
Coordination change, lability and hemilability in metal–organic frameworks
 *a
and
Lee
Brammer
*b
*a
and
Lee
Brammer
*b
Abstract
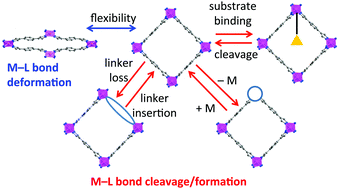
Metal–organic frameworks (MOFs) are some of the most exciting materials in current science. Their utility and diversity of applications depends on a combination of their chemistry, their framework topology and the spatial dimensions of their pores. In this review we concentrate on the chemistry of MOFs. Specifically we bring together many aspects of MOFs that underpin their stability, reactivity and dynamic behaviour within a common theme related to (changes in) metal–ligand bonding. In each area we provide examples to illustrate the behaviour and discuss it in the context of metal lability and coordination changes. Starting with flexible behaviour in which metal–linker bonds undergo deformation rather than cleavage, we then consider coordination changes that lead to open metal sites, changes in framework topology, framework dimensionality or degree of network interpenetration. We show how these changes are linked to development of new properties, including changes in magnetic behaviour, gas adsorption characteristics, construction of composite MOFs and amorphous MOFs, as well as providing new synthetic routes for MOF preparation. We discuss how the lability of the species that make up the MOFs can affect aspects from their synthesis to the possibility of metal and linker exchange reactions that may lead to defects and disorder. The final section reviews hemilability in MOFs, where regions of different chemical behaviour within MOFs can lead to unusual properties, such as self-accelerating and ultraselective adsorption.
- This article is part of the themed collection: Metal-organic frameworks and porous polymers – current and future challenges
 Please wait while we load your content...
Please wait while we load your content...

