
Non-noble metal-catalysed carbonylative transformations
Yahui
Li†
,
Yuya
Hu†
and
Xiao-Feng
Wu
 *
*
Leibniz-Institut für Katalyse e.V. an der Universität Rostock, Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: Xiao-Feng.Wu@catalysis.de
First published on 8th November 2017
Abstract
Transition metal catalysts are formidable tools towards greener chemistry, allowing for low-waste, energy-efficient, and selective reactions. And transition metal-catalysed carbonylation procedures are powerful methodologies for producing carbonyl-containing compounds. The existing reviews/chapters/books are mainly focused on noble metal (Ru, Rh, Ir, Pd, Pt) catalysed carbonylation reactions. In this review, achievements on non-noble metal (Mn, Fe, Cu, Co, Ni) catalysed carbonylative transformations have been summarized and discussed.
 Yahui Li | Yahui Li was born in the Anhui province of China. He studied at Huaibei Normal University, where he received his BSc in 2012. Subsequently, he went to Tongji University and earned his MSc in 2015. Currently he is a PhD student working on metal-catalyzed carbonylation at the Leibniz-Institut für Katalyse (Germany). |
 Yuya Hu | Yuya Hu was born in Gansu, China. She studied chemistry at Lanzhou University. After that she went to France and earned her master's degree from Rennes 1 University. Currently, she is working on organocatalysis for her PhD. |
 Xiao-Feng Wu | Xiao-Feng Wu was born in China. He studied chemistry at Zhejiang Sci-Tech University (China), where he got his bachelor's degree in science (2007). In the same year, he went to Rennes 1 University (France) and earned his master's degree in 2009. Then he joined Matthias Beller's group at Leibniz-Institute for Catalysis (Germany), where he completed his PhD in January 2012. Subsequently he started his independent research at LIKAT and ZSTU where he was promoted to a professor in 2013. In March 2017, Xiao-Feng defended his Habilitation successfully from Rennes 1 University (France). Xiao-Feng has authored >200 publications in international journals, meanwhile he is also the editor or author of 10 books. |
1. Introduction
Since the discovery and identification of carbon monoxide by de Lassone and W. C. Crukshank in the 18th century, exploration of the utilization of this small molecule in organic chemistry has become one of the core topics. And terms ‘carbonylation’ and ‘carbonylative’ are given specially for carbon monoxide related organic transformations. Due to the interest in carbon monoxide related transformations, continuous efforts have been made by generations of organic chemists and many novel procedures have been developed.1 Recently, carbonylation reactions have already become one of the most straightforward choices for the synthesis of carbonyl-containing compounds, and many related procedures have been industrialized.2 In addition to the importance of carbonyl-containing chemicals, through carbonylative transformations, the carbon chain of the parent compounds can be easily extended by introducing one or several molecules of CO which represents one of the cheapest C1 sources. However, concerning the transition metal catalysts applied, most of the efforts have been devoted to noble metal catalysts.1–6 Although these catalyst systems have advantages in reactivity and efficiency, their high costs and toxicity, and the demand of even more expensive but non-reusable phosphine ligands still limit their applications on a large scale.In particular, development of economic and environmentally benign synthetic methods has recently become an important but challenging goal in organic chemistry. And the exploration of non-noble catalysts in organic synthesis has proved to be one of the ideal choices, due to their advantages such as abundance, low price, low toxicity, etc.7 Nevertheless, to the best of our knowledge, no review on non-noble metal catalysed carbonylative reactions has been published so far. Considering the importance of both topics and also based on our ongoing research interests, we became interested to fill this gap. We also hope that this review can intrigue and promote further developments in this interesting area.
This review is organized according to the catalysts applied and the main achievements on non-noble metal (Mn, Fe, Cu, Co, and Ni) catalysed carbonylative transformations are summarized and discussed.
2. Mn-Catalysed carbonylation reactions
Manganese, as the twelfth abundant element and the third most abundant transition metal on the earth, has properties such as low cost and relatively low toxicity. Many novel manganese-catalysed organic reactions have been reported in the past few decades.8a,b As early as in 1965, F. Calderazzo reported a pioneer work on manganese-catalysed carbonylation of amines. Using decacarbonyldimanganese and pentacarbonylmethylmanganese as the catalyst, primary aliphatic amines can be transformed into the corresponding 1,3-dialkylureas in good yields (Scheme 1).8c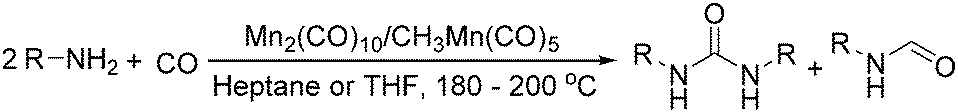 | ||
| Scheme 1 Manganese-catalysed carbonylation of amines. | ||
Later on, Watanabe and co-workers studied the application of manganese catalysts in the carbonylative coupling of alkyl iodides with different nucleophiles.9a,b Alkyl esters and amides can be effectively produced by using alcohols and amines as reaction partners. Additionally, thiols, azides and hydrides can be used as nucleophiles as well (Scheme 2a). Additionally, alkyl bromides can also be applied as the substrates by adding NaI as the additive. Here, high pressure of carbon monoxide and high temperature or irradiation are required. In 1998, Kang and co-workers reported a MnCl2·4H2O (5 mol%)-catalysed carbonylative cross-coupling of organostannanes with hypervalent iodonium salts.9c Under a CO atmosphere (1 bar), good yields of the desired biaryl ketones can be produced (Scheme 2b). Interestingly, biaryls can be formed in good yields with the same substrates under the same conditions just in the absence of CO.
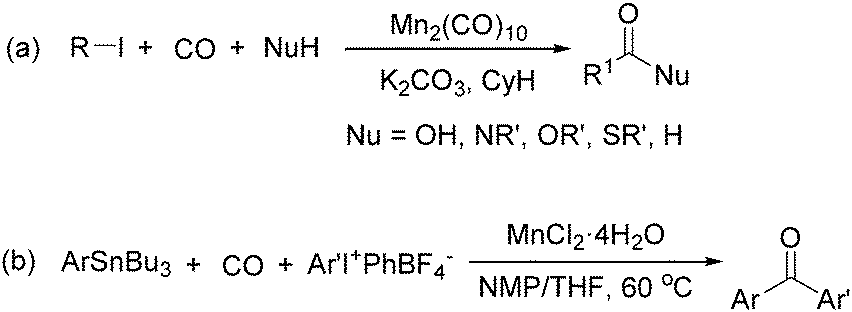 | ||
| Scheme 2 Mn-Catalysed carbonylative coupling of iodides. | ||
More recently, Alexanian and co-workers reported a manganese-catalysed intramolecular carbonylative cyclization of alkenes with alkyl iodides.10 In the presence of 2.5 mol% Mn2(CO)10 and KHCO3 (1 equiv.) under CO (10 bar) pressure in EtOH, five-, six- and seven-membered carbocycles and heterocycles were synthesised in good yields with good diastereoselectivity (Scheme 3). A possible reaction mechanism based on radical nature was provided as well. The reaction starts with the homolysis of a Mn–Mn bond in Mn2(CO)10 dimer to generate the corresponding ˙Mn(CO)5 radical. Then the ˙Mn(CO)5 radical abstracts an iodine atom from the substrates and gives rise to a carbon-centered radical, which undergoes an alkene addition and generates a new carbon radical. The newly formed carbon radical will be trapped by manganese to produce the corresponding acylmanganese intermediate which will provide the final products after a reaction with alcohols.
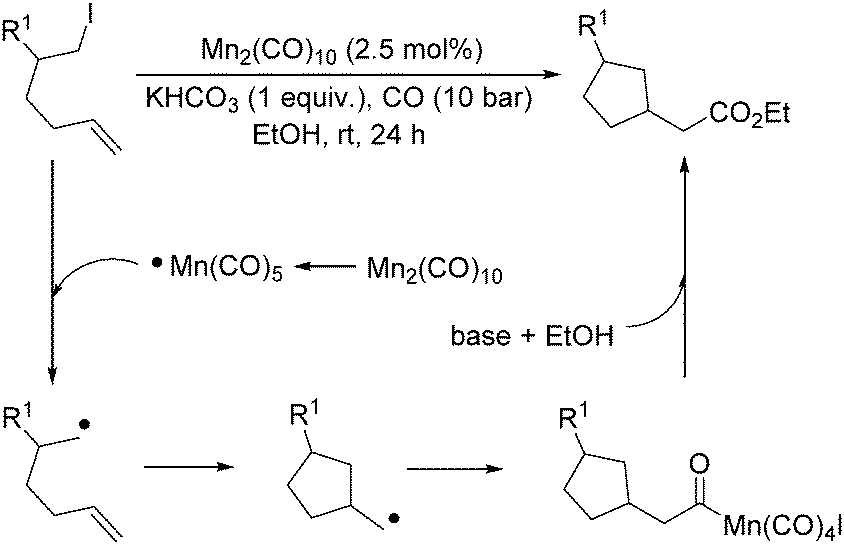 | ||
| Scheme 3 Mn-Catalysed intramolecular carbonylative cyclization of alkenes with alkyl iodides. | ||
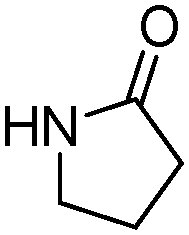
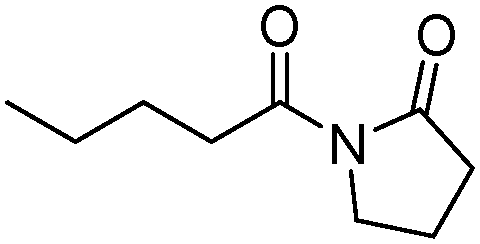
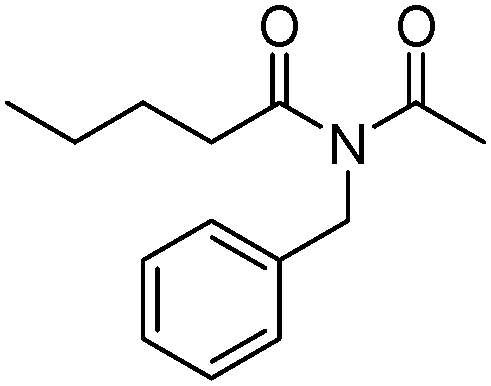
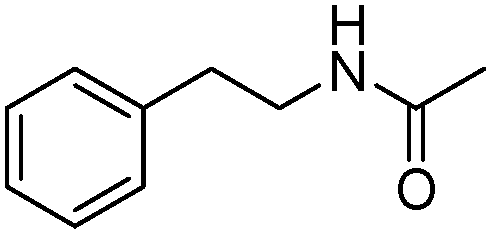
Meanwhile, our group developed a practical manganese-catalysed carbonylation reaction of alkyl halides with amides (Table 1).11 In this work, for the first time, amides were applied as the coupling partner and compared with alcohols and amines; the low nucleophilicity of amides hindered their applications in cross-coupling reactions. By using manganese as the catalyst, the carbonylation of alkyl iodides with amides proceeded and gave the corresponding imides in good yields. Concerning the reaction pathway, a radical mechanism was proposed and also confirmed by EPR investigations.
|
|
|||
|---|---|---|---|
| Entry | 2 | Product | Isolated yield (%) |
| 1 |

|
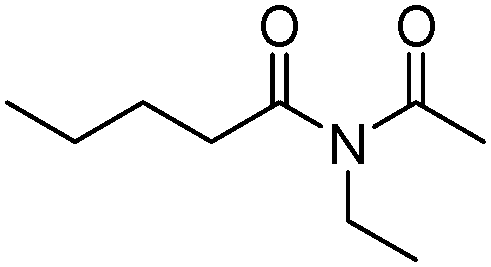
|
58 |
| 2 |
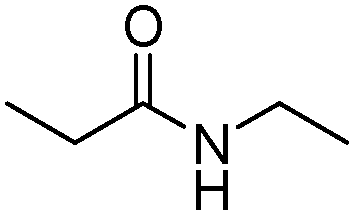
|
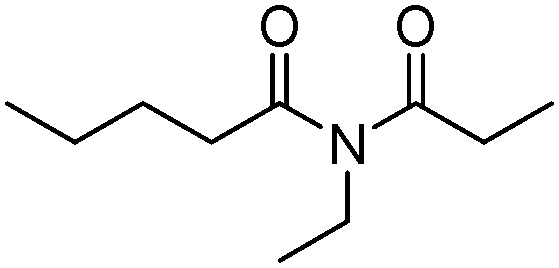
|
60 |
| 3 |
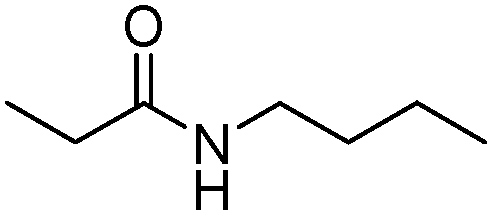
|
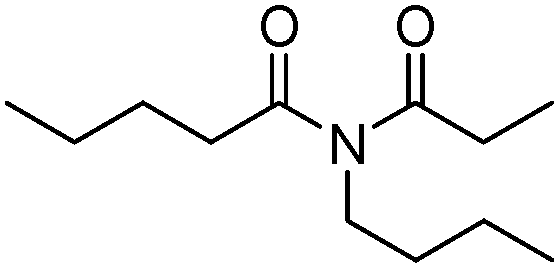
|
75 |
| 4 |
|
|
53 |
| 5 |
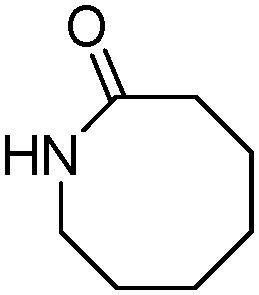
|
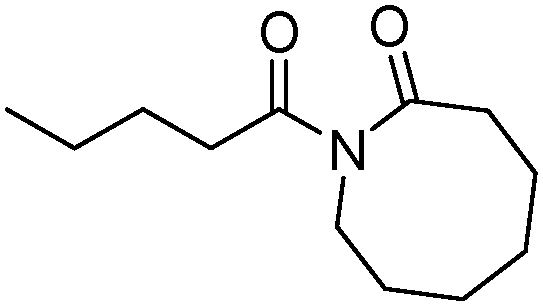
|
83 |
| 6 |
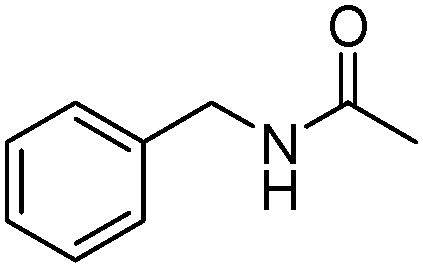
|
|
62 |
| 7 |
|
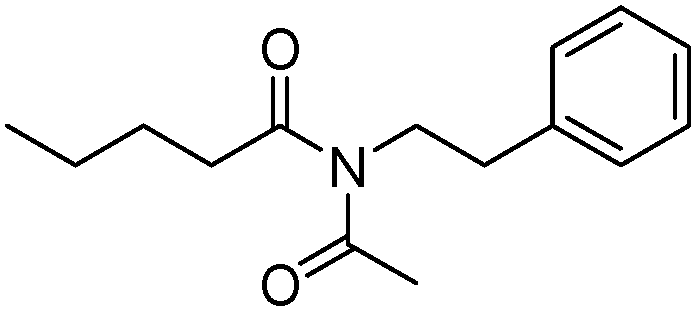
|
66 |
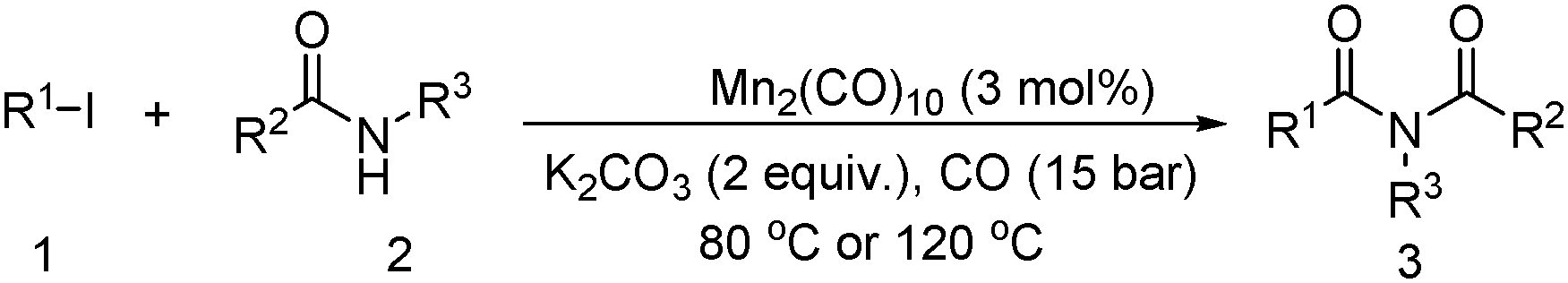
3. Fe-Catalysed carbonylation reactions
The exploration of iron salts as catalysts in organic synthesis has become an attractive topic due to their abundance, low price, high biological compatibility, and also rich oxidation states from −2 to +6.Since the middle of the 20th century, chemists have started to explore iron-promoted carbonylation reactions and the most often used reagents are shown in Fig. 1. Na2Fe(CO)4 which is also known as the Collman reagent was commonly applied as a stoichiometric carbonylation reagent, and several different preparation procedures have been developed.12 The original method needed an expensive sodium–mercury amalgam (Scheme 4, formula (1)) and this factor limited its scale-up, but a more practical procedure was developed later on (Scheme 4, formula (2)). The latest process makes the preparation conditions milder, but the need for bubbling with CO gas makes the manufacture more risky (Scheme 4, formula (3)).
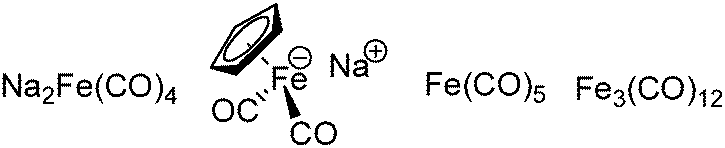 | ||
| Fig. 1 Iron catalysts for carbonylation reactions. | ||
 | ||
| Scheme 4 The synthesis of Na2Fe(CO)4. | ||
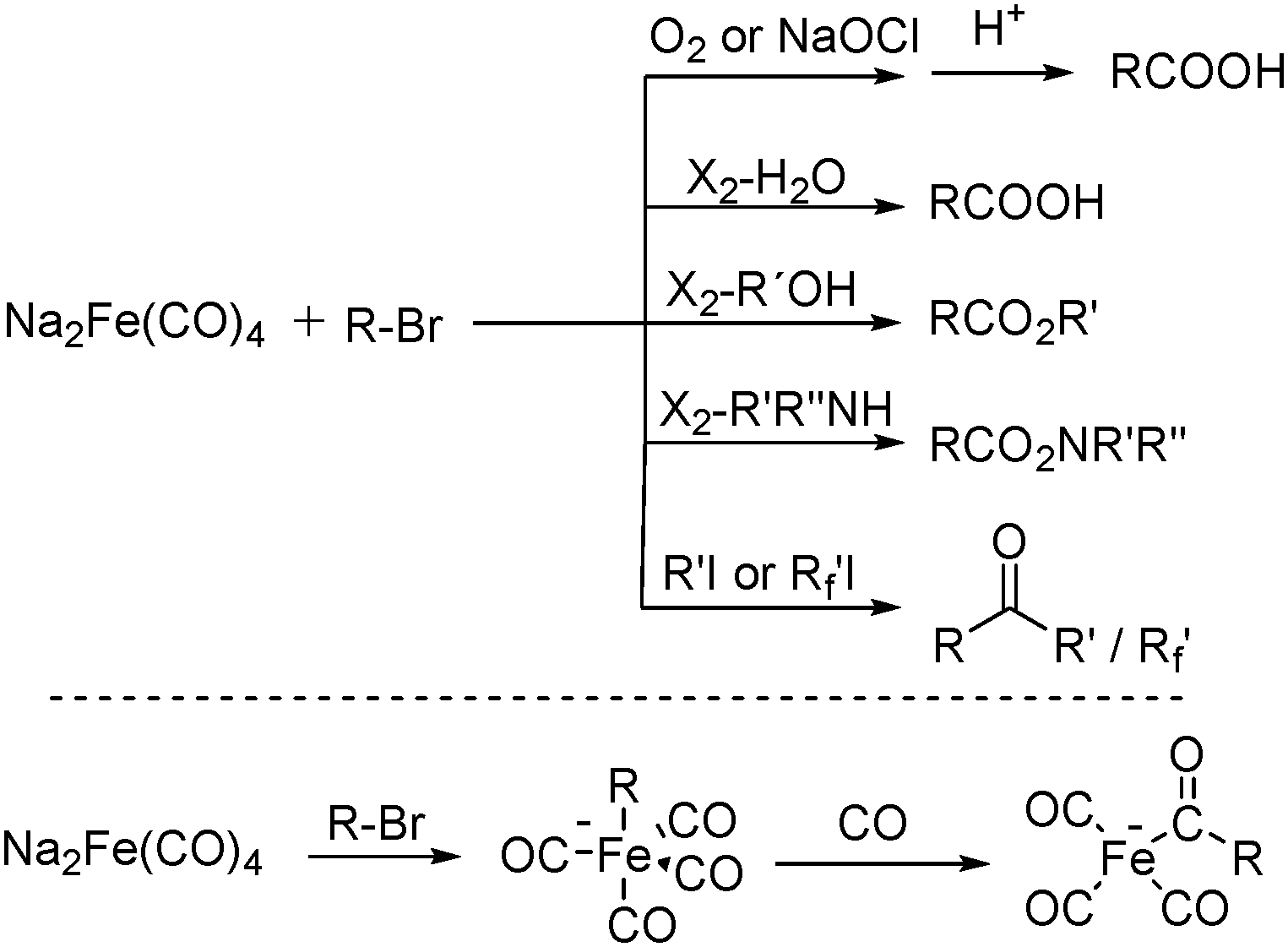
In 1975, Collman found that disodium tetracarbonylferrate can be used in a wide range of useful synthetic reactions (Scheme 5).13 Na2Fe(CO)4 can be considered as a kind of a Grignard reagent. Reactions that use Na2Fe(CO)4 as the catalyst has the advantages of high yields and good tolerance of different functional groups. But it also has some limitations, e.g., tertiary aliphatic halides cannot be used due to its sterical and allylic halides cannot be employed due to the stable 1,3-diene·Fe(CO)3 intermediate.
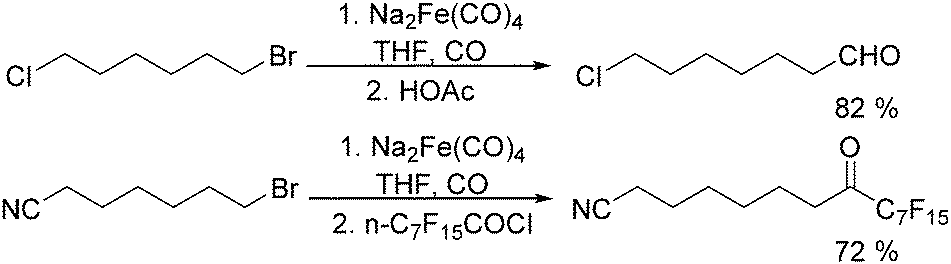 | ||
| Scheme 5 Na2Fe(CO)4-mediated carbonylation reactions. | ||
After that, he also found that Na2Fe(CO)4 can be used for the synthesis of carboxylic acids, esters and amides (Scheme 6). The reaction begins with an SN2 displacement at a carbon atom or an oxidative addition of alkyl bromides to Na2Fe(CO)4 and affords intermediate 2. In the presence of CO, acyl complexes 3 can be generated. Various carboxylic acids,14 esters,14 amides,14 ketones15 and hemifluorinated ketones16 can be produced by reacting the complex with appropriate nucleophiles.
 | ||
| Scheme 6 Na2Fe(CO)4-Mediated carbonylative transformations. | ||
In 1970, Cooke found that using sodium tetracarbonylferrate as the catalyst, alkyl bromides can be transformed into the corresponding aldehydes (Table 2).17 The reaction begins with the oxidative addition of the alkyl bromides to Na2Fe(CO)4 and affords intermediate 2 or 3. Then triphenylphosphine is added to the intermediate and intermediate 4 is obtained. Finally, intermediate 4 will be protonated by acetic acid to give the acyl iron hydride intermediate 5 which undergoes reductive elimination to yield the desired aldehyde (Scheme 7).
|
|
||
|---|---|---|
| Alkyl bromide | Product | Yield (%) |
| CH3(CH2)4Br | CH3(CH2)4CHO | 99 |
| CH3(CH2)8Br | CH3(CH2)8CHO | 91 |
| PhCH2CH2Br | PhCH2CH2CHO | 86 |
| CH3(CH2)5CHBrCH3 | CH3(CH2)5CH(CH3)CHO | 50 |
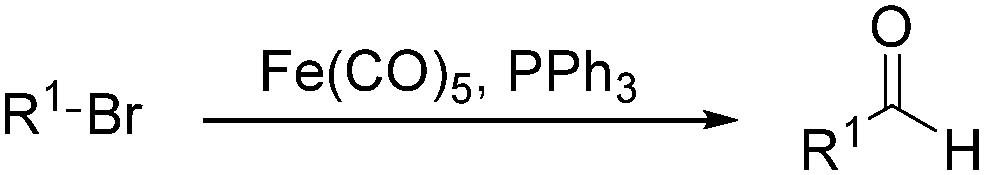
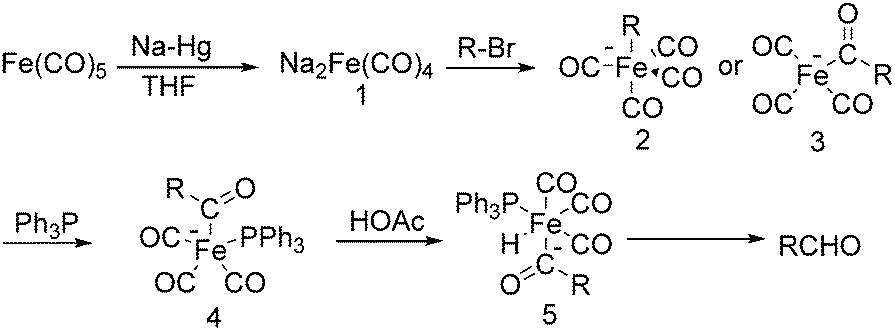 | ||
| Scheme 7 Mechanism of Fe(CO)5-mediated aldehyde synthesis. | ||
Nitro-containing compounds play an important role in organic chemistry. In 1981, a procedure for converting nitro compounds to the corresponding formamides and carbamate esters by using iron and ruthenium carbonyl as the catalyst system was reported by Alper and Hashem.18 These reactions have features of mild conditions (60 °C, atmospheric pressure of CO/H2; Table 3).
|
|
|||
|---|---|---|---|
| 1 | Catalyst | 2 | 3 |
| p-ClC6H4 | Ru3(CO)12 | 59 | — |
| Fe3(CO)12 | — | 35 | |
| p-CH3OC6H4 | Ru3(CO)12 | 56 | — |
| Fe3(CO)12 | — | 62 | |
| p-CH3C6H4 | Ru3(CO)12 | 32 | — |
| Fe3(CO)12 | — | 39 | |
| Ph | Ru3(CO)12 | 69 | — |
| Fe3(CO)12 | — | 61 | |
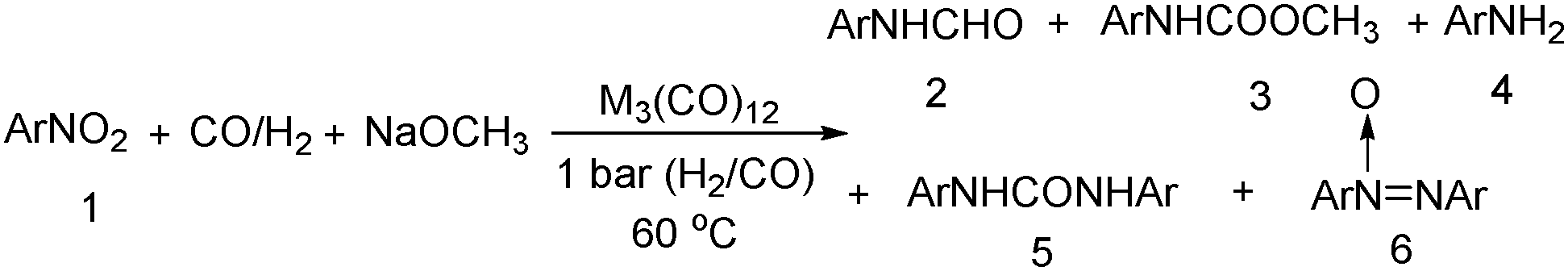
In 1992, Eaton and co-workers reported an interesting iron-catalysed carbonylative [4+1] cycloaddition reaction.19 In the presence of Fe(CO)5 (10 mol%) as the catalyst and CO, conjugated diallenes were transformed into the desired 2,5-diakylidenecyclo-3-pentenones in good yields (Table 4).
|
|
||||
|---|---|---|---|---|
| R1 | R2 | R3 | R4 | Yield (%) |
| C(CH3)3 | CH3 | C(CH3)3 | CH3 | 75 |
| C6H5 | CH3 | C6H5 | CH3 | 81 |
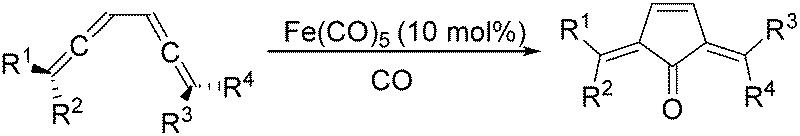
Soon after, the same group also succeeded in extending their substrates to allenyl ketones20a and allenyl imines.20b With iron as the catalyst via carbonylative [4+1] cycloaddition involving the formation of both C–O and C–N bonds, the desired five-membered heterocycles were formed in good yields (Scheme 8a). In their proposed reaction mechanism, irradiation of Fe(CO)5 was necessary to obtain the active species and then a reaction with the starting substrates formed complex A. Complex C was formed through insertion of CO to metallocycle B which gave the final product after reductive elimination.
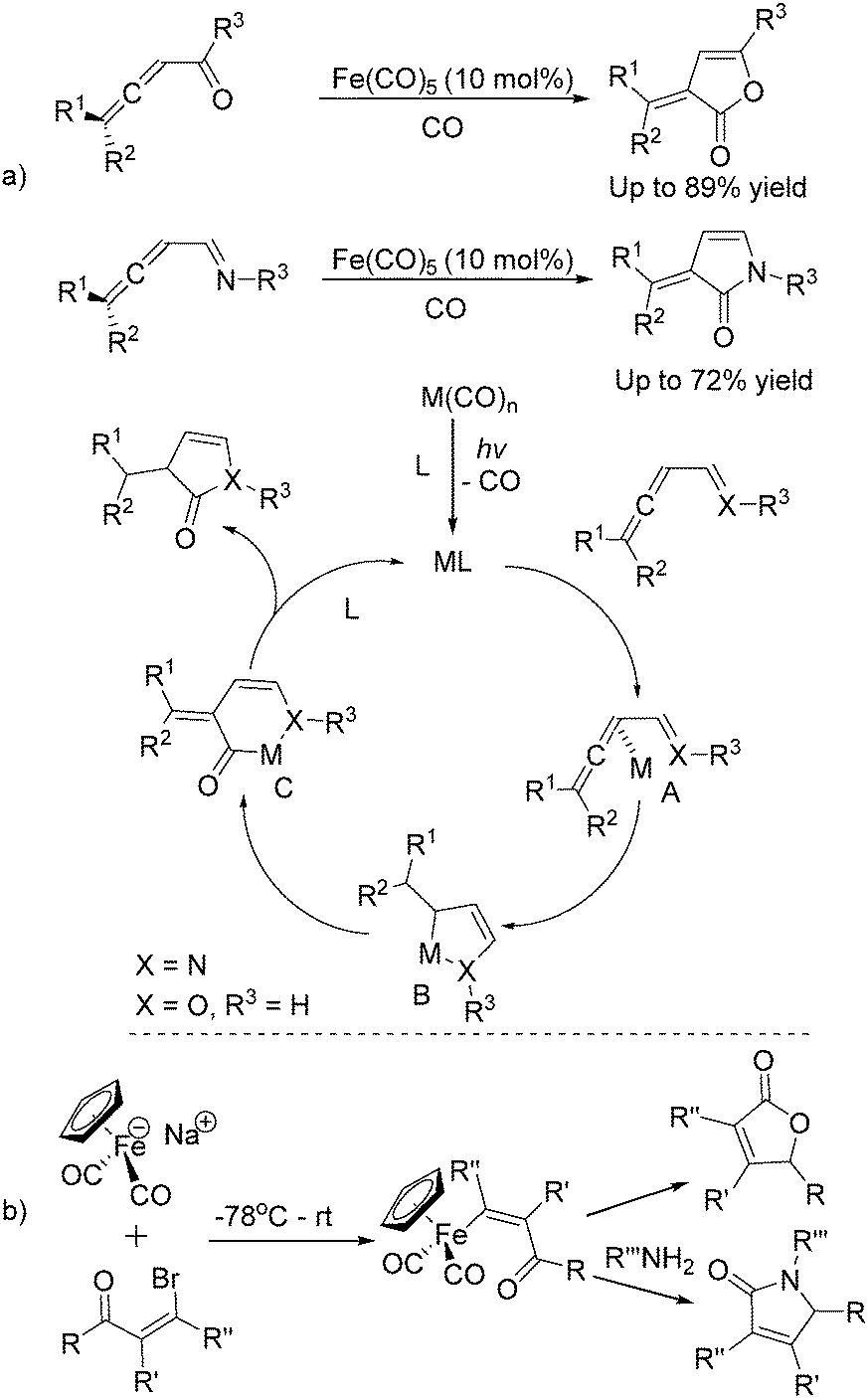 | ||
| Scheme 8 Iron-catalysed [4+1] cycloaddition reactions. | ||
The Rueck-Braun group developed a series of [Cp(CO)2Fe]Na-mediated cyclocarbonylations of β-bromo enals to γ-lactams and γ-lactones (Scheme 8b).21 Iron substituted (Z)-enals were formed as the key intermediates; after being reacted with Grignard reagents or organolithiums the corresponding 5-substituted lactams or γ-lactones were formed in moderate to good yields. These methodologies provide alternative procedures for the preparation of five-membered lactones under mild conditions and some reaction mechanism was also studied, but the necessity of an equivalent amount of iron complex still leaves space for further improvement.
In 1989, Brunet and Taillefer developed a Fe(CO)5–Co2(CO)8 bimetallic system for the carbonylative transformation of aryl iodides (Scheme 9). Using this bimetallic system, iodobenzene can be transformed into benzoic acid22 or benzophenone under 1 bar of carbon monoxide at 60 °C.23 The combination of Bu4NBr and EtOH was found critical. And the function of ethanol was to extract the salt [NBu4]+·[HFe(CO)4]− from water to the organic layer. In the absence of ethanol, iodobenzene can be transformed into benzophenone in fair yield.
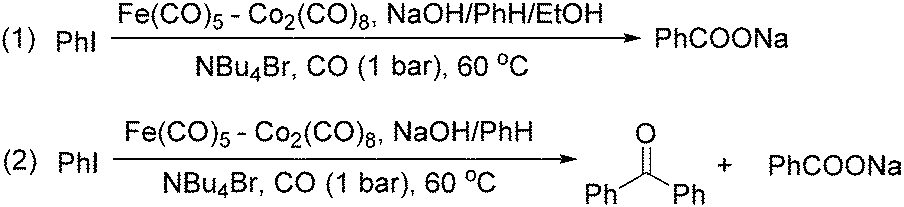 | ||
| Scheme 9 Fe(CO)5–Co2(CO)8 bimetallic-catalysed carbonylation. | ||
Periasamy and co-workers developed a carbonylative transformation of R′2BI in 1991. In the presence of NaCo(CO)4 or Na2Fe(CO)4, symmetric dialkyl ketones can be formed after H2O2/OH− oxidation (Table 5).24 Here the R′2BI applied was prepared in situ by the reaction of alkene with IH2B:N(Et)2Ph at room temperature in benzene. This method provides a straightforward pathway for the synthesis of symmetric dialkyl ketones from alkenes.
|
|
|||
|---|---|---|---|
| Entry | Alkene | Product | Yield |
| 1 | C4H9CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
(C4H9CH2–CH2)2CO |
76 |
| 2 | C6H13CHCH2 |
(C6H13CH2–CH2)2CO |
72 |
| 3 |
|
|
85 |
| 4 |
|
|
75 |
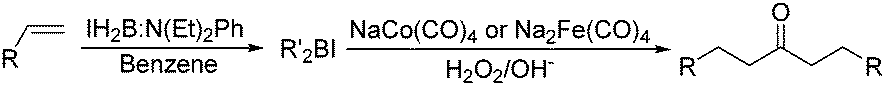
Later on, Periasamy and co-workers reported a homo-coupling reaction of Na(RCO)Fe(CO)4 with CuCl or I2 as the oxidant to produce 1,2-diketones (Scheme 10).25 In this transformation, Na(RCO)Fe(CO)4 reacted with CuCl at 25 °C in THF to form the desired 1,2-diketones in 70–90% yields. In their mechanistic investigations they found that the formation of 1,2-diketone takes place through the decomposition of Cu(RCO)Fe(CO)4 or I(RCO)Fe(CO)4 to RCOFe(CO)4 followed by the formation of η2 complexes of 1,2-diketone intermediates. However, a single-electron reduction pathway cannot be excluded.
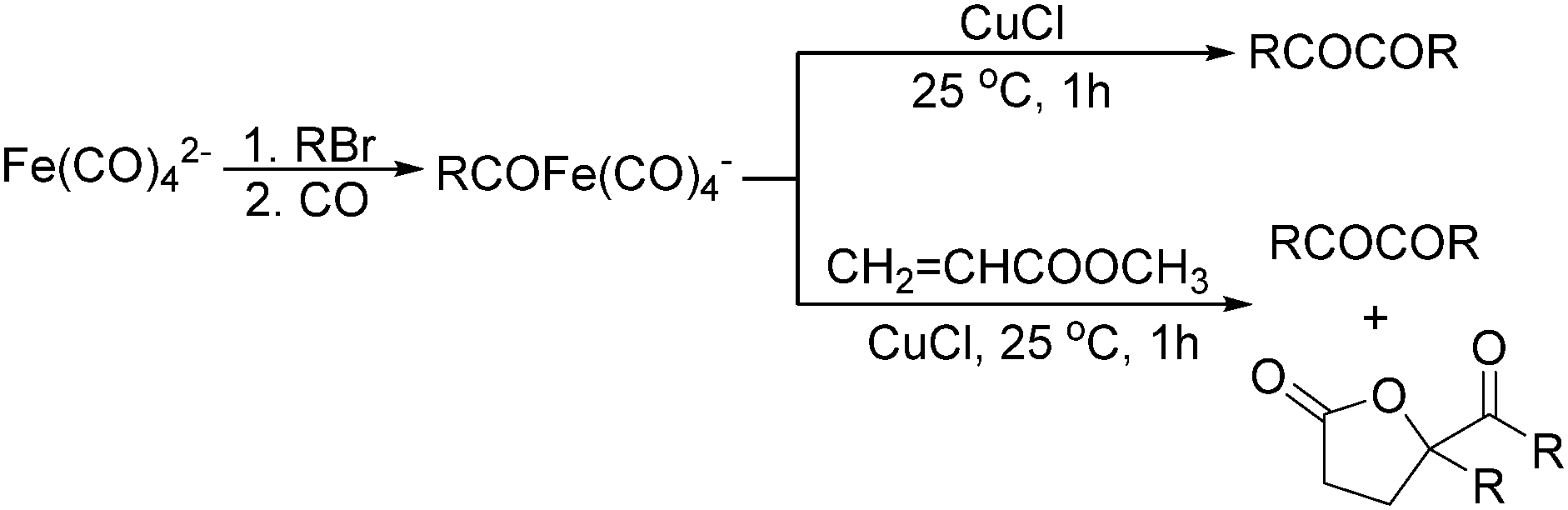 | ||
| Scheme 10 Fe(CO)42−–CuCl mediated carbonylation. | ||
In 1996, Brunet, Periasarny and their co-workers developed a novel method for double carbonylation transformation of alkynes using NaHFe(CO)4 as the catalyst (Table 6).26 This method contained three steps: (i) NaHFe(CO)4 reacted with MeI to generate the active reagent; (ii) the formed reagent reacted with alkynes to form the key intermediate and (iii) it was subsequently oxidized by CuCl2 to give the corresponding cyclobutenediones in 27–42% yields and together with the formation of α,β-unsaturated carboxylic acids.
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yield (%) |
| 1 |
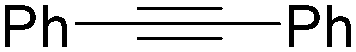
|
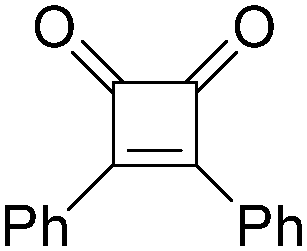
|
42 |
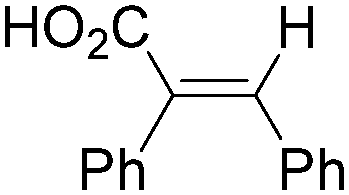
|
22 | ||
| 2 |
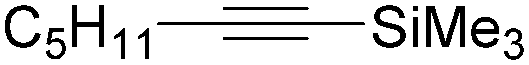
|
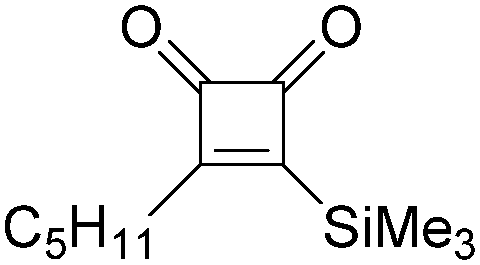
|
37 |
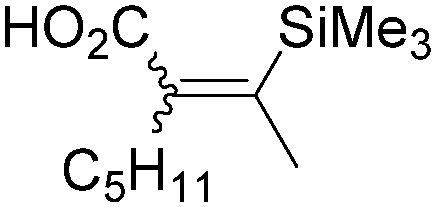
|
12 | ||
| 3 |
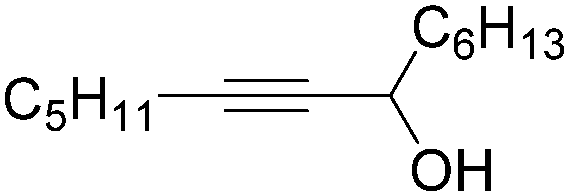
|
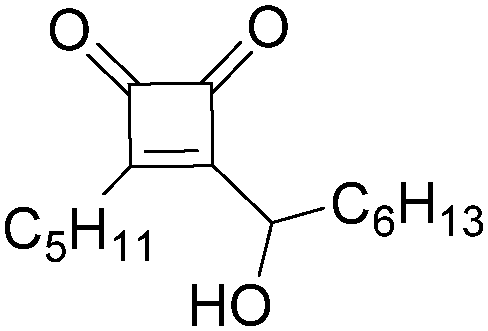
|
27 |
| 4 |
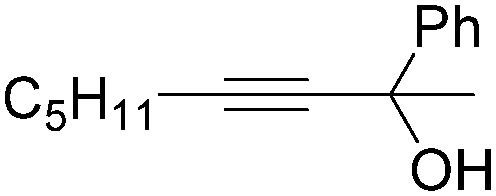
|
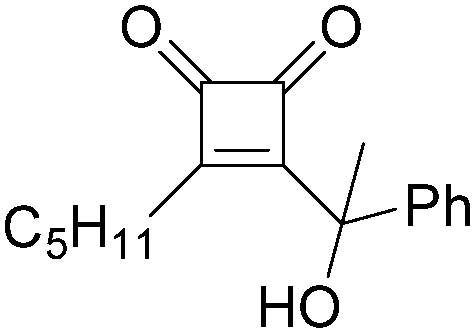
|
31 |
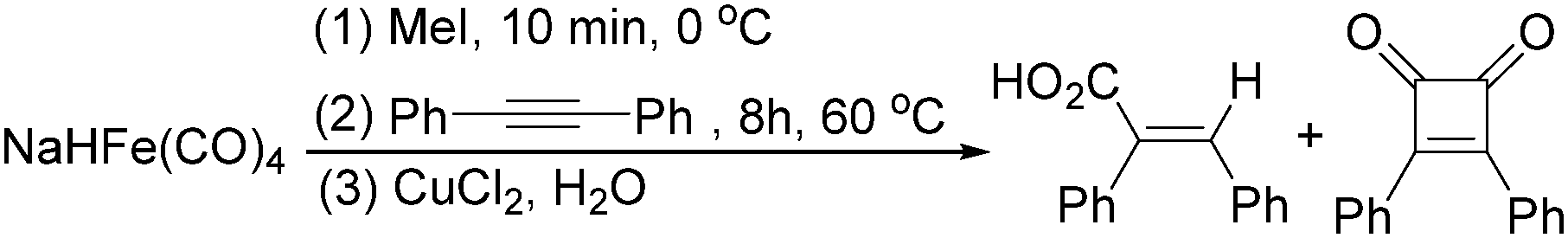
After that, they found that NaH,27 NaBH4,28 amines,29 Me3NO30 and t-BuOK31 can also be used to enhance the reactivity of Fe(CO)5 and Fe3(CO)12 (Scheme 11). Compared with the previous method, the reaction becomes more selective and gives cyclobutenediones as the only product. In the presence of an excess amount of amine oxide, cyclic anhydrides can be observed.
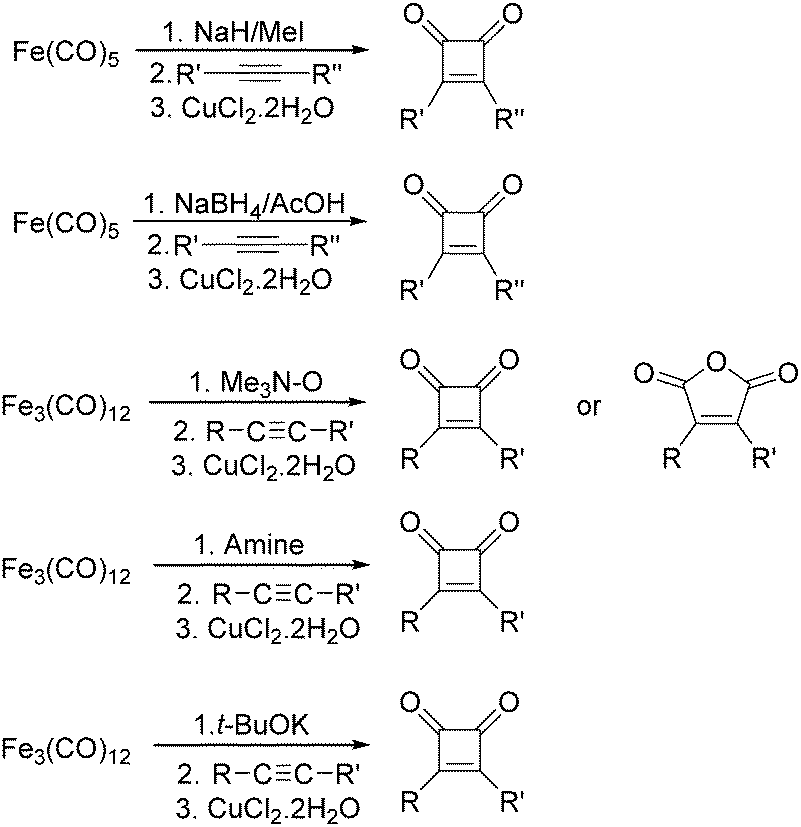 | ||
| Scheme 11 Fe(CO)5 mediated cyclocarbonylation. | ||
Beller and co-workers reported an iron-catalysed carbonylation for the synthesis of succinimides in 2009 (Table 7).32 With 10 mol% of Fe(CO)5 as the catalyst under CO pressure (20 bar) at 120 °C, excellent yields of the desired products can be isolated. This synthetic procedure has also been applied in the synthesis of biologically interesting 3,4-diaryl-substituted maleimides33 and himanimide34 as well.
|
|
||||
|---|---|---|---|---|
| Entry | Alkene | R3NH2 | Product | Yield (%) |
| 1 |
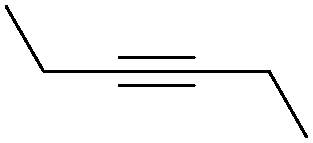
|
NH3 |
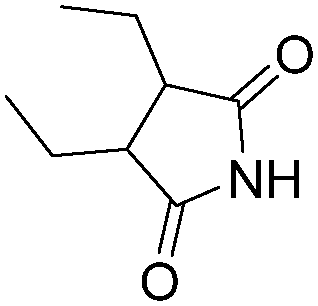
|
84 |
| 2 |
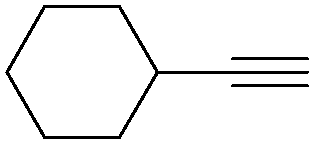
|
NH3 |
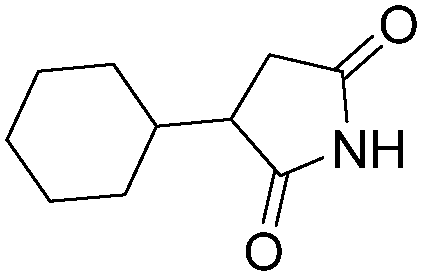
|
83 |
| 3 |
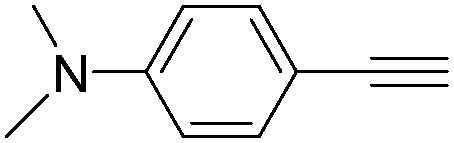
|
NH3 |
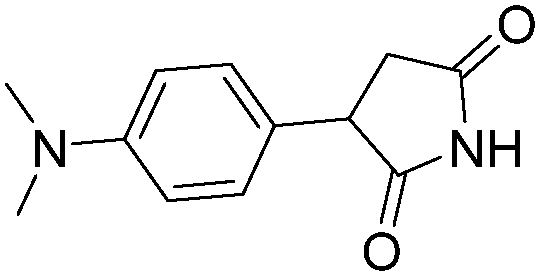
|
55 |
| 4 |
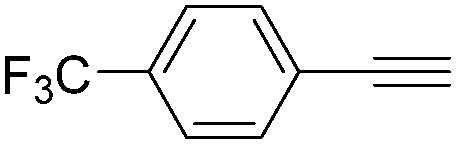
|
NH3 |
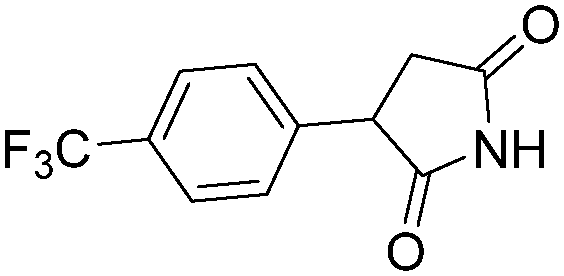
|
37 |
| 5 |
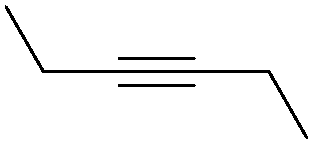
|
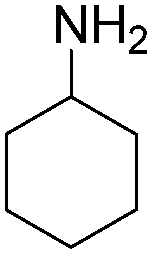
|
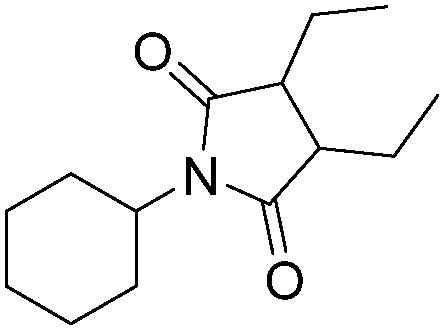
|
56 |
| 6 |
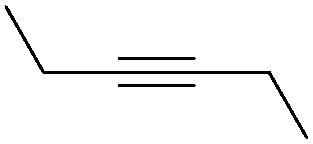
|
|
|
71 |
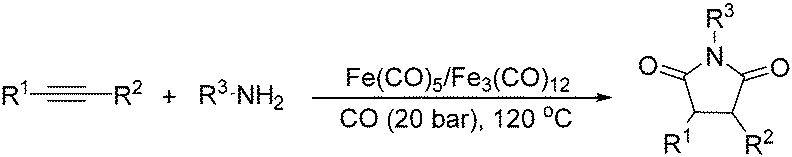
In 2011, Beller and co-workers found that the selectivity can be tuned by the addition of a nitrogen ligand (Table 8).35a α,β-Unsaturated amides can be selectively produced from the same alkynes and amines in the presence of 5 mol% of Fe3(CO)12 and the ligand. With 5 equivalents of NEt3 as the base under CO pressure (10 bar) in THF at 120 °C, 20 different α,β-unsaturated amides were formed in 47–95% yields. Meanwhile, a microwave-assisted aminocarbonylation of ynamides with amines at low pressures of CO (1.3 bar) was reported by Petricci and co-workers.35b (E)-Acrylamides can been regioselectively synthesized after microwave irradiation with [Fe3(CO)12] as the catalyst precursor and triethylamine (TEA) as the ligand.
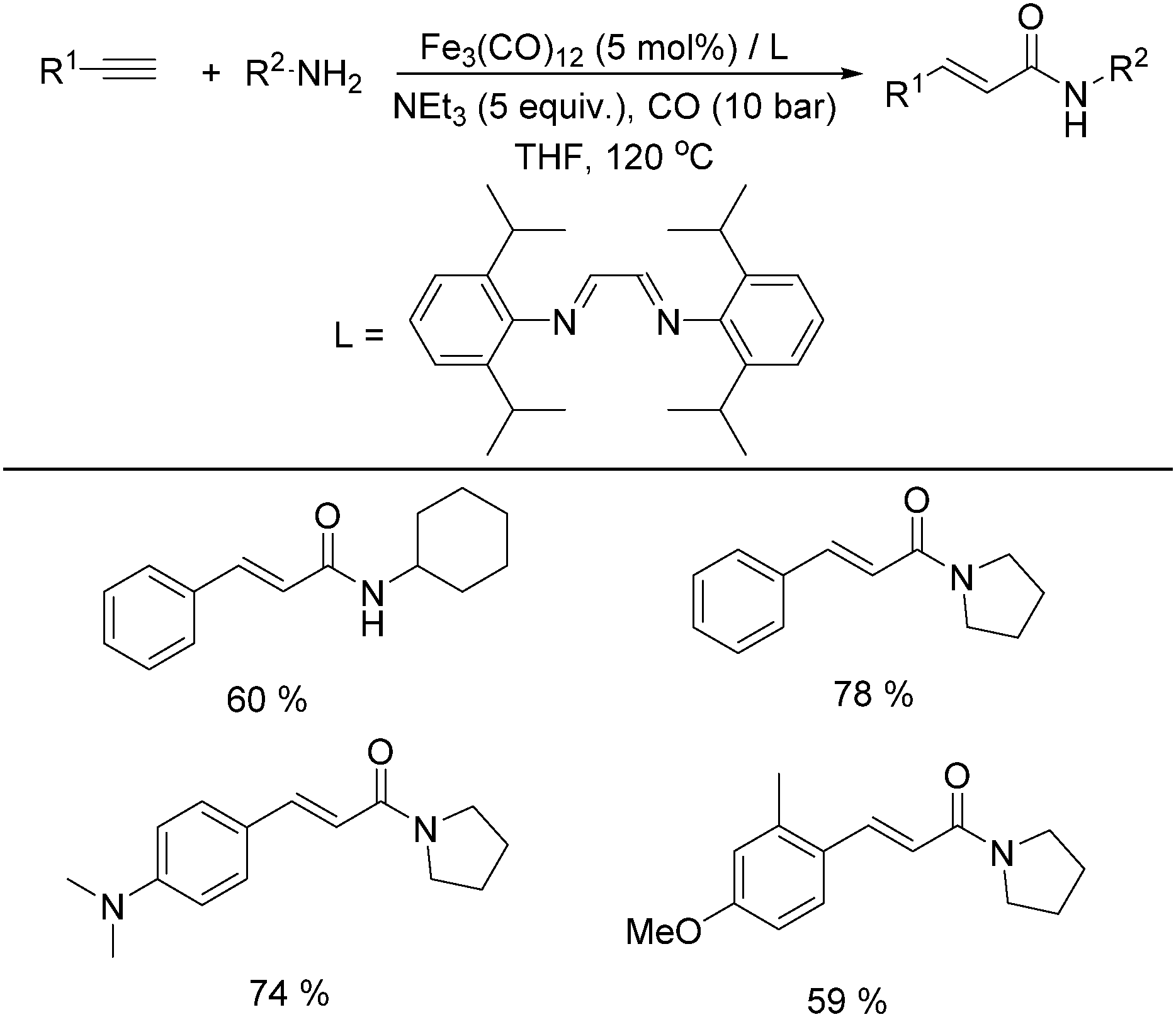
|
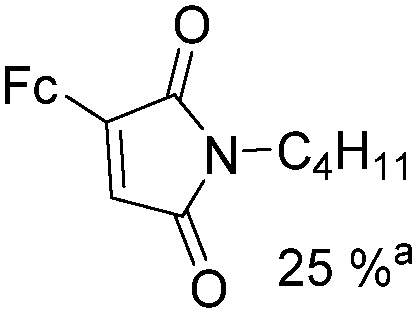
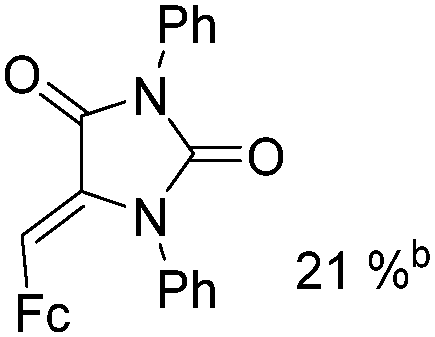
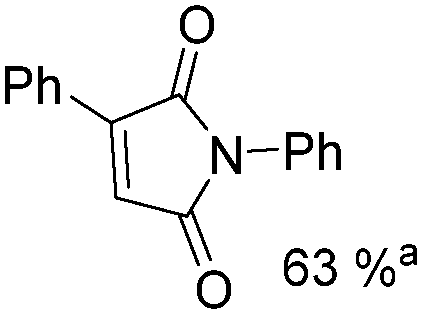
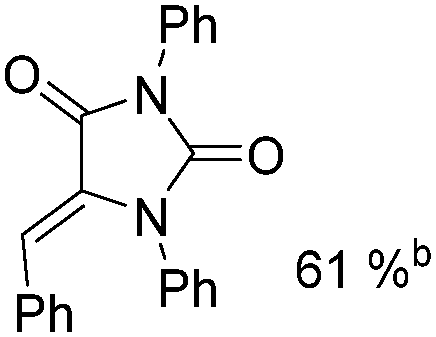
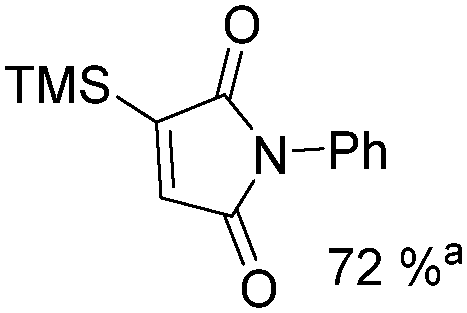
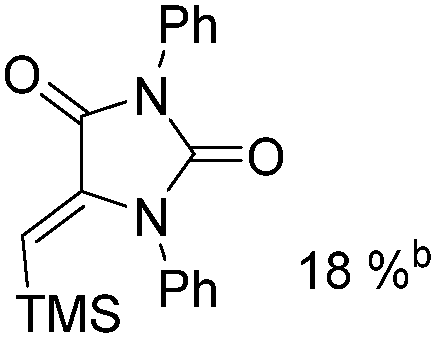
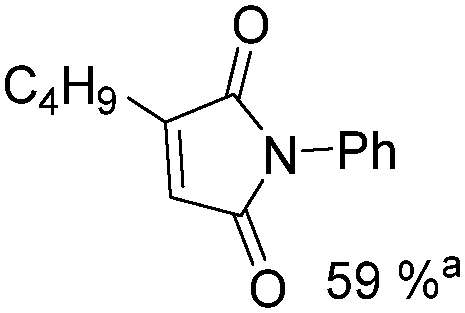
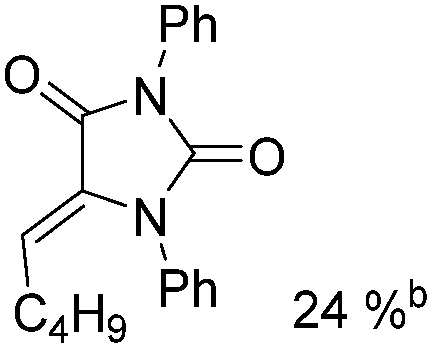
Mathur and co-workers developed a Fe(CO)5-catalysed carbonylative procedure for the synthesis of maleimides and hydantoins in 2012 (Table 9).36 With terminal alkynes and isocyanate as the substrates in presence of CO, the maleimides can be obtained as the major products. Interestingly, hydantoins can be formed in up to 87% yield in the absence of CO.
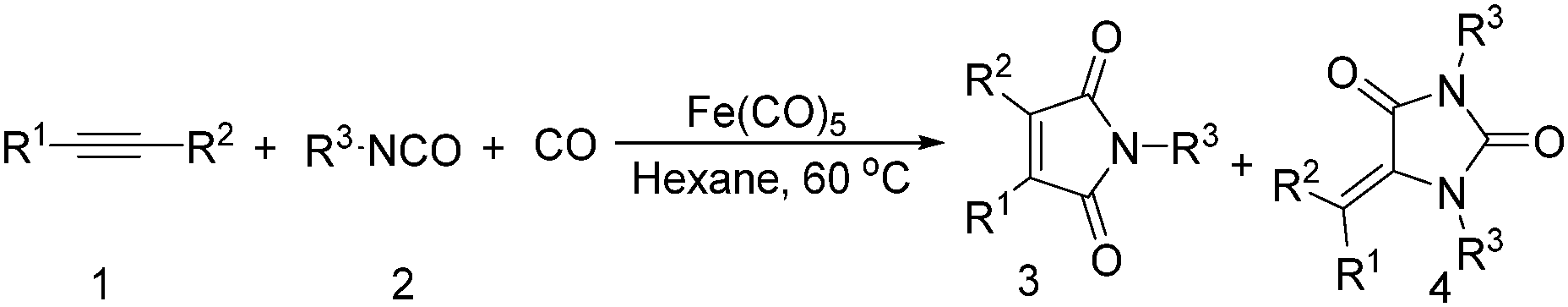
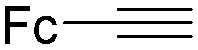
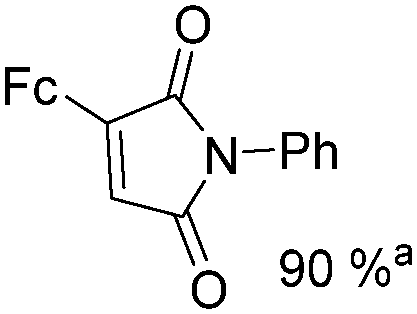
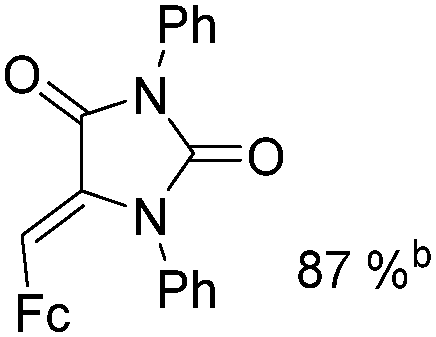
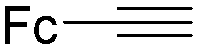
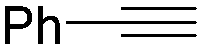
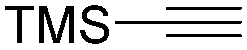
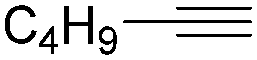
More recently, Iranpoor, Firouzabadi and their co-workers reported a Fe(CO)5 or Mo(CO)6 mediated hydrocarbonylation of phenylacetylene to give α,β-unsaturated esters and thioesters (Table 10a).37a In the presence of 1 equiv. of Fe(CO)5 and 2 equiv. of DABCO in DMF at 100 °C, phenylacetylene reacted with different alcohols and thiols to give the desired products in 87–98%. Notably, a Fe(CO)5/hν catalyst system for producing vinylesters and lactones from alkynes and alcohols was established by Mathur and co-workers in 2010.37b The reactions were carried out at 0 °C, and the selectivity of the products depended on the time of photolysis of the reaction as well as the solvent applied. A stable reaction intermediate ferrole was isolated which produced α,β-vinylester after further photolysis with alcohols.
|
|
|||
|---|---|---|---|
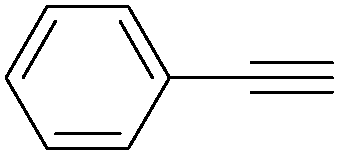
| Entry | Phenylacetylene | Alcohol | Yields (%) |
| 1 |
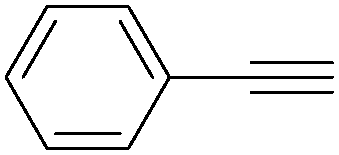
|
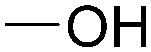
|
96 |
| 2 |
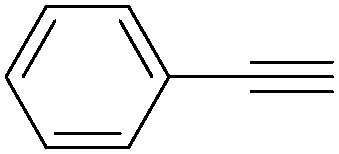
|
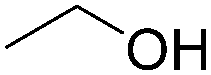
|
95 |
| 3 |
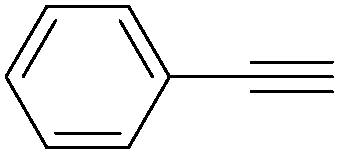
|
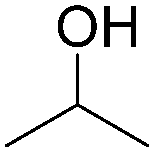
|
98 |
| 4 |
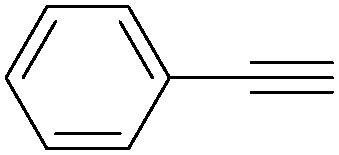
|
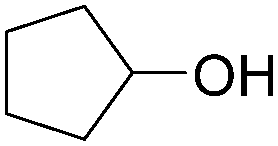
|
90 |
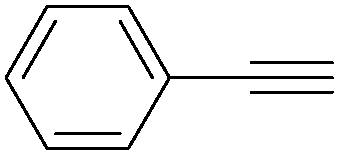
| 5 |
|
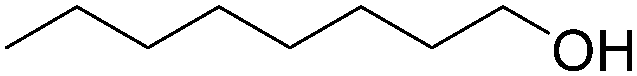
|
96 |
| 6 |
|
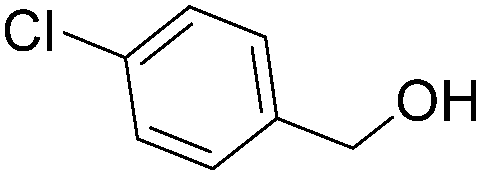
|
87 |
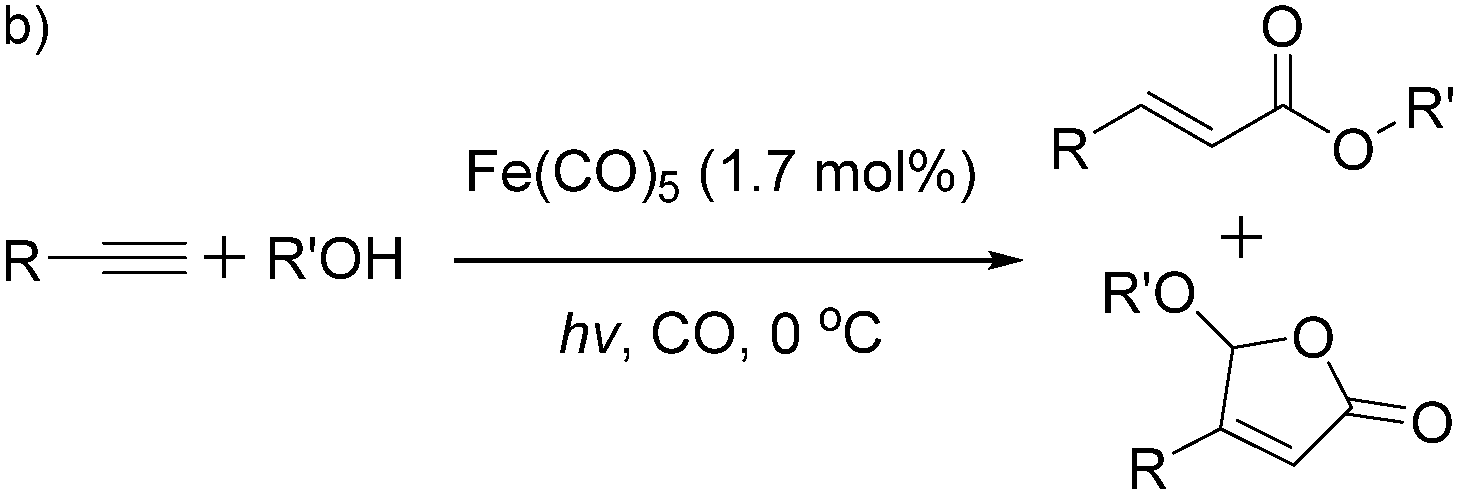
|
|||
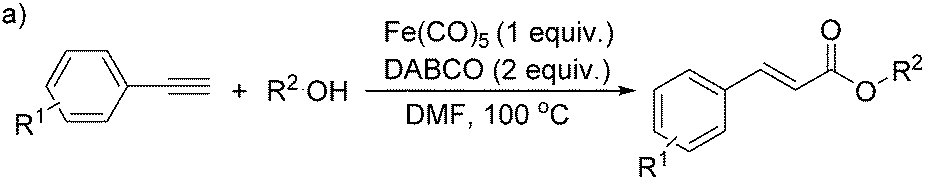
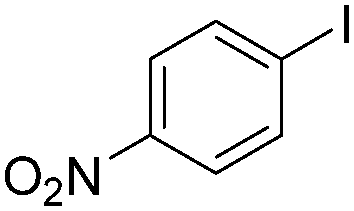
Notably, Han and Zhong developed an iron-catalysed carbonylative Suzuki reaction with PEG-400 as the solvent in 2014 (Table 11).38 In the presence of 4 mol% of FeCl2 and 6 mol% FeCl3 as the catalyst combination with 2 equivalents of NaHCO3 as the base in PEG-400, the desired ketone products can be obtained in 79–94% yields. Good functional group tolerance can be observed, even towards the nitro group. The reactions were performed under atmospheric pressure of carbon monoxide. In their mechanistic studies, they found that an iron carbonyl species was formed as the intermediate and then acted as the CO source to form the product. However, due to the properties of PEG solvents, in situ formed nanoparticles cannot be excluded as the real catalyst.
|
|
||||
|---|---|---|---|---|
| Entry | 1 | 2 | 3 | Yield (%) |
| 1 |
|
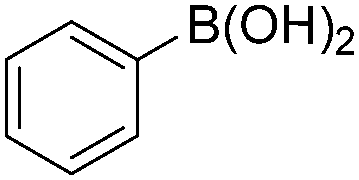
|
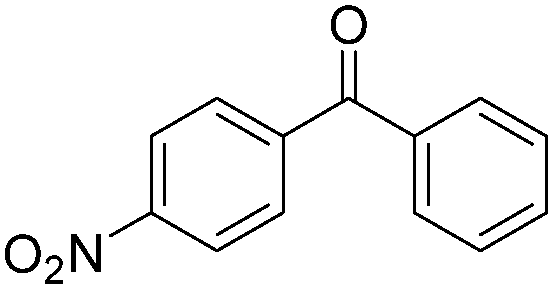
|
92 |
| 2 |
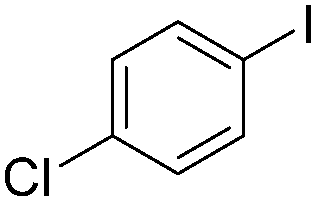
|
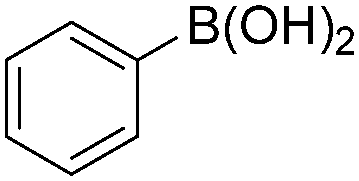
|
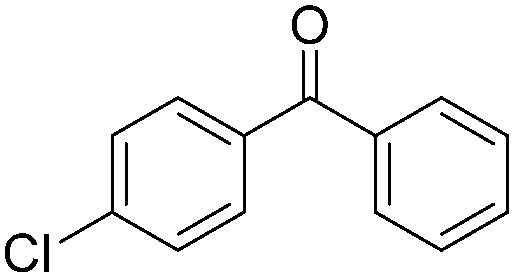
|
90 |
| 3 |
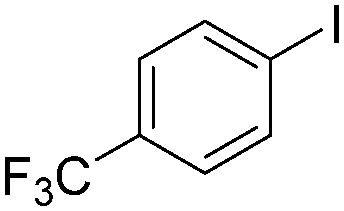
|
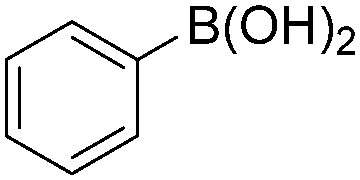
|
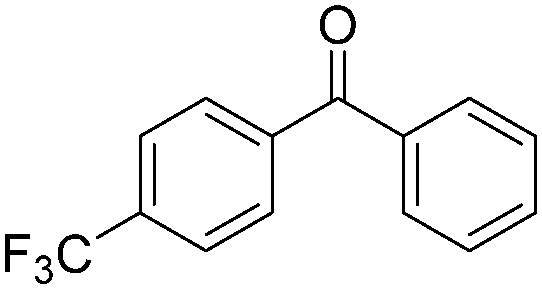
|
75 |
| 4 |
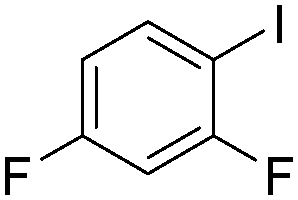
|
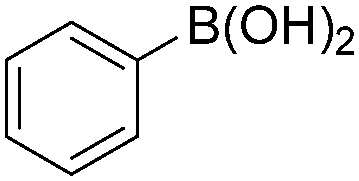
|
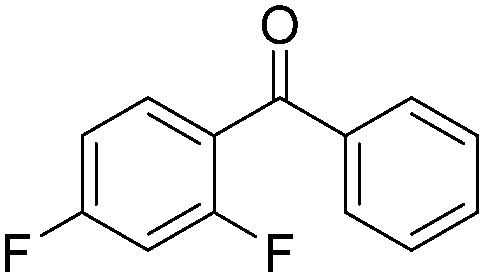
|
71 |
| 5 |
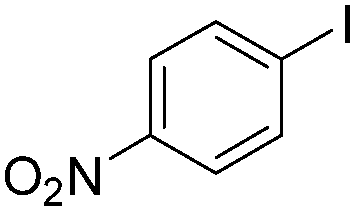
|
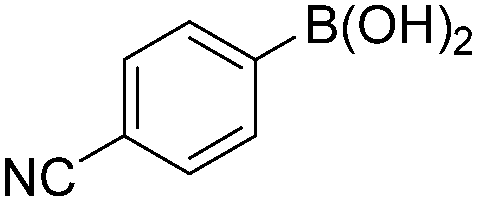
|
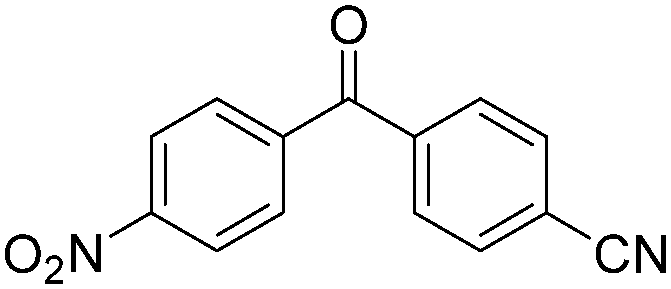
|
91 |
| 6 |
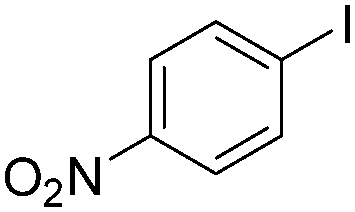
|
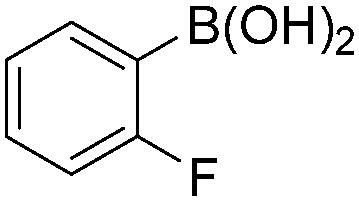
|
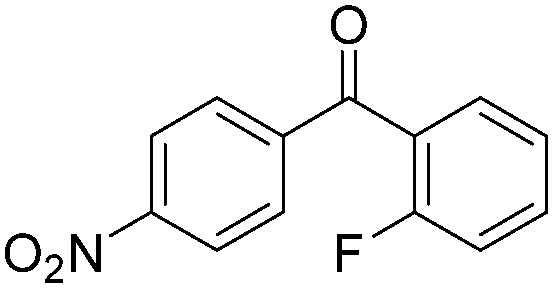
|
90 |
| 7 |
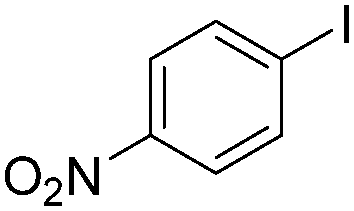
|
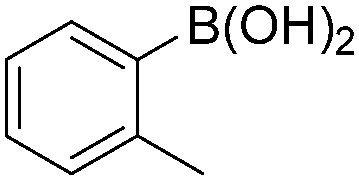
|
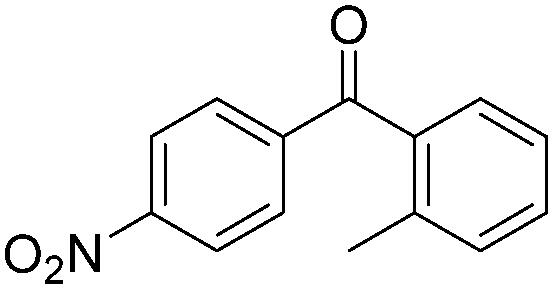
|
79 |
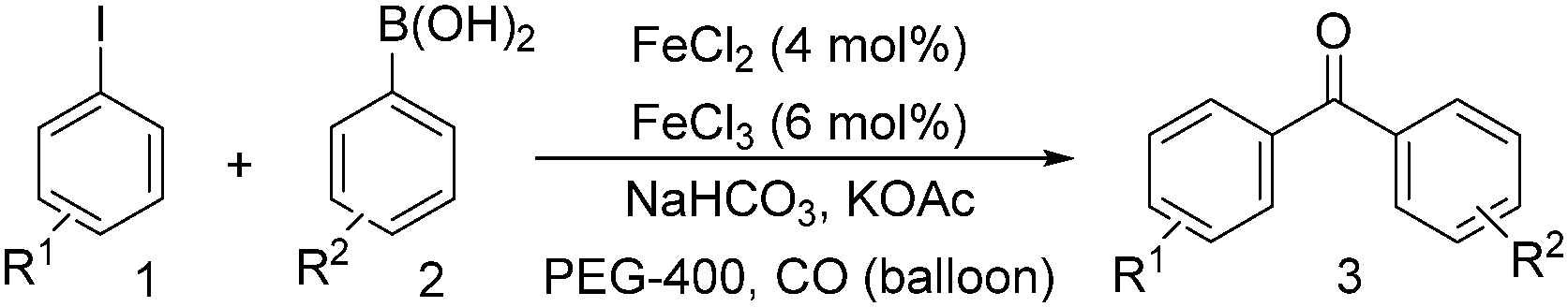
4. Cu-Catalysed carbonylation reactions
Copper catalysts have been extensively employed in oxidation reactions and cross-coupling reactions. Their applications in carbon monoxide insertion related reactions are still very limited. In 1996, Kang and co-workers developed a copper-catalysed cross-coupling and carbonylative coupling reaction of organostannanes (Table 12) and organoboranes (Table 13) with hypervalent iodine compounds.39 These reactions can be achieved under extremely mild conditions (room temperature, 10–120 min).|
|
|||
|---|---|---|---|
| Entry | R1SnBu3 | Product | Yields (%) |
| 1 | CH2CHSnBu3 |
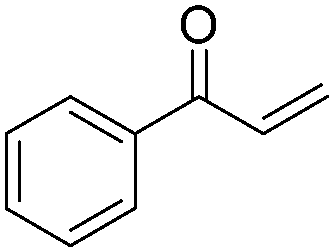
|
85 |
| 2 |
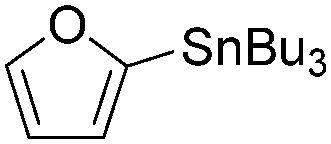
|
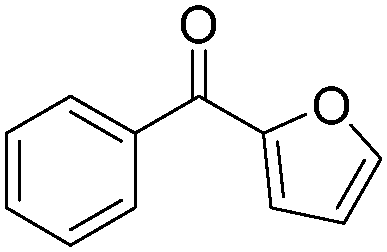
|
94 |
| 3 | Ph-CCSnBu3 |
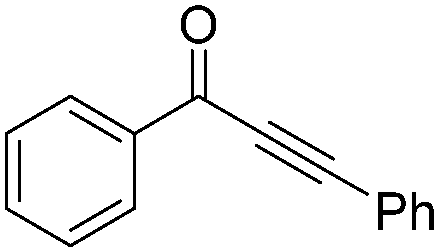
|
84 |
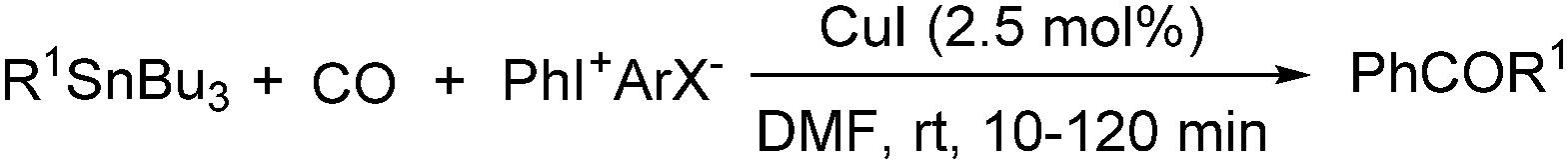
|
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | Product | Yields (%) |
| 1 |
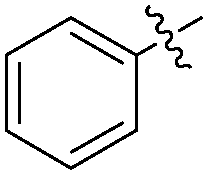
|
H |
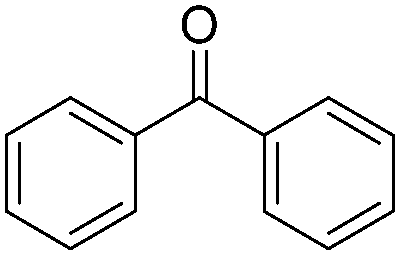
|
78 |
| 2 |
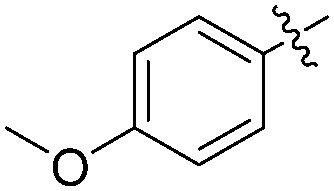
|
H |
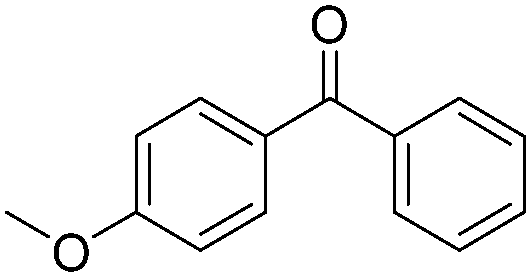
|
63 |
| 3 |
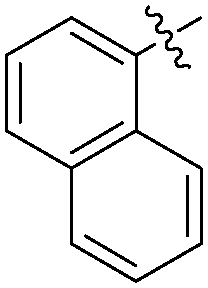
|
H |
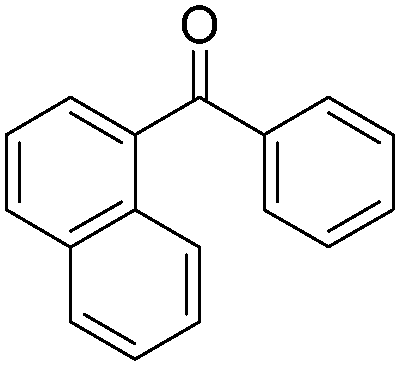
|
68 |
| 4 |
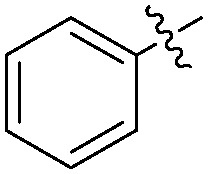
|
CH3 |
|
77 |
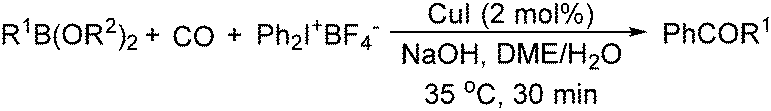
Fujiwara and co-workers developed a transition metal catalysed carboxylation of alkanes and arenes in 1995 (Scheme 12).40 Using palladium and/or copper as the catalyst, with TFA (trifluoroacetic acid) as the solvent, different alkanes can react with CO in TFA to give the corresponding carboxylic acid products. Additionally, a Pd and Cu combined catalyst (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) gives better activity for this transformation.
:1) gives better activity for this transformation.
 | ||
| Scheme 12 Palladium and/or copper-catalysed carboxylation of alkanes and arenes. | ||
Sundermeyer and co-workers developed a copper-catalysed oxidative carbonylation of methanol to produce dimethyl carbonate with N-methylimidazole (NMI) as the ligand in 2001 (Scheme 13).41 After various catalysts testing, (NMI)4CuCl2, (NMI)3CuBr2, and (NMI)4CuI all gave activity and selectivity with DMC (dimethyl carbonate) as the solvent. In 2013, Monopoli, Nacci and their co-workers reported a copper catalysed oxidative carbonylation of glycerol to produce glycerol carbonate (Scheme 14).42 With CuCl2 and pyridine as the catalyst system, using DMAc (N,N-dimethylacetamide) as the solvent under CO/O2 pressure (PCO = 3.3 MPa; PO2 = 0.7 MPa) at 130 °C, excellent conversions (>92%) and selectivities (>93%) can be obtained in a relatively short reaction time (3–4 h).
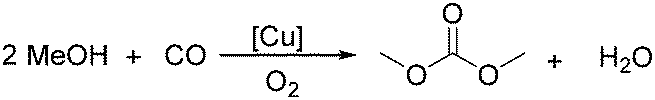 | ||
| Scheme 13 Copper-catalysed synthesis of carbonate. | ||
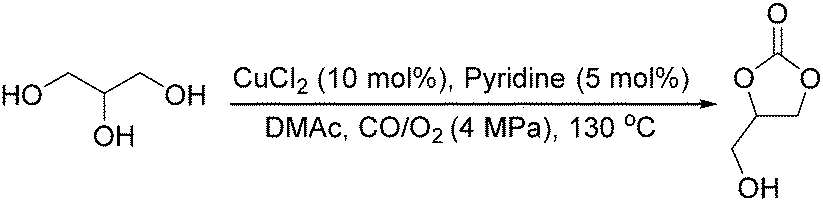 | ||
| Scheme 14 Copper-catalysed oxidative carbonylation. | ||
Recently, NHCs (N-heterocyclic carbenes) have been widely used in transition metal catalysed reactions and have been studied in carbonylation reactions as well. In 2009, Xia, Sun and co-workers developed a novel copper-catalysed double carbonylation reaction of aryl iodides and secondary amines (Table 14).43 High yields and selectivity can be observed for this transformation.
|
|
|||
|---|---|---|---|
| Entry | Aryl-I | Amine | Yields (%) |
| 1 |
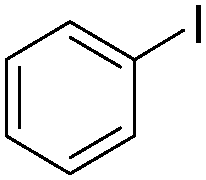
|
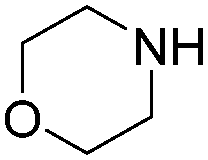
|
92 |
| 2 |
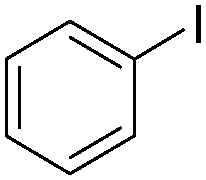
|
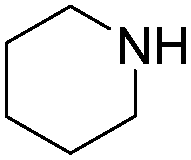
|
89 |
| 3 |
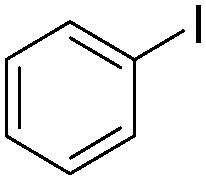
|
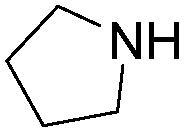
|
83 |
| 4 |
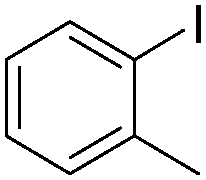
|
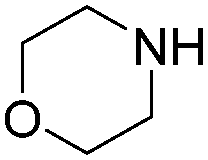
|
80 |
| 5 |
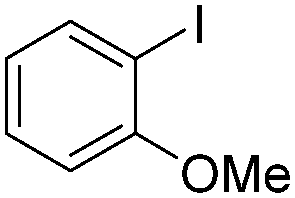
|
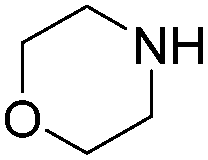
|
93 |
| 6 |
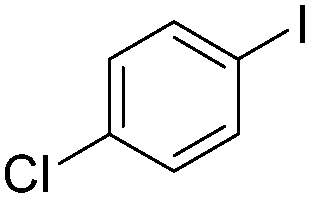
|
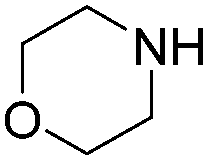
|
93 |
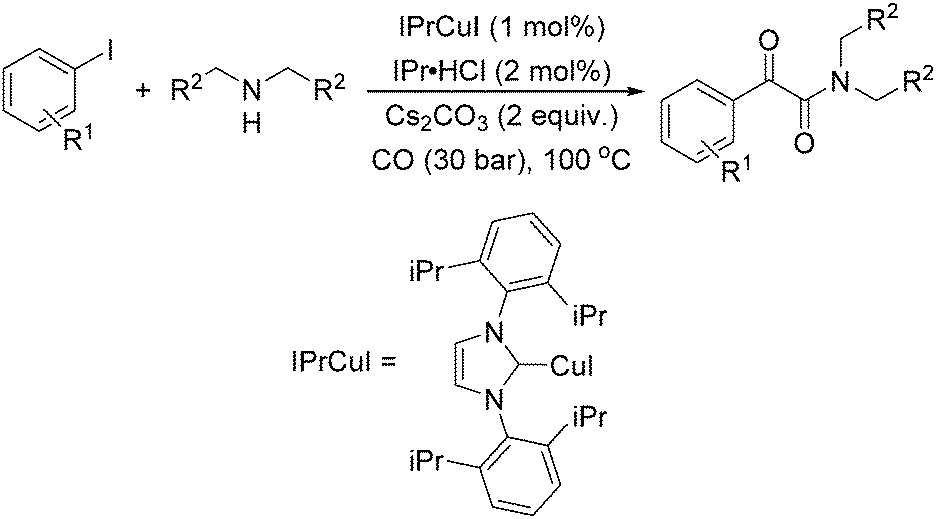
Typically, a carbonylative Sonogashira coupling reaction requires the combination of a palladium catalyst and air-sensitive phosphine ligands as the catalyst system. In 2008, an interesting copper catalysed carbonylative Sonogashira coupling reaction for the synthesis of alkynyl ketones was established by Bhanage and co-workers (Table 15).44 In this procedure, copper bis(2,2,6,6-tetramethyl-3,5-heptanedionate) [Cu(TMHD)2] was used as the catalyst and NEt3 was used as the base. Various alkynones were produced in good yields.
|
|
|||
|---|---|---|---|
| Entry | Ar | R | Yields (%) |
| 1 | Ph | Ph | 80 |
| 2 |
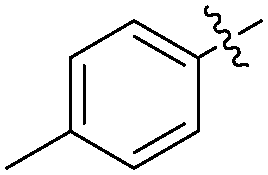
|
Ph | 78 |
| 3 |
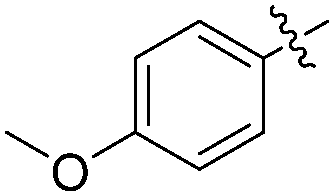
|
Ph | 76 |
| 4 |
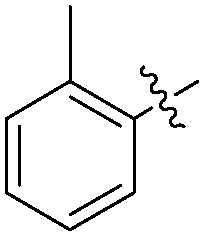
|
Ph | 73 |
| 5 |
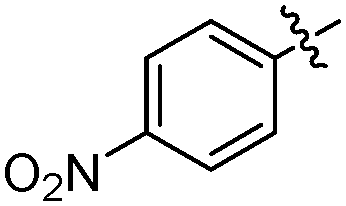
|
Ph | 68 |
| 6 | Ph | n-Hex | 77 |
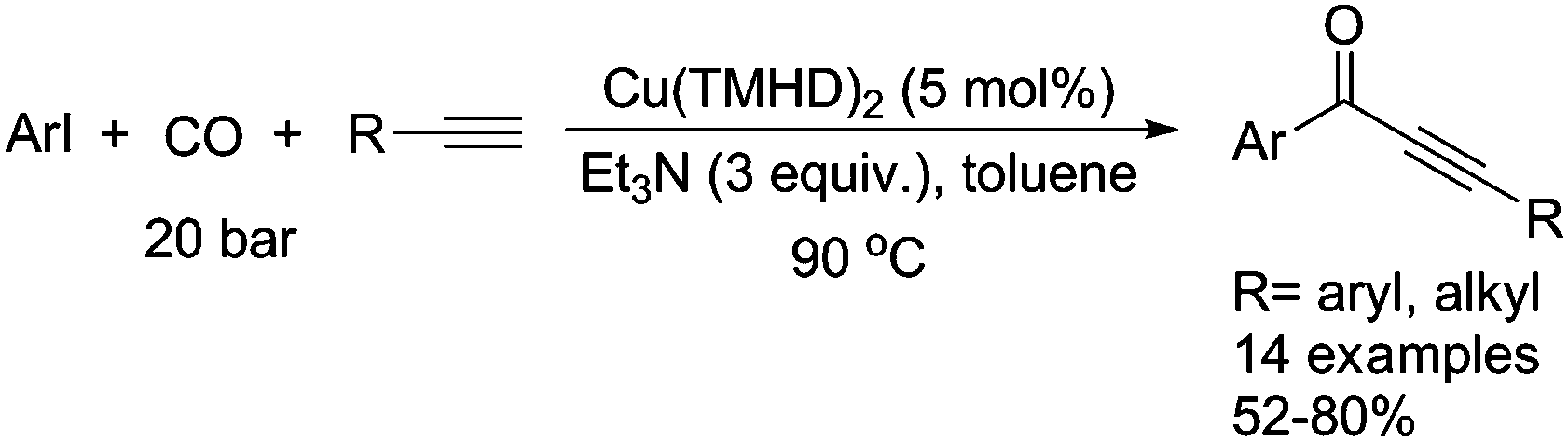
Alternative procedures for the synthesis of carbonyl-containing compounds have been developed as well. Gao and co-workers reported a copper-catalysed dicarbonylation of imidazo[1,2-α]pyridines with acetamides and acetone using molecular oxygen as the terminal oxidant (Scheme 15, formula (1) and (2)).45 The reactions were carried out in the presence of 5 mol% of Cu(OAc)2 in AcOH/t-AmOH under O2 at 120 °C, and the desired 1,2-dicarbonyl imidazo[1,2-α]pyridines were formed regioselectively in good yields. In their 18O-labeling experiments, they proved that the oxygen source of products originated from O2 rather than H2O. Later on, they successfully extended their methodology to 2-methylpyridines (Scheme 15, formula (3)).46
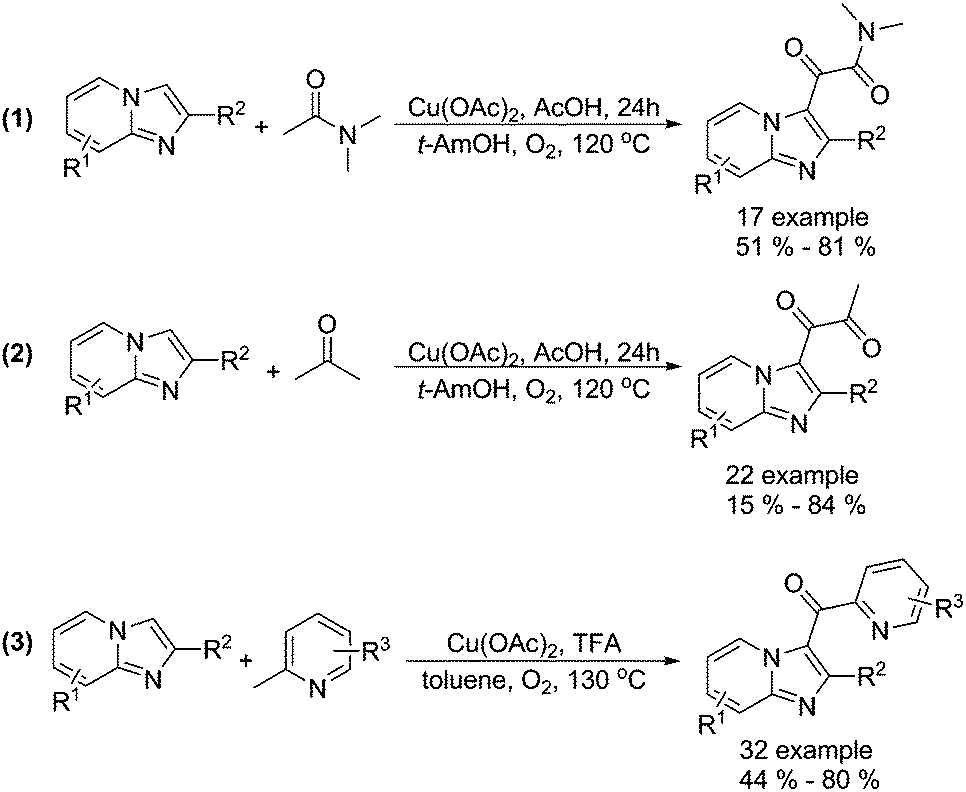 | ||
| Scheme 15 Copper-catalysed acylation of imidazo[1,2-α]pyridines. | ||
The group of Ye and Ke reported a copper-catalysed acylation of terminal alkynes with formamides using TBHP (tert-butyl hydroperoxide) as the oxidant (Scheme 16).47 Terminal alkynes undergo cross-dehydrogenative coupling in formamides at 60 °C and in the presence of catalytic amounts of CuCl2 to give the desired propiolamides in good yields. A competing reaction, the reaction of DMF with phenylacetylene, was performed in the presence of excess diethylamine and no diethylamine product was detected, indicating that the mechanism involving the generation of CO and amine in situ could be ruled out.
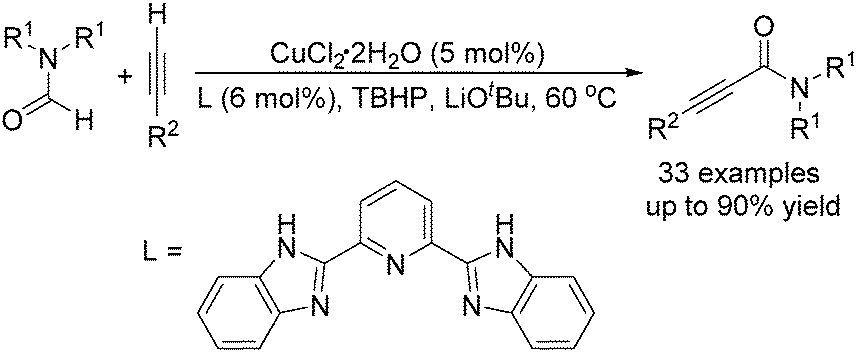 | ||
| Scheme 16 Copper-catalysed acylation of terminal alkynes. | ||
In 2016, a general methodology for copper-catalysed oxidative amidation of terminal alkynes to construct α-ketoamides was reported by Shen, Zhang and their co-workers (Table 16).48 In the presence of 10 mol% of Cu(OTf)2 as the catalyst with 2 equivalents of DBU as the base, terminal alkynes could react with O-benzoyl hydroxylamines in THF and the desired products were obtained in good to excellent yields. It is worth pointing out that O-benzoyl hydroxylamines act as both amination reagent and oxidant here.
|
|
|||
|---|---|---|---|
| Entry | 1 | 2 | Yields (%) |
| 1 |
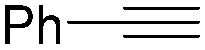
|
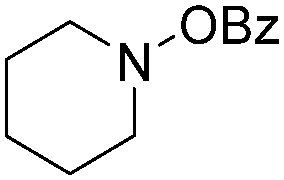
|
84 |
| 2 |
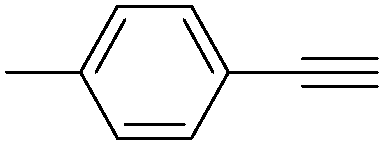
|
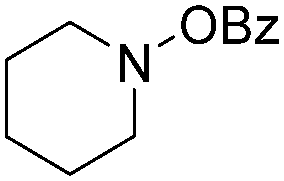
|
82 |
| 3 |
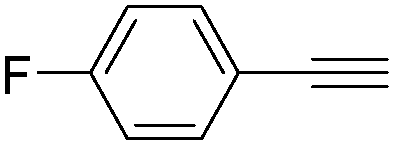
|
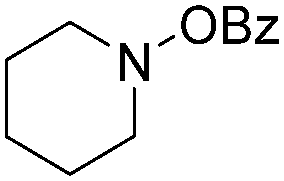
|
80 |
| 4 |
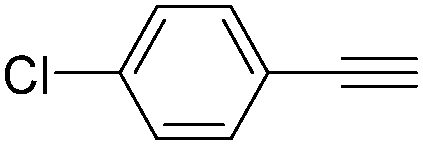
|
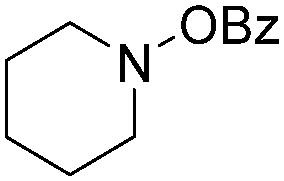
|
78 |
| 5 |
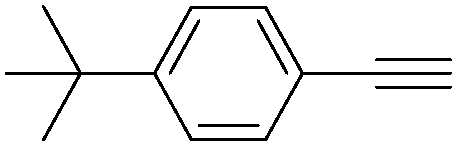
|
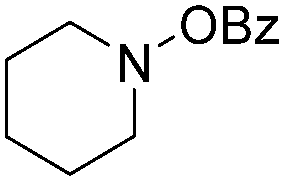
|
72 |
| 6 |
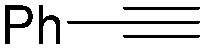
|
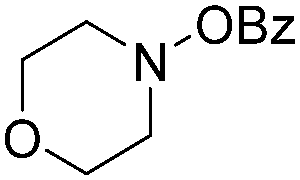
|
72 |
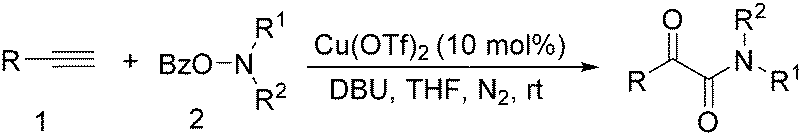
Organic carbamates are important intermediates for the synthesis of pesticides, herbicides and pharmaceutical drugs. An interesting copper-catalysed cross-coupling of anilines with diisopropyl azodicarboxylate was developed by Guan and co-workers in 2016.49 The desired carbamates were isolated in moderate to good yields (Scheme 17). Interestingly, this catalyst protocol was carried out under solvent-free conditions.
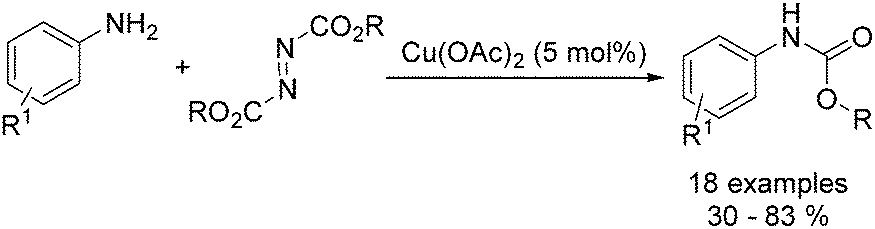 | ||
| Scheme 17 Copper-catalysed carbamate synthesis. | ||
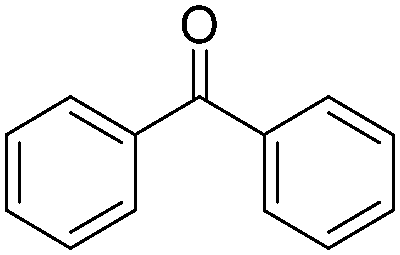
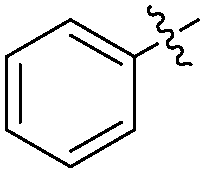
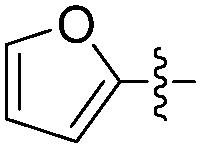
The report from Yu's group in 2005 demonstrated a copper-catalysed carbonylation of alkynyliodonium salts with boronic acids to the corresponding alkynones (Table 17).50 In the presence of 10 mol% of CuI and 1.2 equivalent of K2CO3, alkynyliodonium salts can react with organoboronic acids to form the corresponding α,β-ynone in good yields under a CO atmosphere in DME/H2O.
|
|
|||
|---|---|---|---|
| Entry | R1 | R2 | Yields (%) |
| 1 | Bu |
|
83 |
| 2 | Bu |
|
63 |
| 3 | t-Bu |

|
77 |
| 4 | t-Bu |
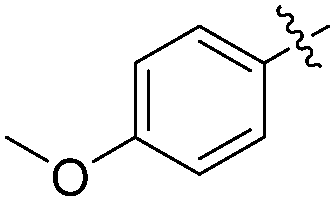
|
88 |
| 5 | Ph |
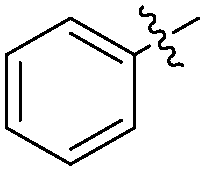
|
77 |
| 6 | Ph |
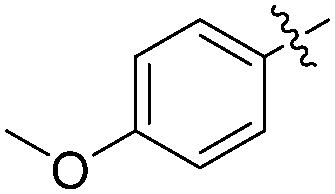
|
79 |
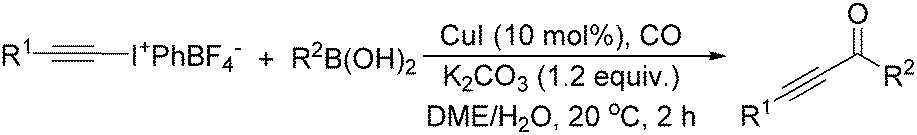
More recently, an efficient method for the synthesis of succinimides via C–H activation was developed by Li, Ge and their co-workers (Scheme 18).51 This protocol is based on the sequential sp3 and sp2 C–H bond activation with nitromethane as the carbonyl source. Using 2-ethyl-2-methylpentanamide bearing a bidentate 8-aminoquinoline directing group as the starting material at 165 °C, with Cu(OAc)2 as the catalyst and K2S2O8 as the oxidant using PhCO2Na as the base, good yields of the desired products can be isolated.
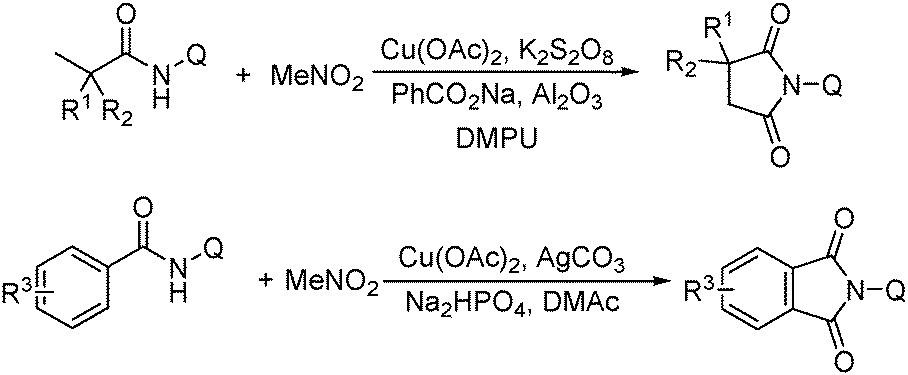 | ||
| Scheme 18 Copper-catalysed synthesis of succinimides. | ||
Our group developed a catalytic system for the carbonylative transformation of alkanes with different nucleophiles more recently (Scheme 19). By applying a copper-1,10-phenanthroline catalytic system, the corresponding products can be obtained in good yields at 120 °C under 20 bar of CO. Compared with traditional radical carbonylations, this catalytic system can reduce the pressure of CO from 80 to 20 bar. By carbonylative coupling of alkanes with amides,52 amines,53 and alcohols,54 imides, amides and esters can be selectively obtained, respectively. Interestingly, under thermal conditions or in the presence of a metal catalyst, peroxides could decompose into the corresponding alkoxy radical, which could give a methyl radical through β-scission of the alkoxy radical. With the awareness of the importance of acetylation reactions and also the academic meaningfulness of using peroxide as a methyl source in carbonylation, we developed an acetylation procedure as well with peroxide as the methyl source.55
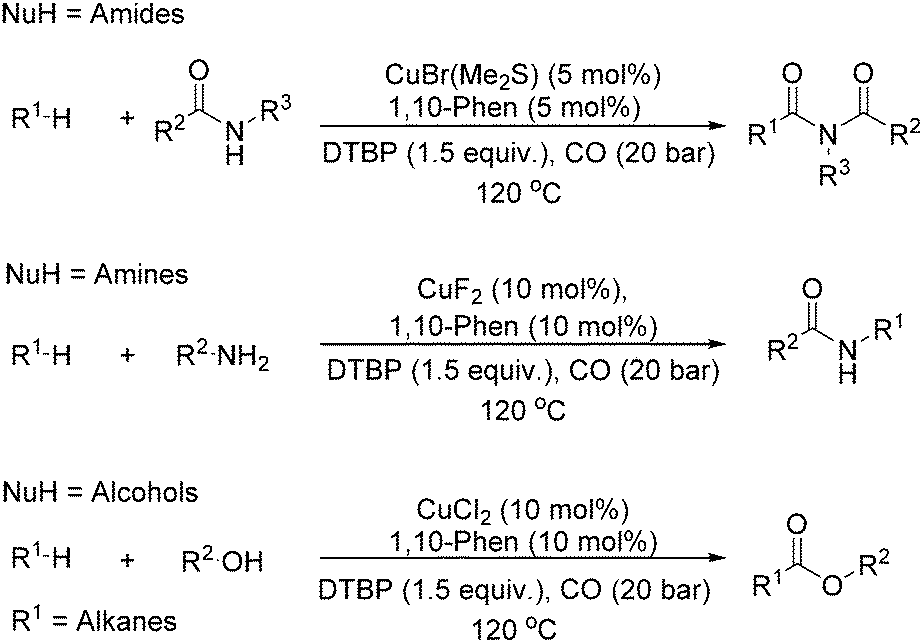 | ||
| Scheme 19 Copper-catalysed carbonylation of alkanes. | ||
5. Co-Catalysed carbonylation reactions
As early as in 1977, Alper's group reported a procedure and demonstrated that carboxylic acids can be synthesized in good to excellent yields by using a catalytic amount of Co2(CO)8 under mild reaction conditions in a phase-transfer system.56 In this procedure, a double carbon monoxide insertion reaction was also described. Then, in 1978, Abbayes and co-workers reported a systematic study on the double carbonylation of substituted benzyl chlorides with cobalt carbonylanion by phase transfer catalysis to give arylpyruvic acids.57 In their study, it was found that substitution on the aromatic ring played an important role in the double carbonylation by phase transfer catalysis: with R = H or m-CF3, which was strongly electron-withdrawing, no keto-acid appeared; while, with R = Me, double carbonylation occurred, regardless of the position of the substituent, suggesting that the electron donating effect of the methyl group was more important than its position. Moreover, further alkylation occurred, giving the substituted arylpyruvic acid. When the aromatic ring bore three methyl groups (in o, o′, and p positions) double carbonylation became an important pathway, giving only the simple arylpyruvic acid (3). In all these cases, the expected arylacetic acid (2) was produced besides the keto-acid (Scheme 20).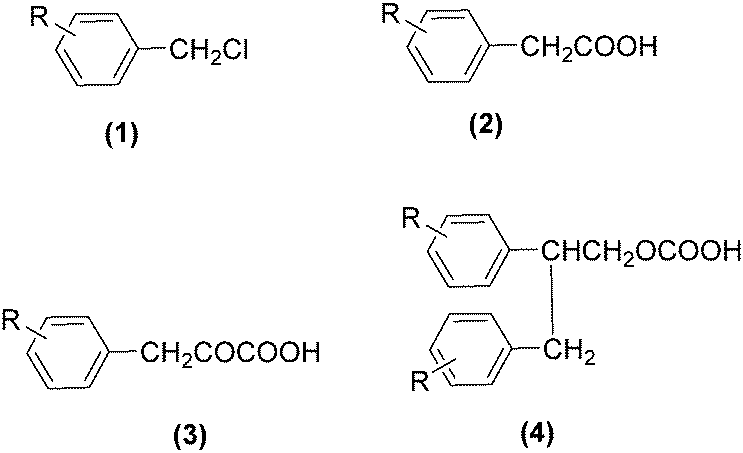 | ||
| Scheme 20 Double carbonylation of benzyl chlorides. | ||
In 1982, Imamoto and co-workers reported a facile method for the conversion of benzyl alcohols into carbon-homologated amides or esters by cobalt carbonyl catalysed carbonylation under mild reaction conditions (Scheme 21).58 In the same year, Foa and Francalanci found that in the phase-transfer catalysis in cobalt catalyzed carbonylation of secondary benzyl halides, the results were highly dependent on the experimental conditions. Alcohols or ethers mainly gave carboxylic acid salts and the use of higher pressure of CO associated with a hydrocarbon organic phase favored the other selectivity.59 Soon after, Foa and co-workers reported a cobalt-catalyzed carbonylation of optically active and α-deuterated phenylethyl halides under phase transfer conditions.60 The stereochemistry of catalytic monocarbonylation of optically active α-deuterated phenylethyl halides, and deuterium-exchange experiments to clarify the origin of double carbonylation were devised.
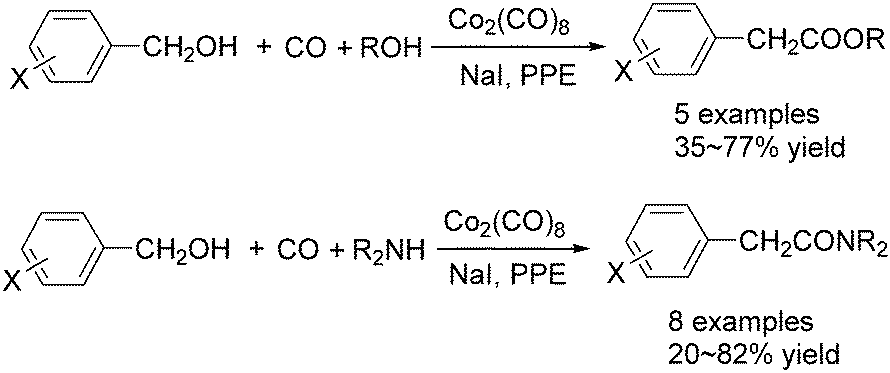 | ||
| Scheme 21 Cobalt-catalysed carbonylation of benzyl alcohols. | ||
In 1985, Alper's group reported the double carbonylation of styrene oxides.61 With Co2(CO)8 as the catalyst, and treatment of substrates with carbon monoxide, NaOH, methyl iodide in benzene, and using cetyltrimethylammonium bromide (CTAB) as the phase transfer agent, good yields of the corresponding products can be obtained (Scheme 22).
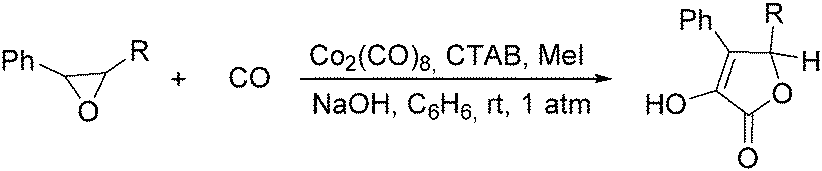 | ||
| Scheme 22 Co-Catalysed double carbonylation of epoxides. | ||
In 1986, a methodology on cobalt-catalysed reductive carbonylation of methyl esters was reported by Wegman and Busby. Acetaldehyde and a carboxylic acid were obtained in high yields via the CoI2–Lil-catalysed reaction of a methyl ester with synthesis gas (Scheme 23).62
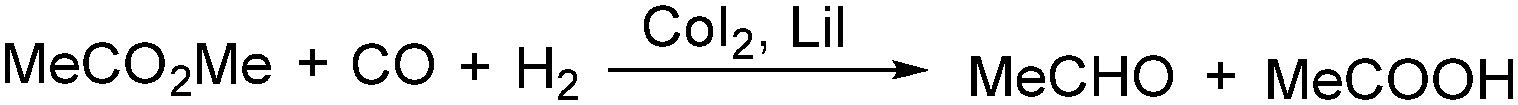 | ||
| Scheme 23 Co-Catalysed carbonylation of methyl ester. | ||
In 1986, Kshimura et al. reported a procedure on cobalt carbonyl catalysed carbonylative transformation of o-halogenated benzoic acids under photo-stimulation (Scheme 24).63 Double carbonylation products can be observed as well under these reaction conditions.
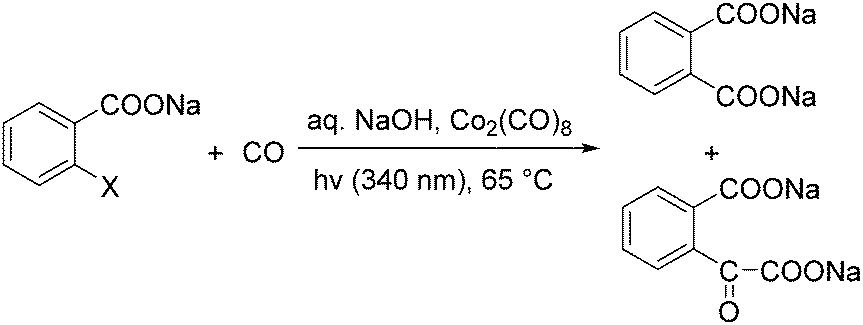 | ||
| Scheme 24 Co-Catalysed carbonylation of o-halogenated benzoic acids. | ||
In 1985, a method for carbonylative transformation of aromatic and heteroaromatic halides was reported by Foa and co-workers (Scheme 25, formula (1)).64 In this mild catalytic system, anionic cobalt complexes were formed and showed great activity towards aromatic halides. One year later, they presented a novel procedure on the double carbonylation of aryl and secondary benzylic halides with Co2(CO)8 as the catalyst.65 The corresponding α-keto acids were generated in moderate to good yields (Scheme 25, formula (2)). In this process, a methyl source (dimethyl sulfate or methyl iodide) was necessary for the carbonylation of aryl halides.
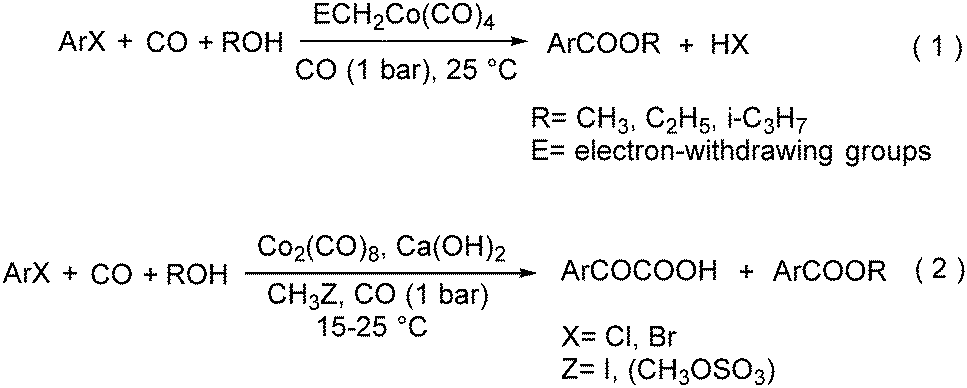 | ||
| Scheme 25 Co-Catalysed carbonylation of organo halides. | ||
An interesting cobalt carbonyl-catalysed carbonylative synthesis of unsaturated carboxylic acids in aqua solutions was developed by Alper and Calet in 1988.66 Under the assistance of a phase transfer catalyst and methyl iodide, carbonylation of vinyl epoxides occurred and gave the desired products in good yields and high regioselectivity (Scheme 26).
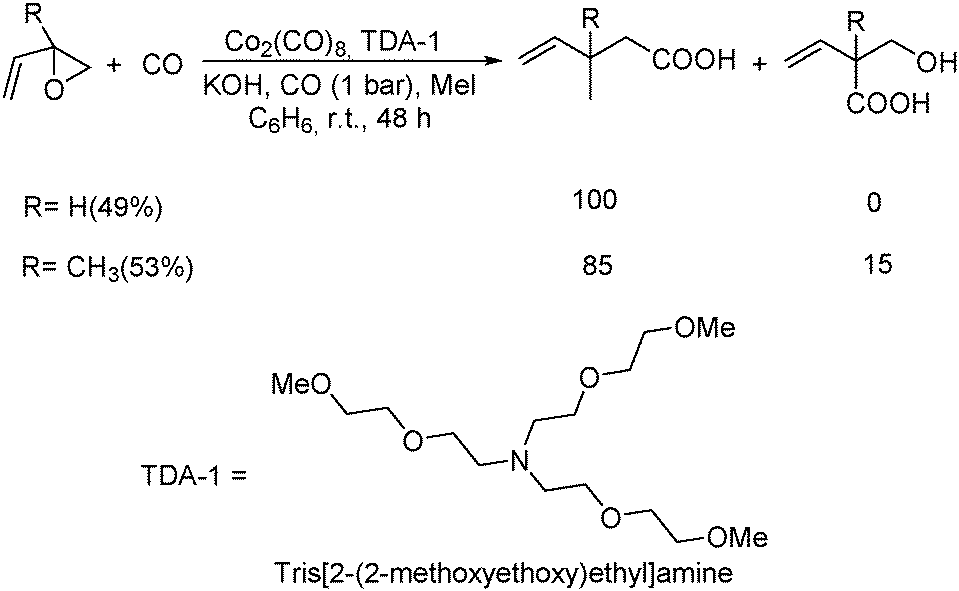 | ||
| Scheme 26 Co-Catalysed carbonylation of vinyl epoxides. | ||
In 1989, Alper and Roberto reported the synthesis of pyrrolidinones by cobalt carbonyl catalysed carbonylation of azetidines. That was the first example of a metal catalysed ring-expansion-carbonylation reaction of azetidines to tetrahydroazepinones (Scheme 27).67 Various pyrrolidinones could be synthesized in high yields and regioselectivity from the parent azetidines. The selectivity between 1,5-disubstituted pyrrolidin-2-ones and 1,3-disubstituted pyrrolidin-2-ones are depends on the properties of the substituent groups and also on the reaction temperature. The reaction displays high regio- and stereoselectivity, and also tolerated functional groups such as esters, ethers, and alcohols.
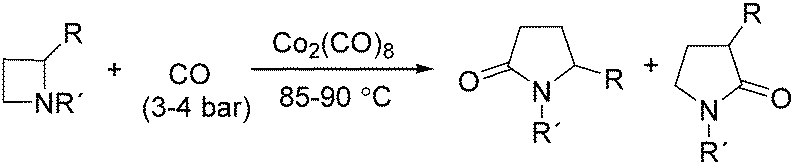 | ||
| Scheme 27 Cobalt-catalysed carbonylative synthesis of pyrrolidinones. | ||
In the same year, Watanabe and co-workers reported a cobalt catalysed ring-opening carbonylation of cyclic ethers using N-(trimethylsilyl)amines as the reaction partner (Scheme 28).68 This method provided an access to the ring-opening carbonylation of a variety of cyclic ethers such as oxiranes, oxetane, tetrahydrofuran, and 1,3-dioxolane. In the presence of a catalytic amount of Co2(CO)8 under carbon monoxide pressure, the corresponding siloxy amides were formed in good yields.
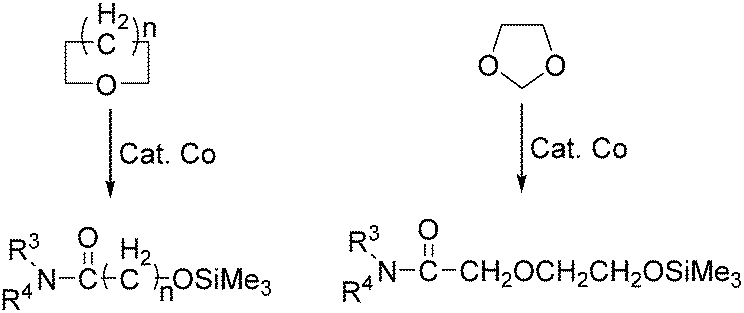 | ||
| Scheme 28 Cobalt-catalysed carbonylation of cyclic ethers. | ||
Remarkably, the first example of the triple carbonylation of epoxy alcohols was developed by Alper's group in 1990.69 With cobalt carbonyl and TDA-1 as the catalyst system, the epoxy alcohols were converted into the corresponding 2-C-(2,5-dihydro-2-oxofur-5-yl)lactic acids in moderate yields, under exceptionally mild conditions (Scheme 29).
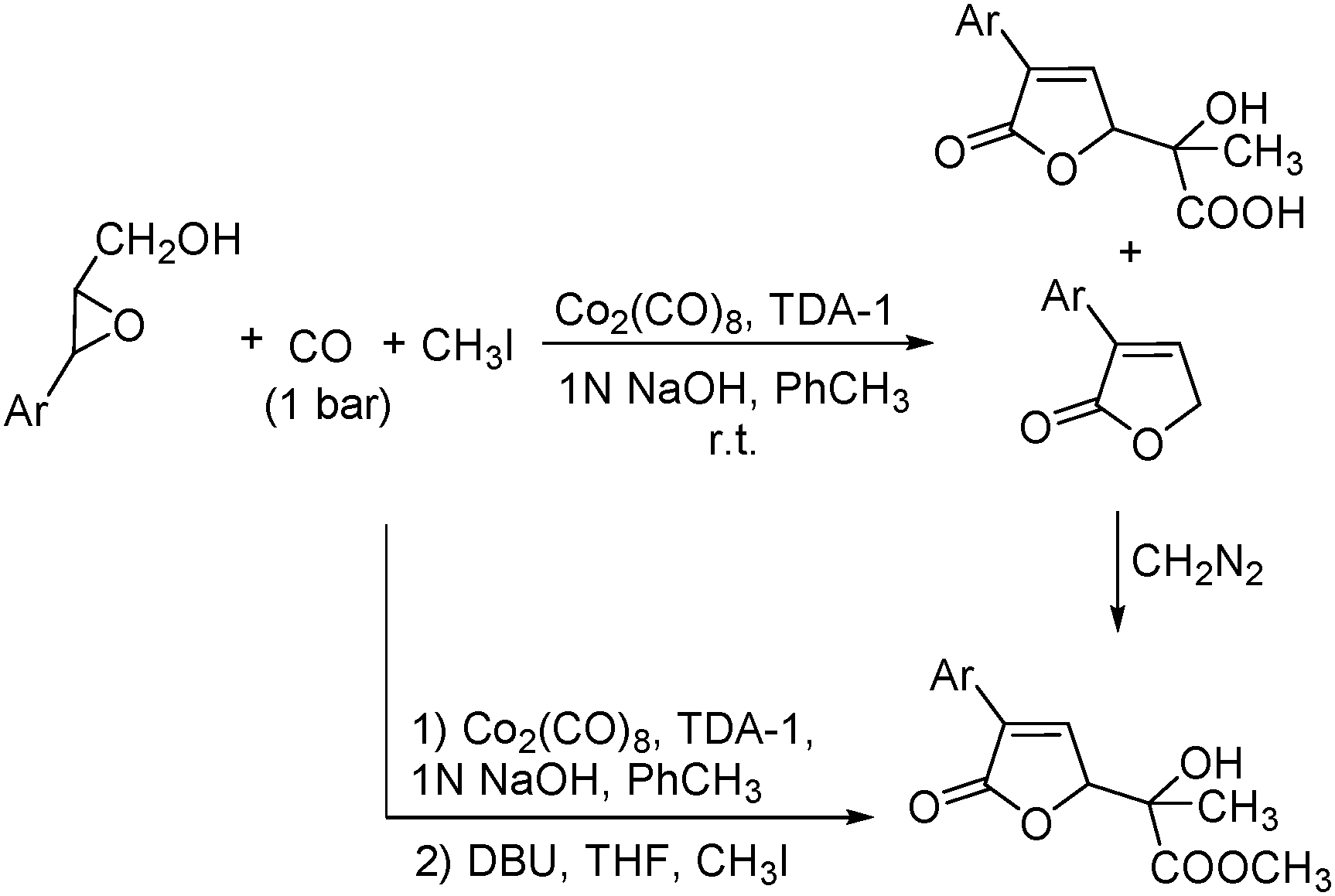 | ||
| Scheme 29 Cobalt catalysed carbonylation of epoxy alcohols. | ||
In 1991, Izawa and co-workers reported a methodology for the synthesis of proline and 2-piperidinecarboxylic acid via cobalt catalysed isomerization–carbonylation of N-acyl unsaturated cyclic amines under hydroformylation conditions in the presence of H2O (Scheme 30).70
 | ||
| Scheme 30 Synthesis of pyrrolidine and 2-piperidinecarboxylic acid. | ||
In 1991, Alper and Grushin discovered the first example of carbonylative transformation of gem-dibromocyclopropanes by using cobalt and nickel salts as catalysts under phase transfer conditions (Scheme 31).71 The best yields were obtained when a combination of nickel and cobalt cyanides was applied in the presence of synthesis gas (CO/H2).
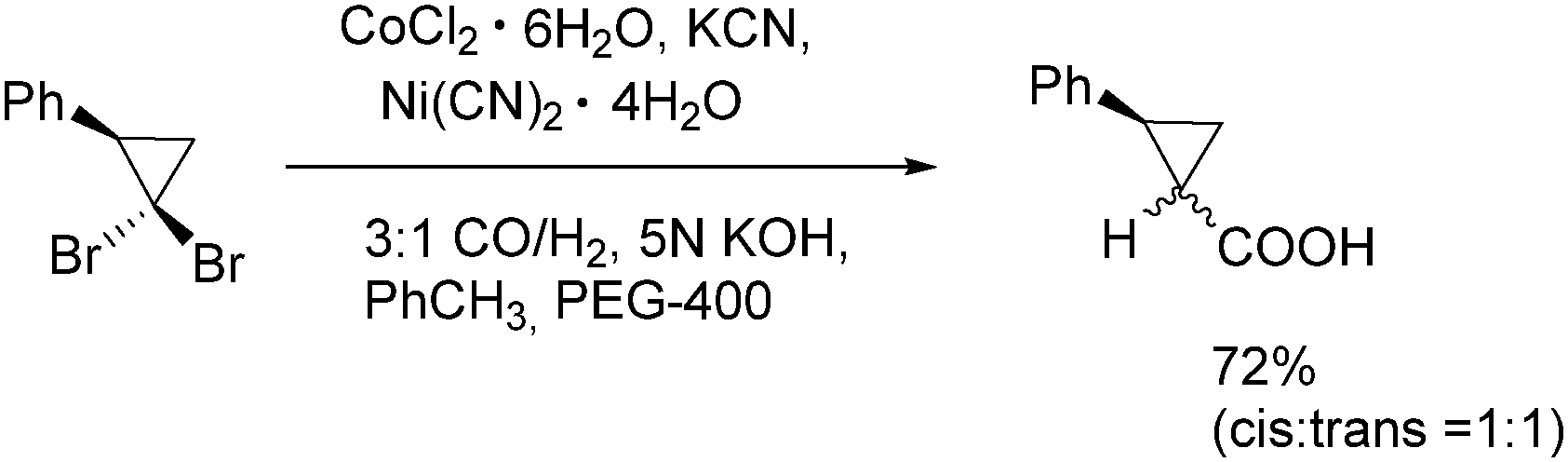 | ||
| Scheme 31 Carbonylation of gem-dibromocyclopropanes. | ||
Fuchikami and co-workers reported a procedure for the carbonylative transformation of alkyl sulfonates in 1991.72 The corresponding esters were produced selectively in the presence of a catalytic amount of Co complex, NaI and alcohols with 1,1,3,3-tetramethylurea (TMU) as the base (Scheme 32).
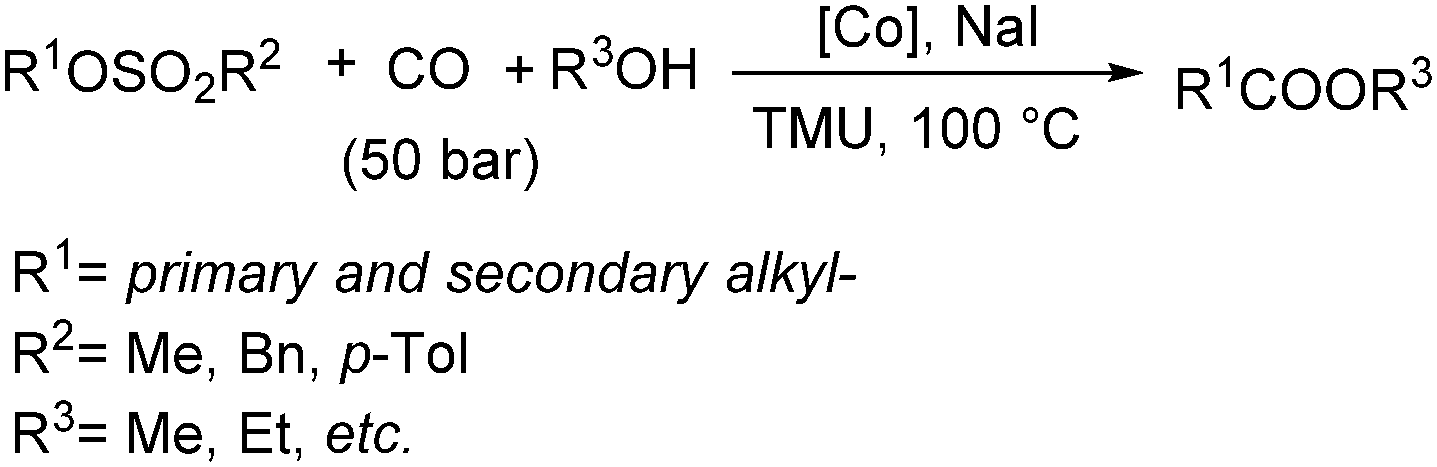 | ||
| Scheme 32 Cobalt-catalysed carbonylation of alkyl sulfonates. | ||
In 1992, Alper and co-workers reported the synthesis of piperidinones, which was formed from the carbonylation of pyrrolidines catalysed by cobalt carbonyl (Scheme 33).73 The reaction was regiospecific in most cases, and the yields of the products were increased in the presence of ruthenium carbonyl as an additive.
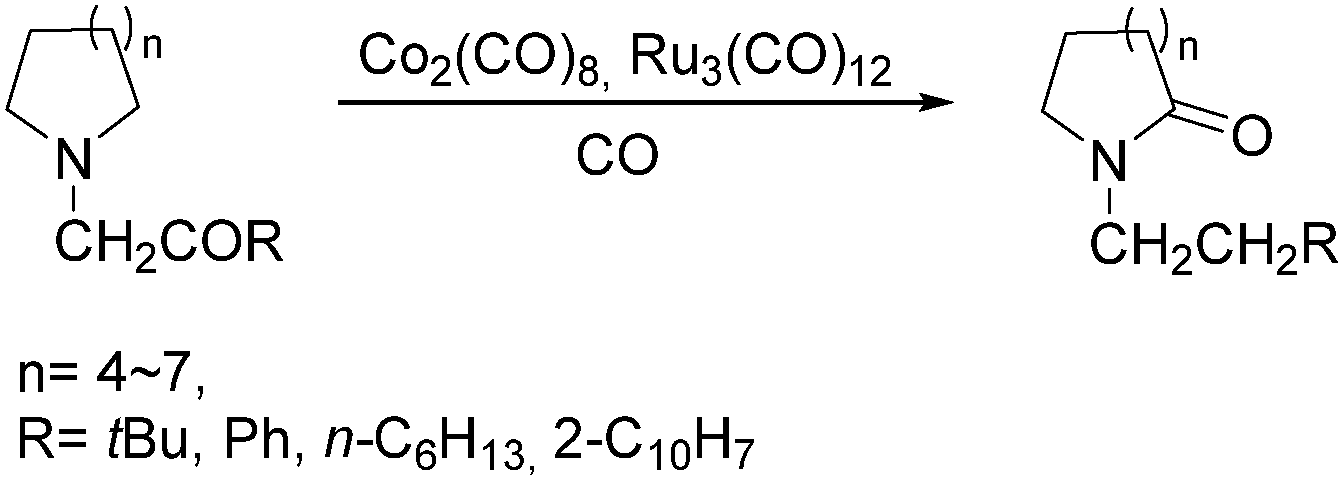 | ||
| Scheme 33 Cobalt-catalysed carbonylation of pyrrolidines. | ||
Rindone and co-workers studied the substituent effects in cobalt-catalyzed oxidative carbonylation of aromatic amines.74 Ureas, isocyanates, carbamates and azo derivatives were generated from the N,N-bis(salicylidene)ethylenediaminocobalt(II)-catalyzed oxidative carbonylation of ortho-, meta- and para-substituted aromatic primary amines in methanol. In their study, it was found that the para-substituted anilines were mainly transformed into the ureas in high yields, since the cobalt(III) intermediates had stability very similar to that of the corresponding cis derivatives, as well as steric effects, which may be suggested to be the intermediates in the formation of ureas.
In the same year, Alper's group reported the carbonylative synthesis of highly strained trans-bicyclic β-lactams. In the presence of Co2(CO)8 as the catalyst, aziridines were transformed into β-lactams in excellent yields (Scheme 34).75 This procedure is valuable in the preparation of highly strained bicyclic-β-lactams containing the trans-7-azabicyclo [4.2.0]octan-8-one nucleus. The reaction began by nucleophilic ring opening of the aziridine by an in situ-generated tetracarbonyl-cobaltate anion.
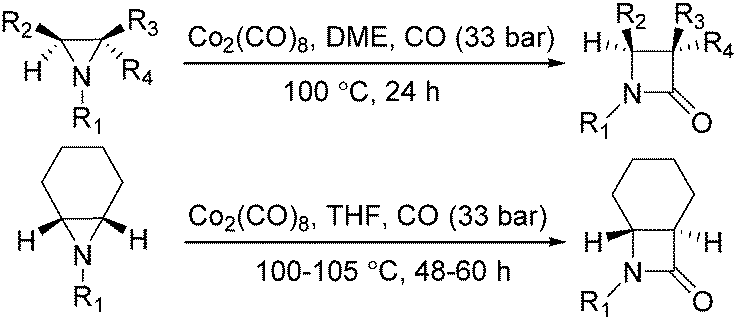 | ||
| Scheme 34 Co2(CO)8-Catalysed carbonylation of aziridines. | ||
In 1996, Pályi, Markó, Alper and their co-workers reported a cobalt-catalysed carbonylation of benzyl halides using polyethylene glycols as the phase-transfer catalyst (Scheme 35).76 Hydroxycarbonylation of benzyl and substituted benzyl chloride and bromide derivatives was achieved in good to excellent yields (up to 97.6%) and quantitative chemoselectivity was obtained in the two-phase system under mild conditions (1 bar CO, room temperature). η1-Benzyl-, η3-benzyl-, and (η1-phenylacetyl) cobalt carbonyls were investigated as intermediates of this carbonylation transformation. It was proposed that: (i) the role of PEG is not simply to transport [Co(CO)4]− from the aqueous to the organic phase but also to accelerate the hydrolysis of the acylcobalt tetracarbonyl complex; (ii) in the presence of the acyl-, alkyl- and (η3-benzyl)cobalt complexes in the hydroxycarbonylation reaction mixtures, (η3-benzyl)cobalt-type complexes and their role as intermediates in phase-transfer reactions were not previously observed.
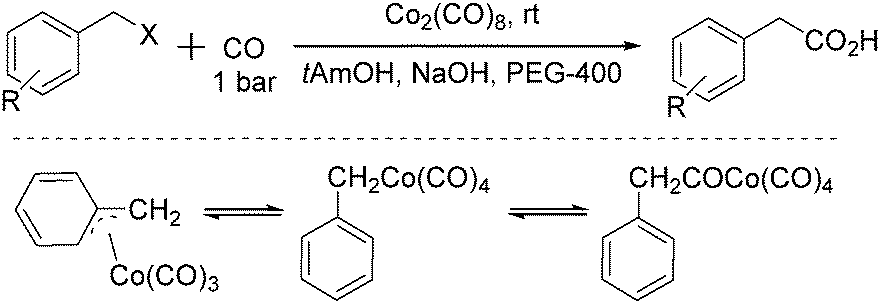 | ||
| Scheme 35 Cobalt-catalyzed carbonylation of benzyl halides. | ||
Lin and Knifton developed a cobalt-catalysed carbonylation of phenylacetaldehyde to N-acetyl-beta-phenylalanine in 1997.77 The reaction was studied both in a batch process and a continuous phase reactor. In this reaction, the addition of a DPPE (1,2-bis(diphenylphosphino)ethane) ligand was compulsory. High yields of the desired products can be achieved and the catalyst can be recovered successfully as well (Scheme 36).
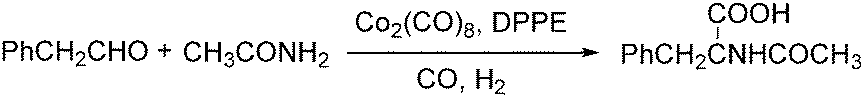 | ||
| Scheme 36 Cobalt-catalyzed carbonylation of phenylacetaldehyde. | ||
In 2001, Alper's group reported a synthetic method for the carbonylative synthesis of β-lactones from epoxides.78 With the cobalt complex (PPNCo(CO)4) (PPN = bis(triphenylphosphine)iminium) as the catalyst and a Lewis acid (BF3·Et2O) as the additive, both simple and functionalized epoxides were regioselectively transformed in good yields (Scheme 37). The carbonylation occurred selectively at the unsubstituted C–O bond of the epoxide ring, and various functional groups were tolerated, such as alkenyl, halide, hydroxyl, and alkyl ether. Interestingly, the stereochemistry of epoxides and aziridines gave completely different results.
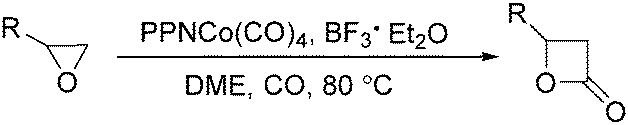 | ||
| Scheme 37 Cobalt-catalysed carbonylation of epoxides. | ||
Dragojlovic et al. reported a cobalt-catalyzed photochemical methoxylcarbonylation of alkenes in the same year.79 The reaction can be successfully carried out on a multigram scale (5–10 g) on a number of alkenes with appropriate choice of light source, filter, concentration of cobalt catalyst and suitable equipment (Scheme 38). In this process, the stability of the cobalt catalyst was crucial for the success of photocarbonylation. For given reaction scale and set of conditions (light source, filter, temperature), there was a narrow range of concentrations of the cobalt catalyst at which the catalyst was stable and the reaction proceeded at a reasonable rate. However, the reaction was limited to mono and disubstituted alkenes (69–85%). Low selectivity was observed with trisubstituted alkenes and dienes.
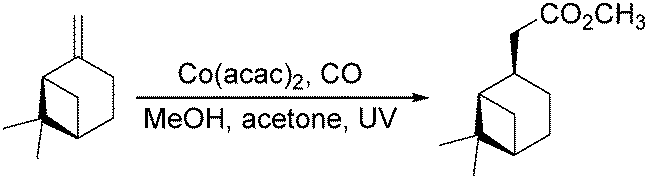 | ||
| Scheme 38 Methoxycarbonylation of alkenes. | ||
In 2002, an efficient and mild carbonylative protocol for the conversion of optically active epoxides to β-hydroxy morpholine amides was developed by Jacobsen and Goodman (Scheme 39).80 The methodology was effective with a variety of epoxides and pure products could easily be isolated by treatment of the crude product mixture with an aqueous acid. Moreover, a valuable new approach for preparing synthetically valuable β-hydroxy-β-ketoesters was discovered by adding aluminum enolate derivatives selectively to the morpholine amides.
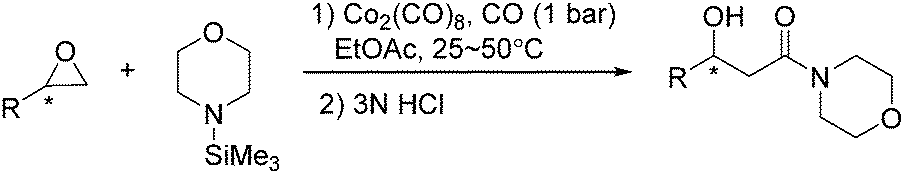 | ||
| Scheme 39 Carbonylative transformation of epoxides. | ||
Jia and Xu reported a methodology for the cobalt-catalysed carbonylative ring-expansion of 2-aryl-2-oxazolines in 2003 (Scheme 40).81 This was the first example of using 2-oxazolines as substrates in transition metal-catalyzed carbonylation reactions. In their mechanistic studies, they proved cobalt radicals as the catalytically active species.
 | ||
| Scheme 40 Cobalt-catalysed carbonylative ring-expansion of 2-aryl-2-oxazolines. | ||
Meanwhile, they succeeded in the carbonylative ring expansion of other heterocycles as well.82 With Co2(CO)8 as the catalyst, a rarely studied exocyclic C–O bond carbonylation proceeded smoothly. Various cycloimino esters could be activated and further carbonylated to afford N-acyllactams in high yields under relatively mild conditions in the absence of HI as the co-promoter (Scheme 41). The reaction opens a possible access to the carbonylation of alcohols without the assistance of HI. In this reaction, an organic amide would replace HI to form the desirable equilibrium that generated the direct reactant for the carbonylation. Later on, they also demonstrated that a catalytic system formed from commercially available Co2(CO)8 and AIBN can work effectively in this type of transformation as well. (4R,5S)-4-Methyl-2,5-diphenyl-2-oxazoline and (4R,5R)-4-methyl-2,5-diphenyl-2-oxazoline can be transformed stereospecifically. The stereoselectivity in all cases favored inversion at the C(5)-position with a diastereomeric excess of up to >97%.83
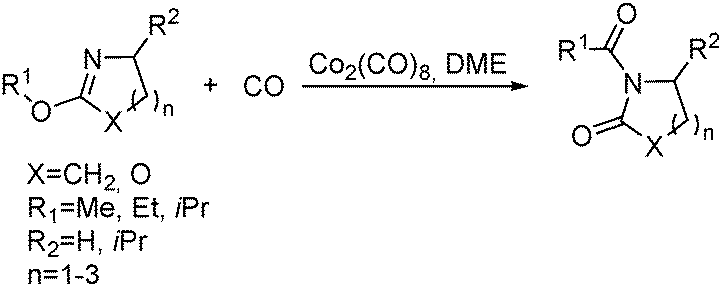 | ||
| Scheme 41 Cobalt-catalysed exocyclic C–O bond carbonylation. | ||
Jia and Liu designed a catalytic procedure for the carbonylative polymerization of epoxides and N-alkylaziridines in 2004.84 Under their conditions, polyesters and amphiphilic poly(amide-block-ester)s can be synthesized effectively (Scheme 42). It had been demonstrated that the combinations of cobalt catalysts and pyridines were versatile catalysts for the carbonylative polymerization of both epoxides and aziridines likely with closely resembling mechanisms. While less active for carbonylative epoxide polymerization than the catalysts generated in situ from Co2(CO)8, the well-defined acid-free catalyst allowed the development of a very convenient synthetic method for diblock copolymers with a degradable block and a nondegradable block. The amphiphilic and degradable diblock copolymers are potentially useful for biomedical applications.
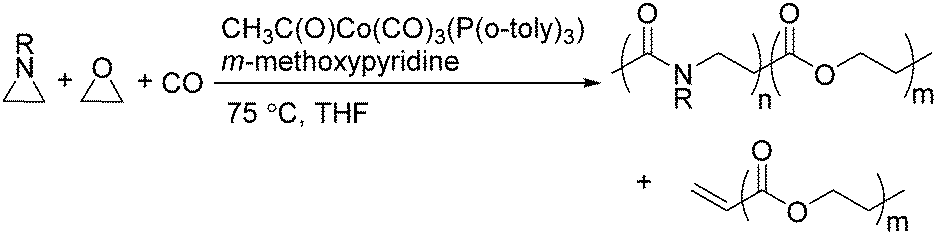 | ||
| Scheme 42 Cobalt-catalysed carbonylative polymerization of heterocycles. | ||
Two years later, Jia and Liu reported a new procedure for the carbonylative polymerization of azetidines.85 With [Co(CH3CO)(CO)3P(o-tolyl)3] as the catalyst and tetrahydrofuran (THF) as both the reagent and solvent, the desired polymers can be selectively produced (Scheme 43). With the use of LiI as a co-catalyst, the γ-lactam byproduct can be eliminated. Both the amount and distribution of the ester units in the polymer backbone were influenced by LiI as well; this finding allowed the synthesis of polymers with alternating amide blocks and ester segments that would undergo a two-stage degradation.
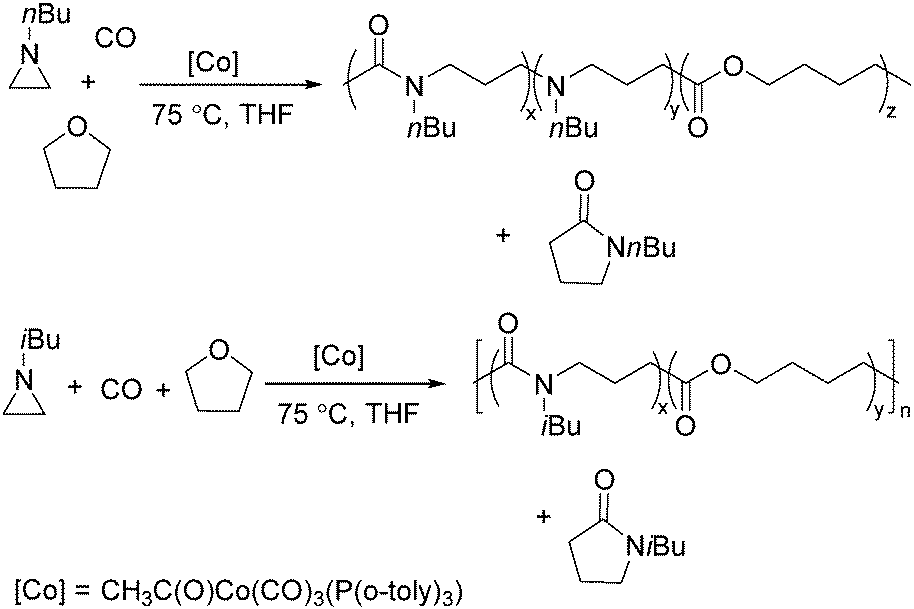 | ||
| Scheme 43 Cobalt-catalysed carbonylative polymerization of azetidines. | ||
In 2008, Jia and co-workers provided a systematic study on the cobalt-catalyzed carbonylative polymerization of N-alkylazetidines involving three representative monomers (Scheme 44).86 The individual N-alkylazetidine monomers displayed different characteristics in the polymerization, which allowed the incorporation of amine and ester units into the amide based polymers. Firstly, the synthesis and characterization of poly(amide-co-amine) with a gradient amine distribution were described. Then, they presented how to control the ester distribution in poly(amide-co-ester)s. Poly(amide-co-ester) containing multiple segments with block or gradient ester distributions could be synthesized by multiple additions of N-isobutylazetidine into the reaction mixture in the presence or absence of LiI. Finally, the discovery of a novel chain transfer pathway via N-benzyl abstraction was made. It was found that N-benyzlazetidine was different from the other two monomers in terms of the chain transfer pathway, which was presented via the nucleophilic abstraction of the N-benzyl group. The remaining acyl-azetidine at the chain end could be utilized for further chemical modifications or polymerization.
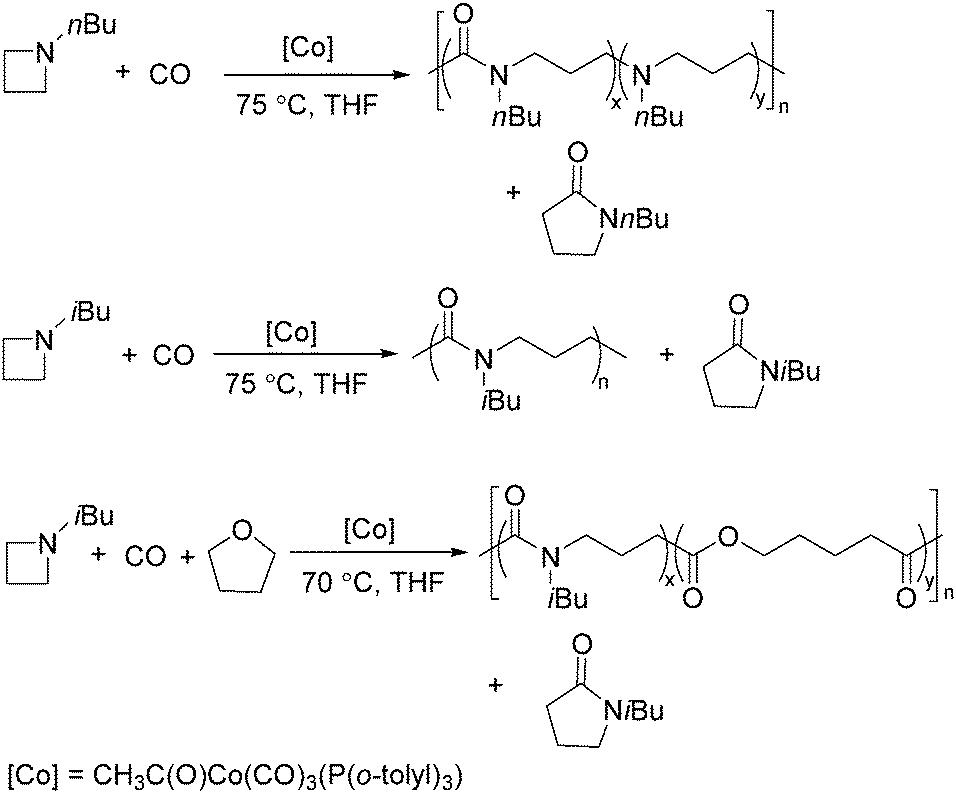 | ||
| Scheme 44 Cobalt-catalysed carbonylative polymerization of N-alkylazetidines. | ||
Sen and co-workers reported a cobalt-catalysed carbonylation of N-alkylbenzaldimines in 2006.87N-Alkylphthalimidines can be produced via tandem C–H activation and cyclocarbonylation (Scheme 45). The resulting formation was proposed to occur by C–H activation of the aryl ring, migratory insertion of the hydride species into the benzaldimine functionality, CO coordination, and insertion into the Co–C bond, followed by reductive elimination of the N-alkylphthalimidine and regeneration of the starting cobalt species.
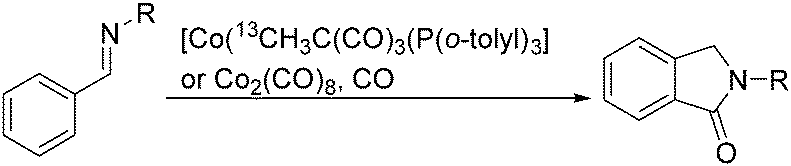 | ||
| Scheme 45 Co-Catalysed carbonylation of N-alkylbenzaldimines. | ||
In 2008, Li and co-workers reported a cobalt-catalyzed oxidative carbonylation of aniline to N,N′-diphenyl urea.88 The catalyst system was composed of a Co(II)-Schiff base complex and a pyridine-type promoter (Scheme 46). The Co(II) complexes of a substituted salen or salophen ligand with an electron-donating hydroxyl group showed good activity. In the catalytic system, a pyridine-type additive as a promoter increased the reactivity of the catalyst. Among the additives, substituted pyridines had better promoting function than pyridine. The promoting effect of substituted pyridines was correlated with their basicity and steric hindrance of substituents.
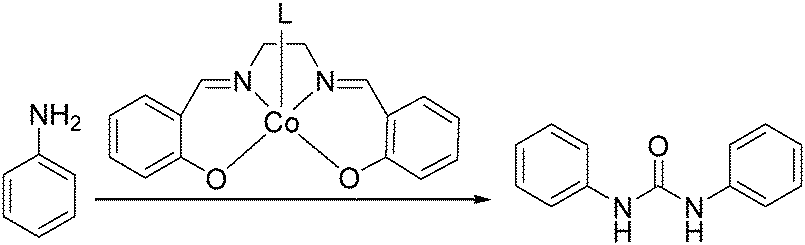 | ||
| Scheme 46 Cobalt-catalysed carbonylation of aniline. | ||
In 2010, Wang's group reported a cobalt-catalyzed photo-promoted carbonylation of chloroalkanes.89 With cobalt compounds [Co(OAc)2,CoCl2] as the catalyst and in the presence of KI, the corresponding esters can be selectively produced (Scheme 47). The reactions carried out under ambient conditions proceeded through two pathways. The role of the iodide ion may be to form more active iodoalkanes via substituting chloride ions in chloroalkanes in situ. Additionally, the activity of this carbonylation can be improved by the addition of NaOAc.
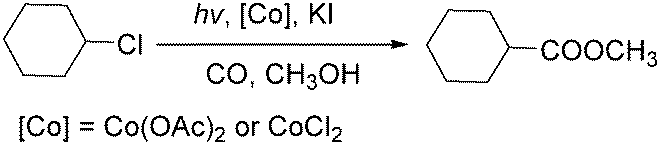 | ||
| Scheme 47 Photopromoted carbonylation of chloroalkanes. | ||
In 2011, an in situ generated catalytic system that allows the ring-expansion carbonylation of epoxides using low precatalyst loadings was developed (Scheme 48).90 The active catalyst, analogous to Coates’ catalyst, was formed in situ from commercially available (TPP)CrCl (TPP = tetraphenylporphyrin) and Co2(CO)8. This practical system circumvented the preparation of air sensitive cobaltate salts, manipulated at low catalyst loadings, and realized the carbonylation of functionalized, sterically demanding and heterocyclic meso-epoxides.
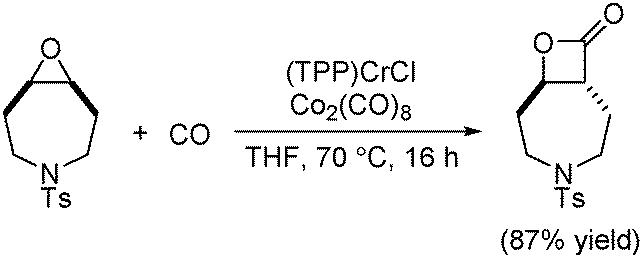 | ||
| Scheme 48 Cobalt-catalysed ring-expansive carbonylation. | ||
In 2011, Ogawa and co-workers reported a novel cobalt carbonyl catalysed thiolative lactonization of alkynes.91 With thiols as the reaction partner by incorporation of two molecules of carbon monoxide, α,β-unsaturated γ-thio-γ-lactones were produced regioselectively (Scheme 49). This process provides a useful method for the catalytic introduction of two molecules of CO into alkynes, regioselectively. Since sulfur compounds were generally believed to be catalyst poisons, these catalytic reactions opened up a new field of transition-metal-catalyzed reactions of organosulfur compounds.
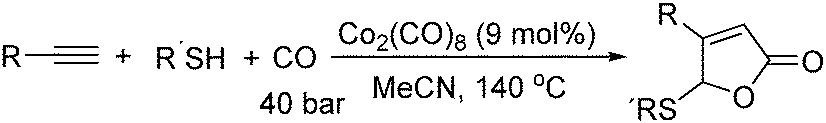 | ||
| Scheme 49 Cobalt-catalysed thiolative lactonization of alkynes. | ||
Later on, they found that internal alkynes can be applied as substrates in this cobalt-catalyzed thiolative double carbonylation with organosulfur as well (Scheme 50).92 This process became a useful tool for preparing the corresponding α,β-unsaturated γ-thio-γ-lactones (butenolide derivatives) in good yields. During the study, it was interestingly found that, for the unsymmetrical alkynes, the thiolative lactonization proceeded with moderate regioselectivity to give the butenolide derivatives on which the carbonyl group preferentially bonded to the less hindered acetylenic carbon.
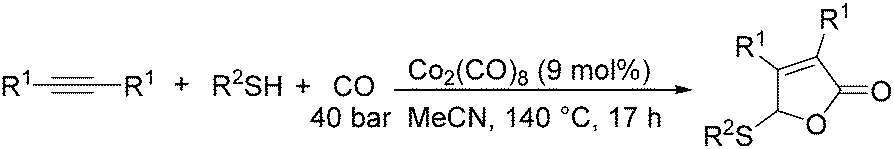 | ||
| Scheme 50 Cobalt-catalysed thiolative double carbonylation of internal alkynes. | ||
Recently, Saliu et al. described an oxidative carbonylation of amine in the presence of a bis(salicylaldehyde)ethylenediimine-cobalt(II) catalyst by using TBD (1,5,7-triazabicyclo[4.4.0]dec-5-ene) as the promoter. Here, the application of TBD would be beneficial and could represent an interesting development in the field of the Co-Schiff base complexes catalyzed oxidative carbonylation, as bicyclic guanidine bases are less toxic and more environmentally friendly than pyridine bases.93
In 2013, Li and co-workers developed a highly efficient cobalt catalysed carbonylation reaction of benzyl chlorides.94 A variety of phenylacetic acid derivatives were produced under atmosphere pressure of carbon monoxide (Scheme 51). In this methodology, benzimidazole was extended as the ligand. Meanwhile, the catalytic mechanism was also demonstrated by computer simulations.
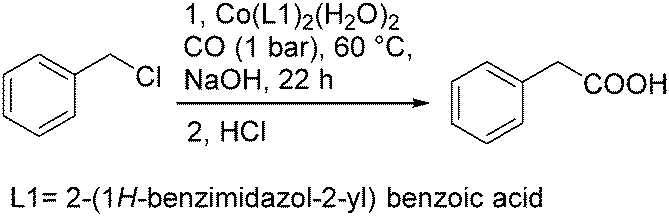 | ||
| Scheme 51 Cobalt-catalysed carbonylative approach to phenylacetic acid derivatives. | ||
In 2014, a convenient catalyst system consisting of Co2(CO)8 and LiCl for catalyzing cyclization of CO, imine, and epoxide was established (Scheme 52).95 This reaction provided an efficient method for the synthesis of substituted 1,3-oxazinan-4-ones from very simple and commercially available starting materials.
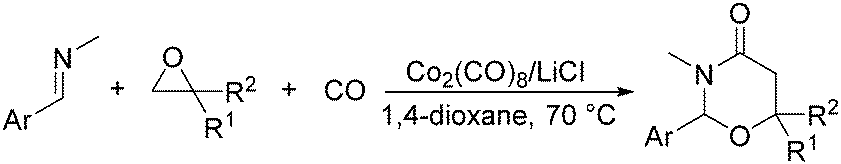 | ||
| Scheme 52 Cobalt-catalysed carbonylation for the synthesis of substituted 1,3-oxazinan-4-ones. | ||
Daugulis and Grigorjeva reported a method for the direct carbonylation of aminoquinoline benzamides in 2014 (Scheme 53).96 Reactions proceeded at room temperature in trifluoroethanol solvent, using oxygen from air as the oxidant and with Mn(OAc)3 as the additive. Imides were obtained by the carbonylation of benzoic and acrylic acid derivatives in good yields. The reaction fully tolerated functional groups such as halogen, nitro, ether, cyano, ester and so on. The removal of the directing group under mild conditions could afford the corresponding phthalimides.
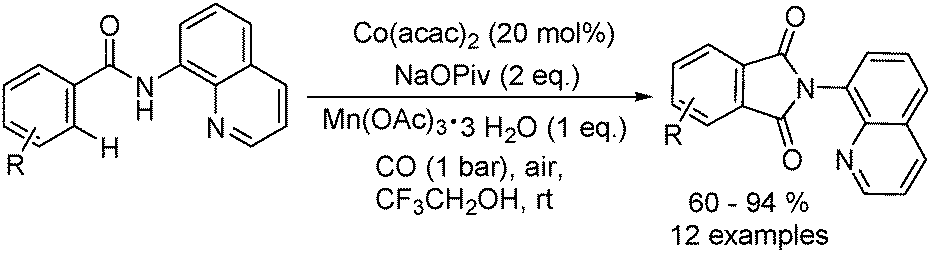 | ||
| Scheme 53 Cobalt-catalysed carbonylation of aminoquinoline benzamides. | ||
More recently, Zhang and co-workers reported an efficient approach for the C(sp2)–H bond carbonylation of benzamides with the use of stable and inexpensive Co(OAc)2·4H2O as the catalyst.97 Interestingly, commercially available and easily handled azodicarboxylates were applied as the nontoxic carbonyl source here (Scheme 54). The reaction revealed a broad scope of substrates bearing diverse functional groups.
 | ||
| Scheme 54 Cobalt-catalysed C(sp2)–H bond carbonylation of benzamides. | ||
In 2017, Gaunt's group reported a cobalt-catalysed carbonylative cyclisation procedure of unactivated aliphatic C–H bonds (Scheme 55).98 The key point of this methodology was the stabilizing effect of the quinolinamide directing group. The process tolerated a wide range of functionalized substrates to generate the corresponding substituted succinimide products. During the manipulation, the operationally simple reaction conditions were complemented by the ability to utilize an atmospheric pressure of carbon monoxide.
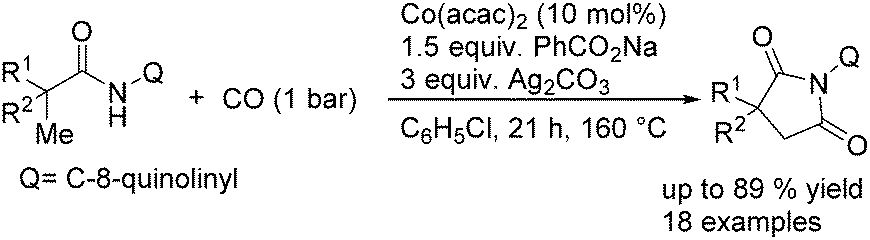 | ||
| Scheme 55 Cobalt-catalysed carbonylative cyclisation of aliphatic C–H bonds. | ||
In the same period, Sundararaju and co-workers reported a similar procedure with trifluorotoluene as the reaction media.99 Under their reaction conditions, a variety of unactivated C(sp3)–H bonds were transformed into the corresponding succinimide derivatives (Scheme 56). The initial mechanistic investigation revealed the involvement of a one electron process operating in the catalytic cycle and it also indicated that the reaction may proceed through a Co(IV) to Co(II) cycle. This straightforward approach provided an easy access to α-spiral succinimides in good yields regioselectively.
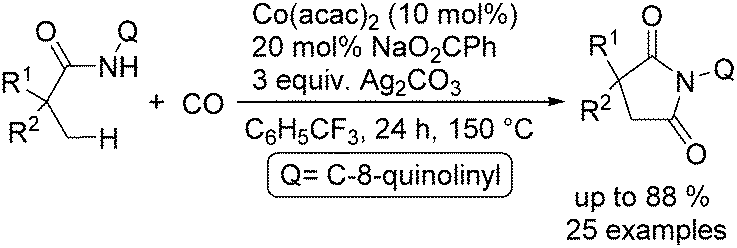 | ||
| Scheme 56 Cobalt-catalysed synthesis of succinimides. | ||
6. Ni-Catalysed carbonylation reactions
Nickel salts are cheap and abundant and have been extensively studied in various organic transformations. However, due to the high risk of the toxic Ni(CO)5 formed from nickel and carbon monoxide, nickel catalysts are studied in CO chemistry with limited cases. As early as in 1969, Corey and Hegedus developed a base-catalysed carboxylation of organic halides with nickel carbonyl as the CO source in protic media.100 Alkyl halides and allylic halides were transformed effectively with alcohols as reaction partners. Later on, Semmelhack and Brickner further studied this methodology and applied it in the synthesis of five- and six-membered lactones in 1981. In the presence of nickel carbonyl reagents via intramolecular two-step carbonylative cyclization, good yields of the desired products can be obtained (Scheme 57).101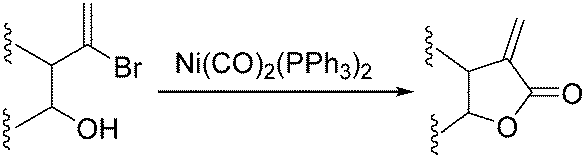 | ||
| Scheme 57 Ni-Mediated synthesis of lactones. | ||
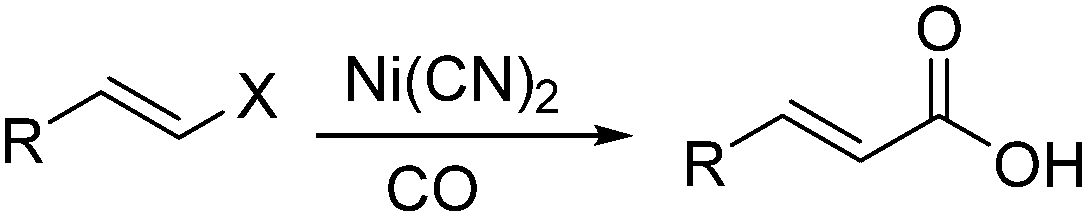
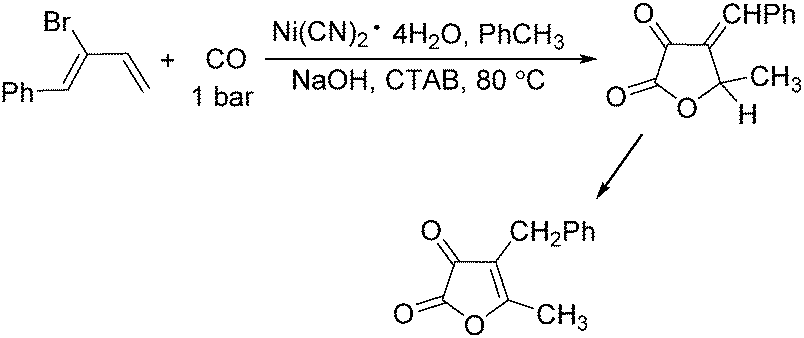
In 1989, Alper and Vasapollo reported the first nickel-catalysed carbonylative transformation of vinyl halides (Scheme 58).102 Nickel cyanide was found to be the best for the carbonylation of vinyl bromides and chlorides under phase transfer conditions. This method was simple both in execution and work-up, and exhibited excellent stereochemical control. In the case with halodienes as the substrates, double carbonylation occurs.103 For example, good yield of α-keto lactone can be produced from 2-bromo-1-phenyl-1,3-butadiene (Scheme 59).
 | ||
| Scheme 58 Nickel-catalysed carbonylation of vinyl halides. | ||
 | ||
| Scheme 59 Double carbonylation of halodienes. | ||
In 1999, Jiang and co-workers reported a procedure for nickel-catalysed carbonylation of methyl acetate to acetic anhydride with methyl iodide as the promoter.104 Different conditions including catalyst precursors, temperatures, pressures, ligands, lithium acetate and methyl iodide concentration were investigated in detail. Additionally, the mechanism and possible side reactions in the carbonylation were studied as well. The in situ reaction monitoring experiments readily enabled the determination of the concentrations of organonickel species as well as the concentration of carbonylation products under fast reaction conditions. In the meantime, an in situ CIR-FTIR investigation of process effects in the nickel catalysed carbonylation of methanol was reported by Okrasinski and co-workers.105 A mechanistic study of the phosphine-modified nickel-catalyzed acetic acid process was also conducted by their group.106
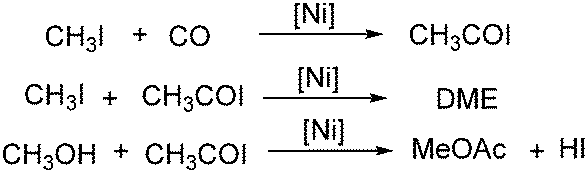
A nickel-aluminum catalysed liquid-phase carbonylation of methanol was reported by Matsumura and Kapoor in 2004 (Scheme 60).107 In this reaction, methanol was converted into methyl acetate in the presence of methyl iodide at 200 °C. Formation of methyl formate depended on the pressure of carbon monoxide applied, which indicated that insertion of carbon monoxide to methyl iodide was a key step of the carbonylation and dimethyl ether reacted with the intermediate to form the final methyl acetate.
 | ||
| Scheme 60 Liquid-phase carbonylation of methanol. | ||
In 2008, Ricart and co-workers reported a nickel-catalysed [2+2+1] carbonylative cycloaddition reaction between norbornenes and allyl halides.108 Catalyzed by a Ni(II) catalyst under very mild conditions (atmospheric pressure and room temperature), the desired products can be produced effectively (Scheme 61). Interestingly, there were two major products whose ratio could be tuned by carefully adding water in different amounts.
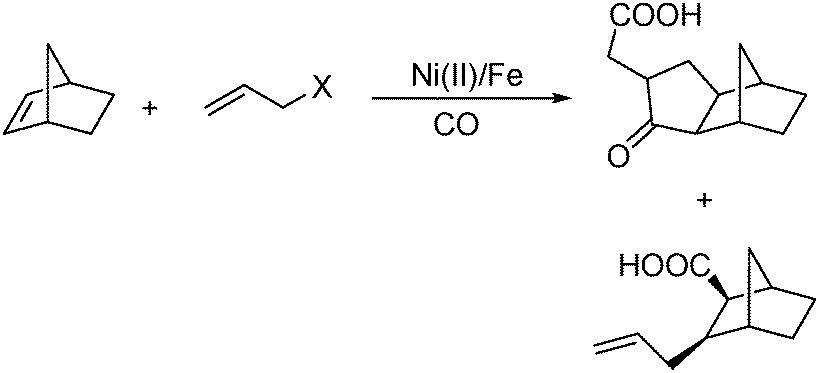 | ||
| Scheme 61 Carbonylative cycloaddition reaction between norbornenes and allyl halides. | ||
Chen and Wang described an interesting nickel-catalysed carbonylative Negishi cross-coupling reaction in the same year (Scheme 62).109 This method readily provided various enones from enol triflates and diorganozinc reagents, with catalytic amounts of nickel(II) chloride-4,4′-dimethoxyl-2,2′-bipyridyl under a carbon monoxide atmosphere. The addition of lithium or magnesium halides, as well as the use of polar solvents facilitated the rate of carbon monoxide insertion. Carbonylative alkenyl–aryl, alkenyl–alkenyl, and alkenyl–alkyl coupling were achieved effectively.
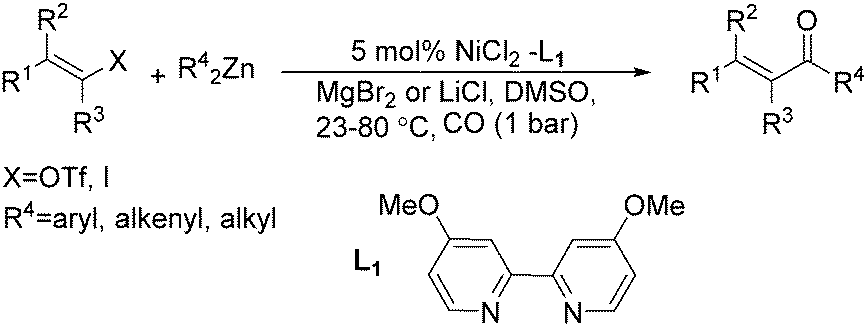 | ||
| Scheme 62 Ni-Catalysed carbonylative Negishi reactions. | ||
Meanwhile, Ogoshi's research group demonstrated the formation of a nickeladihydrofuran by oxidative cyclization of an alkyne and an aldehyde with nickel(0) (Scheme 63).110 The nickeladihydrofuran can be transformed into the corresponding lactone under CO pressure in quantitative yields.
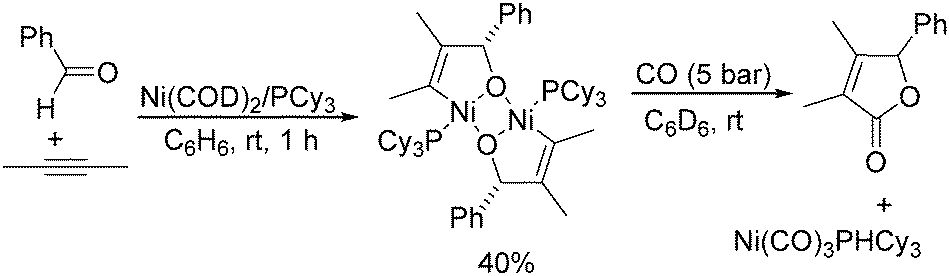 | ||
| Scheme 63 Formation of nickeladihydrofuran and its transformation into lactone. | ||
Interestingly, Lee and co-workers developed a nickel-catalysed coupling reaction of aryl bromides with formamide in 2009 (Scheme 64).111 Using this nickel–phosphite catalytic system, aryl bromides can react with a range of formamides to give the corresponding aryl amides in moderate to good yields. The steric bulkiness of formamides plays a very important role in this reaction. The less hindered formamide required lower catalytic loading for full conversion and produced higher yields than the more hindered one.
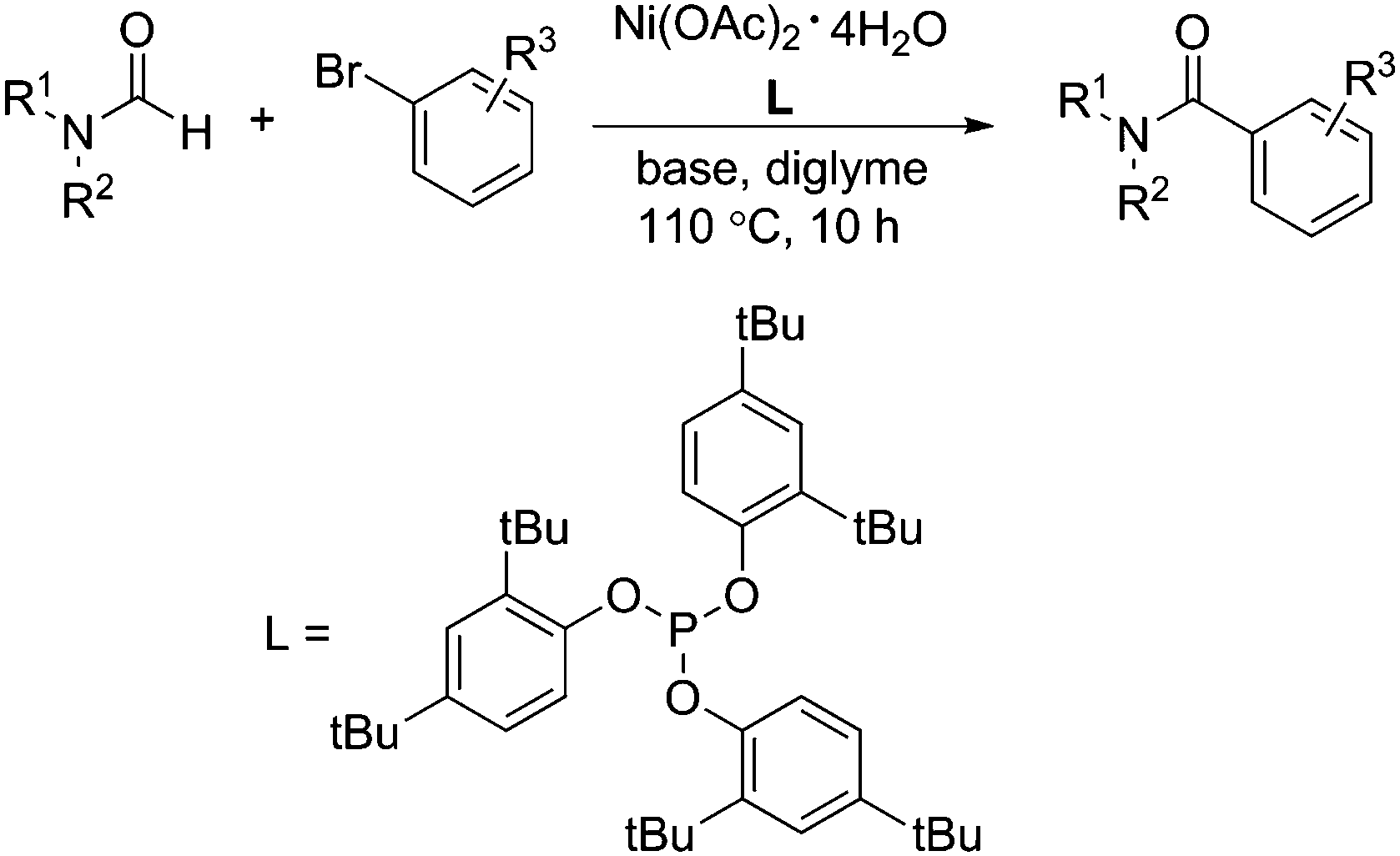 | ||
| Scheme 64 Nickel-catalysed aminocarbonylation of aryl bromides. | ||
In 2009, Ricart, Ventosa and their co-workers reported a catalytic procedure on the selective mono or double carbonylations of alkenes or acetylenes with allyl bromides (Scheme 65).112a Catalyzed by nickel, cyclopentanes, cyclohexanes or plainly-carbonylated adducts can be obtained. And mechanistic studies were performed as well.112b
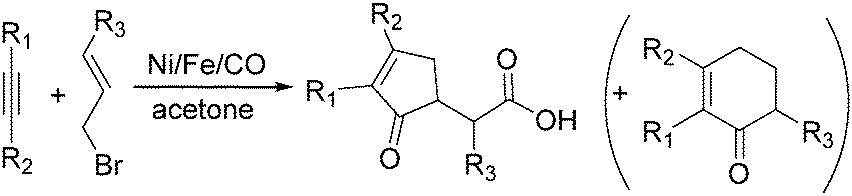 | ||
| Scheme 65 Carbonylative cycloaddition of alkynes and allyl halides in acetone. | ||
Ogosh and co-workers developed a Ni(0)-catalysed [2+2+1] carbonylative cycloaddition of imines and alkynes using phenyl formate as the CO source in 2014 (Scheme 66).113 Using 10 mol% of Ni(cod)2 as the catalyst, 20 mol% of PCy3 as the ligand and 2 equivalents of NEt3 as the base, a variety of lactams can be prepared in good to high yields.
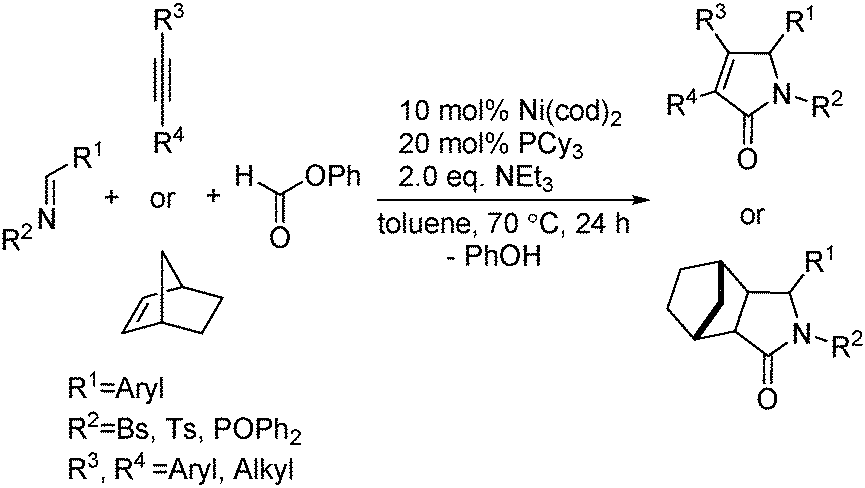 | ||
| Scheme 66 Ni(0)-Catalysed carbonylative cycloaddition of imines. | ||
In 2015, Bengali, Arndtsen and their co-workers described a nickel-catalysed carbonylative procedure for the synthesis of isoindolinones from aryl iodides and imines (Scheme 67).114 In the presence of a catalytic amount of Ni(cod)2, using DiPEA (diisopropylethylamine) as the base and Bu4NCl as the additive, various isoindolinones were isolated in good yields under atmospheric pressure (1 bar) of carbon monoxide.
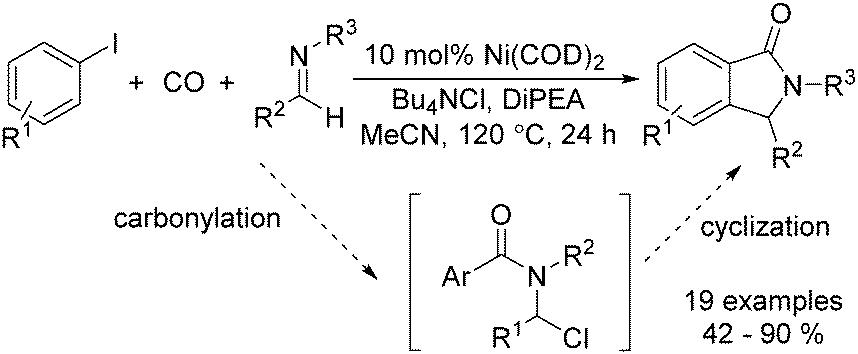 | ||
| Scheme 67 Nickel-catalysed synthesis of isoindolinones. | ||
Recently, Zhou and co-workers developed a nickel-catalysed hydrocarbonylation of terminal alkynes with formic acid as the CO source (Scheme 68).115 The high efficiency and low cost of nickel catalysts made this nickel-catalysed hydrocarboxylation of acetylene a promising process for acrylic acid production. At the same time, Fu and co-workers also developed a nickel-catalysed regio- and stereoselective hydrocarboxylation of alkynes (Scheme 69).116 In the presence of a catalytic 5 mol% Ni(acac)2, 7 mol% dppbz, and 20 mol% Piv2O, alkyne can react with formic acid to afford the desired α,β-unsaturated carboxylic acids.
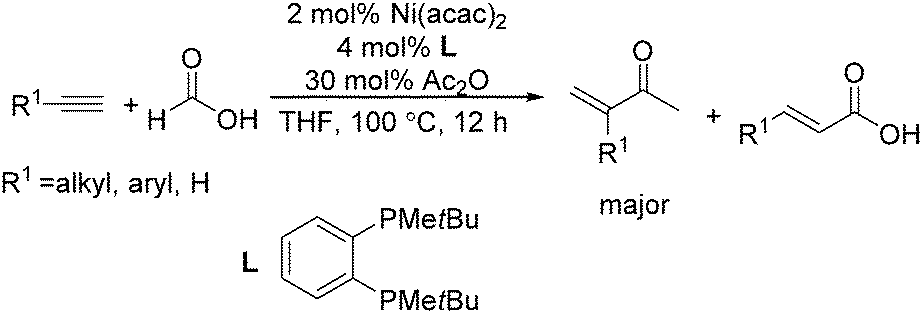 | ||
| Scheme 68 Nickel-catalysed hydrocarbonylation of terminal alkynes. | ||
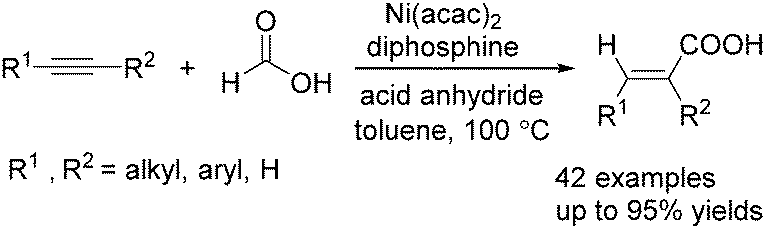 | ||
| Scheme 69 Nickel-catalysed hydrocarbonylation of alkynes with formic acid. | ||
More recently, direct activation of simple C–N bonds via oxidative addition was reported by Huang and co-workers (Scheme 70).117 Through this method, tertiary benzylamines can be transformed into the corresponding amides in moderate to good yields.
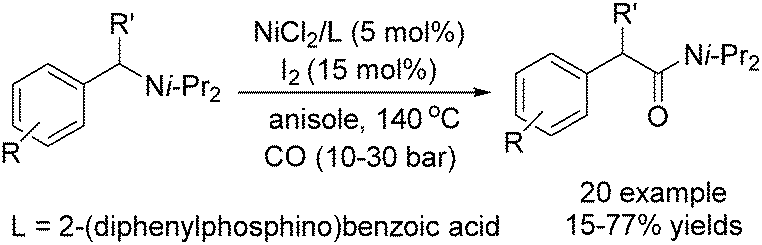 | ||
| Scheme 70 Nickel-catalysed carbonylation of benzyl amines. | ||
7. Electrosynthesis
Electroorganic synthesis is an interesting alternative procedure in modern chemistry.118 And electrochemistry has been applied in carbonylation chemistry as well. The first report in this area was published in 1988 by Petit and co-workers.119 This novel methodology allowed the production of aldehyde from organic halides. From mechanistic studies, Fe(CO)5 and RX must be reduced to Fe(CO)5−˙ and R˙ at the beginning in order to obtain good yields of the desired aldehydes (Scheme 71). The group of Yoshida also devoted their efforts on this topic as well.120,121 Furthermore, they also studied the cell modes, cathode materials and solvent.122 And the best reaction medium for the reaction was acetonitrile containing tetraalkylammonium halides (or OTs-) in a divided cell. The report from Périchon and co-workers in 1995 used a mild reaction condition for the electrochemical synthesis of ketones from organic halides and CO.123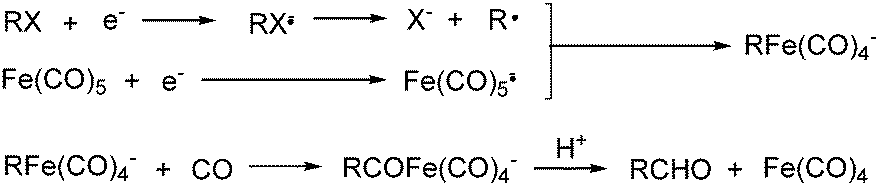 | ||
| Scheme 71 Electro-carbonylation of organic halides. | ||
In 1997, Troupel and co-workers showed that ketones can be formed by electroreduction in an undivided cell of an organic halide–metal carbonyl mixture using a Ni-bpy catalytic system in DMF (Scheme 72, formula (1)).124 Primary benzylic halides can be efficiently converted into the corresponding ketones, but secondary alkyl or benzyl halides did not give the desired product. Another limitation was the failure to obtain unsymmetrical ketones for solutions containing two halides. More recently, the same group also described a successful nickel-catalysed electrosynthesis of unsymmetrical ketones from organic halides and iron pentacarbonyl (Scheme 72, formula (2)).125
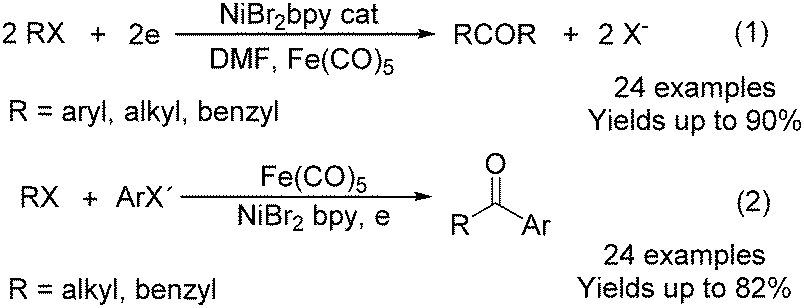 | ||
| Scheme 72 Nickel-catalysed electrosynthesis of unsymmetrical ketones. | ||
Interestingly, Peters and co-workers reported an electrogenerated nickel(I) salen catalysed reduction of primary alkyl monohalides for the formation of aldehydes (Scheme 73, formula (1)).126,127 And the formation of ketones from secondary alkyl monohalides in the presence of oxygen and water was reported by the same group as well (Scheme 73, formula (2)).128 On the basis of experimental results, they gave a possible reaction mechanism. As shown in Scheme 74, nickel(II) salen is converted to nickel(I) salen by controlled-potential reduction (reaction (a)). Reaction (b) shows the interaction of electrogenerated nickel(I) salen with a secondary alkyl monohalide in a two-to-one ratio to give nickel(II) salen, a halide ion, and an alkylnickel(II) species ([RCH2CH(CH3)–Ni(II)L]−). In turn, this organonickel(II) species reacts with O2 (within a solvent cage) to yield a peroxo-bridged complex (reaction (c)), which decomposes to form the desired ketone (reaction (d)).
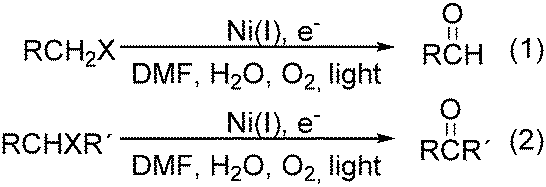 | ||
| Scheme 73 Nickel-catalysed electrosynthesis of aldehydes and ketones. | ||
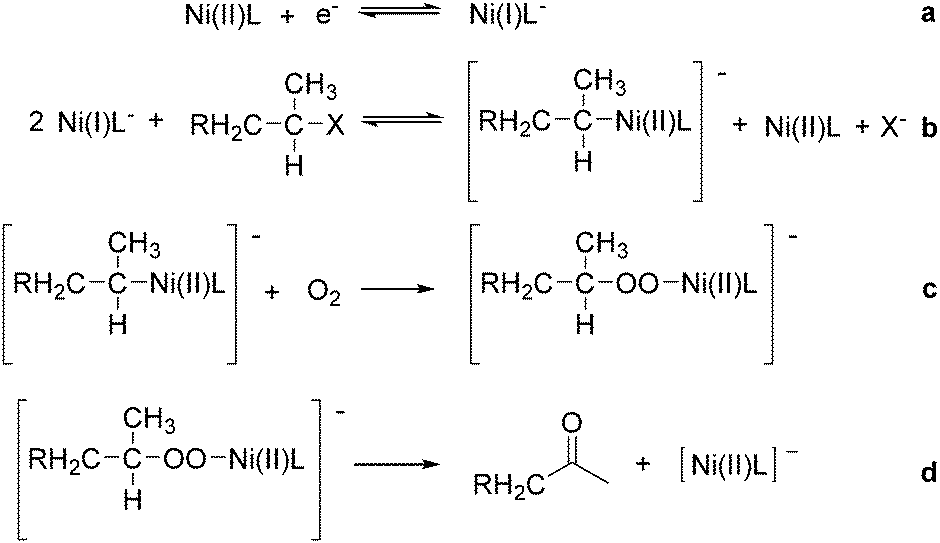 | ||
| Scheme 74 Mechanism of nickel-catalysed electrosynthesis of aldehydes and ketones. | ||
The works by Périchon and co-workers in 1988 led to an efficient procedure for the electrosynthesis of ketones from organic halides and carbon dioxide using nickel–bipyridine complexes (Scheme 75).129 The main advantage of this catalysis protocol lies in the use of CO2 as the source of CO.
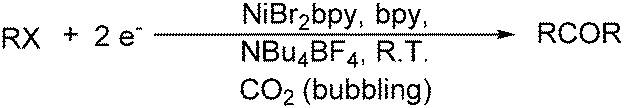 | ||
| Scheme 75 Nickel-catalysed electrosynthesis of ketones using CO2. | ||
In 1993, Hartstock and co-workers developed an electrochemical carbonylation of alkynes to unsaturated diesters using palladium as the catalyst (Scheme 76).130 Experimental results revealed that yields improved as the chain-length of alkynes decreased and cis-unsaturated diesters were the favoured products. Internal alkynes were also found to yield unsaturated diesters in fair to good yields with relatively low conversion.
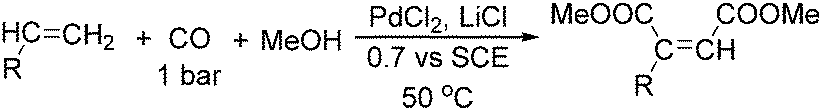 | ||
| Scheme 76 Electrochemical synthesis of unsaturated diesters. | ||
With the growing demand of polycarbonates, developing a new method for synthesis of carbonates remains an important but challenging goal. Recently, Yamanaka and co-workers applied such an electrocatalytic system for the synthesis of diphenyl carbonate (DPC) (Scheme 77).131–134 After that they also studied the influence of various ligands, and N-heterocyclic carbene (NHC) groups were found to be effective for the electrocarbonylation.135
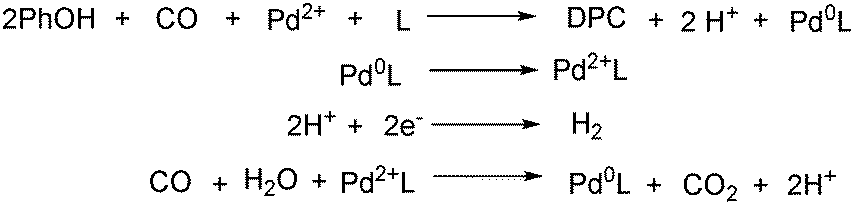 | ||
| Scheme 77 Electrocatalysis for the synthesis of diphenyl carbonate. | ||
8. Summary
In summary, the main achievements on non-noble metal catalysed carbonylative transformations have been summarized and discussed. Cheap metals including manganese, iron, copper, cobalt and nickel are included. Due to the sustainable aspect of electrochemistry, its applications in carbonylation chemistry have been discussed as well. Based on the known obvious advantages of non-noble metal catalysts, it's attractive to explore their activities in carbonylation reactions.However, compared with noble metal catalysts (Pd and Rh), non-noble catalysts are much less studied. Additionally, the catalyst loadings are relatively high in the known procedures and the reaction pathways are still at the ‘proposed’ stage. In the future, more systematic studies should be performed to understand the underlying chemistry. Based on the understanding of reaction mechanisms, the catalyst efficiency can be improved and new reactivity can be further explored.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the generous support from Professor Armin Börner and Professor Matthias Beller in LIKAT.Notes and references
- (a) M. Beller, Catalytic Carbonylation Reactions, Springer, Berlin, 2006 Search PubMed; (b) L. Kollär, Modern Carbonylation Methods, Wiley-VCH, Weinheim, 2008 Search PubMed; (c) B. Gabriele, R. Mancuso and G. Salerno, Eur. J. Org. Chem., 2012, 6825–6839 CrossRef CAS; (d) X.-F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986–5009 RSC; (e) X.-F. Wu, H. Neumann and M. Beller, Chem. Rev., 2012, 113, 1–35 CrossRef PubMed; (f) X.-F. Wu, X. Fang, L. Wu, R. Jackstell, H. Neumann and M. Beller, Acc. Chem. Res., 2014, 47, 1041–1053 CrossRef CAS PubMed; (g) B. Sam, B. Breit and M. J. Krische, Angew. Chem., Int. Ed., 2015, 54, 3267–3274 CrossRef CAS PubMed; (h) R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed; (i) C. F. J. Barnard, Organometallics, 2008, 27, 5402–5422 CrossRef CAS; (j) Q. Liu, H. Zhang and A. Lei, Angew. Chem., Int. Ed., 2011, 50, 10788–10799 CrossRef CAS PubMed; (k) T. Morimoto and K. Kakiuchi, Angew. Chem., Int. Ed., 2004, 43, 5580–5588 CrossRef CAS PubMed; (l) L. R. Odell, F. Russo and M. Larhed, Synlett, 2012, 685–698 CrossRef CAS; (m) H. Konishi and K. Manabe, Synlett, 2014, 1971–1986 CAS; (n) P. Gautam and B. M. Bhanage, Catal. Sci. Technol., 2015, 5, 4663–4702 RSC; (o) S. D. Friis, A. T. Lindhardt and T. Skrydstrup, Acc. Chem. Res., 2016, 49, 594–605 CrossRef CAS PubMed; (p) X.-F. Wu, RSC Adv., 2016, 6, 83831–83837 RSC; (q) J.-B. Peng, X. Qi and X.-F. Wu, ChemSusChem, 2016, 9, 2279–2283 CrossRef CAS PubMed; (r) J.-B. Peng, X. Qi and X.-F. Wu, Synlett, 2017, 175–194 Search PubMed.
- For selected reviews on industrial applications, see: (a) C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef CAS; (b) C. A. Busacca, D. R. Fandrick, J. J. Song and C. H. Senanayaka, Adv. Synth. Catal., 2011, 353, 1815–1864 CrossRef.
- (a) C. H. Schiesser, U. Wille, H. Matsubara and I. Ryu, Acc. Chem. Res., 2007, 40, 303–313 CrossRef CAS PubMed; (b) S. Sumino, A. Fusano, T. Fukuyama and I. Ryu, Acc. Chem. Res., 2014, 47, 1563–1574 CrossRef CAS PubMed; (c) I. Ryu and N. Sonoda, Angew. Chem., Int. Ed. Engl., 1996, 35, 1050–1066 CrossRef; (d) I. Ryu, Chem. Rec., 2002, 2, 249–258 CrossRef CAS PubMed.
- (a) R. Skoda-Földes and L. Kollár, Curr. Org. Chem., 2002, 6, 1097–1119 CrossRef; (b) B. Gabrielr, G. Salerno, M. Costa and G. P. Chiusoli, Curr. Org. Chem., 2004, 8, 919-–946 CrossRef.
- X.-F. Wu and H. Neumann, ChemCatChem, 2012, 4, 447–458 CrossRef CAS.
- For selected examples on iridium-catalyzed carbonylation reactions, see: (a) A. Haynes, P. M. Maitlis, G. E. Morris, G. J. Sunley, H. Adams, P. W. Badger, C. M. Bowers, D. B. Cook, P. I. P. Elliott, T. Ghaffar, H. Green, T. R. Griffin, M. Payne, J. M. Pearson, M. J. Taylor, P. W. Vickers and R. J. Watt, J. Am. Chem. Soc., 2004, 126, 2847–2861 CrossRef CAS PubMed; (b) V. J. Garza and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 3655–3658 CrossRef CAS PubMed; (c) F. Zhu, Y. Li, Z. Wang and X.-F. Wu, Angew. Chem., Int. Ed., 2016, 55, 14151–14154 CrossRef CAS PubMed; (d) F. Zhu, Z. Wang, Y. Li and X.-F. Wu, Chem. – Eur. J., 2017, 23, 3276–3279 CrossRef CAS PubMed.
- For selected reviews on non-noble metal catalysed reactions see: (a) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293–1314 CrossRef CAS PubMed; (b) A. Fürstner, Angew. Chem., Int. Ed., 2009, 48, 1364–1370 CrossRef PubMed; (c) A. Correa, O. Garcia Mancheno and C. Bolm, Chem. Soc. Rev., 2008, 37, 1108–1117 RSC; (d) S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234–6458 CrossRef CAS PubMed; (e) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062–11087 CrossRef CAS PubMed; (f) X.-X. Guo, D.-W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622–1651 CrossRef CAS PubMed.
- (a) W. Liu and L. Ackermann, ACS Catal., 2016, 6, 3743–3752 CrossRef CAS; (b) C. Wang, Synlett, 2013, 1606–1613 CrossRef CAS; (c) F. Calderazzo, Inorg. Chem., 1965, 4, 293–296 CrossRef CAS.
- (a) T. Kondo, Y. Tsuji and Y. Watanabe, Tetrahedron Lett., 1988, 29, 3833–3836 CrossRef CAS; (b) T. Kondo, Y. Sone, Y. Tsuji and Y. Watanabe, J. Organomet. Chem., 1994, 473, 163–173 CrossRef CAS; (c) S.-K. Kang, W.-Y. Kim, Y.-T. Lee, S.-K. Ahn and J.-C. Kim, Tetrahedron Lett., 1998, 39, 2131–2132 CrossRef CAS.
- C. M. McMahon, M. S. Renn and E. J. Alexanian, Org. Lett., 2016, 18, 4148–4150 CrossRef CAS PubMed.
- Y. Li, F. Zhu, Z. Wang, J. Rabeah, A. Brückner and X.-F. Wu, ChemCatChem, 2017, 9, 915–919 CrossRef CAS.
- (a) A. Devasagayaraj and M. Periasamy, Transition Met. Chem., 1991, 16, 503–504 CrossRef CAS; (b) J. J. Brunet, R. Chauvin and D. Neibecker, Synth. Commun., 1997, 27, 1433–1437 CrossRef CAS.
- J. P. Collman, Acc. Chem. Res., 1975, 8, 342–347 CrossRef CAS.
- J. P. Collman, S. R. Winter and R. G. Komoto, J. Am. Chem. Soc., 1973, 95, 249–250 CrossRef CAS.
- J. P. Collman, S. R. Winter and D. R. Clark, J. Am. Chem. Soc., 1972, 94, 1788–1789 CrossRef CAS.
- J. P. Collman and N. W. Hoffman, J. Am. Chem. Soc., 1973, 95, 2689–2691 CrossRef CAS.
- M. P. Cooke, J. Am. Chem. Soc., 1970, 92, 6080–6082 CrossRef CAS.
- H. Alper and K. E. Hashem, J. Am. Chem. Soc., 1981, 103, 6514–6515 CrossRef CAS.
- B. E. Eaton, B. Rollman and J. A. Kaduk, J. Am. Chem. Soc., 1992, 114, 6245–6246 CrossRef CAS.
- (a) M. S. Sigman, C. E. Kerr and B. E. Eaton, J. Am. Chem. Soc., 1993, 115, 7545–7546 CrossRef CAS; (b) M. S. Sigman and B. E. Eaton, J. Org. Chem., 1994, 59, 7488–7491 CrossRef CAS.
- (a) K. Rueck-Braun, Angew. Chem., Int. Ed., 1997, 36, 509–511 CrossRef CAS; (b) K. Rueck-Braun, T. Martin and M. Mikulas, Chem. – Eur. J., 1999, 5, 1028–1037 CrossRef; (c) K. Rueck-Braun and C. Moeller, Chem. – Eur. J., 1999, 5, 1038–1044 CrossRef; (d) P. Amrhein, D. Schollmeyer and K. Rueck-Braun, Organometallics, 2000, 19, 3527–3534 CrossRef CAS; (e) C. Moeller, M. Mikulas, F. Wierschem and K. Rueck-Braun, Synlett, 2000, 182–184 CrossRef.
- J. J. Brunet and M. Taillefer, J. Organomet. Chem., 1988, 361, C1–C4 CrossRef.
- J. J. Brunet and M. Taillefer, J. Organomet. Chem., 1990, 384, 193–197 CrossRef CAS.
- A. Devasagayaraj, M. L. N. Rao and M. Periasamy, J. Organomet. Chem., 1991, 421, 147–150 CrossRef CAS.
- (a) A. Devasagayaraj and M. Periasamy, Tetrahedron Lett., 1992, 33, 1227–1228 CrossRef CAS; (b) M. Periasamy, A. Devasagayaraj and U. Radhakrishnan, Organometallics, 1993, 12, 1424–1428 CrossRef CAS.
- M. Periasamy, U. Radhakrishnan, J. J. Brunet, R. Chauvin and A. W. El Zaizi, Chem. Commun., 1996, 1499–1500 RSC.
- M. Periasamy, M. Beesu and D. S. Raj, J. Organomet. Chem., 2008, 693, 2843–2846 CrossRef CAS.
- M. Periasamy, C. Rameshkumar and U. Radhakrishnan, Tetrahedron Lett., 1997, 38, 7229–7232 CrossRef CAS.
- C. Rameshkumar and M. Periasamy, Tetrahedron Lett., 2000, 2719–2722 CrossRef CAS.
- M. Periasamy, A. Mukkanti and D. S. Raj, Organometallics, 2004, 23, 6323–6326 CrossRef CAS.
- M. Beesu and M. Periasamy, J. Org. Chem., 2011, 76, 543–549 CrossRef CAS PubMed.
- K. M. Driller, H. Klein, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 6041–6044 CrossRef CAS PubMed.
- S. Prateeptongkum, K. M. Driller, R. Jackstell, A. Spannenberg and M. Beller, Chem. – Eur. J., 2010, 16, 9606–9615 CrossRef CAS PubMed.
- S. Prateeptongkum, K. M. Driller, R. Jackstell and M. Beller, Chem. – Asian J., 2010, 5, 2173–2176 CrossRef CAS PubMed.
- (a) K. M. Driller, S. Prateeptongkum, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 537–541 CrossRef CAS PubMed; (b) M. Pizzetti, A. Russo and E. Petricci, Chem. – Eur. J., 2011, 17, 4523–4528 CrossRef CAS PubMed.
- P. Mathur, R. K. Joshi, D. K. Rai, B. Jha and S. M. Mobin, Dalton Trans., 2012, 41, 5045–5054 RSC.
- (a) N. Iranpoor, H. Firouzabadi, A. Riazi and K. Pedrood, J. Organomet. Chem., 2016, 822, 67–73 CrossRef CAS; (b) P. Mathur, R. K. Joshi, B. Jha, A. K. Singh and S. M. Mobin, J. Organomet. Chem., 2010, 695, 2687–2694 CrossRef CAS.
- Y. Zhong and W. Han, Chem. Commun., 2014, 50, 3874–3877 RSC.
- S.-K. Kang, T. Yamaguchi, T.-H. Kim and P.-S. Ho, J. Org. Chem., 1996, 61, 9082–9083 CrossRef CAS.
- Y. Fujiwara, K. Tabaki and Y. Taniguchi, Synlett, 1996, 591–599 CrossRef CAS.
- V. Raab, M. Merz and J. Sundermeyer, J. Mol. Catal. A: Chem., 2001, 175, 51–63 CrossRef CAS.
- M. Casiello, A. Monopoli, P. Cotugno, A. Milella, M. M. Dell’Anna, F. Ciminale and A. Nacci, J. Mol. Catal. A: Chem., 2014, 381, 99–106 CrossRef CAS.
- J. Liu, R. Zhang, S. Wang, W. Sun and C. Xia, Org. Lett., 2009, 11, 1321–1324 CrossRef CAS PubMed.
- P. J. Tambade, Y. P. Patil, N. S. Nandurkar and B. M. Bhanage, Synlett, 2008, 886–888 CAS.
- C. Wang, S. Lei, H. Cao, S. Qiu, J. Liu, H. Deng and C. Yan, J. Org. Chem., 2015, 80, 12725–12732 CrossRef CAS PubMed.
- S. Lei, Y. Mai, C. Yan, J. Mao and H. Cao, Org. Lett., 2016, 18, 3582–3585 CrossRef CAS PubMed.
- J.-J. Wu, Y. Li, H.-Y. Zhou, A.-H. Wen, C.-C. Lun, S.-Y. Yao, Z. Ke and B.-H. Ye, ACS Catal., 2016, 6, 1263–1267 CrossRef CAS.
- G. Shen, L. Zhao, Y. Wang and T. Zhang, RSC Adv., 2016, 6, 78307–78310 RSC.
- M. Usman, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, RSC Adv., 2016, 6, 107542–107546 RSC.
- C.-M. Yu, J.-H. Kweon, P.-S. Ho, S.-C. Kang and G. Y. Lee, Synlett, 2005, 2631–2634 CrossRef CAS.
- X. Wu, J. Miao, Y. Li, G. Li and H. Ge, Chem. Sci., 2016, 7, 5260–5264 RSC.
- Y. Li, K. Dong, F. Zhu, Z. Wang and X.-F. Wu, Angew. Chem., Int. Ed., 2016, 55, 7227–7230 CrossRef CAS PubMed.
- Y. Li, F. Zhu, Z. Wang and X.-F. Wu, ACS Catal., 2016, 6, 5561–5564 CrossRef CAS.
- Y. Li, C. Wang, F. Zhu, Z. Wang, P. H. Dixneuf and X.-F. Wu, ChemSusChem, 2017, 10, 1341–1345 CrossRef CAS PubMed.
- Y. Li, C. Wang, F. Zhu, Z. Wang, J. Francois Soule, P. H. Dixneuf and X.-F. Wu, Chem. Commun., 2017, 53, 142–144 RSC.
- H. Alper and H. des Abbayes, J. Organomet. Chem., 1977, 134, C11–C14 CrossRef CAS.
- H. des Abbayes and A. Buloup, J. Chem. Soc., Chem. Commun., 1978, 1090–1091 RSC.
- T. Imamoto, T. Kusumoto and M. Yokoyama, Bull. Chem. Soc. Jpn., 1982, 55, 643–644 CrossRef CAS.
- F. Francalanci and M. Foa, J. Organomet. Chem., 1982, 232, 59–70 CrossRef CAS.
- F. Francalanci, G. Gardano, L. Abis, T. Fiorani and M. Foa, J. Organomet. Chem., 1983, 243, 87–94 CrossRef CAS.
- H. Alper, H. Arzoumanian, J. F. Petrignani and M. S. Maldonadob, J. Chem. Soc., Chem. Commun., 1985, 340–341 RSC.
- R. W. Wegman and D. C. Busby, J. Chem. Soc., Chem. Commun., 1986, 332–333 RSC.
- T. Kashimura, K. Kudo, S. Mori and N. Sugita, Chem. Lett., 1986, 483–486 CrossRef CAS.
- M. Foa, F. Francalanci, E. Bencini and A. Gardano, J. Organomet. Chem., 1985, 285, 293–303 CrossRef CAS.
- F. Francalanci, E. Bencini, A. Gardano, M. Vincenti and M. Foa, J. Organomet. Chem., 1986, 301, C27–C30 CrossRef CAS.
- H. Alper and S. Calet, Tetrahedron Lett., 1988, 29, 1763–1766 CrossRef CAS.
- D. Roberto and H. Alper, J. Am. Chem. Soc., 1989, 111, 7539–7543 CrossRef CAS.
- Y. Tsuji, M. Kobayashi, F. Okuda and Y. Watanabe, J. Chem. Soc., Chem. Commun., 1989, 1253–1254 RSC.
- H. Alper, A. Eisenstat and N. Satyanarayana, J. Am. Chem. Soc., 1990, 112, 7061–7063 CrossRef.
- Y. Amino, S. Nishi and K. Izawa, Bull. Chem. Soc. Jpn., 1991, 64, 620–623 CrossRef CAS.
- V. V. Grushin and H. Alper, Tetrahedron Lett., 1991, 32, 3349–3352 CrossRef CAS.
- H. Urata, D. Goto and T. Fuchikami, Tetrahedron Lett., 1991, 32, 3091–3094 CrossRef CAS.
- M. D. Wang and H. Alper, J. Am. Chem. Soc., 1992, 114, 7018–7024 CrossRef CAS.
- E. Bolzacchini, S. Meinardi, M. Orlandi and B. Rindone, J. Mol. Catal. A: Chem., 1996, 111, 281–287 CrossRef CAS.
- M. E. Piotti and H. Alper, J. Am. Chem. Soc., 1996, 118, 111–116 CrossRef CAS.
- C. Zucchi, G. Palyi, V. Galamb, E. Sampar-Szerencses, L. Marko, P. Li and H. Alper, Organometallics, 1996, 15, 3222–3231 CrossRef CAS.
- J. Lin and J. F. Knifton, Catal. Lett., 1997, 45, 139–141 CrossRef CAS.
- J. T. Lee, P. J. Thomas and H. Alper, J. Org. Chem., 2001, 66, 5424–5426 CrossRef CAS PubMed.
- V. Dragojlovic, D. B. Gao and Y. L. Chow, J. Mol. Catal. A: Chem., 2001, 171, 43–51 CrossRef CAS.
- S. N. Goodman and E. N. Jacobsen, Angew. Chem., Int. Ed., 2002, 41, 4703–4705 CrossRef CAS PubMed.
- H. Xu and L. Jia, Org. Lett., 2003, 5, 1575–1577 CrossRef CAS PubMed.
- H. Xu and L. Jia, Org. Lett., 2003, 5, 3955–3957 CrossRef CAS PubMed.
- H. Xu, J. A. Gladding and L. Jia, Inorg. Chim. Acta, 2004, 357, 4024–4028 CrossRef CAS.
- G. Liu and L. Jia, J. Am. Chem. Soc., 2004, 126, 14716–14717 CrossRef CAS PubMed.
- G. Liu and L. Jia, Angew. Chem., Int. Ed., 2006, 118, 135–137 CrossRef.
- J. Chai, G. Liu, K. Chaicharoen, C. Wesdemiotis and L. Jia, Macromolecules, 2008, 41, 8980–8985 CrossRef CAS.
- J. K. Funk, H. Yennawar and A. Sen, Helv. Chim. Acta, 2006, 89, 1687–1695 CrossRef CAS.
- L. J. Chen, J. Bao, F. M. Mei and G. X. Li, Catal. Commun., 2008, 9, 658–663 CrossRef CAS.
- Y. P. Jia, Y. N. Cui, J. M. Yin, G. Y. Zhou, S. M. Li, D. B. Gao and X. S. Wang, Chin. Chem. Lett., 2010, 21, 1033–1036 CrossRef CAS.
- P. Ganji, D. J. Doyle and H. Ibrahim, Org. Lett., 2011, 13, 3142–3145 CrossRef CAS PubMed.
- Y. Higuchi, S. Atobe, M. Tanaka, I. Kamiya, T. Yamamoto, A. Nomoto, M. Sonoda and A. Ogawa, Organometallics, 2011, 30, 4539–4543 CrossRef CAS.
- Y. Higuchi, S. Higashimae, T. Tamai and A. Ogawa, Tetrahedron, 2013, 69, 11197–11202 CrossRef CAS.
- F. Saliu, B. Putomatti and B. Rindone, Tetrahedron Lett., 2012, 53, 3590–3593 CrossRef CAS.
- M. She, D. Xiao, B. Yin, Z. Yang, P. Liu, J. Li and Z. Shi, Tetrahedron, 2013, 69, 7264–7268 CrossRef CAS.
- Y. Zhang, J. Ji, X. Zhang, S. Lin, Q. Pan and L. Jia, Org. Lett., 2014, 16, 2130–2133 CrossRef CAS PubMed.
- L. Grigorjeva and O. Daugulis, Org. Lett., 2014, 16, 4688–4690 CrossRef CAS PubMed.
- J. Ni, J. Li, Z. Fan and A. Zhang, Org. Lett., 2016, 18, 5960–5963 CrossRef CAS PubMed.
- P. Williamson, A. Galvan and M. J. Gaunt, Chem. Sci., 2017, 8, 2588–2591 RSC.
- N. Barsu, S. K. Bolli and B. Sundararaju, Chem. Sci., 2017, 8, 2431–2435 RSC.
- E. J. Corey and L. S. Hegedus, J. Am. Chem. Soc., 1969, 91, 1233–1234 CrossRef CAS.
- M. F. Semmelhack and S. J. Brickner, J. Org. Chem., 1981, 46, 1723–1726 CrossRef CAS.
- H. Alper, I. Amer and G. Vasapollo, Tetrahedron Lett., 1989, 30, 2615–2616 CrossRef CAS.
- H. Alper and G. Vasapollo, Tetrahedron Lett., 1989, 30, 2617–2618 CrossRef CAS.
- J. F. Gong, Q. H. Fan and D. Z. Jiang, J. Mol. Catal. A: Chem., 1999, 147, 113–124 CrossRef CAS.
- W. R. Moser, B. J. Marshik-Guerts and S. J. Okrasinski, J. Mol. Catal. A: Chem., 1999, 143, 57–69 CrossRef CAS.
- W. R. Moser, B. J. Marshik-Guerts and S. J. Okrasinski, J. Mol. Catal. A: Chem., 1999, 143, 71–83 CrossRef CAS.
- M. P. Kapoor and Y. Matsumura, Catal. Today, 2004, 93-95, 287–291 CrossRef CAS.
- D. del Moral, J. M. Moreto, E. Molins and S. Ricart, Tetrahedron Lett., 2008, 49, 6947–6950 CrossRef CAS.
- Q. Wang and C. Chen, Tetrahedron Lett., 2008, 49, 2916–2921 CrossRef CAS.
- S. Ogoshi, T. Arai, M. Ohashi and H. Karosawa, Chem. Commun., 2008, 1347–1349 RSC.
- Y. Jo, J. Ju, J. Choe, K. H. Song and S. Lee, J. Org. Chem., 2009, 74, 6358–6361 CrossRef CAS PubMed.
- (a) D. del Moral, A. M. Banet Osuna, A. Cordoba, J. M. Moreto, J. Veciana, S. Ricart and N. Ventosa, Chem. Commun., 2009, 4723–4725 RSC; (b) D. del Moral, S. Ricart and J. M. Moreto, Chem. – Eur. J., 2010, 16, 9193–9203 CrossRef CAS PubMed.
- Y. Hoshimoto, T. Ohata, Y. Sasaoka, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2014, 136, 15877–15880 CrossRef CAS PubMed.
- J. Tjutrins, J. L. Shao, V. Yempally, A. A. Bengali and B. A. Arndtsen, Organometallics, 2015, 34, 1802–1805 CrossRef CAS.
- J. Hou, M. L. Yuan, J. H. Xie and Q. L. Zhou, Green Chem., 2016, 18, 2981–2984 RSC.
- M. C. Fu, R. Shang, W. M. Cheng and Y. Fu, ACS Catal., 2016, 6, 2501–2505 CrossRef CAS.
- H. Yu, B. Gao, B. Hu and H. Huang, Org. Lett., 2017, 19, 3520–3523 CrossRef CAS PubMed.
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- D. Vanhoye, F. Bedioui, A. Mortreux and F. Petit, Tetrahedron Lett., 1988, 29, 6441–6442 CrossRef CAS.
- K. Yoshida, E. I. Kunugita, M. Kobayashi and S. I. Amano, Tetrahedron Lett., 1989, 30, 6371–6374 CrossRef CAS.
- K. Yoshida, M. Kobayashi and S. I. Amano, J. Chem. Soc., Perkin Trans. 1, 1992, 1127–1129 RSC.
- K. Yoshida and H. Kuwata, J. Chem. Soc., Perkin Trans. 1, 1996, 1873–1877 RSC.
- M. Ocafrain, M. Devaud, M. Troupel and J. Perichon, J. Chem. Soc., Chem. Commun., 1995, 2331–2332 RSC.
- E. Dolhem, M. Oçafrain, J. Y. Nédélec and M. Troupel, Tetrahedron, 1997, 53, 17089–17096 CrossRef CAS.
- E. Dolhem, R. Barhdadi, J. C. Folest, J. Y. Nédelec and M. Troupel, Tetrahedron, 2001, 57, 525–529 CrossRef CAS.
- P. Vanalabhpatana, D. G. Peters and J. A. Karty, J. Electroanal. Chem., 2005, 580, 300–312 CrossRef CAS.
- P. Vanalabhpatana and D. G. Peters, Tetrahedron Lett., 2003, 44, 3245–3247 CrossRef CAS.
- P. Vanalabhpatana and D. G. Peters, J. Electroanal. Chem., 2006, 593, 34–42 CrossRef CAS.
- L. Gamier, Y. Rollin and J. Pkrichon, J. Org. Chem., 1989, 367, 347–358 CrossRef.
- F. W. Hartstock, L. B. McMahon and I. P. Tell, Tetrahedron Lett., 1993, 34, 8067–8070 CrossRef CAS.
- T. Murayama, T. Hayashi, Y. Arai and I. Yamanaka, Electrochim. Acta, 2011, 56, 2926–2933 CrossRef CAS.
- R. Kanega and I. Yamanaka, Top. Catal., 2014, 57, 995–999 CrossRef CAS.
- R. Kanega, H. Ogihara and I. Yamanaka, Res. Chem. Intermed., 2015, 41, 9497–9508 CrossRef CAS.
- R. Kanega, H. Ogihara and I. Yamanaka, Catal. Sci. Technol., 2016, 6, 6002–6010 CAS.
- R. Kanega, T. Hayashi and I. Yamanaka, ACS Catal., 2013, 3, 389–392 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |