
Structure-dependent reactivity of Criegee intermediates studied with spectroscopic methods
 *ab
and
Wen
Chao
ab
*ab
and
Wen
Chao
ab
Abstract
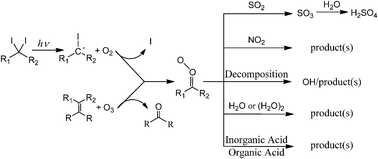
Criegee intermediates are very reactive carbonyl oxides that are formed in reactions of unsaturated hydrocarbons with ozone (ozonolysis). Recently, Criegee intermediates have gained significant attention since a new preparation method has been reported in 2012, which employs the reaction of iodoalkyl radical with molecular oxygen: for instance, CH2I + O2 → CH2OO + I. This new synthesis route can produce Criegee intermediates with a high number density, which allows direct detection of the Criegee intermediate via various spectroscopic tools, including vacuum UV photoionization mass spectrometry, absorption and action spectroscopy in the UV and IR regions, and microwave spectroscopy. Criegee intermediates have been thought to play important roles in atmospheric chemistry, such as in OH radical formation as well as oxidation of atmospheric gases such as SO2, NO2, volatile organic compounds, organic and inorganic acids, and even water. These reactions are relevant to acid rain and aerosol formation. Kinetics data including rate coefficients, product yields and their temperature and pressure dependences are important for understanding and modeling relevant atmospheric chemistry. In fundamental physical chemistry, Criegee intermediates have unique and interesting features, which have been partially revealed through spectroscopic, kinetic, and dynamic investigations. Although previous review articles have discussed Criegee intermediates, new data and knowledge on Criegee intermediates are still being accumulated. In this tutorial review, we have focused on structure-dependent reactivity of Criegee intermediates and various spectroscopic tools that have been utilized to probe the kinetics of Criegee intermediates.
- This article is part of the themed collection: Chemical reaction dynamics
 Please wait while we load your content...
Please wait while we load your content...

