 52, 148 RSC.
- C. Truillet, F. Lux, J. Moreau, M. Four, L. Sancey, S. Chevreux, G. Boeuf, P. Perriat, C. Frochot, R. Antoine, P. Dugourd, C. Portefaix, C. Hoeffel, M. Barberi-Heyob, C. Terryn, L. van Gulick, G. Lemercier and O. Tillement, Dalton Trans., 2013, 42, 12410 RSC.
- M. Frasconi, Z. Liu, J. Lei, Y. Wu, E. Strekalova, D. Malin, M. W. Ambrogio, X. Chen, Y. Y. Botros, V. L. Cryns, J.-P. Sauvage and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 11603 CrossRef CAS PubMed.
- L. He, Y. Huang, H. Zhu, G. Pang, W. Zheng, Y.-S. Wong and T. Chen, Adv. Funct. Mater., 2014, 24, 2754 CrossRef CAS.
- N. Z. Knezevic, V. Stojanovic, A. Chaix, E. Bouffard, K. E. Cheikh, A. Morere, M. Maynadier, G. Lemercier, M. Garcia, M. Gary-Bobo, J.-O. Durand and F. Cunin, J. Mater. Chem. B, 2016, 4, 1337 RSC.
- T. Liu, L. Zeng, W. Jiang, Y. Fu, W. Zheng and T. Chen, Nanomedicine, 2015, 11, 947 CrossRef CAS PubMed.
- D. Sun, Y. Liu, Q. Yu, Y. Zhou, R. Zhang, X. Chen, A. Hong and J. Liu, Biomaterials, 2013, 34, 171 CrossRef CAS PubMed.
- D. Sun, Y. Liu, Q. Yu, X. Qin, L. Yang, Y. Zhou, L. Chen and J. Liu, Biomaterials, 2014, 35, 1572 CrossRef CAS PubMed.
- N. W. Choi, S. S. Verbridge, R. M. Williams, J. Chen, J.-Y. Kim, R. Schmehl, C. E. Farnum, W. R. Zipfel, C. Fischbach and A. D. Stroock, Biomaterials, 2012, 33, 2710 CrossRef CAS PubMed.
- M. Dickerson, B. Howerton, Y. Bae and E. C. Glazer, J. Mater. Chem. B, 2016, 4, 394 RSC.
- G. Bœuf, G. V. Roullin, J. Moreau, L. Van Gulick, N. Zambrano Pineda, C. Terryn, D. Ploton, M. C. Andry, F. Chuburu, S. Dukic, M. Molinari and G. Lemercier, ChemPlusChem, 2014, 79, 171 CrossRef.
- J. Shen, H.-C. Kim, J. Wolfram, C. Mu, W. Zhang, H. Liu, Y. Xie, J. Mai, H. Zhang, Z. Li, M. Guevara, Z.-W. Mao and H. Shen, Nano Lett., 2017, 17, 2913 CrossRef CAS PubMed.
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Pharm. Res., 2016, 33, 2373 CrossRef CAS PubMed.
- C. Zylberberg and S. Matosevic, Drug Delivery, 2016, 23, 3319 CrossRef CAS PubMed.
- W. Sun, S. Li, B. Haupler, J. Liu, S. Jin, W. Steffen, U. S. Schubert, H. J. Butt, X. J. Liang and S. Wu, Adv. Mater., 2017, 29, 1603702 CrossRef PubMed.
- P. Zhang, H. Huang, J. Huang, H. Chen, J. Wang, K. Qiu, D. Zhao, L. Ji and H. Chao, ACS Appl. Mater. Interfaces, 2015, 7, 23278 CAS.
- J. L. Dahlin, J. Inglese and M. A. Walters, Nat. Rev. Drug Discovery, 2015, 14, 279 CrossRef CAS PubMed.
- W. L. Jorgensen, Angew. Chem., Int. Ed., 2012, 51, 11680 CrossRef CAS PubMed.
- B. S. Slusher, P. J. Conn, S. Frye, M. Glicksman and M. Arkin, Nat. Rev. Drug Discovery, 2013, 12, 811 CrossRef CAS PubMed.
- R. Kneller, Nat. Rev. Drug Discovery, 2010, 9, 867 CrossRef CAS PubMed.
- A. J. Stevens, J. J. Jensen, K. Wyller, P. C. Kilgore, S. Chatterjee and M. L. Rohrbaugh, N. Engl. J. Med., 2011, 364, 535 CrossRef CAS PubMed.
- F. Prinz, T. Schlange and K. Asadullah, Nat. Rev. Drug Discovery, 2011, 10, 712 CrossRef CAS PubMed.
- L. P. Freedman, I. M. Cockburn and T. S. Simcoe, PLoS Biol., 2015, 13, e1002165 Search PubMed.
- R. G. Bergman, Perspect. Prof., 1989, 8, 2 Search PubMed.
- M. Baker, Nature, 2016, 533, 452 CrossRef CAS PubMed.
- W. Dekant, Toxicol. Lett., 2016, 263, 76 CrossRef CAS PubMed.
- E. Dolgin, Nat. Rev. Drug Discovery, 2014, 13, 875 CrossRef CAS PubMed.
- http://retractionwatch.com .
- A. Kannt and T. Wieland, Naunyn-Schmiedeberg's Arch. Pharmacol., 2016, 389, 353 CrossRef CAS PubMed.
- A. Mullard, Nat. Rev. Drug Discovery, 2014, 13, 877 Search PubMed.
- S. Mignani, S. Huber, H. Tomás, J. Rodrigues and J.-P. Majoral, Drug Discovery Today, 2016, 21, 239 CrossRef PubMed.
- S. V. Frye, M. R. Arkin, C. H. Arrowsmith, P. J. Conn, M. A. Glicksman, E. A. Hull-Ryde and B. S. Slusher, Nat. Rev. Drug Discovery, 2015, 14, 733 CrossRef CAS PubMed.
- C. G. Begley and L. M. Ellis, Nature, 2012, 483, 531 CrossRef CAS PubMed.
- M. Baker, Nature, 2016, 537, 433 CrossRef CAS PubMed.
- L. P. Freedman, Sci. Transl. Med., 2015, 7, 294ed7 CrossRef PubMed.
- L. P. Freedman, M. C. Gibson and R. M. Neve, BioPharm Int., 2015, 28, 14 Search PubMed.
- A. Torsvik, D. Stieber, P. Ø. Enger, A. Golebiewska, A. Molven, A. Svendsen, B. Westermark, S. P. Niclou, T. K. Olsen, M. Chekenya Enger and R. Bjerkvig, Cancer Med., 2014, 3, 812 CrossRef CAS PubMed.
- The Case for Standards in Life Science Research: Seizing Opportunities at a Time of Critical Need, http://https://www.gbsi.org/publication/the-case-for-biological-standards/, (accessed October 2016).
- N. S. Young, J. P. A. Ioannidis and O. Al-Ubaydli, PLoS Med., 2008, 5, e201 Search PubMed.
- H. G. Drexler, W. G. Dirks, Y. Matsuo and R. A. F. MacLeod, Leukemia, 2003, 17, 416 CrossRef CAS PubMed.
- M. Allen, M. Bjerke, H. Edlund, S. Nelander and B. Westermark, Sci. Transl. Med., 2016, 8, 354re3 CrossRef PubMed.
- A. Castellan, J. Kolc and J. Michl, J. Am. Chem. Soc., 1978, 100, 6687 CrossRef CAS.
- A. Kleensang, M. M. Vantangoli, S. Odwin-DaCosta, M. E. Andersen, K. Boekelheide, M. Bouhifd, A. J. Fornace, H.-H. Li, C. B. Livi, S. Madnick, A. Maertens, M. Rosenberg, J. D. Yager, L. Zhao and T. Hartung, Sci. Rep., 2016, 6, 28994 CrossRef CAS PubMed.
- J. Frearson and P. Wyatt, Expert Opin. Drug Discovery, 2010, 5, 909 CrossRef PubMed.
- S. Mehta, Nat. Biotechnol., 2004, 22, 21 CrossRef CAS PubMed.
- G. Naik, Scientists' Elusive Goal: Reproducing Study Results, Wall Street Journal, 2011.
- M. Rosenblatt, Sci. Transl. Med., 2016, 8, 336ed5 CrossRef PubMed.
- G. Matos, S. Tufik and M. Andersen, Front. Neurol. Neurosci., 2013, 4, 1 Search PubMed.
- Validating key experimental results via independent replication, Science Exchange, http://validation.scienceexchange.com/, (accessed November 2016).
- L. Freedman, G. Venugopalan and R. Wisman, F1000Research, 2017, 6, 1 CrossRef PubMed.
- Announcement: Reducing our irreproducibility, http://https://www.nature.com/news/announcement-reducing-our-irreproducibility-1.12852, (accessed May 2017).
- http://www.figshare.com .
| This journal is © The Royal Society of Chemistry 2017 |