
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From supramolecular polymers to multi-component biomaterials
Olga J. G. M.
Goor†
,
Simone I. S.
Hendrikse†
 ,
Patricia Y. W.
Dankers
* and
E. W.
Meijer
*
,
Patricia Y. W.
Dankers
* and
E. W.
Meijer
*
Institute for Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: e.w.meijer@tue.nl; p.y.w.dankers@tue.nl
First published on 9th October 2017
Abstract
The most striking and general property of the biological fibrous architectures in the extracellular matrix (ECM) is the strong and directional interaction between biologically active protein subunits. These fibers display rich dynamic behavior without losing their architectural integrity. The complexity of the ECM taking care of many essential properties has inspired synthetic chemists to mimic these properties in artificial one-dimensional fibrous structures with the aim to arrive at multi-component biomaterials. Due to the dynamic character required for interaction with natural tissue, supramolecular biomaterials are promising candidates for regenerative medicine. Depending on the application area, and thereby the design criteria of these multi-component fibrous biomaterials, they are used as elastomeric materials or hydrogel systems. Elastomeric materials are designed to have load bearing properties whereas hydrogels are proposed to support in vitro cell culture. Although the chemical structures and systems designed and studied today are rather simple compared to the complexity of the ECM, the first examples of these functional supramolecular biomaterials reaching the clinic have been reported. The basic concept of many of these supramolecular biomaterials is based on their ability to adapt to cell behavior as a result of dynamic non-covalent interactions. In this review, we show the translation of one-dimensional supramolecular polymers into multi-component functional biomaterials for regenerative medicine applications.
 Patricia Y. W. Dankers, Olga J. G. M. Goor, Simone I. S. Hendrikse and E. W. Meijer | Olga J. G. M. Goor is a PhD student in the group of Prof. E. W. Meijer and Dr P. Y. W. Dankers at the TU/e with a strong focus on material design and characterization. She investigates functionalization strategies to modify supramolecular biomaterial surfaces for regenerative medicine applications. |
Simone I. S. Hendrikse is a PhD student under the supervision of Prof. E. W. Meijer and Dr P. Y. W. Dankers. Her research focuses on designing multi-component supramolecular hydrogels as a synthetic extracellular matrix for the culture of organoids. |
Patricia Y. W. Dankers is an Associate Professor Biomedical Materials in the Department of Biomedical Engineering at the Eindhoven University of Technology (TU/e). The goal of her group is to develop materials based on supramolecular chemistry for biomedical applications with a focus on regenerative medicine. |
E. W. Meijer is a Professor in the department of Chemical Engineering and Chemistry, and Biomedical Engineering at the TU/e. The main research focus of his group is the non-covalent synthesis of complex molecular systems where the ‘engineering of adaptive complex molecular systems’ is recognized as a major challenge. |
Introduction
Since the cellular environment orchestrates cell behaviour in a dynamic yet spatiotemporal manner,1 it is an enormous inspiration to many of us, who are intrigued to mimic these systems for regenerative medicine. The interwoven fibrous network of the extracellular matrix (ECM) has long been thought to be impossible to mimic because of its extreme complexity and cell type dependent properties (Fig. 1). So far major progress is being made to arrive at simplified yet functional materials guiding cell behaviour similar to the in vivo ECM. Unfortunately, one material does not fit every requirement since every cell type is distinct, and contains unique protein compositions and isoforms, requiring cell type specific material properties. Another important property of the ECM is its structure. The ECM consists mainly of fibrous proteins, like collagen, laminin, elastin, and proteoglycans containing glycosaminoglycan (GAG) chains.2,3 Collagens are either self-assembled into fibrils, associate with fibrils, or form networks and provide structural support, whereas laminins polymerize upon activation through binding certain integrins and are important in cell adhesion. Also fibronectins are known to assemble into fibrils upon activation by integrins and align with intracellular actin stress fibers. In addition, elastin provides elasticity due to cross-linking and nidogen-1 connects laminin to collagen type IV. In contrast, glycosaminoglycans are carbohydrate-based polymers, which are highly negatively charged due to hydroxylate, carboxylate and sulphate groups and are able to bind many water molecules and different proteins. This strong solvability and the high water content facilitate high resistance to compressive forces, allow diffusion of various bioactive compounds, and stabilize and present growth factors.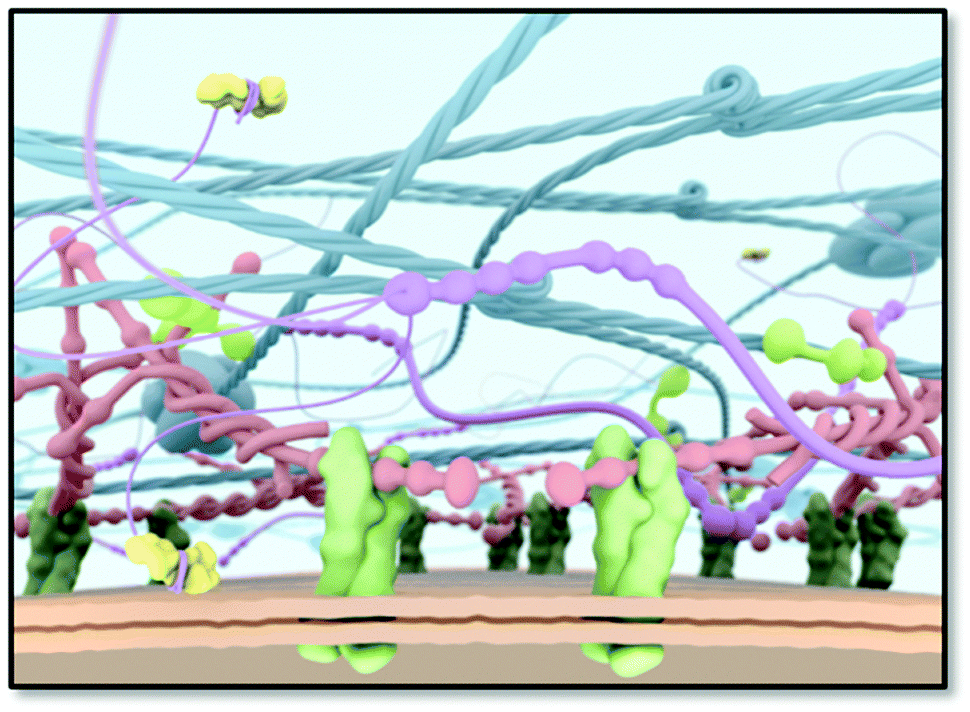 | ||
| Fig. 1 A schematic scheme of the ECM as a target for an artificial ECM mimic. Cell surface receptors (e.g. integrins; in green) bind to ECM proteins, thereby anchoring the cell to the ECM. | ||
All ECM proteins can exist in different isoforms that have slightly different functions. However, the common property is their ability to bind other ECM proteins by non-covalent interactions. Therefore, the ECM is an interconnecting network that links various ECM proteins together and to the cell surface via specific receptors such as the integrin receptors. The ECM can in general be divided in various types that are tissue and/or organ specific. Furthermore, the ECM is different in structure and composition at the cell surface (pericellular matrix), in epithelial and endothelial tissues (basement membrane), and in connective tissues (interstitial ECM). The basement membrane is a thin, dense area at the cell–ECM interface that is highly enriched in collagen type IV, laminin, perlecan and nidogen.3 It is known as the specialized ECM region which also regulates the cell behaviour. Integrin receptors present in the cell membrane connect the ECM with the intracellular cytoskeleton.4,5 Since integrins have the remarkable ability to signal bidirectionally, they are able to induce both intracellular and extracellular changes due to ECM or intracellular stimuli, respectively. In order to tightly bind to ECM components, multiple activated integrins must cluster together to form focal adhesions.6 The subsequent signal transduction conducted by integrins also occurs in response of physical forces, known as mechanotransduction.7 Mechanical forces, between cells, cells and the ECM, and the ECM itself determine the subsequent cell response. A high cytoskeletal tension causes differentiation, whereas a low tension maintains the undifferentiated state.8 Inspired by the ECM, many systems, both natural and synthetic, have been developed for the culture of various cells in vitro, and as scaffolds for guiding regeneration in vivo.
As the natural ECM is based on non-covalent interactions between the individual components and these fibrous structures are formed by the self-assembly of polypeptide chains, it is very logical that a synthetic mimic of the ECM is based on a multi-component supramolecular material in general and supramolecular polymers more specifically.9 These synthetic supramolecular polymers formed by the self-assembly of monomeric building blocks can be subsequently transformed into either hydrogels or elastomeric materials where biological functions can be integrated in a modular fashion. In this review we first discuss the development of supramolecular polymers, followed by their use as a synthetic ECM for in vivo function as load bearing scaffolds for valves and vessels, and subsequently as hydrogels for in vitro culturing of cells. The review concludes with a perspective on supramolecular materials for use in biomedical applications.
About supramolecular polymers
During the time that Jöns Jakob Berzelius (1779–1848) introduced the term polymers in 1832,10 it was thought that these substances consisted of ill-defined colloidal aggregates of small particles. It was not until 1920 that Hermann Staudinger (1881–1963) coined the term macromolecules – originally “Hochmolekulare Verbindungen” – in his famous article in the Berichte der Deutschen Chemischen Gesellschaft.11 Although it took time before the macromolecular concept was generally accepted, it dominated the field of polymer materials ever since. Many of the material properties of polymers are due to the entanglements of these covalently linked monomers in a long macromolecule. The idea of polymers made by molecular association, however, never really disappeared, although no claims were made to arrive at useful material properties. In a Chemical Review of 1929, G. G. Longinescu wrote “More than fifty years ago, Louis Henry first proposed the hypothesis of molecular polymerization, now called molecular association (Annales de la Société scientifique de Bruxelles 1878, 3, 267)”.12 Also some of the dynamic properties of imidazole in organic solvents were often explained by the formation of one-dimensional aggregates.After the introduction of the concept of supramolecular chemistry, chemists tried to design and study larger aggregates from small molecules, while some older studies were revisited. The recent developments for one-dimensional aggregates started in 1988 with the disclosure of the double hydrogen-bonded polymeric assembly by Wuest (Fig. 2).13 Although this first example was only a polymer in the crystalline state, similar to the crystals of terephthalic acid, it showed the option to design a polymeric array by multiple hydrogen bonds. The first supramolecular polymer in the liquid crystalline phase was published by Lehn in 1990 using a triple hydrogen bonded analogue.14 Here, two complementary monomeric units (A–A and B–B) were synthesized and assembled in (A–A:B–B)n polymeric structures and chiral fibre-like architectures were observed with electron microscopy. This important finding was followed by the mixing of a bis-acid and a bis-pyridine by Griffin and in this case the supramolecular complexes could be drawn into fibres displaying mechanical properties.15,16
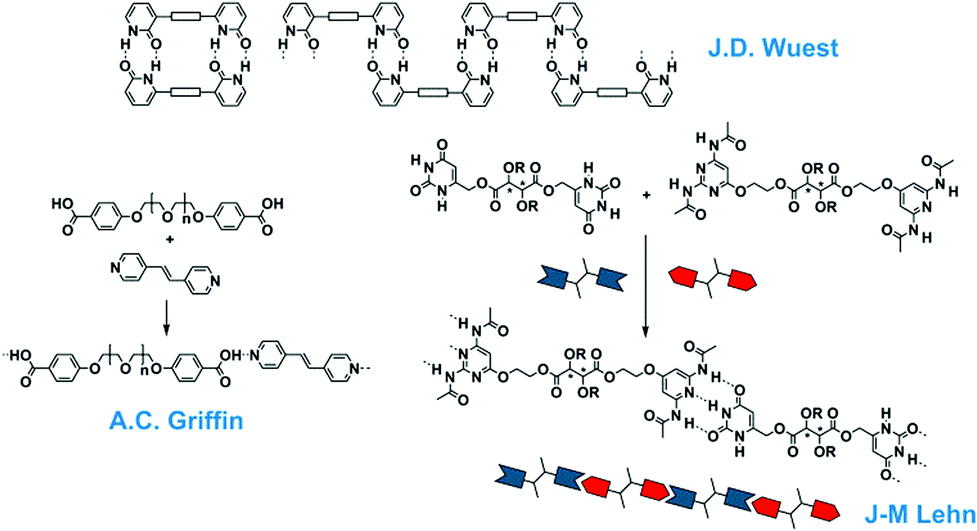 | ||
| Fig. 2 One-dimensional supramolecular aggregates with single-, double- and triple hydrogen bonding motifs. | ||
Although these beautiful first examples represent the birth of supramolecular polymers, the association constants between the repeating building blocks are actually too low to give high virtual molecular weights of these polymers in the solution, melt or amorphous state and hence these structures do not possess the typical material properties of synthetic macromolecules. Hence the idea to make mechanically strong polymeric materials by the linear association of small molecules remained in the realm of fantasy. That all changed when the easy to synthesize ureidopyrimidinone (UPy) self-complementary quadruple hydrogen bonding unit was introduced having a dimerization constant of Kdim = 10−7–10−8 M−1 and a life time of 0.1–1 second, both properties depending on the dielectric constant of the solvent.17 This UPy-group when coupled to both sides of a spacer yielded strong supramolecular polymers with macroscopic properties that were traditionally only reserved for macromolecules (Fig. 3). Films and fibres could be prepared, that at low temperatures, resemble in many ways the bulk properties of macromolecules. Obviously, the dynamic nature of the connection between the repeating units provided new options for processing; e.g. at higher temperatures the life time became shorter, and the viscosity was significantly reduced. Through the years the use of the UPy-motif expanded and a large number of different spacers, oligomers and even macromolecules were decorated with the UPy-motif. It became a central motif in the field of supramolecular polymer materials.18,19 Several additional supramolecular interactions were introduced and in some cases materials with combined interactions were prepared. A very successful combination of interactions yielded supramolecular thermoplastic elastomers (TPEs),20 a class of molecules that are used as temporary biomaterials for regenerative medicine (vide infra). Next to new options for processing, also unique self-healing properties of supramolecular materials were discovered that created novel applications.21 Finally, the modular approach, where different structures all modified with the same supramolecular motif, created unlimited modifications of the supramolecular polymers by just mixing.
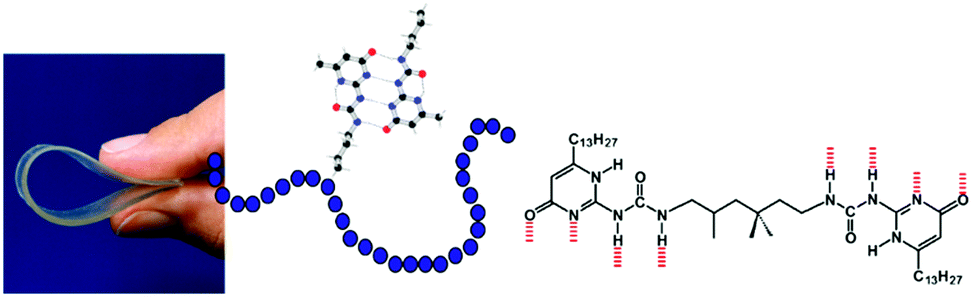 | ||
| Fig. 3 Supramolecular polymer materials based on the quadruple hydrogen bonded ureidopyrimidinone motif. | ||
Next to using supramolecular interactions between small molecules in making polymeric materials in bulk, supramolecular polymers in solution attracted considerable interest. These one-dimensional aggregates possess many of the properties of synthetic macromolecules in solution and in addition, they mimic many properties of natural filaments, like collagen and actin. Most of the polymers used as materials are random-coil polymers in solution and again show a strong similarity with macromolecules in dilute and concentrated solutions. However, where ordered polymers are formed in solution, the bulk properties of these materials are characterized by their liquid crystalline behaviour. Historically, J- and H-aggregates of many dye molecules can be regarded as the first examples of these one-dimensional aggregates in solution. However, in most of the early days, the aggregates were not very soluble and aggregated in the second and third dimension or even precipitated. The breakthrough came when discotic liquid crystals were assembled in dilute apolar solvents.22 The assembly process was based on solvophobic effects and resulted in micrometer-long one-dimensional supramolecular polymers.
Through the years, an enormous number of supramolecular polymers have been disclosed in a range of solvents. In particular, the self-assembly in organic solvents showed the strong similarity between these one-dimensional aggregates and macromolecules. For details the reader is referred to one of the many reviews written on this topic.23–26 Of more recent date is the study to understand the mechanism of formation of these supramolecular polymers, with an equal K-model for random coil polymers and a nucleation–elongation mechanism for ordered filaments. In the latter case, issues like pathway complexity, amplification of chirality, as well as the use of these polymers in electronic applications have been studied (Fig. 4).25
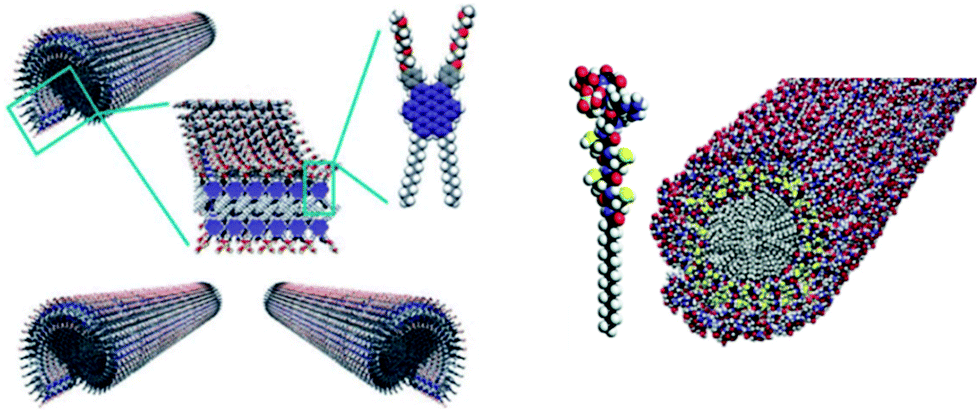 | ||
| Fig. 4 Functional supramolecular polymers with intriguing electronic properties originating from aromatic tubular amphiphiles (left) and biological applications originating from the peptide amphiphiles (right). | ||
A special class of one-dimensional supramolecular polymers is the one that is self-assembled in water, with the peptide amphiphile assemblies of Stupp as one of the most remarkable examples. These biologically relevant supramolecular polymers are studied in great detail for their applications in tissue engineering and regenerative medicine.27 They are studied both as isolated fibres and as part of hydrogels when the concentration of the peptide amphiphiles is increased. Many more supramolecular polymers in water are disclosed to form dynamic supramolecular hydrogels, and it became a very active field of research for biomaterials as artificial extracellular matrices; an important part of this review.
In the 25 years after the first publications on supramolecular polymers based on the one-dimensional assembly of small molecules, the field has grown to an important new branch of polymer science, where the modular approach in the assembly of multiple building blocks has opened new avenues to arrive at functional materials, most notably in the field of biomaterials.
Supramolecular elastomeric materials
Supramolecular polymers can greatly contribute in the development of mechanically strong TPE materials to be applied as elastic biomaterials. The modular character of these materials allows the introduction of complex (bio)functionality. Moreover, as a result of the interesting properties of the TPE polymer materials, they are envisioned to be applied in a broad range of regenerative medicine applications, i.e. in the cardiovascular field. Supramolecular TPEs are noncovalent analogues of block copolymers, composed of high molecular weight, low polarity segments (soft blocks) and low molecular weight, high polarity segments (hard blocks).28 The hard segments are composed of self-assembling motifs that are able to form (semi)-crystalline domains upon assembly, resulting in phase separation and endowing thermoplastic material properties. Supramolecular TPE materials contain reversible interactions, such as hydrogen bonds, in the main chain of the polymer.29 The mechanical properties of these polymers are a direct result of these secondary interactions. A glass transition temperature or a melt temperature above room temperature improves the mechanical properties of the materials since the hydrogen bonds act as crosslinks. However, these hydrogen bonds have limited mobility to rearrange when the glass transition temperature is above room temperature, which limits the self-healing capacity of the materials. When designing biomaterials for regenerative medicine applications, TPEs with glass transition temperatures well above 37 °C would ideally facilitate mechanical support at the site of implantation.The design criteria of a synthetic biomaterial that is able to support load bearing tissues, should meet a range of requirements including biodegradability, biocompatibility, elasticity, matching mechanical properties, processability, adaptability and modularity. Generally, these materials are processed using harsh conditions (i.e. organic solvent or high temperatures) but act at the interface in an aqueous environment. Evaporation of the organic solvent in which a supramolecular polymer is formed results in the formation of supramolecular bulk materials. Many processing methods have been reported in order to prepare porous scaffolds composed of micrometer fibers, including electrospinning, melt spinning, solvent casting, thermally induced phase separation and phase inversion methods. As a result of both the mechanical and physical strength as well as the elastomeric properties of supramolecular TPEs, these materials are applied in a broad range of regenerative medicine applications, including small-diameter vascular grafts, cardiac patches, blood vessels and renal living membranes.
In this section, we first describe the design criteria to enable the applications of supramolecular TPEs as fibrous multi-component biomaterials. Subsequently, three classes of supramolecular TPEs (polyurethanes, bisurea-based and UPy-based materials) are highlighted that form fibre-like structures in bulk. Moreover, we elaborate on their potential as supramolecular biomaterials in the field of regenerative medicine.
From supramolecular polymers to supramolecular materials
Two classes of supramolecular TPE can be distinguished: materials prepared from one-dimensional assemblies of supramolecular building blocks (i.e. supramolecular polymers), and materials prepared through chain extension of oligomers or via crosslinking of polymeric precursors by supramolecular interactions (i.e. supramolecular materials). Ordered, noncovalent interactions, i.e. hydrogen bonding, host–guest interactions and electrostatic interactions, contribute to the design of the material and account for the order of dynamics that is involved in the supramolecular assembly of these materials. Within supramolecular biomaterials, a complex interplay between material property parameters determines the ultimate material properties.9 These materials should be able to mimic and adapt to the native environment of the cell while at the same time biocompatible, biodegradable and mechanical properties are desired.30 Advances in the field of synthetic polymers have increased the use of biomaterials in biomedical applications.31 The intrinsic dynamic behaviour of supramolecular assemblies has received significant interest in the past decades. The development of life-like materials dictates many design criteria in synthesizing supramolecular TPEs. Among others, we propose the following criteria to be important when supramolecular TPE materials are used in vivo, in an in situ tissue engineering approach:1. The mechanical properties should meet the requirements of the native environment
2. The biomaterial should display bioactive and biomimetic modules
3. The material should exhibit adaptive properties
Supramolecular polymers based on the poly-urethane motif
Thermoplastic polyurethanes (TPUs) were first developed in the late 1930s and to date are widely used in industrial applications due to their interesting properties. In the late 1960s polyurethane (PU) elastomers were first considered as potential biomaterials by Boretos and coworkers.32 These are TPE materials composed of hard and soft segments that can be synthesized via the reaction between diisocyanate and a diol. The soft blocks are formed by a polyol and an isocyanate, and are responsible for the flexibility and elastomeric character of the materials. The hard block, constructed from a chain extender and an isocyanate results in the toughness and physical performance of the TPU material. High polarity segments in the hard phase result in crystalline domains whereas low polarity segments determine the soft flexible matrix phase. The elastic behaviour of TPU arises from the crystalline domains that act as physical crosslinks in the material and the elongation behaviour is determined by the flexible chains. These properties result in high elongation, tensile strength and elastic behaviour. Moreover, at increased temperatures, TPU becomes soft and easily processable, and upon cooling the material hardens.The introduction of fibrous assemblies is achieved via supramolecular TPU materials, which benefit from reversible and stimuli-responsive behaviour arising from noncovalent links between the repeating units for chain extension that rely on intermolecular recognition to form complex architectures. In contrast to covalent polymers, temperature increase in supramolecular TPU leads to dissociation of the noncovalent bond, resulting in a decrease in viscosity and mechanical properties allowing for easy processability, self-healing and shape memory properties.33 Fiber-like morphologies in supramolecular TPU materials are formed as a result of both lateral and end-to-end interactions between the polymer chains. This demonstrates a cooperative behaviour between the end-to-end and lateral supramolecular interactions.20
Linear polyurethanes with a soft segment of poly(tetramethylene oxide) (PTMO) and toluene diisocyanate diamide (TDI) as a urethane segment were prepared to study structural, morphological, thermal and mechanical properties of the materials. Monodisperse TDI with discrete supramolecular units exhibits high crystallinity, which results in a curved nano-ribbon morphology. The effect of the hard segment concentration and the TDI units in the soft segment play an important role in determining the copolymer properties.34 Both thermomechanical and morphological properties were investigated for their impact on bioinspired hierarchical ordering in segmented polyurethane/urea (PUU).35 The polymers contained either a peptidic, triblock soft segment or an amorphous, nonpeptidic block soft segment with an amorphous or a crystalline hard segment. The peptidic soft segment results in the formation of intermolecular hydrogen-bonded β-sheet conformation, at low molecular weight (<10 peptide residues). Hard segments were composed of either a crystalline 1,6-hexamethylene diisocyanate or an amorphous isophorone diisocyanate and chain extended with 1,4-butanediol. The PUU showed phase separation behaviour as was observed by thermal and morphological characterization. Moreover, including the peptide segment resulted in an increase of the long spacing between the domains and retained β-sheet organization, which demonstrated that additional ordering in segmented PUU enables tunability in mechanical properties. These insights can be applicable in the development of advanced biomaterials. Interestingly, small variations in the binding constant of the end groups in polyurethane-based TPEs provides important insights into the design of polyurethane-based materials in which weak noncovalent interactions can be used to tune the self-assembly of the materials.36 It turned out that the hydrogen bonding end-group could influence the binding constants in solution and bulk as well as mechanical properties when changing from dibutyl to morpholine to dial in combination with urea groups. These results signify the importance of material design in the advance of supramolecular polymeric materials.
The main classes of TPU materials that find applications in the biomedical field consist of polyester TPU, polyether TPU and polycaprolactone TPU. TPU biomaterials are of particular interest as materials for tissue engineering, where the mechanical properties are the most important design criteria. Wagner and coworkers developed a biodegradable electrospun TPE based on poly(ester urethane) urea (PEUU). In a recent study they report on biodegradable elastomeric coatings with drug-eluting properties for application in degradable vascular stents based on magnesium.37 The performance of coatings based on poly(carbonate urethane) urea (PCUU) and poly(ester urethane) urea (PEUU) coated magnesium alloys were compared. It was shown that the PCUU coating effectively slowed down the magnesium alloy corrosion in dynamic degradation testing compared to the PEUU coated stents. Moreover, a significant reduction in platelet adhesion was observed and the release of an antiproliferative agent showed effective inhibition of rat smooth muscle cell proliferation. In another study, electrospun PEUU constructs were used to elucidate the role of biaxial strain on ECM secretion by vascular smooth muscle cells (VSMCs).38 This was one of the first reports which considered the effects of physiologically relevant deformations in terms of ECM synthesis and culminates the understanding of developmental processes of the ECM. This broadens the scope in terms of engineering materials that can be applied in highly demanding mechanical environments. In the design and development of small diameter tissue engineered vascular graft (TEVG) scaffolds, the mechanical and structural properties should closely mimic those of the native vessel. Moreover, cell integration, adhesion and growth should be facilitated. A PEUU small diameter, bilayered, biodegradable, elastomeric scaffold was developed with a highly porous inner layer.39 The inner layer was produced via thermally induced phase separation (TIPS), which allows cell integration and growth, and an external microfibrous reinforcing layer (∼50 μm) was produced via electrospinning. Morphological and mechanical characterization showed similar properties compared to native arterial structure, strength and elasticity. Moreover, the scaffold showed firm integration of the two polymer layers and no delamination. This approach is envisioned to represent a step forward towards future clinical translation.
In another study, Wagner and coworkers investigated the effect of a bi-layered polyurethane-based extracellular matrix cardiac patch in ventricular wall remodelling in a rat model.40 In the material design, the ventricular mechanical support was taken into account via electrospinning of the poly(ester carbonate urethane)urea (PECUU) material (Fig. 5a) as well as the incorporation of ECM-based hydrogel components (Fig. 5b). The cardiac patches were spicardially implanted in rats and evaluated 10 weeks after infarction. The results showed a favourable remodelling response and improved functional outcome when the ECM hydrogel part was integrated into the patch. The amount of host cell infiltration in these patches was higher compared to PECUU patches that have a different fibre orientation (Fig. 5c). This study shows the benefit of a cardiac patch design that combines both mechanical properties and ECM-based bioactive components.
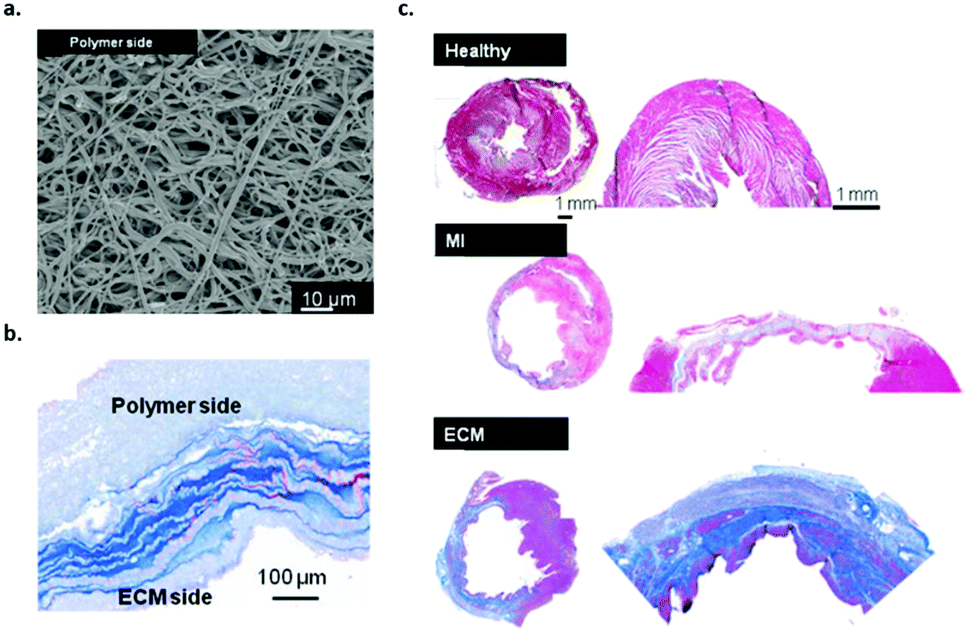 | ||
| Fig. 5 Bilayered polyurethane-based cardiac patch based on PECUU, (a) SEM image of the material pre-implantation, (b) Masson's staining of the bilayered scaffold patch cross-section showing both polymer and ECM rich layers and (c) MT staining of the whole heart and infarct/patch regions after 8 weeks, healthy muscle and scar tissue (upper image), after MI (middle image) and the bilayered PECUU materials as a heart valve implant (lower image). Modified from ref. 40 with permission from Elsevier, copyright 2016. | ||
The materials based on supramolecular TPU described here all display mechanical properties that meet the requirements of cardiovascular applications, where not only mechanical strength but also elasticity of the materials is important. Moreover, bioactive properties can be introduced via the incorporation of bioactive urethane-based moieties, which could direct specific behaviour in vivo. As far as biomimetic properties are concerned, the TPU materials show similar mechanical properties compared to native arterial structures. Adaptive behaviour is envisioned to be easily incorporated. Upon implantation, cardiovascular biomaterials are expected to execute their function immediately, and should respond to the native environment.
Supramolecular thermoplastic elastomers based on the bisurea motif
Polyamide and polyurethane TPE materials can be processed at elevated temperatures. Upon hydrogen bonding between the polymer chains noncovalent crosslinks are formed that are able to induce crystallization. As there are noncovalent crosslinks present in these materials, these materials could be regarded as supramolecular polymers, but since the entanglements of the high molecular weight polymer chains have an influence on the macroscopic properties, they do not account for true supramolecular polymers (i.e. polymers in which the supramolecular monomers are held together via directed, non-covalent interactions). Nevertheless, the mechanical properties as well as the processability of polyamides and polyurethanes have been an inspiration in the synthesis and development of new polymers where amide and urethane functionalities are replaced with urea motifs, which form bifurcated hydrogen bonds with higher binding energies. Although the N,N′-dimethylurea and N,N′-diethylurea motifs are well known to self-assemble in nonpolar solvents, the introduction of branching increases solubility. Lortie and coworkers showed that by choosing adequate substituents, an increased solubility of urea compounds in nonpolar solvents can be obtained. These types of A–B monomers self-assemble to form supramolecular polymers.41 The effect of soft segment molecular weight on the structure–property relationship of polyurea was investigated in copolymers composed of poly(tetramethylene oxide) (PTMO) soft segments and diisocyanate (DI) (either 1,6-hexamethylene DI (HDI), 1,4-phenylene DI (pPDI) or 1,4-trans-cyclohexyl DI (CHDI)). The polyurea showed microphase separated structures with fibre-like hard segments randomly distributed throughout the soft segment. Upon deformation beyond the yield point, the fibres breakup into smaller fibre-like structures, which appeared to be partially reversible and time dependent. Both 1k and 2k PTMO HDI polyurea showed thermally stable behavior and would potentially be melt-processible.42Upon the reaction of amine-functionalized oligomers with diisocyanates, bisurea TPE could be synthesized that shows nanofiber morphology. The mechanism at which the urea motif aggregates is cooperative since the formation of dimers is less favourable due to the alignment of the dipole moments. In addition, the bisurea motifs bundle together and crystallize into long nanofibers that act as supramolecular crosslinks. This material behaviour results in reinforcement properties of the materials and provides enhanced mechanical properties. Moreover, the hydrogen bond strength exceeds that of amides and urethanes. Co-poly(ether urea)s have been synthesized with poly(tetrahydrofuran) (pTHF) soft segments and hard blocks consisting of a controlled amount of 1–4 urea groups. Material properties range from a viscous liquid (for 1 urea group) to insoluble gel-forming polymers (for 3 or 4 urea groups). Materials composed of 2 urea groups in the hard block showed satisfying mechanical and processing properties, since the materials are both elastic and soluble.43 Elucidating the morphology of these TPEs composed of 2 urea groups as the hard block revealed that fibres were formed.44 The incorporation of a tris-urea motif into poly-dimethylsiloxane (PDMS) chains gave rise to the formation of materials that display self-repairing properties.45
In order to investigate molecular recognition in bisurea TPEs, bisurea-pyrene probes were synthesized and mixed with the bisurea pTHF block copolymers with matching or nonmatching bisurea blocks.46 It was shown that the bisurea-pyrene probes randomly dispersed in the hard blocks of the matching bisurea blocks. Upon mixing with the nonmatching bisurea motif phase separation was observed. Supramolecular recognition between bisurea additives in a TPE host polymer results in selective modulation of mechanical properties.47 Moreover, when the bisurea additives were equipped with matching bisurea groups, these additives were randomly dispersed into the hard blocks of the TPE whereas non-matching bisurea additives phase separate from the polymers.46 This supramolecular self-sorting mechanism can be applied in order to selectively incorporate matching bisurea motifs into the material and thereby introduce functionality, i.e. bioactivity in order to improve cell adhesion to the TPE materials,48 or mechanical fillers to improve the mechanical properties of the materials.49 As a result of the high melting temperature, the mobility of the bisurea hydrogen bonding motifs is reduced and therefore these TPEs do not exhibit self-healing properties. The crystallization properties of the bisurea based TPE provides them with favourable mechanical material properties.
Leibler and coworkers developed a rubber-like system with recoverable extensibility.21 Network formation occurs after mixing ditopic and multitopic molecules with (amidoethyl)-imidazolidone, bis(amidoethyl)urea and diamidotetraethyltriurea side moieties which act as multiple-hydrogen-bonding crosslinkers. Upon fracturing, the materials self-heal when the two surfaces were brought together at room temperature. This behavior was shown to be reproducible many times. At low temperatures hydrogen bonds act as crosslinks and result in soft rubber-like material properties, whereas at elevated temperatures these hydrogen bonds were broken yielding a viscoelastic liquid.
Recently, Bouten and coworkers developed a bioresorbable TPE implant material based on bisurea modified polycarbonate (PC-BU) (Fig. 6a) that was processed into a heart valve and implanted in vivo in sheep.50 As a proof-of-concept study, an electrospun cell-free valvular implant (Fig. 6b) was implanted to form new valvular tissue inside the heart using an in situ tissue engineering approach. The materials showed sustained functionality up to 12 months and upon degradation of the material a layered collagen and elastic matrix was produced to replace the implant (Fig. 6).
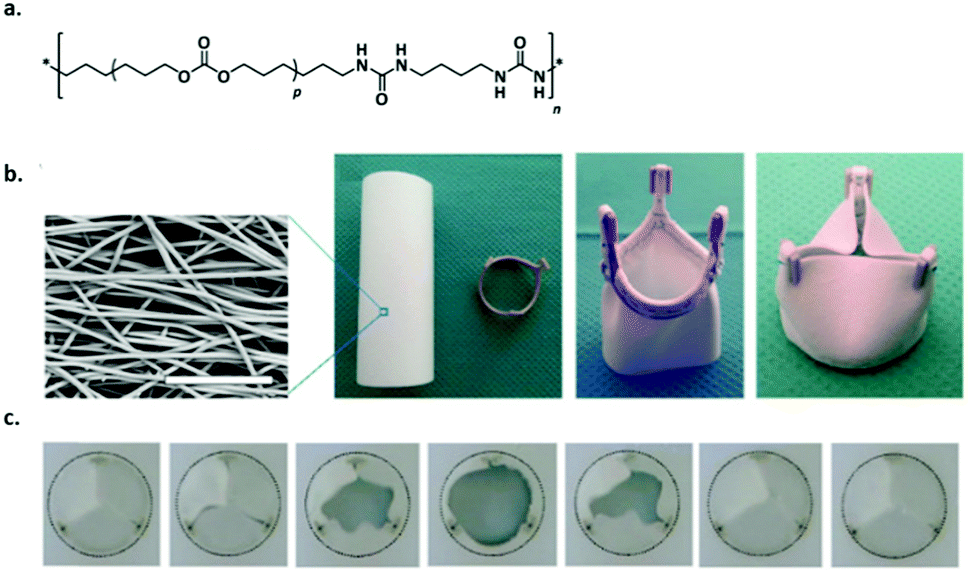 | ||
| Fig. 6 Supramolecular polymer materials based on (PC-BU) as heart valve implants, (a) chemical structure of the supramolecular polymer (p is on average 16–17), (b) SEM image of the fibrous microstructure of the valve, which is composed of a tube that is sutured onto a reinforcement crown (scale bar represents 50 μm) and (c) movie stills of an in vitro valve functionality test to demonstrate proper opening and closure of the leaflets. Modified from ref. 50 with permission from Elsevier, copyright 2017. | ||
As outlined in this section, bisurea based supramolecular TPEs exhibit excellent mechanical properties due to bundling and crosslinking of the bisurea motifs, and subsequently crystallize into long nanofibers that exhibit enhanced mechanical properties. This endorses bisurea based supramolecular TPE materials to be used as scaffold materials in regenerative medicine, in particular in the cardiovascular field. The modularity of the bisurea motifs allows for the incorporation of functionality that is required by the site of implantation in the native environment. Along these lines, bioactivity can easily be incorporated via this modular platform. Moreover, the adaptive material properties can be tuned, i.e. material composition, and responsiveness to the native environment can easily be altered.
Supramolecular polymers based on the ureido-pyrimidinone (UPy) motif
The main advantage of supramolecular polymers is their strong dependence of the melt viscosity on temperature. Above the materials melting point, small increases in temperature lead to large reduction in viscosity, which ensures easy processability into a variety of different materials.51 The development of the self-complementary quadruple hydrogen bonding motif based on 2-ureido-4[1H]-pyrimidinone (UPy) has led to the development of a new class of supramolecular polymers (vide supra). Upon end-functionalization of polymers with UPy-moieties, difunctional supramolecular polymers can be generated, resulting in the formation of stable and long polymer chains both in solution and in bulk. UPy-functionalization as chain extension in telechelic polymers improves the material properties, i.e. combining the mechanical properties of conventional macromolecules and low melt viscosity of low molecular weight organic compounds. Moreover, reversibility adds interesting properties to the materials giving rise to copolymers with different compositions and ensures self-healing supramolecular network formation. The nanofiber formation involves a hierarchical process, which starts from the phase-separated melt followed by dimerization of UPy-units. In order to laterally aggregate into high aspect ratio nanofibers, urea functionalities as well as a non-substituted five position are required. Nanofibers form as a result of 1D stack formation along with secondary nucleation of multiple stacks. Moreover, stack formation and the formation of nanofibers can be suppressed upon the introduction of branching at the six position of the UPy-motif. These details provide useful insights into the kinetic behaviour of nanofiber formation and therewith can be used to design adaptable supramolecular materials.52The ease of processing of these supramolecular polymers in the melt or in solution, the excellent properties in the solid state, their self-assembling compatibility and the reversibility of the supramolecular motif yields materials that are perfectly suited as supramolecular biomaterials.53 It has already been shown that via a modular approach, functionality can be introduced into these materials. Upon the functionalization of peptides with a UPy-moiety, these UPy-peptides can be mixed with a UPy-polymer and easily processed into a material. The introduction of bioactivity via this modular approach has shown great potential, both in in vitro as well as in vivo analyses (Fig. 7).54 Upon mixing chain extended UPy-modified polycaprolactone (CE-UPy-PCL) with bifunctional UPy-modified poly(ethylene glycol) (PEGdiUPy) and subsequent electrospinning into vascular grafts, it was shown that cell infiltration in vivo could significantly be reduced (Fig. 7b and c).55 Bilayered electrospun scaffolds composed of UPy-modified UPy (PCLdiUPy) on one side and a mixture of PCLdiUPy and PEGdiUPy at the other side (Fig. 7d) successfully suppressed cell adhesion on the PCLdiUPy mixed with the PEGdiUPy side of the scaffold. Upon the introduction of 4 mol% UPy-modified GGRGDS-peptide, the scaffolds could be reactivated and cells were able to attach (Fig. 7e). This unique approach demonstrates the modularity of the UPy-based biomaterials.56 The successful incorporation of either bioactive cues or non-cell adhesive moieties intro supramolecular TPE materials based on the UPy-motif via a modular approach provides a platform for a multitude of regenerative medicine applications, i.e. renal and cardiovascular applications.57–61
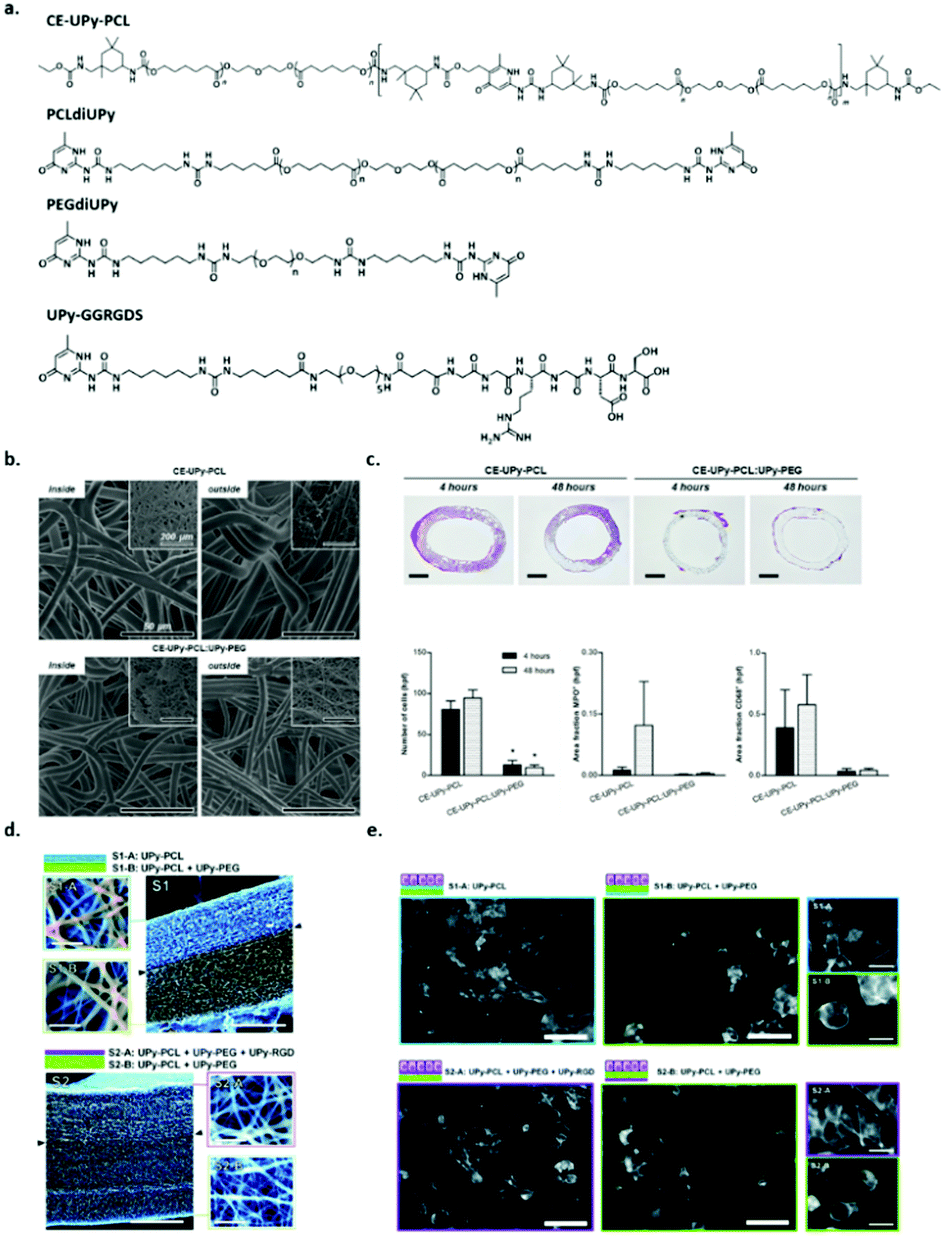 | ||
Fig. 7 Supramolecular polymer materials based on ureido-pyrimidinone (UPy), (a) chemical structures of CE-UPy-PCL, PCLdiUPy, PEGdiUPy and UPy-GGRGDS, (b) SEM images of the morphology of both the inside (lumen) and the outside of the electrospun vascular grafts of CE-UPy-PCL (top) and CE-UPy-PCL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :UPy-PEG (90:10) (bottom) and an inset of the final construct (top left), scale bars represent 50 μm, (c) cross sectional slices of the grafts both 4 and 48 hours after implantation, scale bars represent 500 μm. Adapted from ref. 55 with permission from Wiley, copyright 2015. (d) Quantitative analysis of histological data showing reduced cellularity in CE-UPy-PCL:UPy-PEG vascular grafts (left), MPO positive granulocytes (middle) and CD68+ macrophages (right) relative to tissue area. Bar graphs represent infiltrating cell numbers from four representative high power fields (hpf) per tissue section analyzed at 400× magnification. The data are represented as mean ± S.E.M., (e) SEM micrographs of the electrospun bilayered scaffolds S1 and S2, composed of PCLdiUPy, PEGdiUPy and UPy-GGRGDS building blocks (scale bars represent 200 μm). Top and bottom views show fiber diameters and pore sizes of the scaffolds (scale bars represent 5 μm) and (f) fluorescent microscopy images of HK-2 cells on the different scaffolds 14 h after seeding, scale bars represent 200 μm, in the enlarged views (right) scale bars represent 25 μm, the morphological differences between the cells on the different scaffolds are clearly present. Adapted from ref. 56 – published by the Royal Society of Chemistry. :UPy-PEG (90:10) (bottom) and an inset of the final construct (top left), scale bars represent 50 μm, (c) cross sectional slices of the grafts both 4 and 48 hours after implantation, scale bars represent 500 μm. Adapted from ref. 55 with permission from Wiley, copyright 2015. (d) Quantitative analysis of histological data showing reduced cellularity in CE-UPy-PCL:UPy-PEG vascular grafts (left), MPO positive granulocytes (middle) and CD68+ macrophages (right) relative to tissue area. Bar graphs represent infiltrating cell numbers from four representative high power fields (hpf) per tissue section analyzed at 400× magnification. The data are represented as mean ± S.E.M., (e) SEM micrographs of the electrospun bilayered scaffolds S1 and S2, composed of PCLdiUPy, PEGdiUPy and UPy-GGRGDS building blocks (scale bars represent 200 μm). Top and bottom views show fiber diameters and pore sizes of the scaffolds (scale bars represent 5 μm) and (f) fluorescent microscopy images of HK-2 cells on the different scaffolds 14 h after seeding, scale bars represent 200 μm, in the enlarged views (right) scale bars represent 25 μm, the morphological differences between the cells on the different scaffolds are clearly present. Adapted from ref. 56 – published by the Royal Society of Chemistry. | ||
The use of UPy-based supramolecular TPEs has enormous potential in a broad range of applications, including cardiovascular grafts, renal membranes and blood vessels. Due to the strong hydrogen bonding affinity between the UPy-motifs, networks based on nanofibers are formed and the mechanical performance as well as the responsive properties of the materials can be tuned by the choice of prepolymer that can be modified with the UPy-motif. Moreover, due to the modular character of these materials, functionality can easily be introduced. Upon modification of functional cues with a UPy-motif, bioactive or antifouling behaviour can be introduced. By the introduction of a reactive additive, more complex structures can be introduced at the surface of the material using a post-modification approach, which further expands the applicability of these materials, as mimicking the native environment can be improved. Supramolecular UPy-based TPEs were developed in which a reactive UPy-based additive was incorporated, that could enable post-modification of the surface via click chemistry after material preparation (Fig. 8a). Via this strategy, the processing conditions of both the materials and complex analytes (i.e. proteins and growth factors) could be decoupled, as they are highly incompatible. The post-modification approach is based on the efficient and selective inverse Diels–Alder cycloaddition between tetrazine and trans-cyclooctene, which is reported as the fastest bioorthogonal ligation strategy, with a k2 of 103–106 M−1 s−1.62–64Via the modular approach, UPy-functionalized tetrazine (UPy-Tz) moieties are incorporated into the supramolecular TPE material that incorporate into the polymer nanofibers (Fig. 8b). Surface functionalization is proposed via a selective reaction of UPy-Tz with trans-cyclo-octene modified bioactives (Fig. 8c–e). This decoupled modification strategy greatly expands the scope of biomolecules that can be introduced at the surface of the supramolecular TPE. This approach has been shown to be suitable for the selective introduction of an anti-fouling coating at the supramolecular TPE surface as well.65 Moreover, a variety of material preparation methods (i.e. 3D-printing, melt spinning and electrospinning) can be realized, which hold great promise for the production of functional supramolecular biomaterials that can be applied in the field of regenerative medicine.66
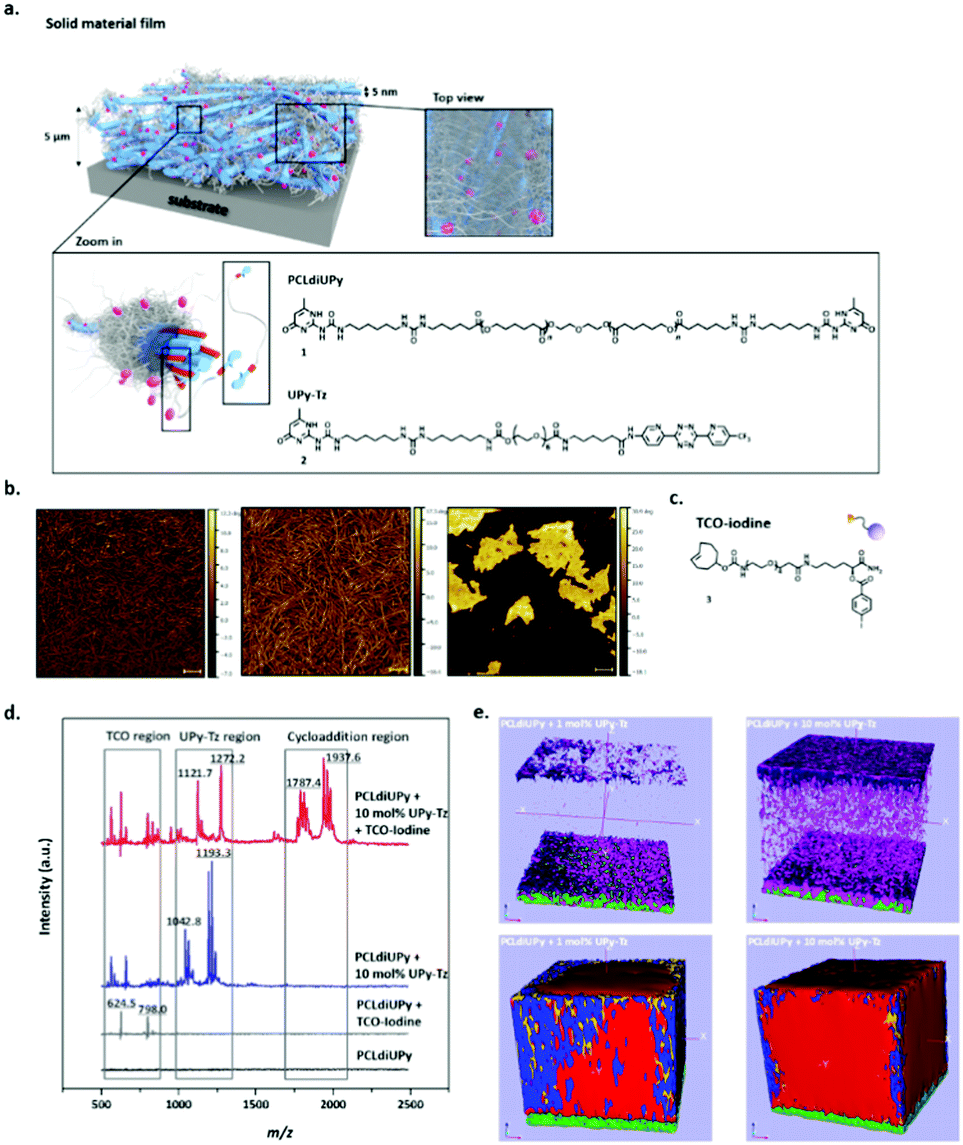 | ||
| Fig. 8 Schematic representations of the surface modified supramolecular materials, (a) the fibers within this solid network (depicted in blue) are composed of bundles of stacked UPy–UPy moieties in the lateral direction, forming a hard phase of ∼5 nm in diameter. The soft phase is composed of the PCL polymer chains (depicted in grey) filling up the total space in between the hard phase (as is common for traditional TPE). In a modular fashion, UPy-functionalized tetrazine guest moieties (UPy-Tz, (2), pink dots) can be incorporated in the core of these nanofibers formed by the PCLdiUPy (1) molecules. The top view provides an overview of the solid film from above, demonstrating the dense characteristic of the films, (b) AFM phase micrographs of PCLdiUPy with 1, 5 and 10 mol% UPy-Tz incorporated, respectively, (c) chemical structure of a TCO-iodine that can react at the surface of the supramolecular film, (d) surface MALDI-ToF MS analysis of PCLdiUPy (black line), PCLdiUPy incubated with TCO-iodine (grey line), PCLdiUPy with 10 mol% UPy-Tz (blue line) and PCLdiUPy with 10 mol% UPy-Tz incubated with TCO-iodine (red line) and (e) ToF-SIMS depth profile of PCLdiUPy and PCLdiUPy with 10 mol% UPy-Tz, relevant mass fragments are depicted in different colors: iodine = purple, fluorine = pink, UPy-fragment m/z 124 = red, UPy-fragment m/z 150 = blue, PCL-fragment = yellow, ITO = green. Adapted from ref. 66 with permission from Wiley, copyright 2016. | ||
Advances of supramolecular thermoplastic elastomers to supramolecular biomaterials
Ideal biomaterials effectively combine tunable mechanical performance, regulation of degradability, ease of bioactivity incorporation, and the ability to mimic the natural environment of the implantation site in the patient. Biological systems are exceptionally complex, and there remains a great need to develop synthetic materials capable of recapitulating the structural and functional complexity of biological materials in order to create truly mimetic systems for enhanced therapeutic function. Supramolecular biomaterials can replicate aspects of structural and/or functional features of biological signal transduction. Biomaterials that can replace or recapitulate deficient native materials or signalling pathways could be especially useful for applications in regenerative medicine or tissue engineering. As synthetic scaffolds, supramolecular biomaterials can act as structural mimics of fibrous matrix components.9Tailoring the supramolecular polymer chemistry to develop next generation biomaterials requires new fabrication technologies in order to more closely mimic the native environment in vivo. The benefits of supramolecular polymers over conventional polymers arise from their inherently dynamic nature and the corresponding beneficial material properties. More specifically, the fibrous supramolecular assemblies that have been disclosed here benefit from their modular character to enable additive incorporation into the supramolecular polymer fibres. While the mechanical performance is hardly influenced by additive incorporation, this strategy enables the introduction of specific function into the material.
Supramolecular polymers as hydrogelators
Hydrogels form an attractive class of biomaterials since their aqueous environment can mimic the ECM. The hydrophilic polymers are able to absorb up to 99% of water, allowing for the encapsulation of cells under physiological conditions. Next to the water molecules bound to the polymer chains, free water molecules filling up the space in the network are able to allow for nutrient diffusion.67 This provides a supportive 3D environment closely resembling the in vivo situation. Hydrogel networks are formed by cross-linking and/or physical chain entanglements, of which the cross-linking mechanism can be mainly categorized based on covalent or physical interactions. Covalent networks are generally permanent whereas physical networks are reversible. Hence, the polymer concentration and/or the number of cross-links can be regulated to tune the macroscopic properties.Since the ECM is highly dynamic, hydrogels based on reversible cross-links better recapitulate the native environment.9,68 Non-covalent supramolecular interactions are highly dynamic where the non-covalent bond is in equilibrium. Several types of supramolecular interactions exist, usually in the form of directional hydrogen bonds, electrostatic interactions, π–π interactions and hydrophobic effects. Networks can be formed where supramolecular moieties are presented along a polymer backbone, however, fibre-like nanostructures upon self-assembly of amphiphilic monomeric units closer mimic the fibrous structures found in nature e.g. the basement membrane. Particularly collagen type IV, which is known as the network forming collagen solely present in the basement membrane, is only a few nanometers in diameter due to the entanglement of only three peptide chains around each other, rather than bundled fibrils.3,69 Also laminin (∼2–7 nm) and fibronectin (∼2 nm) consist of long peptide chains of only a few nanometers in diameter.70
In one-dimensional nanofibers formed by self-assembly in aqueous solutions, the amphiphilic character of the monomeric units is essential. The hydrophilic part allows for water solubility, whereas the non-covalent interactions should be protected from water penetration. Tight packing, due to stronger hydrophobic effects and/or hydrogen bonding results in stiffer macroscopic material properties, whereas disordered packing results in softer hydrogels due to increased water penetration. Therefore the internal dynamics and macroscopic physical properties of supramolecular hydrogels can be tuned by varying the packing of the monomers within the fibre.71,72
The ECM provides a niche to the embedded cell, regulating the behaviour and the survival of cells. Inspired by this phenomenon, naturally derived or synthetic ECM mimics provide an excellent support for the culture of cells and stem cells. These platforms play important roles in e.g. elucidating the role of specific compounds in cell behaviour, directing the differentiation of stem cells into specific cell lineages, and up-scaling of specific cells or multi-cellular structures for regenerative medicine purposes.73,74 In particular, organoids, which are in vitro cultured multi-cellular structures from pluripotent or adult stem cells, are becoming increasingly important in the biomedical field since they closely recapitulate the in vivo organs and can possibly be used to regenerate diseased tissue.75,76 Hence these organoid platforms can be used to obtain valuable insights into tissue development, homeostasis and disease progression. So far, the culture of organoids is optimized in animal derived culture media, like Matrigel (which is explained in the natural hydrogels section), which gives rise to organoids which are variable in size and viability, therefore lacking the reproducibility properties of in vivo organs. By using biomaterials, a more precise control of the environment can be obtained which might lead to more consistent organoid structures.
With the purpose to culture stem cells in a 3D environment for biomedical applications, it is important that the hydrogel meets certain design criteria. Here, we propose a few design criteria that are considered important for the design of hydrogels, where the key factors of the ECM – ECM–cell interactions, physical properties and growth factor presentation are included:
1. Cells should be encapsulated under physiological conditions.
2. Control of the physical properties of the hydrogel is important to match the in vivo situation.
3. Cell adhesion sites should be incorporated in the hydrogel.
4. The hydrogel should allow dynamics, like degradation, remodelling, and capturing of secreted ECM components.
5. High cell viability and long-term expansion should be supported as well as the maintenance of a specific phenotype.
6. The hydrogel should be able to capture, stabilize and release tissue specific growth factors.
Natural hydrogels
The most frequently used naturally derived hydrogel for the expansion of stem cells is Matrigel. Matrigel contains extracted extracellular matrix proteins from Englebreth-Holm Swarm sarcoma cells which provide an optimal and highly biofunctional natural environment for stem cells, hence maintaining the self-renewal and pluripotency of the embedded stem cells.77,78 Despite being very useful for the long term expansion of stem cells, Matrigel has several disadvantages: (1) the composition is highly heterogeneous, with a batch-to-batch similarity of only 50–60%, causing reproducibility problems; (2) it is extracted from tumor cells preventing clinical use and (3) the stiffness of the hydrogel is extremely soft which cannot be tuned to match the tissue of origin.To overcome the last two challenges, tissues have been isolated and decellularized to obtain tissue specific ECMs. Upon the removal of the cells, the shape, structure and components can be maintained and have shown promising results in vitro and in vivo.79 However, decellularization methods can damage the composition, resulting in dysfunction or even a loss of important ECM components. Since the composition is hard to control, synthetic alternatives are proposed to provide a better solution.
Synthetic covalent hydrogels
Many covalent hydrogels are developed that show great promise for the 3D culture of stem cells. Here we would like to highlight a few breaking developments also important for the advances in supramolecular hydrogels. Compared to many research studies performed on 2D substrates, where the physical properties and epitope presentation direct cell lineage differentiation and spreading behaviour, in 3D systems, the micro-environmental cues have a significantly different impact on the cell response.80 To better recapitulate the in vivo situation, a 3D environment where the cell should be able to remodel its environment is required. Both the groups of Mooney81 and Chen82 showed that the traction forces applied by the cells are able to reorganize the RGD ligands into clusters, which is not possible on extremely rigid substrates. Although the crosslink density can be tuned to match the mechanical properties of the in vivo situation, Lutolf and coworkers showed that when increasing the crosslink density, not only was cell spreading and proliferation restricted, but also 3D cell migration was hampered.83,84 In contrast, cell spreading and migration were promoted when enzymatic cleavable crosslinks were incorporated into the design allowing the cell to locally degrade the surrounding environment. Next to enzymatic degradation of hydrogels, also hydrolytic degradable hydrogels were employed to control the degree of degradation, for example by Burdick and coworkers.85Finally, we would like to highlight the importance of growth factor sequestering and stabilization to protect growth factors from denaturation. Maynard and coworkers screened several covalent polymers for their ability to stabilize proteins, showing the importance of installing sulfonated, zwitterionic or trehalose side chains on polymers.86,87 By immobilizing growth factors using electrostatic interactions, significantly lower amounts of growth factors are required, and a prolonged presentation to growth factor receptors is achieved.
The lessons learned from covalent systems indicate that there is an increasing need for dynamic systems, allowing clustering of epitopes and remodelling of the ECM, local degradation of the hydrogel to allow expansion and/or migration, and shielding of growth factors from inactivation yet presenting them to the cell. Supramolecular hydrogels have the advantage over these covalent systems that they are intrinsically dynamic, responsive, adaptive, modular and tunable.9 Furthermore, as stated before, fibrous structures closer resemble the ECM environment, therefore, here we focus on hydrogel systems formed by the assembly of fibrous structures.
From covalent to supramolecular hydrogels
In order to allow local adaptable properties yet maintaining long-term stability of a hydrogel, several hybrid systems have been developed.88 Incorporation of reversible bonds requires the use of non-covalent interactions or dynamic covalent interactions. In dynamic covalent chemistry, covalent bonds can be temporarily broken and reformed again.89 Although interesting self-healing materials were developed under physiological conditions,90 reversibility is usually slower than non-covalent interactions and might require the addition of a catalyst. Moreover, when crosslinking kinetics are too slow, cellular adhesion might be hampered, which results in sedimentation of cells to the bottom of the culture plate causing unfavourable 2D growth instead of 3D. Non-covalent moieties have been attached to covalent polymers to induce crosslinking via directional host–guest interactions, or via introducing secondary interactions by hydrophobic, hydrogen bonding or electrostatic interactions. For example, Scherman et al. are using host–guest interactions based on cucurbit[n]uril,91 and have recently developed a non-fibrous dual network with a trace amount of covalent crosslinks forming elastic and stiff hydrogels.92 Moreover, Rowan and coworkers developed strain stiffening polymers by having a polyisocyanide backbone enriched with reversible hydrogen bonds between the amino acids in the side chains.93 Although the hybrid hydrogels bring along highly stretchable materials with good mechanical properties, these systems lack the ability to reposition functional epitopes along a fibrous backbone, in order to enable clustering and optimal integrin spacing as compared to fully non-covalently assembled systems. Only a few examples exist that are able to form nanofibrous structures capable of repositioning functional epitopes, such as self-assembling peptides, peptide amphiphiles and supramolecular amphiphiles. More detailed excellent reviews about peptide based supramolecular hydrogels are found elsewhere (e.g.ref. 94–96).Self-assembling peptides
Self-assembling peptides have the advantage over synthetic polymers that they are intrinsically dynamic and naturally derived, which provides them with biodegradable and non-toxic properties. A few self-assembling peptides have been discovered, which are mainly ionically complementary. This means that by alternating positive and negative charges, the peptides can be stacked. The mostly used self-assembling peptide is RADA16-I,97 commercialized as PuraMatrix. These peptides self-assemble in stable β-sheets via electrostatic interactions between the positively charged arginine (R) and negatively charged aspartic acid (D), with additional hydrophobic interactions between the alanine units (A) (see Fig. 9a and b). Fibres are formed with a diameter of about 10 nm (Fig. 9c) and were investigated in several in vitro and in vivo studies. Recently, Chen and coworkers attached a brain derived neurotrophic factor (BDNF) peptide onto RADA16 and showed that the co-culture of human umbilical cord mesenchymal stem cells and activated astrocytes promoted the proliferation and differentiation of neural cells in lesions after traumatic brain injury (Fig. 9d).98 Unfortunately, apoptosis was observed in larger, about 5 mm diameter, injured cavities. Neuro-regeneration was also observed when induced pluripotent stem cells were encapsulated in RADA16-I scaffolds and after neuronal induction transplanted into mouse brains.99 When the cells were encapsulated after neural induction, the survival in vitro was only limited. Despite major steps being made in in vivo studies, long term viability of the cells remains a concern.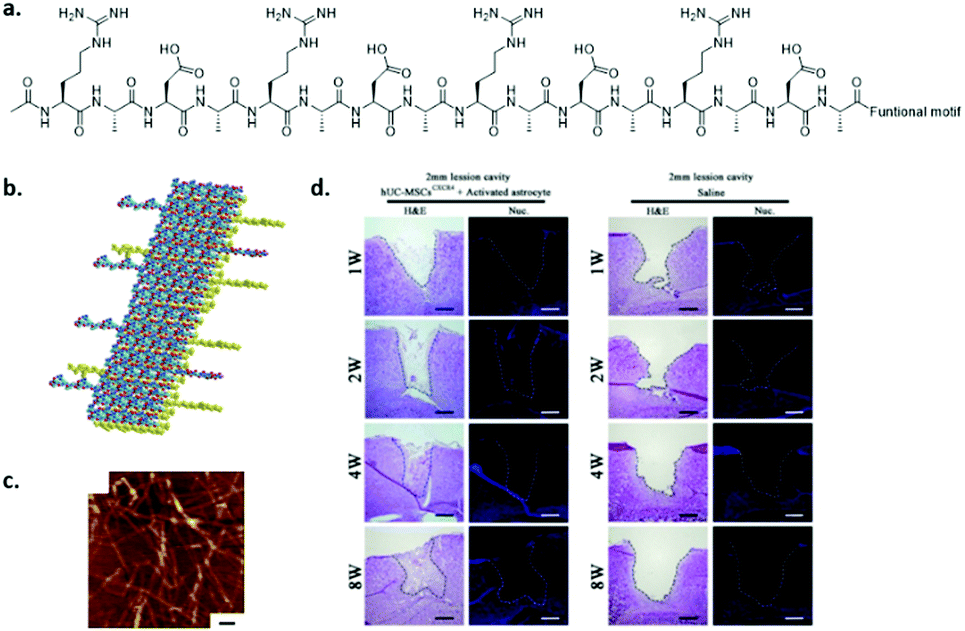 | ||
| Fig. 9 The self-assembling peptide RADA16-I. (a) Chemical structure, (b) schematic representation of stacking, and (c) AFM (tapping mode) measurement showing fibres (scale bar indicates 100 nm). Modified from ref. 97, PLoS One, copyright 2007. (d) Brain regeneration in a 2 mm lesion cavity when seeding activated astrocytes and human umbilical cord mesenchymal stem cells embedded in a BDNF functionalized RADA16 hydrogel (left) or when injecting only saline (right) after 1–8 weeks. Modified from ref. 98 with permission from Elsevier, copyright 2016. | ||
The peptide P11-4 (Ac-QQRFEWEFEQQ-NH2), commercialized as Curodont™, self-assembles into β-sheets containing fibrous structures, depending on the concentration and pH, using a combination of electrostatic interactions between the arginine and glutamic acid, hydrophobic interactions between the aromatic rings and hydrogen bonding between glutamines.100 The anionic groups of the glutamic acids were proposed to attract calcium ions, inducing mineralization in dental caries-like lesions.101 In 2013, P11-4 was shown to repair early dental lesions in patients by inducing remineralization 30 days post-treatment,102 and currently, this peptide is in clinical trials.103 Although promising results were obtained and the way to clinical trials is paved, the structures used here can be further optimized to accelerate remineralization. Since only a small amount of negative charges was included in the system, the system might benefit from including proteoglycan mimics or phosphorylated groups.
Nanofibrous structures based on Fmoc protected dipeptides have also been developed and have been shown to support the culture of chondrocytes in 2D and 3D environments.104 Self-assembly is induced by a combination of hydrogen bonding and π–π interactions, and upon using a pH switch, self-supporting hydrogels were observed at <1 wt% concentration with a fiber diameter in the range of 19–68 nm. Chemical functionalities, i.e. amine, carboxylic acid and hydroxyl, were incorporated at the end of the dipeptides and were shown to assemble into 1D fibres as well, however the mechanical properties at the hydrogel level were significantly influenced.105 Although the tested gels supported the culture of bovine chondrocytes, only the hydroxyl terminated peptide nanofibers supported other cell lines. Dalby et al. investigated the influence of hydrogel stiffness and the incorporation of metabolites into Fmoc-peptide nanofibers on the differentiation of perivascular stem cells.106 It was shown that specific lipids were consumed during the differentiation into chondrocytes and osteocytes, which demonstrates the importance of metabolites in directing the differentiation into a specific cell lineage. Yet, incorporating functional epitopes, like cell adhesive peptides, might further enhance cell viability.
Self-assembling peptide amphiphiles
Peptide amphiphiles (PA) consist of a peptide part and a long hydrophobic tail which initiates hydrophobic collapse into cylindrical micelles (Fig. 10). Several functional amino acids as well as adhesive peptides have been incorporated into PAs to introduce biofunctionality, and were shown to form 1D fibres despite the chemical modifications.107 The fibronectin derived peptide RGDS was attached to the periphery of the monomers and was shown to induce the adhesion of several cells and stem cells.108 In contrast to covalent networks, where the spacing of the RGD needs to be tightly regulated109 or multivalent RGD display is required,110 in supramolecular fibres, bioactive cues can migrate along the fibre backbone allowing adaptation and optimal integrin spacing. Stupp and coworkers demonstrated that a significantly lower amount of bioactive guest presentation is required (i.e. about 5 mol%) to achieve cell attachment corroborating this hypothesis.111 The main question is whether these highly dynamic systems are able to resist the pulling forces applied by the cells. In principle, the non-covalent interactions that induce supramolecular polymer formation are relatively weak and can be broken. However, due to multiple hydrogen bonds, hydrophobic effects and additional β-sheet or π–π stacking, the packing of these structures can be regulated. For example, Stupp et al. also showed that disordered fibre packing can be obtained by substituting valine for alanine. As a consequence, β-sheet formation was decreased and disordered, which reduced the physical properties of the gel at the macroscopic level.72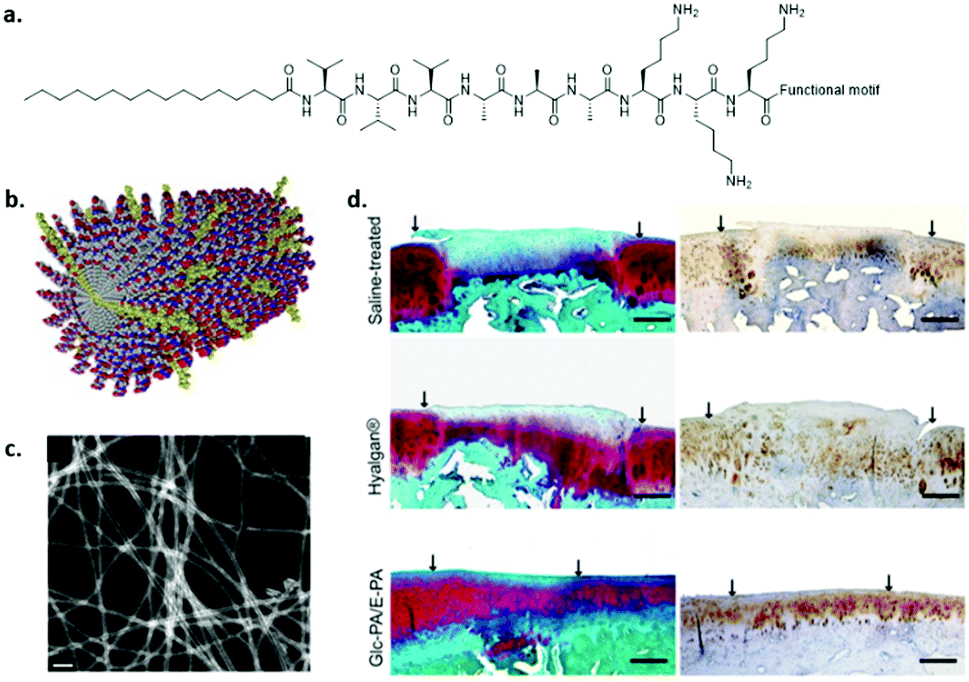 | ||
| Fig. 10 An example of a self-assembled peptide amphiphile. (a) Chemical structure, (b) cartoon illustration of assembly. Modified from ref. 116 with permission from Elsevier, copyright 2009. (c) STEM image of co-assembled glucose-PA and carboxylic acid-PA, (d) MSC encapsulated in glucose functionalized PA co-assembled with glutamic acid end functionalized PAs (bottom) showed articular cartilage regeneration within 12 weeks after transplantation as compared to saline (top) and Hyalagan (middle). Modified with permission from ref. 112 copyright 2016 American Chemical Society. | ||
By introducing a glucose functionalized amino acid in PAs, Guler and coworkers aimed to mimic hyaluronic acid by combining the glucose-PA with carboxylic acid terminated PAs (Fig. 10c and d).112 It was shown that mesenchymal stem cells differentiated into chondrocytes without the use of exogenous growth factors by targeting the CD44 receptor. Chondrogenic differentiation of MSCs was also studied by Tekinay and coworkers by utilizing a heparin mimicking PA.113 By co-assembling a sulfonate functionalized PA, a carboxylate and an amine end-functionalized PA, the importance of epitope presentation on chondrogenesis was shown.
Although promising results are obtained, functional epitopes on peptide amphiphiles are closely displayed to the fibre backbone, thereby inducing steric hindrance of the backbone when binding to a target. Spacing of functional cues on RADA16-I was elucidated by investigating both the self-assembly behaviour and the epitope presentation. It was demonstrated that 4 glycines improved both the stability and the presentation.114 Improved cell spreading was also observed when 5 glycines were used to space RGD epitopes on PAs.115 While a longer spacer length decreases steric effects of the backbone improving epitope availability, a spacer which is too long might have a decreased binding strength due to gained flexibility.
Supramolecular self-assembling amphiphiles
Although peptides are non-toxic and biodegradable, peptide synthesis on a large scale is quite costly. Therefore, there is an increasing need for cheap, large scale alternatives. As a consequence, self-assembling amphiphilic monomers are required which have a self-assembling core, and a water soluble periphery.The self-complementary UPy molecule was modified with PEG to enable water solubility, a urea moiety to allow lateral hydrogen bonding, and hydrophobic linkers to protect the inner hydrogen bonding core from water (Fig. 11).117 A combination of hydrophobic effects, hydrogen bonding and π–π stacking enables the self-assembly of these monomeric units into 1D fibres. Moreover, telechelic (bivalent) UPy molecules spaced by different lengths of PEG were developed and were shown to form hydrogels above a critical concentration. By changing the hydrophobic to hydrophilic ratio of the molecule, different fibre lengths were observed, and a difference in internal dynamics was elucidated.57 By varying the packing of the monomers in the fibre, functional epitopes can be presented in a highly dynamic fashion, or frozen in the backbone. A few examples exist where UPy hydrogels were shown to be useful in biomedical applications. For example, UPy hydrogels have been loaded with growth factors by physical entanglement and have been shown to reduce scar collagen in a myocardial infarction pig model (Fig. 10d).118 However, fast release of growth factors was observed, which can be improved by including functional epitopes in the fibres. By incorporating growth factor binding peptides into supramolecular fibres, growth factors can be captured via electrostatic interactions and released in a more sustained fashion. Although other functional UPy hydrogels have been developed,119 the culture of stem cells has not been reported yet.
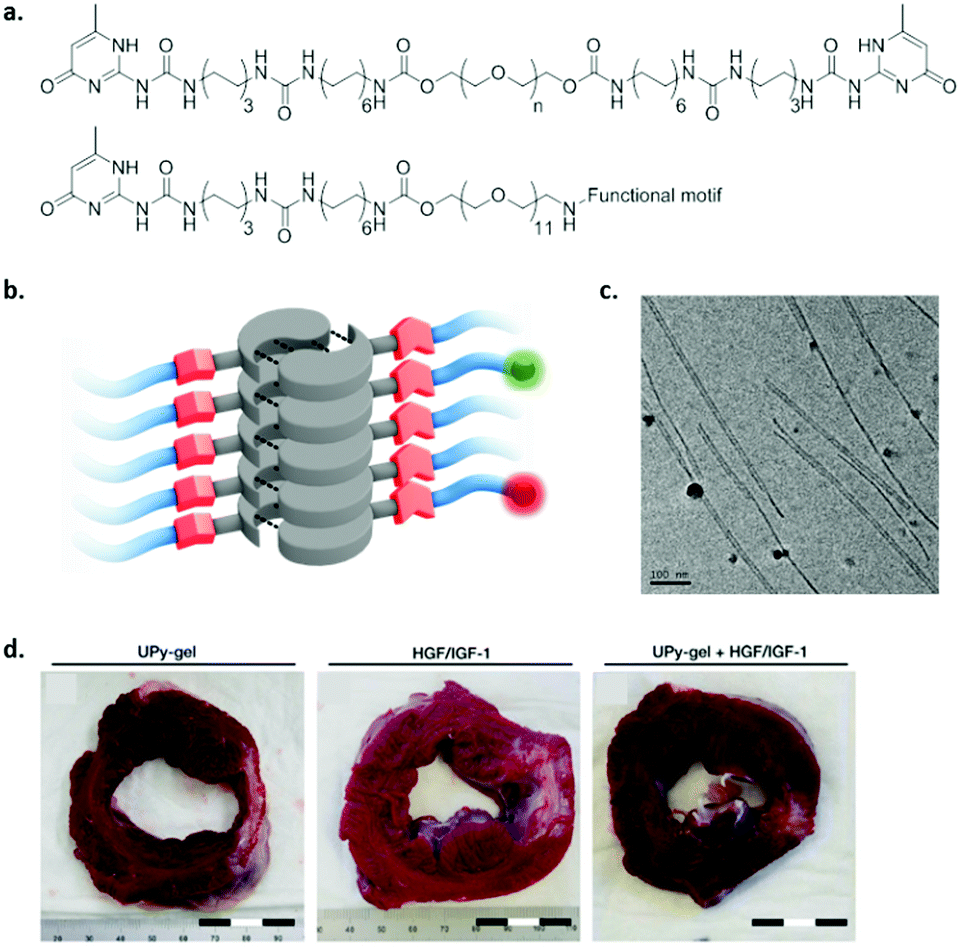 | ||
| Fig. 11 An example of a UPy based self-assembling amphiphile. (a) Chemical structure, top; bivalent UPy (n = 20 kDa), bottom; functional UPy monomer, (b) schematic representation of fibre formation, and (c) cryo-TEM of UPy based fibres (scale bar represents 100 nm). Adapted from ref. 71 – published by the Royal Society of Chemistry. (d) Cardiac repair upon treatment with a pristine UPy hydrogel (left), growth factors HGF and IGF-1 in saline (middle) or physical entangled growth factors in UPy hydrogel (right). Scar tissue (white stain) was reduced, 4 weeks after treatment with a growth factor loaded UPy-PEG hydrogel. Adapted from ref. 118 with permission from Wiley, copyright 2014. | ||
Benzene-1,3,5-tricarboxamide (BTA) based molecules have also been modified to allow water solubility and were shown to form fibres in water due to a combination of 3-fold hydrogen bonding, a hydrophobic pocket protecting the inner core from water penetration and π–π interactions between the benzene rings.120 A subtle change in the length of the aliphatic spacer was shown to have a tremendous effect on the internal dynamics in the fibre, also showing the importance of the hydrophobic to hydrophilic ratio.121 Similar to the UPy, BTA based hydrogels were formed at higher concentrations, and cross-linking was induced by introducing telechelic BTAs.122 Functional BTA hydrogels were developed where positively charged BTAs were incorporated in the fibres and were shown to capture siRNA by electrostatic interactions.123 While these systems are highly investigated for their structural and dynamic properties, they need to be investigated in vitro to examine their suitability for the culture of cells and stem cells.
Considerations of supramolecular hydrogels towards supramolecular biomaterials
Supramolecular hydrogels show great promise in mimicking the extracellular matrix. Their fibrous morphology is able to recapitulate dynamic processes found in nature and they are capable of supporting the culture of cells and stem cells in a 3D fashion. Next to gaining fundamental understanding into tissue development, homeostasis and disease progression, cell, or even organoid culture, can be used to regenerate diseased tissue.From the described examples it is clear that the design of the supramolecular monomers remains very important in directing both the mechanical properties as well as the dynamic behaviour of the material. Next to favourable dynamic properties to induce clustering formation, the supramolecular fibres are also prone to breakage by pulling forces exerted by cells. Therefore, the fibres need to be stable enough to resist these pulling forces and should preserve the incorporation of cell adhesive epitopes to maintain cell support.
Although cell adhesive peptides or other functional peptides were incorporated in supramolecular hydrogels, more complex multi-component systems should be developed in order to induce simultaneous events. For example, next to integrin binding, targeting growth factor receptors should allow for synergistic effects. Moreover, functional cues need to be presented in a multivalent and spatiotemporal fashion. Due to the modularity of supramolecular systems, different functional epitopes can be simply incorporated into hydrogels which allows for excellent suitability for high throughput screening.
Different growth factors should also be stabilized and released at different time intervals in order to support the formation of functional tissue. For instance, during the regeneration of liver tissue, initially vascularization should be initiated for a short period of time, by VEGF and bFGF, and the release of HGF is necessary for a longer period of time in order to support the long-term proliferation of hepatocytes.124 Therefore, the stimuli-responsiveness of the hydrogel is highly important, where cells should be able to actively remodel its environment to release certain factors.
The ultimate challenge for hydrogels remains the capability to support long-term viability, and the maintenance or induction into specific cell types. The hydrogel needs to be disassembled upon a certain trigger, in order to transfer or split cells, and should be able to maintain specific (stem) cell markers for multiple passages. Moreover, every cell type is unique and as a consequence requires its own set of optimized design parameters to induce the desired cell response. Although these parameters need to be tuned to bring supramolecular hydrogels to the next level, major steps towards in vitro and in vivo applications have already been made. However, the culture of enough cells for regenerative medicine applications remains a challenge in current research.
Perspective
Regenerative medicine is positioned as an innovative field that will provide large advancements for healthcare, through convergence, i.e. combining expertise from various disciplines, such as chemistry, medicine, biology, physics, engineering, informatics and mathematics.125 The promises are large and the expectations high. This field classically started as a tissue engineering approach;126 an in vitro method that made it possible to culture patient's own cells on degradable variants of biomaterials in bioreactors in the lab, after which implantation of these partly in vitro formed tissues in a patient has shown to be beneficial for inducing repair.127,128 Nowadays the field has emerged into either an in situ approach solely using ‘regenerative’ materials, or a completely in vitro approach primarily depending on the expansion of stem cells and organoids with temporary supportive materials (through ECM components, with a synthetic or artificial ECM as the holy grail).In this review we discussed the development of supramolecular biomaterials based on one-dimensional fibrous assemblies. Our proposal is that such systems are eminently suitable to be applied as biomaterials in the regenerative medicine field owing to their resemblance with the natural ECM. In our body the ECM is a large aggregate of an enormous amount of different molecules varying from structural and signalling proteins to polysaccharides, held together via directed non-covalent interactions. The ECM can be seen as a large fibrous, supramolecular hydrogel-like structure. The ECM interacts with cells via reciprocal interactions resulting in feedback–response mechanisms that control tissue homeostasis. We showed that the synthetic fibrous supramolecular structures can be developed into two classes of materials that serve either of the new emerging approaches, i.e. TPEs for in situ engineering of load bearing tissues, and hydrogels as artificial matrices for stem cell and organoid expansion in vitro.
Supramolecular TPEs for load bearing applications have been shown to meet the mechanical property criteria e.g. for cardiovascular implants, while bio-activation can be performed via a modular approach using supramolecular additives. This shows that changing the biochemical material properties solely depends on the mixture of the individual ingredients, without the need for synthesizing a new material/polymer for every wish to change the biochemical composition. As discussed, in situ regeneration of load bearing tissues is within reach, reflected by the exceptional example of successful performance in a clinical study by Xeltis on synthetic heart valve implantations in humans (launched press release, October 2016). An important message is that the balance and/or trade-off between simplicity, which is necessary to provide clinical translation, and complexity, in order to mimic the natural ECM, needs to be carefully sought. It might be contingent that in patients with high regenerative potential in situ engineering with simple, pristine, non-active materials is effective, while in patients with impaired regeneration, e.g. suffering from diabetes, renal failure and/or immunological disorders, the introduction of complexity in the form of bioactivity is a necessity.
The second class of materials comprises supramolecular hydrogels for in vitro stem cell and organoid expansion. For complex (internal) organs, such as the kidney, liver and pancreas an in situ engineering approach seems to be beyond reach. Via an in vitro approach relying on the internal regenerative capacity of organoid cultures, we and others propose to develop synthetic mimics of the ECM to be applied as a 3D culture in vitro platform with the requirement to be conveniently removable after culturing in order to achieve safe in vivo transplantation of the expanded organoids. Also in this approach the balance between mimicking the complex structure of the ECM, and the need for a simple system for clinical translation is an important factor. However, we believe that the complexity of the hydrogel systems in vitro is allowed to be larger than the TPE for in situ engineering (that primarily have a mechanical function). Here, the challenge is to bring functionality in these systems in such a way that they are able to adapt and respond to the biological entities, proteins, cells and tissues, they encounter. This bi-directional dynamic behaviour, also referred to as dynamic reciprocity,129 is of utmost importance to achieve robust, sustainable organoid cultures. The introduction of a feedback-response mechanism in biomaterials design130,131 is a challenge and only a few examples exist.132 We propose to do this via a top down or bottom up engineering approach, in which either the cells create their own niche via secretion of ECM molecules, or by synthetically reconstructing the ECM in a minimally synthetic way using various bioactive entities, respectively, en route to meeting nature's complexity using supramolecular fibrous assemblies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the European Research Council (FP7/2007–2013) ERC Grant Agreement 308045 and the Ministry of Education, Culture and Science (Gravity program 024.001.03) and funded by the NWO/DPI program NEWPOL (project #731.015.503), partially financed with TKI (Topconsortia for Knowledge Innovation) allowance provided by the Dutch Ministry of Economic Affairs.Notes and references
- T. Rozario and D. W. DeSimone, Dev. Biol., 2010, 341, 126–140 CrossRef CAS PubMed.
- C. Frantz, K. M. Stewart and V. M. Weaver, J. Cell Sci., 2010, 123, 4195–4200 CrossRef CAS PubMed.
- R. Kalluri, Nat. Rev. Cancer, 2003, 3, 422–433 CrossRef CAS PubMed.
- K. R. Legate, S. A. Wickström and R. Fässler, Genes Dev., 2009, 23, 397–418 CrossRef CAS PubMed.
- R. O. Hynes, Cell, 2002, 110, 673–687 CrossRef CAS PubMed.
- C. K. Choi, M. Vicente-Manzanares, J. Zareno, L. A. Whitmore, A. Mogilner and A. R. Horwitz, Nat. Cell Biol., 2008, 10, 1039–1050 CrossRef CAS PubMed.
- C. C. DuFort, M. J. Paszek and V. M. Weaver, Nat. Rev. Mol. Cell Biol., 2011, 12, 308–319 CrossRef CAS PubMed.
- P. M. Tsimbouri, R. J. McMurray, K. V. Burgess, E. V. Alakpa, P. M. Reynolds, K. Murawski, E. Kingham, R. O. C. Oreffo, N. Gadegaard and M. J. Dalby, ACS Nano, 2012, 6, 10239–10249 CrossRef CAS PubMed.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS PubMed.
- J. J. Berzelius, Jahres-Ber., 1832, 11, 44–48 Search PubMed.
- H. Staudinger, Berichte Dtsch. Chem. Ges. B Ser., 1920, 53, 1073–1085 CrossRef.
- G. G. Longinescu, Chem. Rev., 1929, 6, 381–418 CrossRef CAS.
- Y. Ducharme and J. D. Wuest, J. Org. Chem., 1988, 53, 5787–5789 CrossRef CAS.
- C. Fouquey, J.-M. Lehn and A.-M. Levelut, Adv. Mater., 1990, 2, 254–257 CrossRef CAS.
- C.-M. Lee, C. P. Jariwala and A. C. Griffin, Polymer, 1994, 35, 4550–4554 CrossRef CAS.
- C. B. St. Pourcain and A. C. Griffin, Macromolecules, 1995, 28, 4116–4121 CrossRef CAS.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. Folmer, J. H. Hirschberg, R. F. Lange, J. K. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- L. Brunsveld, B. J. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS PubMed.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- H. Kautz, D. J. M. van Beek, R. P. Sijbesma and E. W. Meijer, Macromolecules, 2006, 39, 4265–4267 CrossRef CAS.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- A. R. A. Palmans, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Angew. Chem., Int. Ed. Engl., 1997, 36, 2648–2651 CrossRef CAS.
- J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1990, 29, 1304–1319 CrossRef.
- J.-M. Lehn, Polym. Int., 2002, 51, 825–839 CrossRef CAS.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- H. Cui, M. J. Webber and S. I. Stupp, Biopolymers, 2010, 94, 1–18 CrossRef CAS PubMed.
- W. P. J. Appel, M. M. L. Nieuwenhuizen and E. W. Meijer, in Supramolecular Polymer Chemistry, ed. A. Harada, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, pp. 1–28 Search PubMed.
- S. H. M. Söntjens, R. A. E. Renken, G. M. L. van Gemert, T. A. P. Engels, A. W. Bosman, H. M. Janssen, L. E. Govaert and F. P. T. Baaijens, Macromolecules, 2008, 41, 5703–5708 CrossRef.
- F. Edalat, I. Sheu, S. Manoucheri and A. Khademhosseini, Curr. Opin. Biotechnol, 2012, 23, 820–825 CrossRef CAS PubMed.
- R. Langer and D. A. Tirrell, Nature, 2004, 428, 487–492 CrossRef CAS PubMed.
- J. W. Boretos and W. S. Pierce, J. Biomed. Mater. Res., 1968, 2, 121–130 CrossRef CAS PubMed.
- K. A. Houton and A. J. Wilson, Polym. Int., 2015, 64, 165–173 CrossRef CAS.
- D. De and R. J. Gaymans, Macromol. Mater. Eng., 2009, 294, 405–413 CrossRef CAS.
- J. C. Johnson, N. D. Wanasekara and L. T. J. Korley, Biomacromolecules, 2012, 13, 1279–1286 CrossRef CAS PubMed.
- P. J. Woodward, D. Hermida Merino, B. W. Greenland, I. W. Hamley, Z. Light, A. T. Slark and W. Hayes, Macromolecules, 2010, 43, 2512–2517 CrossRef CAS.
- X. Gu, Z. Mao, S.-H. Ye, Y. Koo, Y. Yun, T. R. Tiasha, V. Shanov and W. R. Wagner, Colloids Surf., B, 2016, 144, 170–179 CrossRef CAS PubMed.
- A. D’Amore, J. S. Soares, J. A. Stella, W. Zhang, N. J. Amoroso, J. E. Mayer John, W. R. Wagner and M. S. Sacks, J. Mech. Behav. Biomed. Mater., 2016, 62, 619–635 CrossRef PubMed.
- L. Soletti, Y. Hong, J. Guan, J. J. Stankus, M. S. El-Kurdi, W. R. Wagner and D. A. Vorp, Acta Biomater., 2010, 6, 110–122 CrossRef CAS PubMed.
- A. D’Amore, T. Yoshizumi, S. K. Luketich, M. T. Wolf, X. Gu, M. Cammarata, R. Hoff, S. F. Badylak and W. R. Wagner, Biomaterials, 2016, 107, 1–14 CrossRef PubMed.
- S. Boileau, L. Bouteiller, F. Lauprêtre and F. Lortie, New J. Chem., 2000, 24, 845–848 RSC.
- S. Das, I. Yilgor, E. Yilgor, B. Inci, O. Tezgel, F. L. Beyer and G. L. Wilkes, Polymer, 2007, 48, 290–301 CrossRef CAS.
- R. M. Versteegen, R. P. Sijbesma and E. W. Meijer, Macromolecules, 2005, 38, 3176–3184 CrossRef CAS.
- R. M. Versteegen, R. Kleppinger, R. P. Sijbesma and E. W. Meijer, Macromolecules, 2006, 39, 772–783 CrossRef CAS.
- N. Roy, E. Buhler and J.-M. Lehn, Chem. – Eur. J., 2013, 19, 8814–8820 CrossRef CAS PubMed.
- N. E. Botterhuis, S. Karthikeyan, D. Veldman, S. C. J. Meskers and R. P. Sijbesma, Chem. Commun., 2008, 3915–3917 RSC.
- R. A. Koevoets, R. M. Versteegen, H. Kooijman, A. L. Spek, R. P. Sijbesma and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 2999–3003 CrossRef CAS PubMed.
- E. Wisse, A. J. H. Spiering, E. N. M. van Leeuwen, R. A. E. Renken, P. Y. W. Dankers, L. A. Brouwer, M. J. A. van Luyn, M. C. Harmsen, N. A. J. M. Sommerdijk and E. W. Meijer, Biomacromolecules, 2006, 7, 3385–3395 CrossRef CAS PubMed.
- E. Wisse, L. E. Govaert, H. E. H. Meijer and E. W. Meijer, Macromolecules, 2006, 39, 7425–7432 CrossRef CAS.
- J. Kluin, H. Talacua, A. I. P. M. Smits, M. Y. Emmert, M. C. P. Brugmans, E. S. Fioretta, P. E. Dijkman, S. H. M. Söntjens, R. Duijvelshoff, S. Dekker, M. W. J. T. Janssen-vanden Broek, V. Lintas, A. Vink, S. P. Hoerstrup, H. M. Janssen, P. Y. W. Dankers, F. P. T. Baaijens and C. V. C. Bouten, Biomaterials, 2017, 125, 101–117 CrossRef CAS PubMed.
- T. F. A. de Greef and E. W. Meijer, Nature, 2008, 453, 171–173 CrossRef CAS PubMed.
- W. P. J. Appel, G. Portale, E. Wisse, P. Y. W. Dankers and E. W. Meijer, Macromolecules, 2011, 44, 6776–6784 CrossRef CAS.
- A. W. Bosman, R. P. Sijbesma and E. W. Meijer, Mater. Today, 2004, 7, 34–39 CrossRef CAS.
- P. Y. W. Dankers, M. C. Harmsen, L. A. Brouwer, M. J. A. Van Luyn and E. W. Meijer, Nat. Mater., 2005, 4, 568–574 CrossRef CAS PubMed.
- G. C. van Almen, H. Talacua, B. D. Ippel, B. B. Mollet, M. Ramaekers, M. Simonet, A. I. P. M. Smits, C. V. C. Bouten, J. Kluin and P. Y. W. Dankers, Macromol. Biosci., 2016, 16, 350–362 CrossRef CAS PubMed.
- B. B. Mollet, M. Comellas-Aragonès, A. J. H. Spiering, S. H. M. Söntjens, E. W. Meijer and P. Y. W. Dankers, J. Mater. Chem. B, 2014, 2, 2483–2493 RSC.
- D. E. P. Muylaert, G. C. van Almen, H. Talacua, J. O. Fledderus, J. Kluin, S. I. S. Hendrikse, J. L. J. van Dongen, E. Sijbesma, A. W. Bosman, T. Mes, S. H. Thakkar, A. I. P. M. Smits, C. V. C. Bouten, P. Y. W. Dankers and M. C. Verhaar, Biomaterials, 2016, 76, 187–195 CrossRef CAS PubMed.
- P. Y. W. Dankers, J. M. Boomker, A. H. der Vlag, F. M. M. Smedts, M. C. Harmsen and M. J. A. van Luyn, Macromol. Biosci., 2010, 10, 1345–1354 CrossRef CAS PubMed.
- W. P. J. Appel, E. W. Meijer and P. Y. W. Dankers, Macromol. Biosci., 2011, 11, 1706–1712 CrossRef CAS PubMed.
- E. Wisse, A. J. H. Spiering, P. Y. W. Dankers, B. Mezari, P. C. M. M. Magusin and E. W. Meijer, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1764–1771 CrossRef CAS.
- P. Y. W. Dankers, E. N. M. van Leeuwen, G. M. L. van Gemert, A. J. H. Spiering, M. C. Harmsen, L. A. Brouwer, H. M. Janssen, A. W. Bosman, M. J. A. van Luyn and E. W. Meijer, Biomaterials, 2006, 27, 5490–5501 CrossRef CAS PubMed.
- R. Selvaraj and J. M. Fox, Curr. Opin. Chem. Biol., 2013, 17, 753–760 CrossRef CAS PubMed.
- J. M. Fox and M. S. Robillard, Curr. Opin. Chem. Biol., 2014, 21, v–vii CrossRef CAS PubMed.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605 CrossRef CAS PubMed.
- O. J. G. M. Goor, J. E. P. Brouns and P. Y. W. Dankers, Polym. Chem., 2017, 8, 5228–5238 RSC.
- O. J. G. M. Goor, H. M. Keizer, A. L. Bruinen, M. G. J. Schmitz, R. M. Versteegen, H. M. Janssen, R. M. A. Heeren and P. Y. W. Dankers, Adv. Mater., 2017, 29, 1604652 CrossRef PubMed.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2012, 64, 18–23 CrossRef.
- E. A. Appel, J. del Barrio, X. Jun Loh and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 6195–6214 RSC.
- A. Boutaud, D. B. Borza, O. Bondar, S. Gunwar, K. O. Netzer, N. Singh, Y. Ninomiya, Y. Sado, M. E. Noelken and B. G. Hudson, J. Biol. Chem., 2000, 275, 30716–30724 CrossRef CAS PubMed.
- J. Engel, E. Odermatt, A. Engel, J. A. Madri, H. Furthmayr, H. Rohde and R. Timpl, J. Mol. Biol., 1981, 150, 97–120 CrossRef CAS PubMed.
- S. I. S. Hendrikse, S. P. W. Wijnands, R. P. M. Lafleur, M. J. Pouderoijen, H. M. Janssen, P. Y. W. Dankers and E. W. Meijer, Chem. Commun., 2017, 53, 2279–2282 RSC.
- E. T. Pashuck, H. Cui and S. I. Stupp, J. Am. Chem. Soc., 2010, 132, 6041–6046 CrossRef CAS PubMed.
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS PubMed.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS PubMed.
- S. B. Shah and A. Singh, Acta Biomater., 2017, 53, 29–45 CrossRef CAS PubMed.
- M. A. Lancaster and J. A. Knoblich, Science, 2014, 345, 1247125 CrossRef PubMed.
- H. K. Kleinman and G. R. Martin, Semin. Cancer Biol., 2005, 15, 378–386 CrossRef CAS PubMed.
- C. S. Hughes, L. M. Postovit and G. A. Lajoie, Proteomics, 2010, 10, 1886–1890 CrossRef CAS PubMed.
- N. Nakamura, T. Kimura and A. Kishida, ACS Biomater. Sci. Eng., 2017, 3, 1236–1244 CrossRef CAS.
- B. M. Baker and C. S. Chen, J. Cell Sci., 2012, 125, 3015–3024 CrossRef CAS PubMed.
- N. Huebsch, P. R. Arany, A. S. Mao, D. Shvartsman, O. A. Ali, S. A. Bencherif, J. Rivera-Feliciano and D. J. Mooney, Nat. Mater., 2010, 9, 518–526 CrossRef CAS PubMed.
- B. M. Baker, B. Trappmann, W. Y. Wang, M. S. Sakar, I. L. Kim, V. B. Shenoy, J. A. Burdick and C. S. Chen, Nat. Mater., 2015, 14, 1262–1268 CrossRef CAS PubMed.
- M. Ehrbar, S. C. Rizzi, R. G. Schoenmakers, B. S. Miguel, J. A. Hubbell, F. E. Weber and M. P. Lutolf, Biomacromolecules, 2007, 8, 3000–3007 CrossRef CAS PubMed.
- M. Ehrbar, A. Sala, P. Lienemann, A. Ranga, K. Mosiewicz, A. Bittermann, S. C. Rizzi, F. E. Weber and M. P. Lutolf, Biophys. J., 2011, 100, 284–293 CrossRef CAS PubMed.
- C. Chung, M. Beecham, R. L. Mauck and J. A. Burdick, Biomaterials, 2009, 30, 4287–4296 CrossRef CAS PubMed.
- S. J. Paluck, T. H. Nguyen, J. P. Lee and H. D. Maynard, Biomacromolecules, 2016, 17, 3386–3395 CrossRef CAS PubMed.
- E. M. Pelegri-O’Day, S. J. Paluck and H. D. Maynard, J. Am. Chem. Soc., 2017, 139, 1145–1154 CrossRef PubMed.
- H. Wang and S. C. Heilshorn, Adv. Mater., 2015, 27, 3717–3736 CrossRef CAS PubMed.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- D. D. McKinnon, D. W. Domaille, J. N. Cha and K. S. Anseth, Adv. Mater., 2014, 26, 865–872 CrossRef CAS PubMed.
- J. Liu, C. S. Y. Tan, Y. Lan and O. A. Scherman, Macromol. Chem. Phys., 2016, 217, 319–332 CrossRef CAS.
- J. Liu, C. S. Y. Tan, Z. Yu, Y. Lan, C. Abell and O. A. Scherman, Adv. Mater., 2017, 29, 1604951 CrossRef PubMed.
- P. H. J. Kouwer, M. Koepf, V. A. A. Le Sage, M. Jaspers, A. M. van Buul, Z. H. Eksteen-Akeroyd, T. Woltinge, E. Schwartz, H. J. Kitto, R. Hoogenboom, S. J. Picken, R. J. M. Nolte, E. Mendes and A. E. Rowan, Nature, 2013, 493, 651–655 CrossRef CAS PubMed.
- R. Ravichandran, M. Griffith and J. Phopase, J. Mater. Chem. B, 2014, 2, 8466–8478 RSC.
- R. Pugliese and F. Gelain, Trends Biotechnol., 2017, 35, 145–158 CrossRef CAS PubMed.
- X.-Q. Dou and C.-L. Feng, Adv. Mater., 2017, 29, 1604062 CrossRef PubMed.
- A. Horii, X. Wang, F. Gelain and S. Zhang, PLoS ONE, 2007, 2, e190 Search PubMed.
- W. Shi, C. J. Huang, X. D. Xu, G. H. Jin, R. Q. Huang, J. F. Huang, Y. N. Chen, S. Q. Ju, Y. Wang, Y. W. Shi, J. B. Qin, Y. Q. Zhang, Q. Q. Liu, X. B. Wang, X. H. Zhang and J. Chen, Acta Biomater., 2016, 45, 247–261 CrossRef CAS PubMed.
- N. L. Francis, N. K. Bennett, A. Halikere, Z. P. Pang and P. V. Moghe, ACS Biomater. Sci. Eng., 2016, 2, 1030–1038 CrossRef CAS.
- A. Aggeli, M. Bell, L. M. Carrick, C. W. G. Fishwick, R. Harding, P. J. Mawer, S. E. Radford, A. E. Strong and N. Boden, J. Am. Chem. Soc., 2003, 125, 9619–9628 CrossRef CAS PubMed.
- J. Kirkham, A. Firth, D. Vernals, N. Boden, C. Robinson, R. C. Shore, S. J. Brookes and A. Aggeli, J. Dent. Res., 2007, 86, 426–430 CrossRef CAS PubMed.
- P. A. Brunton, R. P. W. Davies, J. L. Burke, A. Smith, A. Aggeli, S. J. Brookes and J. Kirkham, Br. Dent. J., 2013, 215, E6 CrossRef CAS PubMed.
- Self Assembling Peptide P11-4 in Patients With Early Occlusal Carious Lesions – Full Text View – http://ClinicalTrials.gov, http://https://clinicaltrials.gov/ct2/show/study/NCT02724592, accessed 10 July 2017.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- V. Jayawarna, S. M. Richardson, A. R. Hirst, N. W. Hodson, A. Saiani, J. E. Gough and R. V. Ulijn, Acta Biomater., 2009, 5, 934–943 CrossRef CAS PubMed.
- E. V. Alakpa, V. Jayawarna, A. Lampel, K. V. Burgess, C. C. West, S. C. J. Bakker, S. Roy, N. Javid, S. Fleming, D. A. Lamprou, J. Yang, A. Miller, A. J. Urquhart, P. W. J. M. Frederix, N. T. Hunt, B. Péault, R. V. Ulijn and M. J. Dalby, Chem., 2016, 1, 298–319 CAS.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5133–5138 CrossRef CAS PubMed.
- J. Boekhoven and S. I. Stupp, Adv. Mater., 2014, 26, 1642–1659 CrossRef CAS PubMed.
- J. Huang, S. V. Grater, F. Corbellini, S. Rinck, E. Bock, R. Kemkemer, H. Kessler, J. Ding and J. P. Spatz, Nano Lett., 2009, 9, 1111–1116 CrossRef CAS PubMed.
- J. Lam and T. Segura, Biomaterials, 2013, 34, 3938–3947 CrossRef CAS PubMed.
- H. Storrie, M. O. Guler, S. N. Abu-Amara, T. Volberg, M. Rao, B. Geiger and S. I. Stupp, Biomaterials, 2007, 28, 4608–4618 CrossRef CAS PubMed.
- S. Ustun Yaylaci, M. Sardan Ekiz, E. Arslan, N. Can, E. Kilic, H. Ozkan, I. Orujalipoor, S. Ide, A. B. Tekinay and M. O. Guler, Biomacromolecules, 2016, 17, 679–689 CrossRef CAS PubMed.
- S. U. Yaylaci, M. Sen, O. Bulut, E. Arslan, M. O. Guler and A. B. Tekinay, ACS Biomater. Sci. Eng., 2016, 2, 871–878 CrossRef CAS.
- F. Taraballi, A. Natalello, M. Campione, O. Villa, S. M. Doglia, A. Paleari and F. Gelain, Front. Neuroeng., 2010, 3, 1 CAS.
- S. Sur, F. Tantakitti, J. B. Matson and S. I. Stupp, Biomater. Sci., 2015, 3, 520–532 RSC.
- M. J. Webber, J. Tongers, M.-A. Renault, J. G. Roncalli, D. W. Losordo and S. I. Stupp, Acta Biomater., 2010, 6, 3–11 CrossRef CAS PubMed.
- P. Y. W. Dankers, T. M. Hermans, T. W. Baughman, Y. Kamikawa, R. E. Kieltyka, M. M. C. Bastings, H. M. Janssen, N. A. J. M. Sommerdijk, A. Larsen, M. J. A. van Luyn, A. W. Bosman, E. R. Popa, G. Fytas and E. W. Meijer, Adv. Mater., 2012, 24, 2703–2709 CrossRef CAS PubMed.
- M. M. C. Bastings, S. Koudstaal, R. E. Kieltyka, Y. Nakano, A. C. H. Pape, D. A. M. Feyen, F. J. van Slochteren, P. A. Doevendans, J. P. G. Sluijter, E. W. Meijer, S. A. J. Chamuleau and P. Y. W. Dankers, Adv. Healthcare Mater., 2014, 3, 70–78 CrossRef CAS PubMed.
- R. E. Kieltyka, M. M. C. Bastings, G. C. van Almen, P. Besenius, E. W. L. Kemps and P. Y. W. Dankers, Chem. Commun., 2012, 48, 1452–1454 RSC.
- C. M. A. Leenders, L. Albertazzi, T. Mes, M. M. E. Koenigs, A. R. A. Palmans and E. W. Meijer, Chem. Commun., 2013, 49, 1963–1965 RSC.
- X. Lou, R. P. M. Lafleur, C. M. A. Leenders, S. M. C. Schoenmakers, N. M. Matsumoto, M. B. Baker, J. L. J. van Dongen, A. R. A. Palmans and E. W. Meijer, Nat. Commun., 2017, 8, 15420 CrossRef CAS PubMed.
- C. M. A. Leenders, T. Mes, M. B. Baker, M. M. E. Koenigs, P. Besenius, A. R. A. Palmans and E. W. Meijer, Mater. Horiz., 2013, 1, 116–120 RSC.
- M. H. Bakker, C. C. Lee, E. W. Meijer, P. Y. W. Dankers and L. Albertazzi, ACS Nano, 2016, 10, 1845–1852 CrossRef CAS PubMed.
- R. A. Perez, C.-R. Jung and H.-W. Kim, Adv. Healthcare Mater., 2017, 6, 1600791 CrossRef PubMed.
- Report by MIT, 2016.
- R. Langer and J. P. Vacanti, Science, 1993, 260, 920–926 CAS.
- E. S. Place, N. D. Evans and M. M. Stevens, Nat. Mater., 2009, 8, 457–470 CrossRef CAS PubMed.
- L. Zhang and T. J. Webster, Nano Today, 2009, 4, 66–80 CrossRef CAS.
- R. Xu, A. Boudreau and M. J. Bissell, Cancer Metastasis Rev., 2009, 28, 167–176 CrossRef PubMed.
- E. Morris, M. Chavez and C. Tan, Curr. Opin. Biotechnol, 2016, 39, 97–104 CrossRef CAS PubMed.
- H. J. Wagner, A. Sprenger, B. Rebmann and W. Weber, Adv. Drug Delivery Rev., 2016, 105, 77–95 CrossRef CAS PubMed.
- M. F. Maitz, U. Freudenberg, M. V. Tsurkan, M. Fischer, T. Beyrich and C. Werner, Nat. Commun., 2013, 4, 2168 Search PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |