
Direct growth of graphene on rigid and flexible substrates: progress, applications, and challenges
 *abc
*abc
Abstract
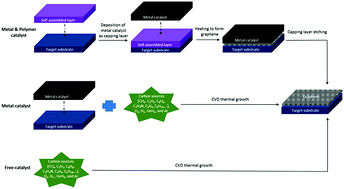
Graphene has recently been attracting considerable interest because of its exceptional conductivity, mechanical strength, thermal stability, etc. Graphene-based devices exhibit high potential for applications in electronics, optoelectronics, and energy harvesting. In this paper, we review various growth strategies including metal-catalyzed transfer-free growth and direct-growth of graphene on flexible and rigid insulating substrates which are “major issues” for avoiding the complicated transfer processes that cause graphene defects, residues, tears and performance degradation in graphene-based functional devices. Recent advances in practical applications based on “direct-grown graphene” are discussed. Finally, several important directions, challenges and perspectives in the commercialization of ‘direct growth of graphene’ are also discussed and addressed.
- This article is part of the themed collection: Graphene Chemistry
 Please wait while we load your content...
Please wait while we load your content...

