
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium-catalysed σ-activation for remote meta-selective C–H functionalisation
Jamie A.
Leitch
 and
Christopher G.
Frost
*
and
Christopher G.
Frost
*
Department of Chemistry, University of Bath, Claverton Down, Somerset, BA2 7AY, UK. E-mail: c.g.frost@bath.ac.uk
First published on 17th October 2017
Abstract
The search for selective C–H functionalisation has enabled some of the most elegant techniques in modern catalysis. Herein, we review the rapidly expanding field of ruthenium catalysed σ-activation as a tool in the selective meta-C–H functionalisation of arenes.
 Jamie A. Leitch | Jamie Leitch studied chemistry at the University of Bath as an undergraduate. He completed his MChem project on developing copper catalysis for the synthesis of antibacterials. He then stayed at the University of Bath to undertake his PhD studies under the supervision of Professor Christopher G. Frost, in collaboration with Syngenta as a CASE student. His research has focused on the control of site selectivity in catalytic C–H functionalisation, with a main interest in the derivatisation of biologically interesting molecules. |
 Christopher G. Frost | Chris Frost completed his BSc(Hons) and PhD (with Jonathan M. J. Williams) at the University of Loughborough. After a postdoctoral fellowship with Prof. Philip D. Magnus at the University of Texas at Austin, he joined the Chemistry Department at the University of Bath and in 2011 was promoted to Full Professor in Organic Chemistry. Research in the Frost group involves creating new chemical tools for catalysis and biosensing. Specifically, the group are developing effective tandem or sequential catalytic processes to modify the structural and physicochemical properties of molecules. The realisation of new tandem synthetic methodology for drug discovery or new materials for recognition and amplification in molecular sensors is their goal. Current projects involve electrochemical detection of DNA, catalytic signal amplification, post-synthetic modification of oligonucleotides and exploring new concepts and selectivities in C–H activation. Frost has published over 100 papers and has presented more than 50 invited lectures at conferences, universities and companies in the UK and internationally. |
Key learning points1. Introduction to key challenges in C–H activation.2. Ruthenium-catalyzed meta-selective functionalisation achieved by ortho-cyclometalation activating remote positions. 3. Remote functionalisation often enabled by radical coupling partners. 4. Examination of the inception and key first findings in a bustling new field in C–H activation. |
I Introduction
The ability to transform inert C–H bonds into reactive functional groups has come to the forefront of modern synthetic developments in recent years. A range of elegant methods have been developed using a multitude of transition metal catalysts to enable the formation of a variety of C–X and C–C bonds.1,2 One of the major assets of C–H functionalisation is in the potential it holds for late stage functionalisation of bioactive structures in inter alia drug discovery.3The major challenge in C–H functionalisation is the ubiquity and similarity of many C–H bonds in a complex organic molecule vis-à-vis steric and electronic influences. To this end, the utilisation of a Lewis basic directing group has become commonplace in the C–H activation toolbox. Here a directing group (DG) coordinates a metal centre, which then facilitates C–H activation selectively in the ortho position to the directing group (Scheme 1a). This reduces the thermodynamic cost of C–H activation and enables the specific activation of certain C–H bonds and, most importantly, with predictability. This cyclometalated species can subsequently enable coordination of a coupling partner and reductive elimination to give the C–H functionalised product and regenerate the metal centre (Scheme 1b).
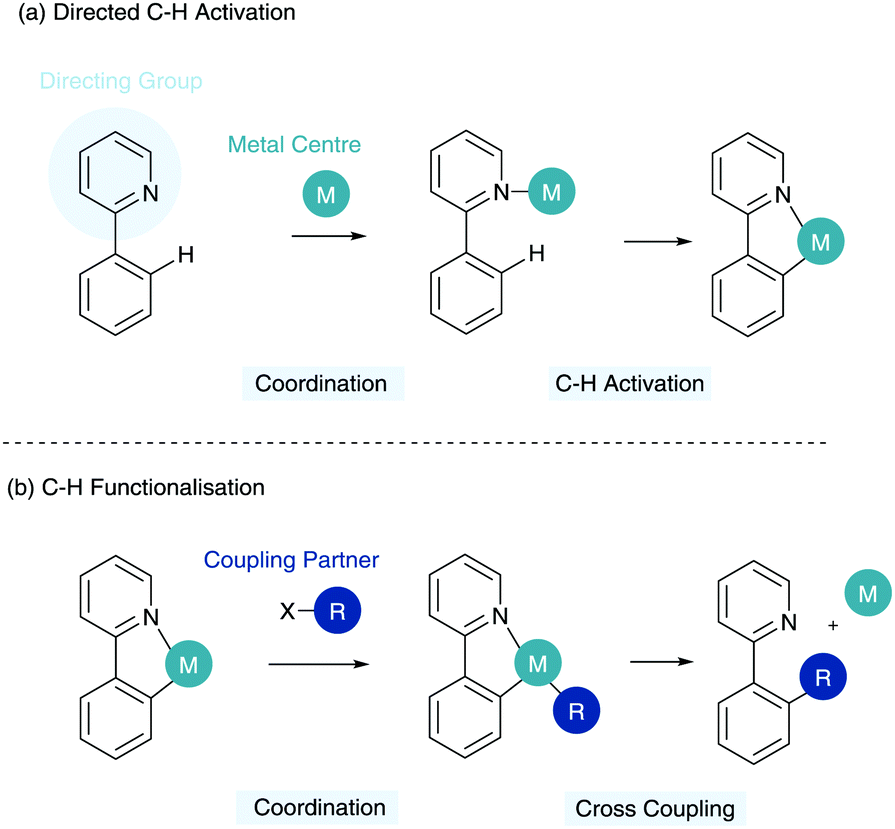 | ||
| Scheme 1 Directed C–H functionalisation methodology. | ||
This has led to an influx of catalytic systems utilising a plethora of different Lewis basic directing groups, covered in depth in a recent review by Liu and Zhang.4 However, in order to access more remote C–H bonds in an arene (meta and para-selective C–H functionalisation)5,6 specialised techniques have been developed.7,8 In the arena of meta-selective C–H functionalisation three primary techniques have been established. The first of these is the use of meticulously designed templated directing groups, pioneered by Yu (Scheme 2a).9,10 Here an organic linker “template” places a directing group proximal to the meta C–H bond. Traditional metal coordination, cyclometalation and functionalisation then occurs at the meta position. The second is the use of a transient mediator (Scheme 2b) where an ortho/ortho functionalisation pattern where the first insertion is then removed in situ.11,12 The final of these is ruthenium-catalysed σ-activation. σ-Activation benefits from the use of strong cyclometalated species as described in Scheme 1, however instead of partaking in standard oxidative addition/reductive elimination chemistry, it uses the cyclometalate to influence the ring electronically enabling para functionalisation to the metal centre. This ortho/para relationship gives a net meta-selective C–H functionalisation with respect to the directing group (Scheme 2c). This functionalisation can only take place in an ortho/ortho fashion under sterically biased substrates, however the meta-substituted structure is still formed. This tutorial review is based on the last of these techniques, and hopes to educate and excite readers about σ-activation, from its genesis at the stoichiometric organometallic level, the transfer to synthetically viable catalysis and the possibilities available for the future of this chemistry in site-selective synthesis.
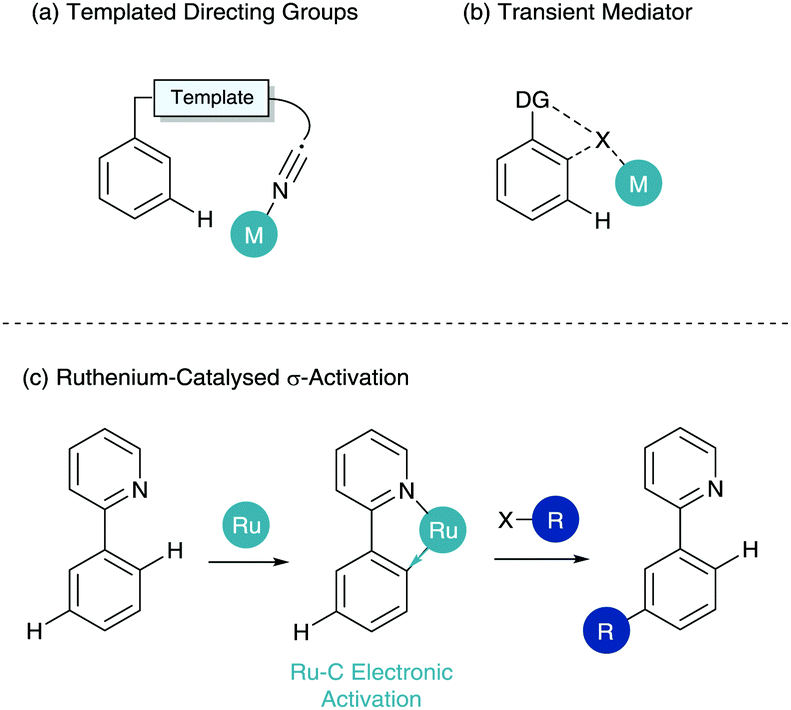 | ||
| Scheme 2 Techniques for meta-selective C–H functionalisation. | ||
II Stoichiometric work
The concept of utilising a ruthenium metal centre in this kind of methodology was first investigated by Roper in 1994, where benzene and toluene complexes of ruthenium were shown to undergo nitration para to the ruthenium centre (Scheme 3).13 The reaction methodology was shown to give rise to multiple products based on the ruthenium core, however only the most relevant structure is shown. This shows that the location of the ruthenium centre was actively dictating the regioselectivity of C–H functionalisation via electronic influence on the aromatic ring.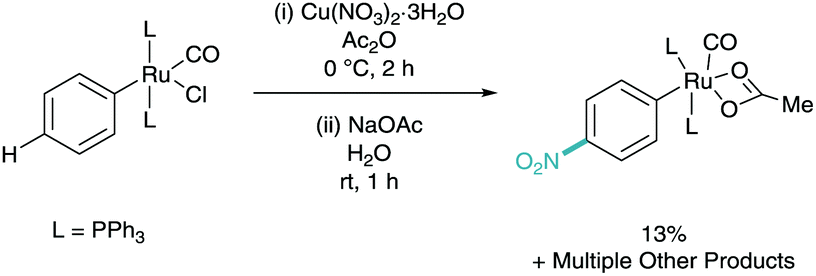 | ||
| Scheme 3 C–H halogenation of cyclometalated ruthenium complexes. | ||
In 1998 Coudret reported that phenylpyridine complexes of ruthenium underwent electrophilic C–H halogenations at the para position to the metal–carbon σ-bond in impressive yields.14 In this case N-bromosuccinimide (NBS) and iodine were used as the halogenating agents (Scheme 4). They then used these aryl halide structures in palladium catalysed cross coupling methodology.
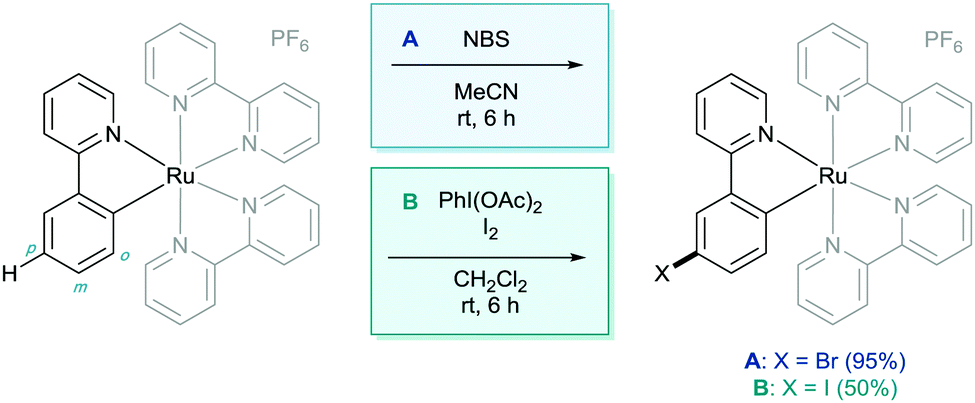 | ||
| Scheme 4 C–H halogenation of cyclometalated ruthenium complexes. | ||
This was followed the subsequent year by the work carried out by Roper and Wright. Here they found, again, that cyclometalated phenylpyridine complexes of ruthenium (and osmium) underwent electrophilic aromatic substitution (SEAr) on the phenylpyridine ligand (Scheme 5).15 They showed that under nitration conditions that the di-nitrated substrate ortho and para to the metal centre was isolated in low yields (Scheme 4A). They also demonstrated that electrophilic bromination took place in much higher yields and with selectivity solely for the para position (Scheme 4B). These two products indicate that the selectivity has been dictated by the metal centre acting as a Friedel–Crafts type ortho/para directing group, through the Ru–C σ-bond. It is this activation through a σ-bond which has since given its name to this field of catalysis.
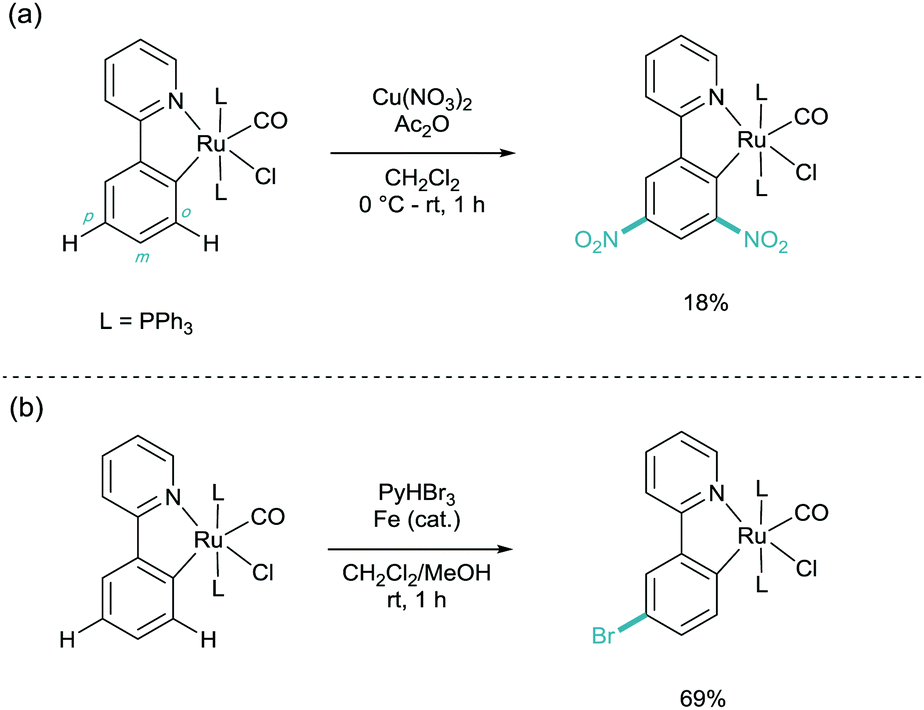 | ||
| Scheme 5 Electrophilic substitution of cyclometalated ruthenium species. | ||
III From organometallic to catalytic
This initial organometallic work and concept that σ-activation could be used as a tool in selective synthesis then lay dormant until 2011. In a report on the ortho-C–H alkylation of ketimine derivatives using primary alkyl bromides as coupling partners, Ackermann disclosed meta-substituted by-products in yields up to 7% (Scheme 6). Whilst this was not ratified as σ-activation mechanistically at the time, it has been noted as the first example of this methodology reported in the literature.16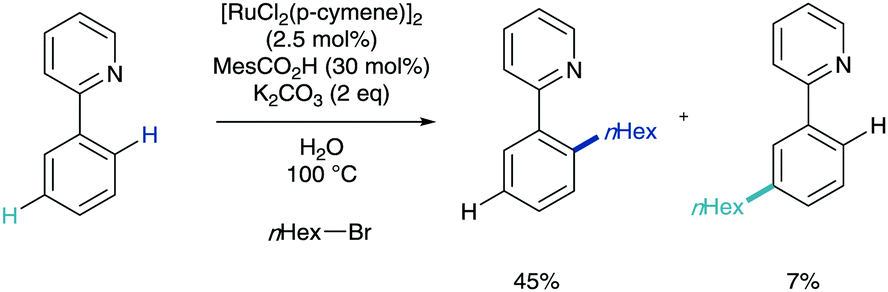 | ||
| Scheme 6 First observation of meta-C–H functionalisation in a catalytic process. | ||
Later that year, Frost disclosed the first catalytic process to enable the selective ruthenium catalysed meta-sulfonation of phenylpyridine (Scheme 7a).17 Here yields of up to 80% were reported with no ortho-selective impurities which would take place via traditional pathways. The report also contained key information about the potential mechanism at play. The ortho-cyclometalated phenylpyridine species was synthesised and submitted to the reaction conditions enabling quantitative access to the meta-sulfonated product (Scheme 7b). This ruthenacycle was also shown to be a catalytically competent organometallic in the reaction methodology. These findings, along with the initial stoichiometric work led the authors to propose that the cyclometalated species could direct SEAr C–H sulfonation para to the metal centre giving the net meta-sulfonated phenylpyridine. This presented the first synthetically useful catalytic development in σ-activation and set the stage for further investigation.
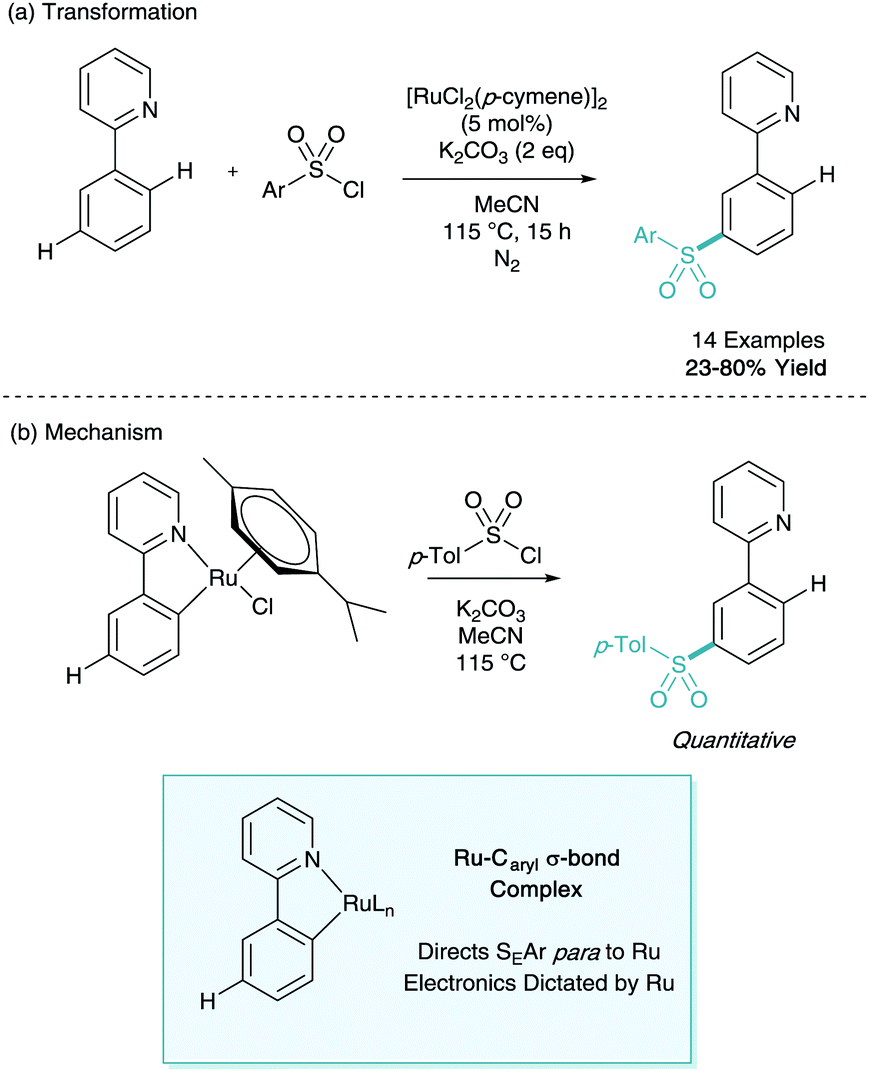 | ||
| Scheme 7 Ruthenium catalysed meta-sulfonation of phenylpyridine. | ||
This was followed in 2013 with a report from Ackermann on the meta-alkylation of phenylpyridine.18 Moving on from his previous report (Scheme 6), here he established that employing secondary alkyl halides enabled solely meta-selective C–H functionalisation in synthetically useful yields (Scheme 8a). In this report, they detailed the vital use of the sterically demanding benzoic acid, 1,4,6-trimethyl(mesityl)benzoic acid (MesCO2H). The authors also present a much wider scope of this methodology, using pyrazole, pyrimidine, benzimidazole and imidazole directing groups. This methodology was also reported to proceed via a cyclometalation, σ-activation, SEAr-type pathway and a full plausible mechanism was proposed (Scheme 8b). They also observed complete racemization of a enantiomerically enriched coupling partner which reinforced up this electrophilic proposal. In a footnote, the authors did note that the interesting find that the addition of radical scavenger TEMPO suppressed reactivity, which foreshadowed subsequent developments.
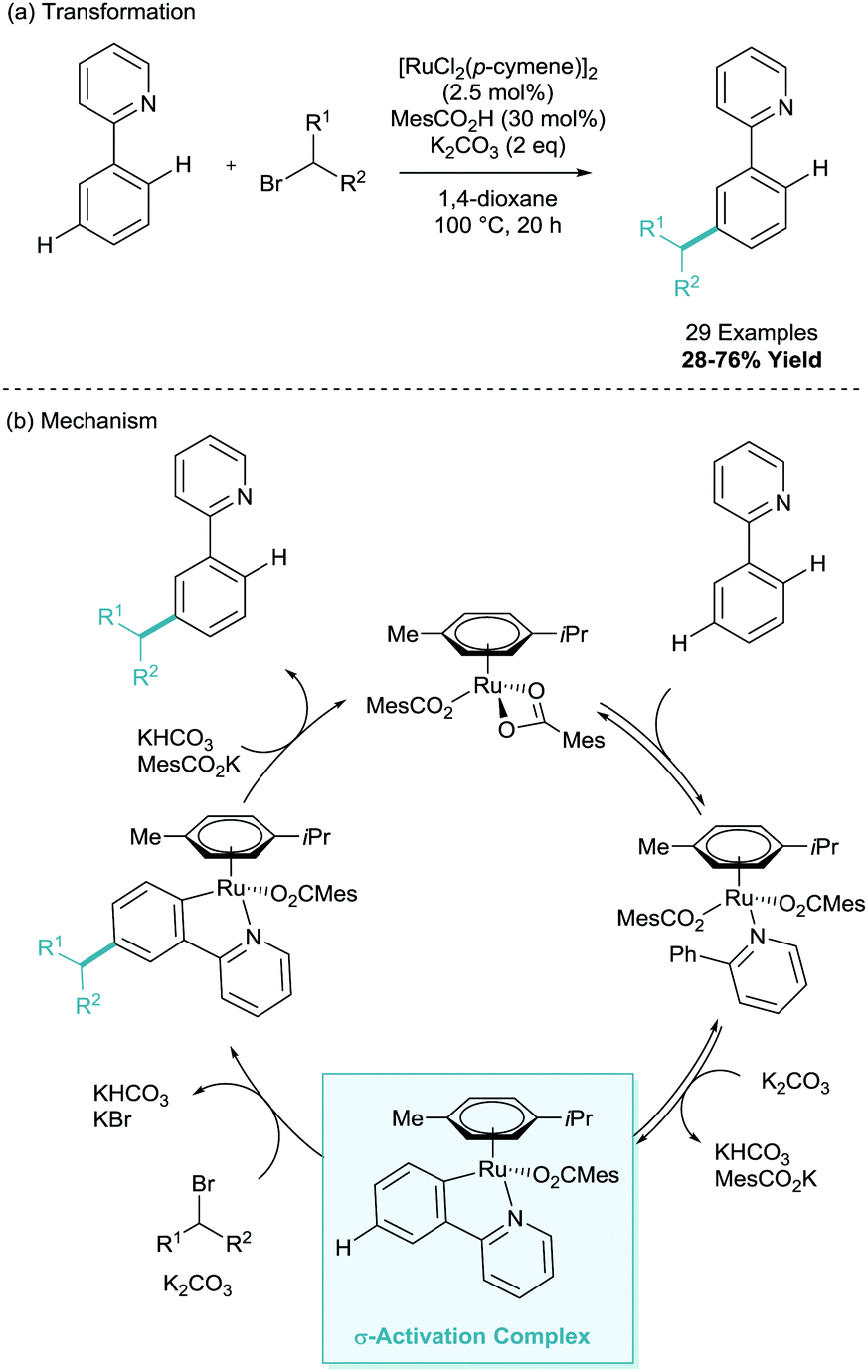 | ||
| Scheme 8 Ruthenium catalysed meta-alkylation of phenylpyridine using secondary alkyl halides. | ||
IV The radical revolution
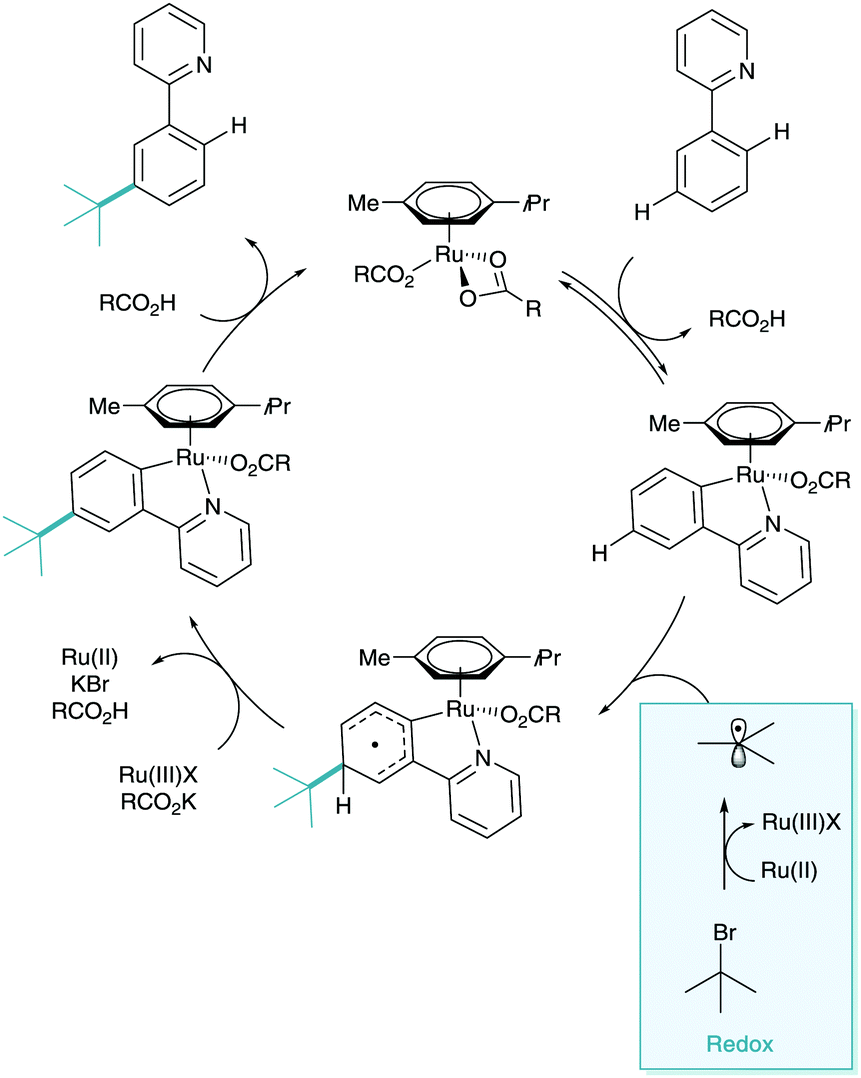
Quiet dawned upon the world of σ-activation like a simmering pot as other classic electrophiles were explored with little success.19 It wasn’t until 2015 that a full alternative radical mechanism was proposed resulting in a flood of new methodologies to the area. The groups of Frost and Ackermann simultaneously reported the meta-selective alkylation using tertiary alkyl halides (Scheme 9a).20,21 Whilst the transformation itself was inherently iterative of the meta-alkylation methodology above, the two new reports contained game-changing insights into the mechanistic nature of the functionalisation. The two groups suggested that the coupling partner interaction with the cyclometalated arene were through a radical addition rather than SEAr proposed beforehand (Scheme 9b).22 Frost evidenced this radical functionalisation using radical trapping experiments (TEMPO), by using coupling partners with a strongly disfavoured carbocation such as 1-bromoadamantane and tertiary α-halo carbonyls, and also by the isolation and characterisation of polymeric by-products. Ackermann explored radical clock experiments, racemisation, and SEAr precursor studies. Between the two reports on the same transformation, one could argue there was now unequivocal proof of a radical functionalisation. Both of the mechanisms proposed suggest a dual role ruthenium which enables both the traditional cyclometalation but also as a redox catalyst (Scheme 10). This secondary role is capable of donating an electron to the coupling partner via single electron transfer (SET), facilitating homolytic cleavage, leaving the stabilised tertiary alkyl radical. This radical then interacts with the para position of the cyclometalate. Redox rearomatisation, proton abstraction and protodemetalation gives the meta-substituted product. Both groups did not specify the nature of this “Ru(II)” species, however kinetic investigations by Ackermann suggest the reaction is second order in catalyst, suggesting a secondary outer sphere ruthenium species.23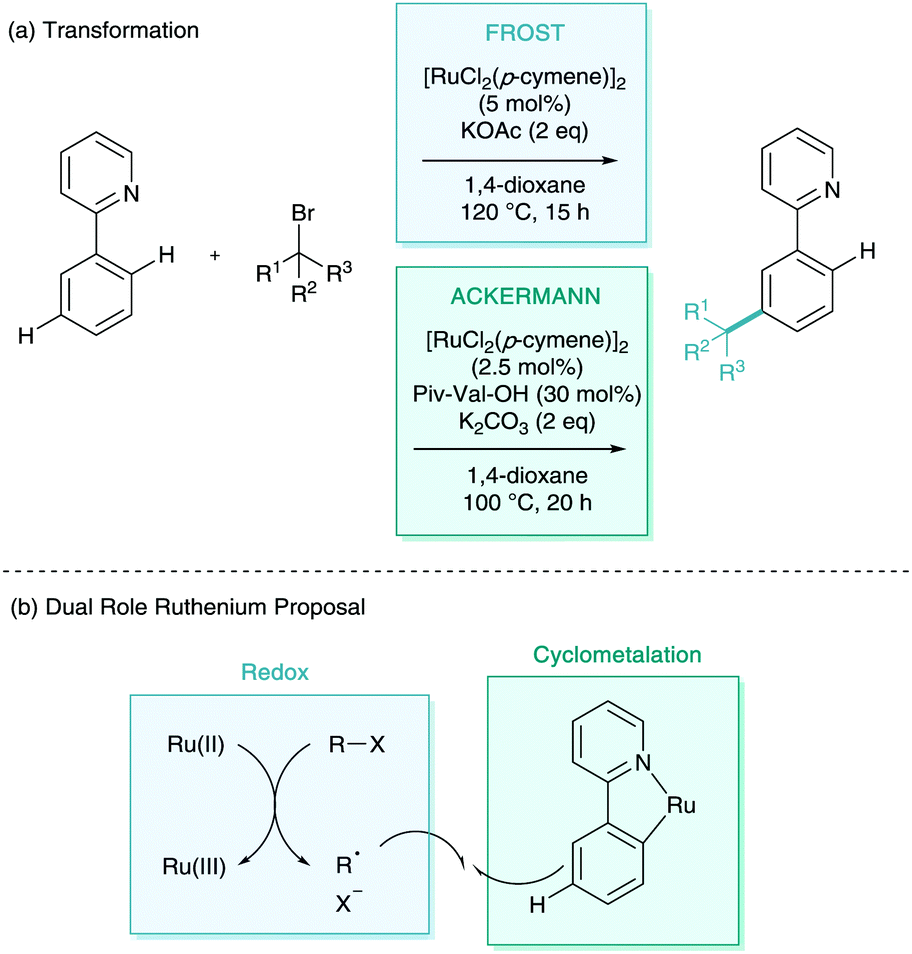 | ||
| Scheme 9 Ruthenium catalysed meta-alkylation of phenylpyridine using tertiary alkyl halides and radical proposal. | ||
 | ||
| Scheme 10 Mechanism for meta-alkylation of phenylpyridine. | ||
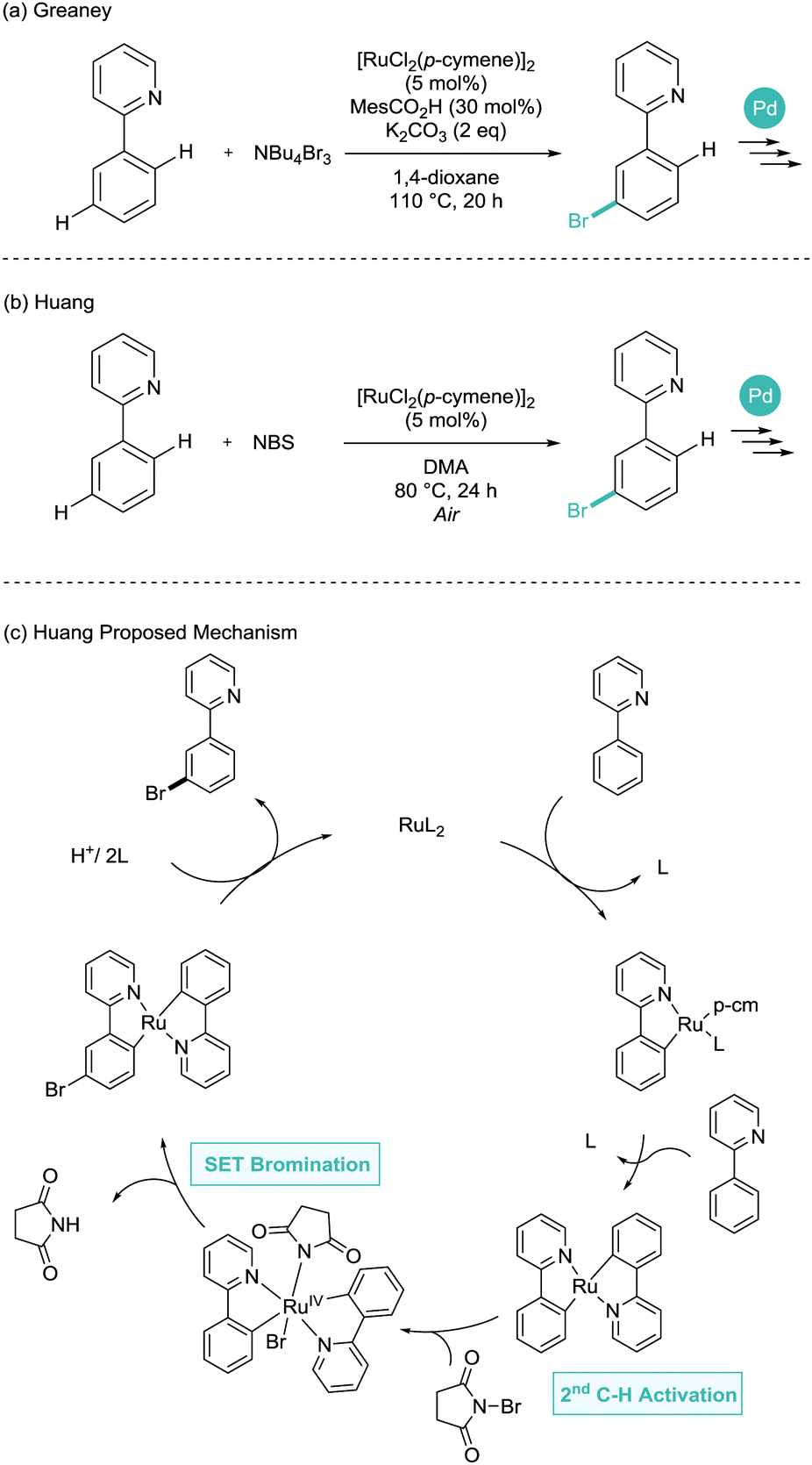
Later in 2015, both the Greaney and Huang groups proposed, again almost simultaneously, the meta-bromination of phenylpyridine derivatives.24,25 Greaney and Huang utilised tetrabutylammonium tribromide and N-bromosuccinimide as the bromine source respectively (Scheme 11). Huang suggested an SET process similar to those shown above. However, here involving a second C–H activation and with a defined inner sphere ruthenium catalyst generating bromine radicals. Despite the necessity for an air atmosphere (uncommon in σ-activation), this is not commented on with regards to the mechanism. Both reports made great use of the key benefit of installing aryl bromides, where subsequent palladium catalysed cross coupling enabled the formation of meta-arylated, borylated, alkenylated, alkylated and alkynylated arenes.
 | ||
| Scheme 11 Ruthenium catalysed meta-bromination of phenylpyridine. | ||
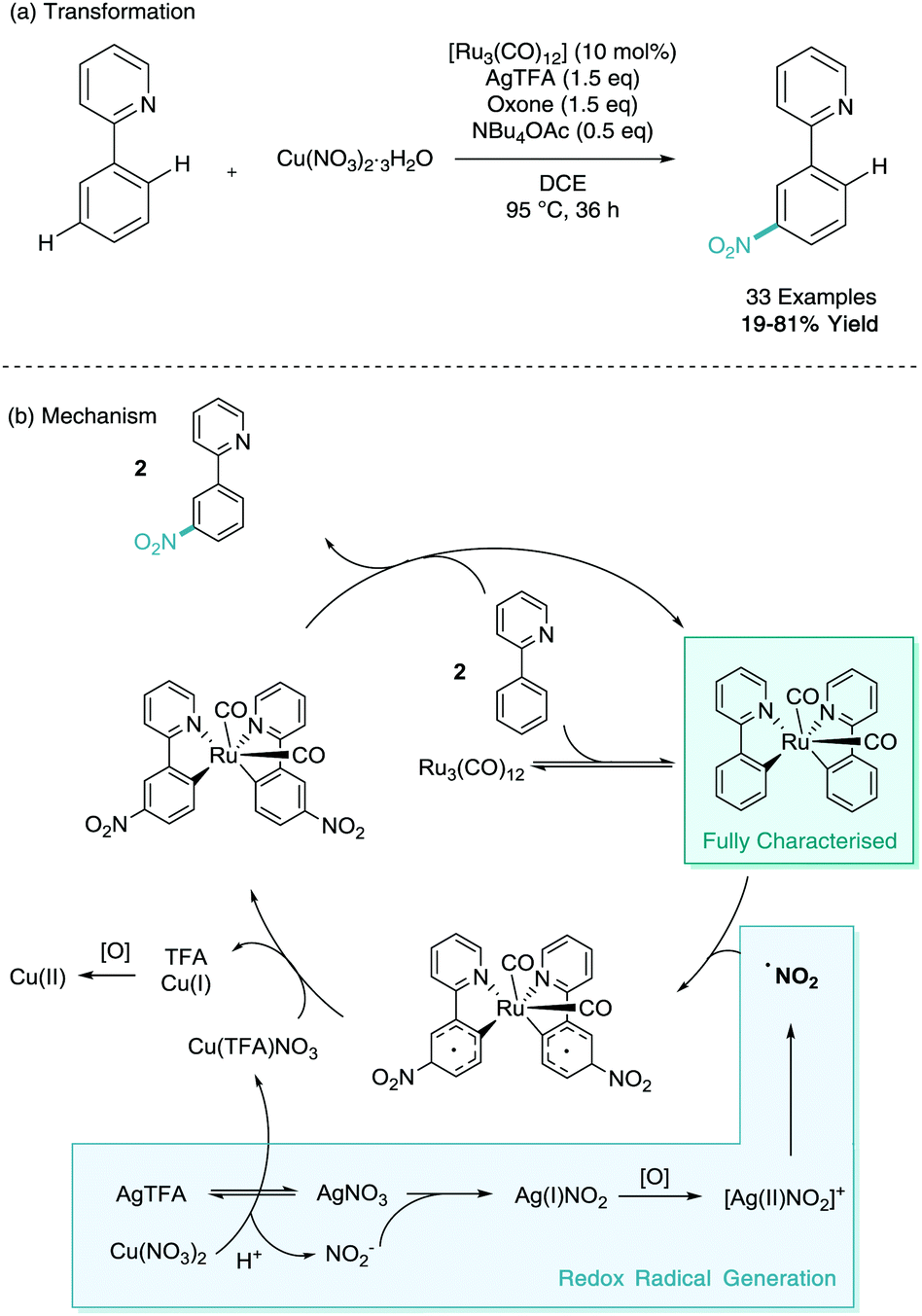
Despite a C–H nitration reaction being the first example of stoichiometric ruthenium σ-activation, it took until 2016 for ruthenium catalysed meta-selective C–H nitration to be developed.26 Here, Zhang employed [Ru3(CO)]12 as the ruthenium catalyst, as well as a silver salt (silver trifluoroacetate, AgTFA), oxidant (oxone), base (tetrabutylammonium acetate, NBu4OAc), and nitro source (copper nitrate trihydrate). Despite the cocktail of reagents, this has enabled efficient and selective meta-nitration of an array of arenes (Scheme 12). Zhang also proposed a mechanism involving two C–H activated phenylpyridine structures. In this instance this bi-pyridyl complex was isolated from the mixture, characterised via single crystal X-ray diffraction and proven to be catalytically competent in the reaction mixture. They also propose a dual copper/silver oxidation cascade which enables the formation of the nitro radical. The meta-nitrated arenes were also shown to be widely diversifiable in a variety of post-synthetic functional group interconversions (FGI).
 | ||
| Scheme 12 Ruthenium(0)-catalysed meta-nitration of phenylpyridine. | ||
Following these mechanistic insights, Frost also reported a revised radical mechanism in 2016 for the initial meta-sulfonation methodology. This was proposed to undergo a similar mechanism where SET enables formation of the tosyl radical, after similar mechanistic results were discovered with that methodology.27
V Recent advances
The mechanistic foundations had now been set for σ-activation methodology, and these insights inspired a catalogue of different applications building on the established transformations. The following part of this review is split into three subsections entailing the expansion of this technique: structural template expansion, new C–X bond formations, and catalyst development.V.I Structural template expansion
One of the key drawbacks in σ-activation, which the reader may have identified, is the over reliance on phenylpyridine. Despite being an excellent model substrate employed by multiple groups, phenylpyridine has limited biological activity and limited scope for further derivation. To expand the scope of this synthetic technique, an auxiliary approach can be used. Here one can furnish a motif with a directing group, carry out the remote functionalisation, and then cleave the directing group to reveal the more useful functionality.This technique was first employed by Ackermann in his 2015 tertiary alkylation report. Here they use N-pyrimidinyl aniline as a model substrate in the meta-alkylation methodology (Scheme 13a).21 After acidic cleavage of this directing group in aqueous media, this now led to the meta-functionalised aniline. It should be noted that the regioselectivity of functionalisation is complementary to the natural ortho/para directing selectivity of an aniline. The same group also expanded on this in a recent report using ketimine directing groups, which when treated with aqueous acid after functionalisation, reveals the acetophenone (Scheme 13b).28 They reported a vast scope of 40 examples of different structures utilising both secondary and tertiary alkyl halide coupling partners. They also demonstrated a vast array of post-synthetic modifications of the ketimine group to give anilines, phenols, indoles and benzylamines, as well as in situ ortho-C–H functionalisation to give di-substituted arenes.
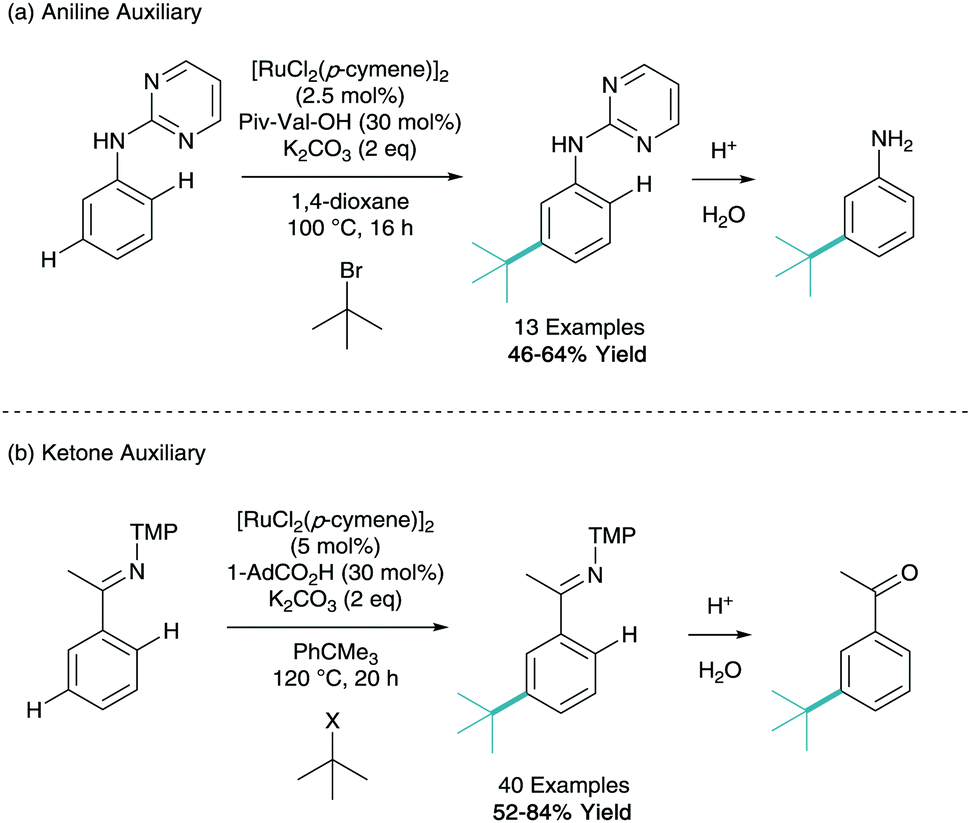 | ||
| Scheme 13 Use of cleavable auxiliaries in meta-selective C–H functionalisation. TMP = 3,4,5-trimethoxyphenyl. X = Cl, Br or I. | ||
Following a similar concept of creating the meta substituted aniline building block, Li has reported the meta-alkylation and sulfonation of diazobenzenes (Scheme 14a).29,30 Both methodologies were shown to be selective for functionalisation on only one of the aryl rings and using catalytic iron powder in acetic acid, the diazo could be transformed into the free aniline. The same group have also demonstrated the meta-alkylation of phenol furnished with a pyridine directing group (Scheme 14b). This can subsequently be removed under forcing conditions to give the meta-alkyl phenol.31
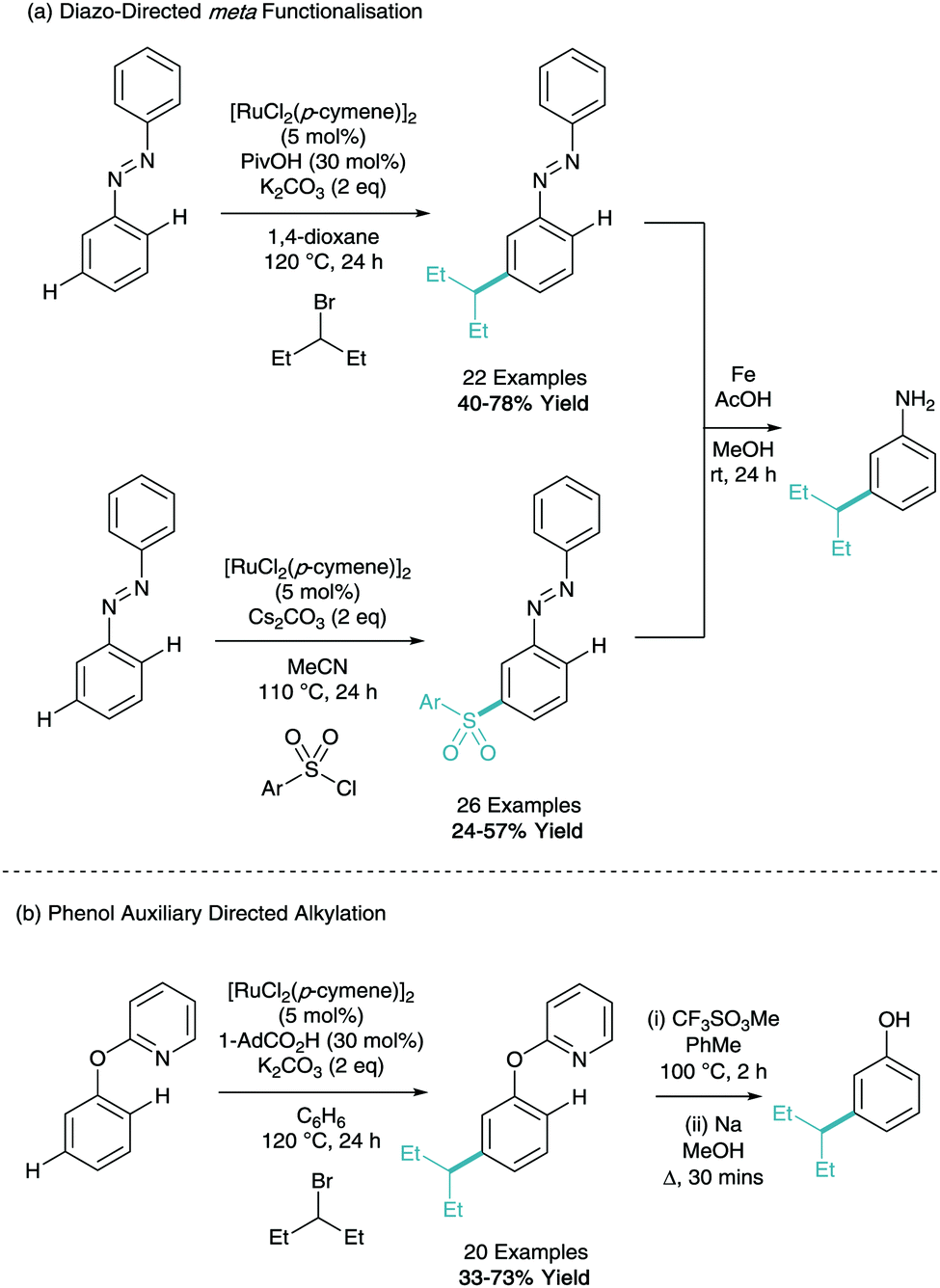 | ||
| Scheme 14 Ruthenium catalysed meta-functionalisation of aniline and phenol auxiliaries. | ||
C–H activation is a powerful tool that can be employed to derivatise biologically relevant structures32 such as indoles. Frost reported the remote C–H alkylation of indole derivatives in 2017 (Scheme 15).33 The difficulty of selectively accessing the benzenoid ring (C4–7) of an indole has been highlighted in a recent review.34 This functionalisation was shown to utilise a primary directing group at N1 and an ancillary directing group at C3 to enable remote C6 alkylation on the benzenoid ring. This methodology also benefited from the first input from computational chemistry in σ-activation, where calculated Fukui indices on organic and inorganic structures helped elucidate that cyclometalation at C2 would in turn place electron density on the C6 position.
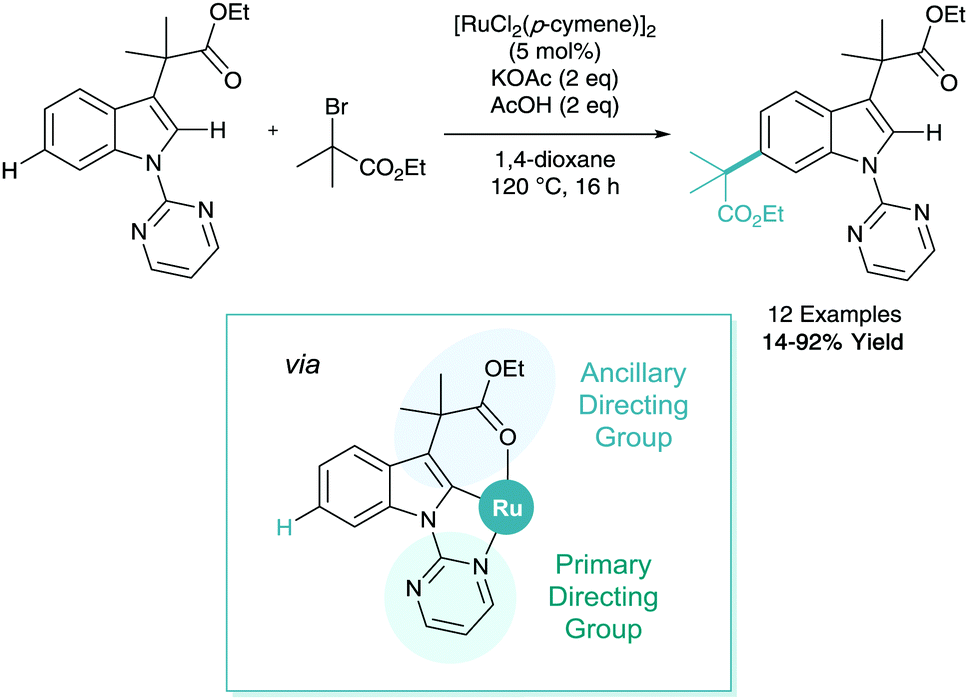 | ||
| Scheme 15 Remote C6-selective ruthenium catalysed C–H alkylation of indole derivatives. | ||
As a follow up to their work on the meta-nitration of phenylpyridines (and related heteroaromatics), Zhang reported a further iteration of this methodology in 2017.35 Here, they focused on the use of the oxime directing group as a removal auxiliary. In this process, they also reported a variation on their catalytic system to utilise silver nitrate as the nitro source and O2 as a co-oxidant (Scheme 16).
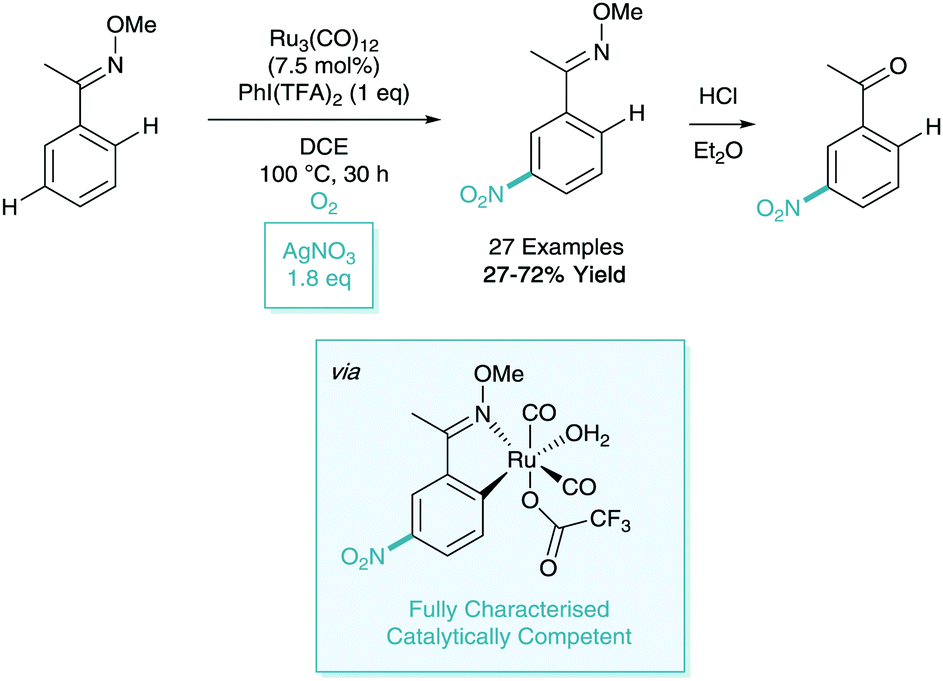 | ||
| Scheme 16 Ruthenium(0) catalysed meta-nitration of oximes. | ||
V.II New C–X bond formations
One of the other avenues which is necessary to develop σ-activation is the search for new C–X or C–C bond formation reactions to expand the arsenal of transformations available.In early 2017, both Ackermann and Wang reported the meta-selective C–H difluoroalkylation and monofluoroalkylation of heteroarenes (Scheme 17).36,37 Ackermann proposed a triarylphosphine additive as a co-catalyst, along with a wide scope of arenes, directed by pyridine, pyrimidine, pyrazole, and purine directing groups (Scheme 17a). A radical process was also suggested although the exact role of the nature of phosphine in the mechanism was not presented. Despite this, they did demonstrate both carboxylic acid (MesCO2H) and phosphine co-catalysts were vital to catalysis. Wang's report employs a palladium(0) co-catalyst (Pd(PPh3)4) in the same transformation. They propose a Pd(0)/Pd(I) redox cycle for the generation of the difluoroalkyl radicals (Scheme 17b). Zhao has also since proposed a ruthenium catalysed meta-difluoroalkylation of similar substrates with an alternative additive of silver triflimide (AgNTf2) and, surprisingly, in many cases without an additive at all.38
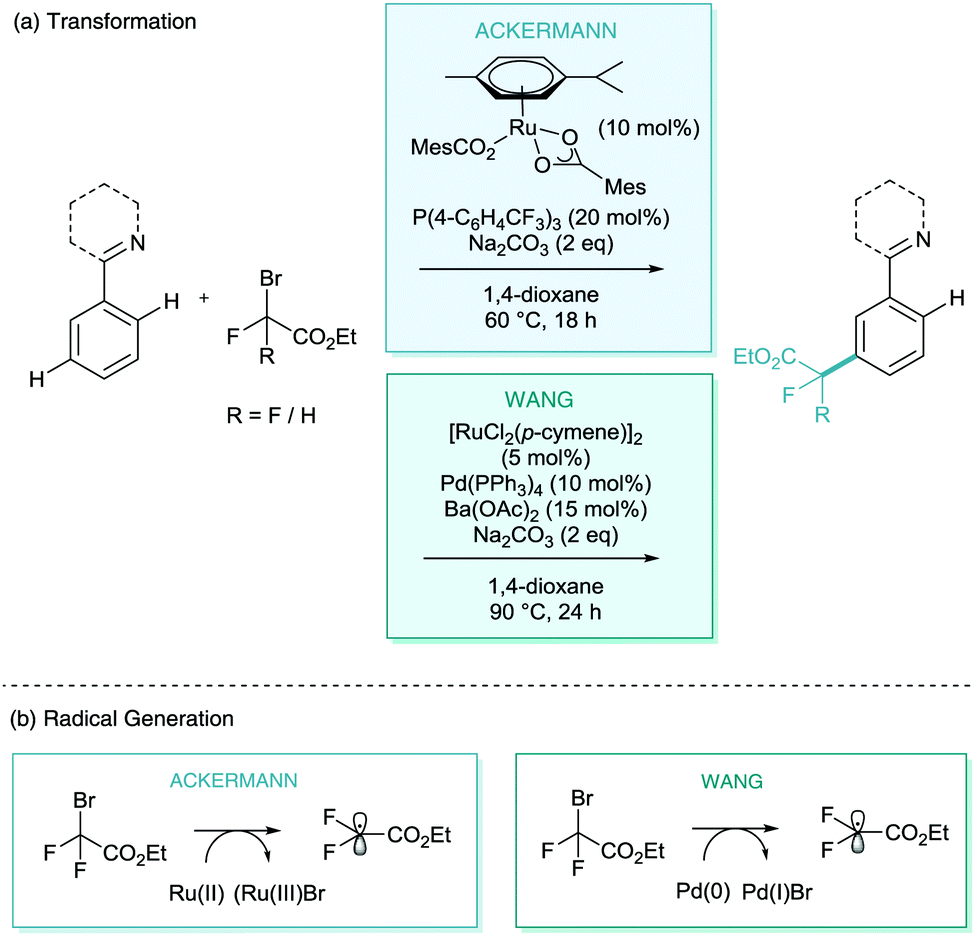 | ||
| Scheme 17 Ruthenium catalysed meta difluoro- and monofluoroalkylation of heteroarenes. | ||
Since Ackermann's first observation of a meta-C–H alkylation using primary alkyl halides in small quantities, σ-activation methodology has utilised secondary and tertiary alkyl halides much more frequently as they are less likely to undergo tradition oxidative addition/reductive elimination pathways. In 2017, Frost reported the meta-alkylation of phenylpyridine derivatives utilising primary α-halocarbonyls. They found that similar co-catalysts as above (triarylphosphines and Pd(PPh3)4) drive the reaction towards meta-selectivity over competing ortho-selectivity (Scheme 18). A majority of the examples led to exclusive meta-selectivity, however with some substituted arenes some selectivity issues did remain.39
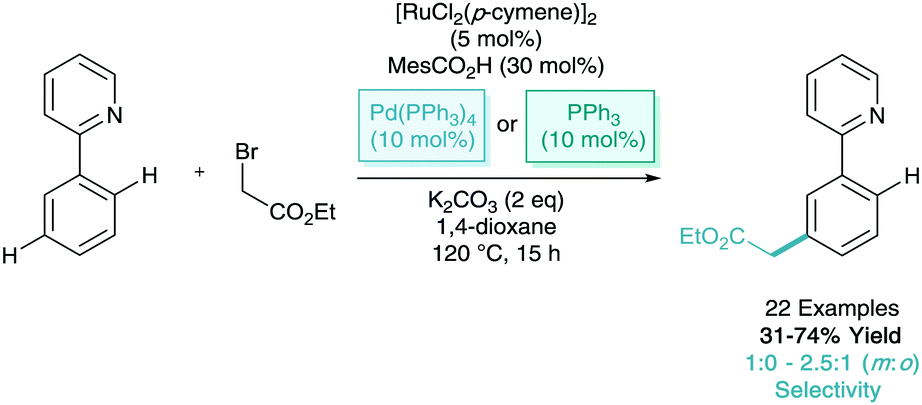 | ||
| Scheme 18 Ruthenium catalysed alkylation of phenylpyridine with primary alkyl halides. | ||
In early 2017, Shi and Zhao reported the meta-benzylation of phenylpyridine.40 This was achieved using RuCl3 as the ruthenium source, ferrocene and di-tert-butyl peroxide (DTBP) in radical generation, toluenes as benzyl precursors, and a binol derived phosphate ligand (L1, Scheme 19a). The use of bulky phosphate was shown to suppress competing ortho-functionalisation. Through in-depth investigation into the role of the additives they proposed a mechanism (Scheme 19b), they proposed the formation of a di-phenylpyridine structure similar to that reported by Zhang,26 however with the bulky phosphate ligand bound. They then suggest that through SET from ferrocene and hydrogen abstraction from DTBP give the tolyl radical. This radical then interacts with the cyclometalated phenylpyridine at the para position to the metal, redox rearomatisation with Fe(III) and proton abstraction, and finally protodemetalation gives the meta-benzylated substrate.41
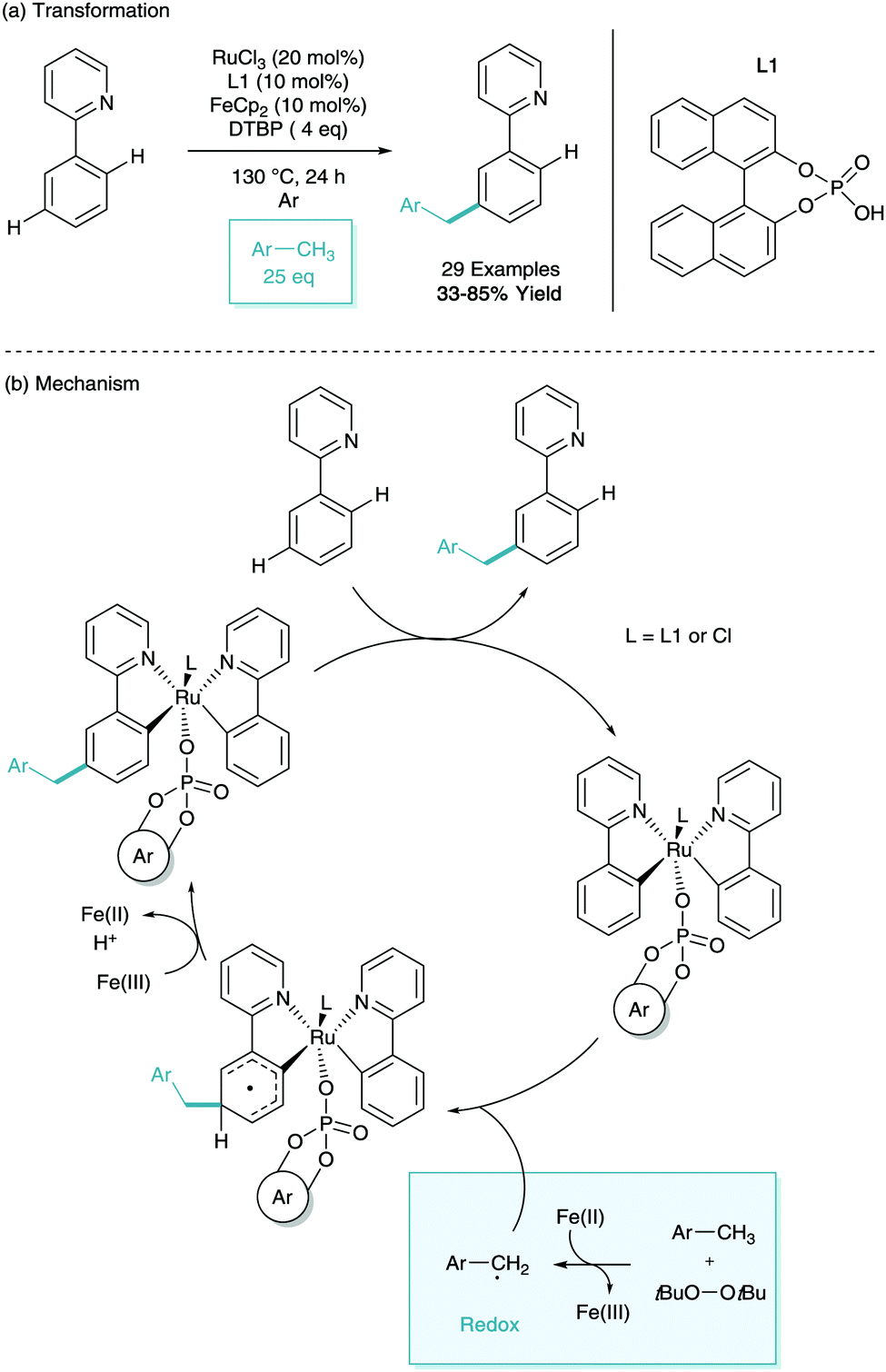 | ||
| Scheme 19 Ruthenium catalysed meta benzylation of phenylpyridine. | ||
The methodologies described in this subsection not only permit the introduction of different C–C bonds but also allow the use of varying redox active metals, such as iron and silver, and more importantly show they are compatible with the σ-activation system. These developments set the stage for further investigation into redox partners.
V.III Catalyst development
As shown throughout this tutorial review, catalytic σ-activation relies on the use of the platinum-group transition metal, ruthenium. The use of high loadings (<30 mol% Ru by weight) is also a common feature of many processes. Due to this there will be should be a focus in the coming years on performing these methodologies in a more sustainable manner.In early 2017, Ackermann was the first (and to date, only) to tackle this problem.42 Here they adapted the meta-bromination methodology developed by Huang and set out to investigate the use of a recyclable heterogeneous ruthenium catalyst (Scheme 20). They showed that the use of a user-friendly silica-derived catalysts Ru@SiO2 outperformed its homogeneous counterparts. The substrate of focus of this report also demonstrated the first use of a purine directing in σ-activation methodology. The catalyst was shown to be amenable to recycling, and was used as many as seven times with an impressive yield drop of less than 20%.
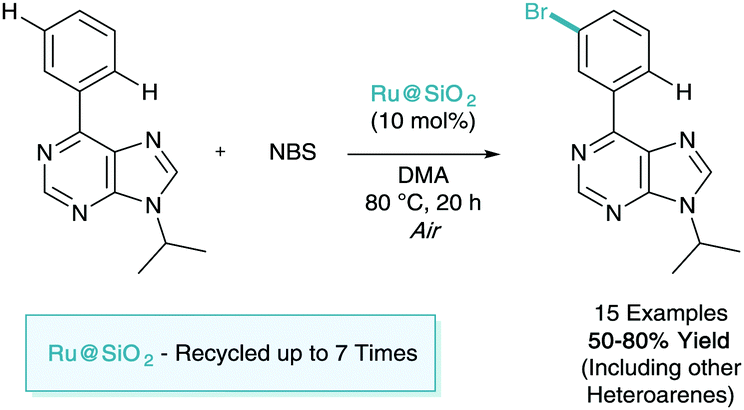 | ||
| Scheme 20 meta-Bromination using a recyclable heterogeneous ruthenium catalyst. | ||
VI Conclusions
Ruthenium catalysed σ-activation has been shown as a key tool in accessing remote meta-selective functionalisation, moving from an organometallic to catalytic synthetic methodology. We have seen the radical revolution which has enabled the development of many new methodologies seen post-2015 in this field. We have seen that either sterically crowded coupling partners or ligand sets have suppressed traditional ortho-functionalisation pathways and promoted complementary meta-functionalisation. These insights will only lead to a stronger influx of reports in this field. As there have already been as many methodologies published in 2017 as the previous six years combined, the stage is now set for ruthenium-catalyzed σ-activation to move from proof of concept research to a broadly applicable methodology in the synthetic toolbox. We now stare into the horizon of ruthenium catalysed σ-activation where we expect to see the continuing rapid expansion in scaffolds, new C–C/C–X bond forming processes and well-defined bespoke catalysts. These will no doubt be applied in the late stage functionalisation of biologically relevant molecules, in drug development, and in the creation of novel building blocks.Conflicts of interest
There are no conflicts to declare.Notes and references
- L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed.
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed.
- J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed.
- Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC.
- A. Dey, S. Agasti and D. Maiti, Org. Biomol. Chem., 2016, 14, 5440 CAS and references therein.
- A. Dey, S. Maity and D. Maiti, Chem. Commun., 2016, 52, 12398 RSC and references therein.
- J. Schrank, A. Tlili and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 9426 CrossRef PubMed.
- J. Li, S. De Sarkar and L. Ackermann, Top. Organomet. Chem., 2016, 55, 217 CrossRef.
- D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 416, 518 CrossRef PubMed and citing references.
- S. Das, C. D. Incarvito, R. H. Crabtree and G. W. Brudwig, Science, 2006, 312, 1941 CrossRef CAS PubMed.
- X.-C. Wang, W. Gong, L.-Z. Fang, R.-Y. Zhu, S. Li, K. M. Engle and J.-Q. Yu, Nature, 2015, 519, 334 CrossRef CAS PubMed and citing references.
- Z. Dong, J. Wang and G. Dong, J. Am. Chem. Soc., 2015, 137, 5887 CrossRef CAS PubMed.
- G. R. Clark, C. E. L. Headford, W. R. Roper, L. J. Wright and V. P. D. Yap, Inorg. Chim. Acta, 1994, 220, 261 CrossRef CAS.
- C. Coudret, S. Frayasse and J.-P. Launay, Chem. Commun., 1998, 663 RSC.
- A. M. Clark, C. E. F. Rickard, W. R. Roper and L. J. Wright, Organometallics, 1999, 18, 2813 CrossRef CAS.
- L. Ackermann, N. Hofmann and R. Vicente, Org. Lett., 2011, 13, 1875 CrossRef CAS PubMed.
- O. Saidi, J. Marafie, A. E. W. Ledger, P. M. Liu, M. F. Mahon, G. Kociok-Kohn, M. K. Whittlesey and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298 CrossRef CAS PubMed.
- N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877 CrossRef CAS PubMed.
- P. M. Liu and C. G. Frost, Org. Lett., 2013, 15, 5862 CrossRef CAS PubMed.
- A. J. Paterson, S. St John-Campbell, M. F. Mahon, N. J. Press and C. G. Frost, Chem. Commun., 2015, 51, 12807 RSC.
- J. Li, S. Warratz, D. Zell, S. De Sarkar, E. E. Ishikawa and L. Ackermann, J. Am. Chem. Soc., 2015, 137, 13894 CrossRef CAS PubMed.
- G. B. Boursalian, W. S. Ham, A. R. Mazzotti and T. Ritter, Nat. Chem., 2016, 8, 810 CrossRef CAS PubMed and references therein.
- Ackermann and co-workers have since proposed a first order dependence on ruthenium catalyst in ref. 28.
- C. J. Teskey, A. Y. W. Lui and M. F. Greaney, Angew. Chem., Int. Ed., 2015, 54, 11677 CrossRef CAS PubMed.
- Q. Yu, L. Hu, Y. Wang, S. Zheng and J. Huang, Angew. Chem., Int. Ed., 2015, 54, 15284 CrossRef CAS PubMed.
- Z. Fan, J. Ni and A. Zhang, J. Am. Chem. Soc., 2016, 138, 8470 CrossRef CAS PubMed.
- P. Marcé, A. J. Paterson, M. F. Mahon and C. G. Frost, Catal. Sci. Technol., 2016, 6, 7068 Search PubMed.
- J. Li, K. Korvorapun, S. De Sarkar, T. Rogge, D. J. Burns, S. Warratz and L. Ackermann, Nat. Commun., 2017, 8, 15430 CrossRef PubMed.
- G. Li, X. Ma, C. Jia, Q. Han, Y. Wang, J. Wang, L. Yu and S. Yang, Chem. Commun., 2017, 53, 1261 RSC.
- G. Li, X. Lv, K. Gao, Y. Wang, S. Yang, L. Yu, Y. Yu and J. Wang, Org. Chem. Front., 2017, 4, 1145 RSC.
- G. Li, P. Gao, X. Lv, C. Qu, Q. Yan, Y. Wang, S. Yang and J. Wang, Org. Lett., 2017, 19, 2682 CrossRef CAS PubMed.
- J. A. Leitch, P. B. Wilson, C. L. McMullin, M. F. Mahon, Y. Bhonoah, I. H. Williams and C. G. Frost, ACS Catal., 2016, 6, 5520 CrossRef CAS and references therein.
- J. A. Leitch, C. L. McMullin, M. F. Mahon, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 2616 CrossRef CAS.
- J. A. Leitch, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 5618 CrossRef CAS.
- Z. Fan, J. Li, H. Lu, D.-Y. Wang, C. Wang, M. Uchiyama and A. Zhang, Org. Lett., 2017, 19, 3199 CrossRef CAS PubMed.
- Z. Ruan, S.-K. Zhang, C. Zhu, P. N. Ruth, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 2045 CrossRef CAS PubMed.
- Z.-Y. Li, L. Li, Q.-L. Li, K. Jing, H. Xu and G.-W. Wang, Chem. – Eur. J., 2017, 23, 3285 CrossRef CAS PubMed.
- C. C. Yuan, X. L. Chen, J. Y. Zhang and Y. S. Zhao, Org. Chem. Front., 2017, 4, 1867 RSC.
- A. J. Paterson, C. J. Heron, C. L. McMullin, M. F. Mahon, N. J. Press and C. G. Frost, Org. Biomol. Chem., 2017, 15, 5993 CAS.
- G. Li, D. Li, J. Zhang, D.-Q. Shi and Y. Zhao, ACS Catal., 2017, 7, 4138 CrossRef CAS.
- Prior to submission of this manuscript, a second meta-benzylation was published: B. Li, S.-L. Fang, D.-Y. Huang and B.-F. Shi, Org. Lett., 2017, 19, 3950 CrossRef CAS PubMed.
- S. Warratz, D. J. Burns, C. Zhu, K. Korvorapun, T. Rogge, J. Scholz, C. Jooss, D. Gelman and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 1557 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |