
Dissipative out-of-equilibrium assembly of man-made supramolecular materials

Abstract
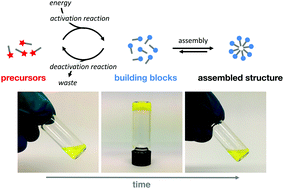
The use of dissipative self-assembly driven by chemical reaction networks for the creation of unique structures is gaining in popularity. In dissipative self-assembly, precursors are converted into self-assembling building blocks by the conversion of a source of energy, typically a photon or a fuel molecule. The self-assembling building block is intrinsically unstable and spontaneously reverts to its original precursor, thus giving the building block a limited lifetime. As a result, its presence is kinetically controlled, which gives the associated supramolecular material unique properties. For instance, formation and properties of these materials can be controlled over space and time by the kinetics of the coupled reaction network, they are autonomously self-healing and they are highly adaptive to small changes in their environment. By means of an example of a biological dissipative self-assembled material, the unique concepts at the basis of these supramolecular materials will be discussed. We then review recent efforts towards man-made dissipative assembly of structures and how their unique material properties have been characterized. In order to help further the field, we close with loosely defined design rules that are at the basis of the discussed examples.
- This article is part of the themed collection: Chemical systems out of equilibrium
 Please wait while we load your content...
Please wait while we load your content...

