
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inverse electron demand Diels–Alder reactions in chemical biology
B. L.
Oliveira†
*a,
Z.
Guo†
a and
G. J. L.
Bernardes
 *ab
*ab
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: bljp2@cam.ac.uk; gb453@cam.ac.uk
bInstituto de Medicina Molecular, Faculdade de Medicina, Universidade de Lisboa, Avenida Professor Egas Moniz, Lisboa, 1649-028, Portugal. E-mail: gbernardes@medicina.ulisboa.pt
First published on 29th June 2017
Abstract
The emerging inverse electron demand Diels–Alder (IEDDA) reaction stands out from other bioorthogonal reactions by virtue of its unmatchable kinetics, excellent orthogonality and biocompatibility. With the recent discovery of novel dienophiles and optimal tetrazine coupling partners, attention has now been turned to the use of IEDDA approaches in basic biology, imaging and therapeutics. Here we review this bioorthogonal reaction and its promising applications for live cell and animal studies. We first discuss the key factors that contribute to the fast IEDDA kinetics and describe the most recent advances in the synthesis of tetrazine and dienophile coupling partners. Both coupling partners have been incorporated into proteins for tracking and imaging by use of fluorogenic tetrazines that become strongly fluorescent upon reaction. Selected notable examples of such applications are presented. The exceptional fast kinetics of this catalyst-free reaction, even using low concentrations of coupling partners, make it amenable for in vivo radiolabelling using pretargeting methodologies, which are also discussed. Finally, IEDDA reactions have recently found use in bioorthogonal decaging to activate proteins or drugs in gain-of-function strategies. We conclude by showing applications of the IEDDA reaction in the construction of biomaterials that are used for drug delivery and multimodal imaging, among others. The use and utility of the IEDDA reaction is interdisciplinary and promises to revolutionize chemical biology, radiochemistry and materials science.
 B. L. Oliveira | Dr Bruno Oliveira received his PhD from the University of Lisbon in 2012 under the guidance of Profs João Galamba and Isabel Santos. His PhD work involved collaborative visits to the groups of Prof. Roger Alberto (University of Zurich, Switzerland), Prof. Maria João Ramos (University of Porto, Portugal) and Prof. Francisco Blanco (CNIO, Spain). In 2013 he moved to Boston, USA to work with Prof. Peter Caravan (Massachusetts General Hospital/Harvard Medical School) in the development of PET/SPECT nuclear probes for thrombus detection. Since 2015 he has been working with Dr Gonçalo Bernardes, University of Cambridge, where he develops new bioorthogonal methods for protein modification and imaging. |
 Z. Guo | Zijian Guo received his Bachelor of Science degree in Fundamental Science (Chemistry and Biology) from Tsinghua University (Beijing, China) in 2014. He is currently a PhD candidate under the supervision of Dr Gonçalo Bernardes in the Department of Chemistry, University of Cambridge (Cambridge, UK). His research is focusing on developing novel bioorthogonal chemistry methodologies for protein and cell labelling. |
 G. J. L. Bernardes | Dr Gonçalo Bernardes is a Group Leader at the Department of Chemistry, University of Cambridge, U.K. He is also the Director of the Chemical Biology and Pharmaceutical Biotechnology Unit at the Instituto de Medicina Molecular, Portugal. After completing his DPhil degree in 2008 at the University of Oxford, U.K., he undertook postdoctoral work at the Max-Planck Institute of Colloids and Interfaces, Germany, and the ETH Zürich, Switzerland, and worked as a Group Leader at Alfama Lda in Portugal. He started his independent research career in 2013, and his research group interests focus on the development of site-selective chemical protein modification for basic biology and drug development. He is a Royal Society University Research Fellow and the awardee of a Starting Grant from the European Research Council (TagIt). |
Introduction
Chemical site-selective protein modification has become increasingly popular for probing and controlling protein functions in vitro and in living systems. The progressive developments of the last decade in genetic encoding1 and aqueous chemoselective reactions for protein modification2 have provided tools to “decorate” biomolecules with the desired functionality (affinity probes, fluorophores, reactive tags, post-translational protein modifications – PTMs, etc.) without significantly perturbing their native functions (Fig. 1).3–6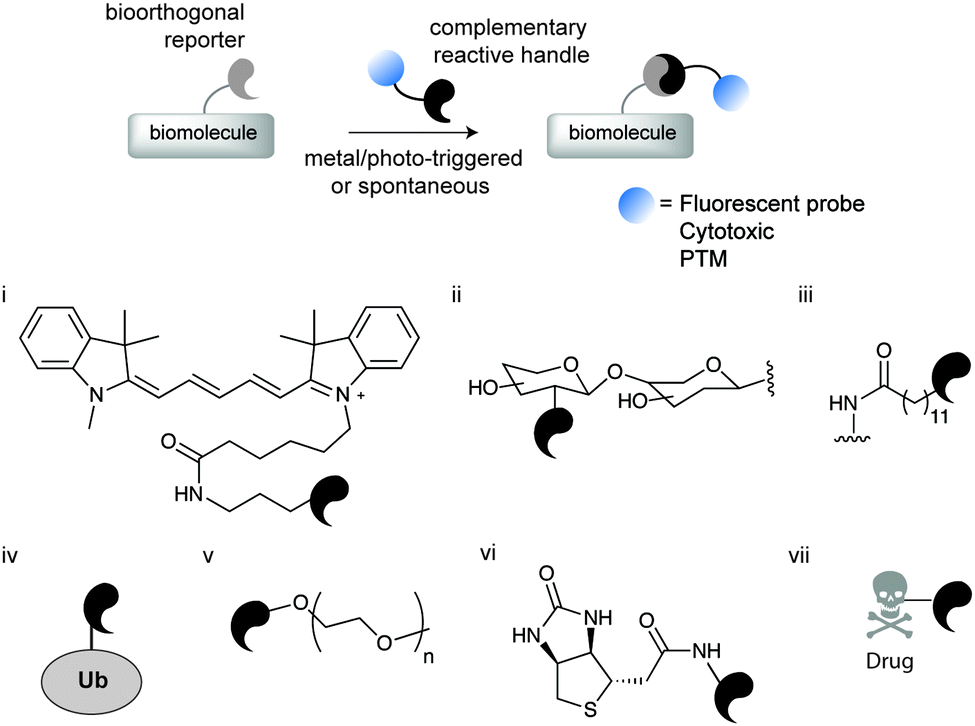 | ||
| Fig. 1 Schematic representation of chemical biology approaches for protein labelling and/or engineering through bioorthogonal chemistry. The first step involves the introduction of a bioorthogonal reporter on a protein either by genetic, enzymatic or chemical methods. In a second step the bioorthogonal reporter reacts with a molecule of interest bearing a complementary reactive handle. This approach has been used to install a diverse array of molecules for (i) fluorescent labelling,8 (ii) glycosylation,10 (iii) lipidation,11 (iv) ubiquitination,12 (v) PEGylation,13 (vi) biotinylation,14 and (vii) drugs.15 | ||
These transformations have been used, for example, for the spatial and temporal control of biomolecules in vivo,7 for super-resolution imaging,8 and for the elucidation of the role of post-translational modifications,9 among others. Ideally, such bioorthogonal reactions must be (1) selective over other potential reactive functional groups present on biomolecules, (2) proceed in aqueous media at (3) near physiological pH and (4) have fast reaction rates at room temperature (or up to 37 °C) using low reactant concentrations, all to ensure high modification efficiency. However, the existing bioorthogonal reactions are not yet able to meet all the bioorthogonal requirements simultaneously, and thus, the specific labelling of biomolecules in their native environment using chemical reactions is still a very challenging task.1,2
For instance, the requirement of Cu(I) to catalyse the 1,3 dipolar cycloaddition between azides and alkynes raises toxicity issues for its direct utilization in living cells due to Cu(I)-mediated generation of reactive oxygen species (ROS) from O2.16,17 To circumvent this issue, a strain-promoted azide–alkyne cycloaddition version that does not require the use of copper was developed.18 However, the bioorthogonality of this approach remains an issue, since the cyclooctyne reagents may undergo side reactions with cellular and plasma nucleophiles (e.g. the sulphydril side chain of free Cys) limiting the number of targets this approach may be used for.19,20 Amongst all bioorthogonal reactions developed to date, the [4+2] cycloaddition of 1,2,4,5-tetrazines (s-tetrazines, Tz) and various dienophiles, referred as inverse electron demand Diels–Alder (IEDDA) reaction, is the one that satisfies most of the bioorthogonal criteria (e.g. fast, selective, biocompatible and catalyst-free) necessary for use in applications from protein labelling to cancer imaging or materials science.
In this review, we present recent developments of this fast and robust bioorthogonal reaction and highlight its recent applications in the abovementioned fields. We begin in Section 1 by presenting a general overview of the most representative bioorthogonal reactions with a focus on their general utilities and challenges. The IEDDA reaction has emerged as an important tool for probing the mechanism and function of bioactive molecules in living systems. For such applications, the rate constant is of critical importance as very fast kinetics are required for labelling cellular processes that occur on biological time scales. In this regard, the exceptional speed of the IEDDA reaction revolutionised our ability to explore such demanding applications. In Section 2 the key factors that are behind the fast kinetics of the reaction will be discussed. In addition, the increased use of the IEDDA reaction has triggered the development of new tetrazine and dienophile precursors to create more stable and reactive partners. As such, a brief summary of the methodologies recently described for the synthesis of “IEDDA precursors” will be also presented. As the reaction rates depend directly on the reaction partners, Section 3 presents an overview of the most relevant tetrazines and dienophiles, their stability and their reactivity. Another beneficial aspect of using tetrazines as coupling partners is the increase of fluorescence observed after bioorthogonal reaction, resulting in a “turn-on system” useful for example for cell imaging applications. In this section, we also provide some representative examples of highly fluorogenic tetrazines for IEDDA labelling. Over the years numerous tetrazine and dienophile functionalities have been incorporated into proteins via genetic, enzymatic and chemical approaches. These methods are presented and discussed in Section 4. In Section 5 we present some selected examples where the appealing features of the tetrazine ligation provide a step-change in biological applications beyond the reach of other bioorthogonal chemical reactions. One of the most challenging and important applications of the tetrazine ligation is the selective labelling of biomolecules inside living organisms for imaging and therapy using pretargeting approaches. These applications, which are extensively explored by radiochemists, are summarized in Section 6. The major focus of bioorthogonal chemistry in the past two decades has been largely centred on 'bond formation' reactions for protein modification. Just very recently, focus has been placed on reactions that can instead cleave specific bonds under bioorthogonal conditions. Section 7 will focus on recent developments in IEDDA elimination reactions for accurate spatiotemporal control over protein function and drug activity. Section 8 outlines the potential of the IEDDA reactions in the development of biomaterials for applications in biology and medicine such as 3-D cell culture, drug delivery, multimodal imaging and clinical diagnostics. Finally, we draw conclusions and present a brief prospect of the role of IEDDA reactions in the field of bioorthogonal chemistry.
1. Meeting the candidates: a brief introduction to bioorthogonal chemistry
Arguably the most quoted example of a click reaction is the Cu(I)-catalysed [3+2] azido–alkyne cycloaddition (CuAAC) between an azide and alkyne, which was discovered independently by Sharpless et al. and Meldal et al. (Fig. 2i).16,17 Since these initial findings the CuAAC has been shown to be suitable for protein labelling in living systems, however, the wide use of CuAAC chemistry has been hindered by the potential toxicity induced by copper. In fact, Bertozzi et al. reported that mammalian cells could only survive to low concentrations (<500 μM) of Cu(I), leading to very slow CuAAC reaction rates at such concentrations that are not compatible with biological time scales. In addition, Zebrafish embryos also exhibited a similar sensitivity to Cu(I) limiting its use for in vivo applications.21 To improve the biocompatibility of the CuAAC reactions, the toxicity of Cu(I) was addressed by use of water-soluble Cu(I) ligands that stabilize the metal oxidation state and so, prevent the release of free copper ions that generate toxic reactive oxygen species.22,23 Notably, these biocompatible ligands have allowed the labelling of glycans in developing zebrafish embryos.23 The Cu(I) catalyst toxicity could be also obviated by use of strained alkyne substrates. The strain-promoted [3+2] azide–alkyne cycloaddition (SPAAC) developed by Bertozzi et al. in 2004 proceeds under physiological conditions without the need for a catalyst, however, in relatively slower kinetics when compared to the Cu(I) catalysed reaction (Fig. 2ii).18 Further enhancements in the rate constant of SPAAC additions have been reported using novel cyclooctyne analogues (e.g. DIBO, DIFO and BCN).24 The use of azides as chemical reporters has been also explored by the Staudinger ligation with phosphines (Fig. 2iii).25 Whereas azides produce a stable triazole linkage in CuAAC and SPAAC reactions, the Staudinger ligation generates a stable amide bond. Soon after the publication on the Staudinger ligation, Raines et al.26 and Bertozzi et al.27 simultaneously reported the so-called traceless Staudinger ligation (Fig. 2iv). Although this click reaction has sufficient biocompatibility to be performed in living systems it suffers from phosphine oxidation and very slow kinetics.28 A wider list of bioorthogonal reactions includes other chemical transformations such as aldehyde/ketone-amine ligations29 (Fig. 2v), photoclick 1,3-dipolar cycloadditions between tetrazoles and substituted alkenes30,31 (Fig. 2vi) and, strain-promoted cycloadditions involving nitrones and alkynes32 (Fig. 2vii). These bioorthogonal reactions, however, have been used mainly for in vitro bioconjugation because of the reaction conditions (e.g. requirement of an acidic pH and a light source in the case of ketones/aldehydes and tetrazoles, respectively). Recently, transition metal catalysis has been considered a powerful tool for the continued expansion of the bioconjugation toolkit (Fig. 2viii). Several emerging examples for forming new carbon–carbon bonds have been described including Suzuki33 and Sonagashira couplings34 or olefin metathesis.35 This “new class” of bioorthogonal reagents have been explored for labelling of proteins in vitro and on the cell surface.36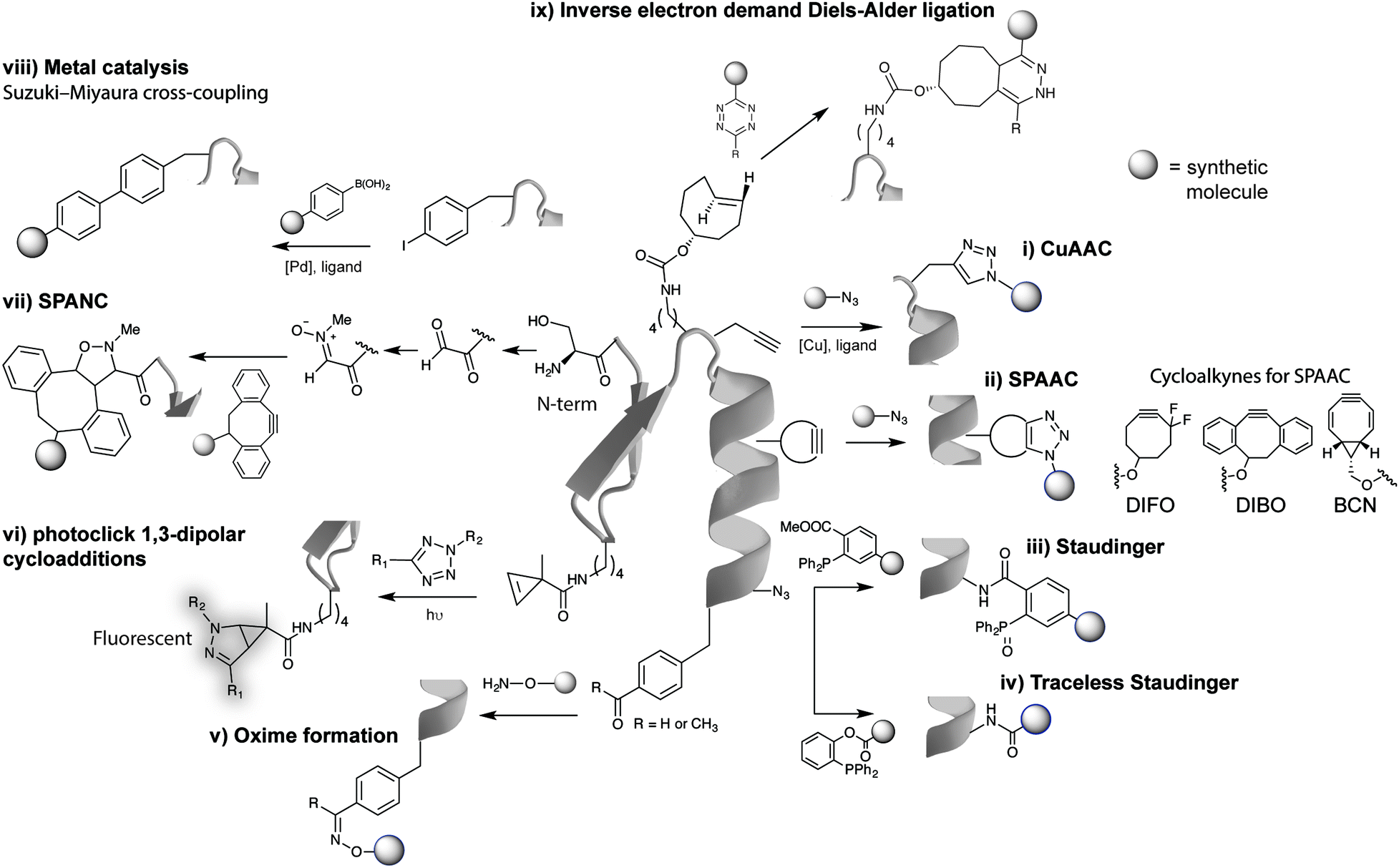 | ||
| Fig. 2 Representative examples of bioorthogonal reactions for protein modification. (i) CuAAC,22,23 (ii) SPAAC,24 (iii) Staudinger,25 (iv) traceless Staudinger,26,27 (v) hydrazones and oximes formation,29 (vi) photoclick reactions,30,31 (vii) strain-promoted alkyne-nitrone cycloaddition (SPANC) reactions,32 (viii) transition metal catalysis,33 and (ix) IEDDA reactions. Reactive handles installed site selectively into proteins by genetic encoding techniques except for (vii). | ||
A breakthrough was achieved with the IEDDA reaction between tetrazines and strained dienophiles (Fig. 2ix), arguably the most optimal bioorthogonal reaction developed to date. A general overview of the features of the abovementioned bioorthogonal chemical reactions is presented in Fig. 3 with a focus on the general utility of each reported bioorthogonal reaction in terms of their reaction rates and labelling efficiency but also their various biological applications. The non-metal nature and physiological pH makes IEDDA more cell-friendly than CuAAC and oxime ligation. Superfast kinetics (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold faster than CuAAC) allows IEDDA to proceed with low concentrations, similar to the concentrations of intracellular proteins. This click reaction endows itself an irreplaceable tool to study cellular functions and dynamic processes. Indeed, this reaction meets the need for demanding biological applications, i.e., has high selectivity, is compatible with mild reaction conditions in water and has impressive fast kinetics. Such assets have never been achieved with other site-selective reactions, highlighting the importance of IEDDA ligations in bioorthogonal chemistry.
000-fold faster than CuAAC) allows IEDDA to proceed with low concentrations, similar to the concentrations of intracellular proteins. This click reaction endows itself an irreplaceable tool to study cellular functions and dynamic processes. Indeed, this reaction meets the need for demanding biological applications, i.e., has high selectivity, is compatible with mild reaction conditions in water and has impressive fast kinetics. Such assets have never been achieved with other site-selective reactions, highlighting the importance of IEDDA ligations in bioorthogonal chemistry.
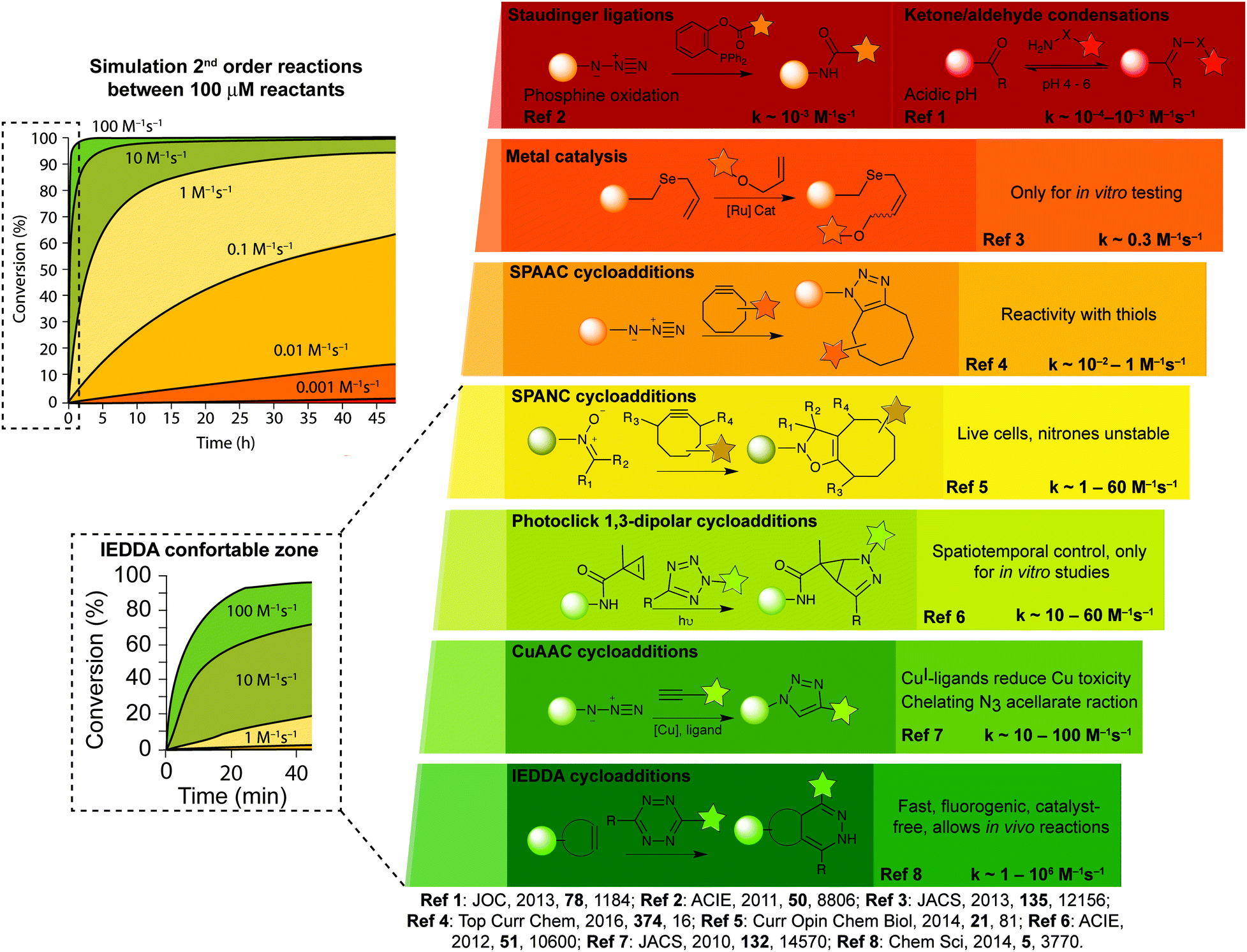 | ||
| Fig. 3 Examples of bioorthogonal reactions useful for bioconjugation and general comments about their utility and challenges. | ||
2. Principles of IEDDA reaction
2.1 Mechanism
Diels–Alder [4+2]-cycloaddition describes the reaction between a diene (e.g. 1,2,4,5-tetrazines) and a dienophile (alkene or alkyne) to form a six-membered ring in a π4s + π2s fashion (Fig. 4a), via suprafacial/suprafacial interaction of 4π-electrons of the diene with the 2π-electrons of the dienophile (Fig. 4b). In contrast to a normal electron demand Diels–Alder reaction, where an electron-rich diene reacts with an electron-poor dienophile, in an inverse-electron-demand Diels–Alder reaction (IEDDA), an electron-rich dienophile reacts with an electron-poor diene (Fig. 4b). The ability of tetrazines to react with unsaturated compounds was first revealed in 1959 by Lindsey et al.37 This Diels–Alder reaction proceeds via the 1,4-addition of the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NC– diene system of the tetrazine to an appropriate alkene, yielding a highly strained bicyclic intermediate (Fig. 4a). Upon the evolution of 1 equivalent of nitrogen, the adduct undergoes a retro-Diels–Alder reaction to afford the corresponding 4,5-dihydropyridazine, which either isomerises to the corresponding 1,4-dihydro-isomers or is oxidized to give a pyridazine product (Fig. 4a). Alkyne dienophiles directly yield the respective pyridazine upon reaction.
N–NC– diene system of the tetrazine to an appropriate alkene, yielding a highly strained bicyclic intermediate (Fig. 4a). Upon the evolution of 1 equivalent of nitrogen, the adduct undergoes a retro-Diels–Alder reaction to afford the corresponding 4,5-dihydropyridazine, which either isomerises to the corresponding 1,4-dihydro-isomers or is oxidized to give a pyridazine product (Fig. 4a). Alkyne dienophiles directly yield the respective pyridazine upon reaction.
 | ||
| Fig. 4 Mechanism of IEDDA reaction. (a) Schematic representation of the reaction between a dienophile and a tetrazine. (b) Frontier molecular orbital of normal and inverse electron demand Diels–Alder reaction. EDG = electron-donating group, EWG = electron-withdrawing group. | ||
2.2 Features for reactivity of IEDDA reactants
Based on the frontier molecular orbital theory (FMO), the IEDDA reaction kinetics is governed by the energy gap between the corresponding HOMO and LUMO of the reactants (Fig. 4b). In particular, any pairs of diene/dienophile with a smaller HOMOdienophile–LUMOdiene energy difference would react faster in IEDDA reactions. Since the earlier comprehensive kinetics studies in organic solvents performed by Sauer and Boger, efforts have been made to find fast IEDDA reactants for rapid bioorthogonal reaction in aqueous media. Indeed, by fine-tuning the tetrazine and dienophile pairs considerable rate enhancements and bioorthogonality have been achieved. The different factors tuning the IEDDA kinetics are discussed as follows.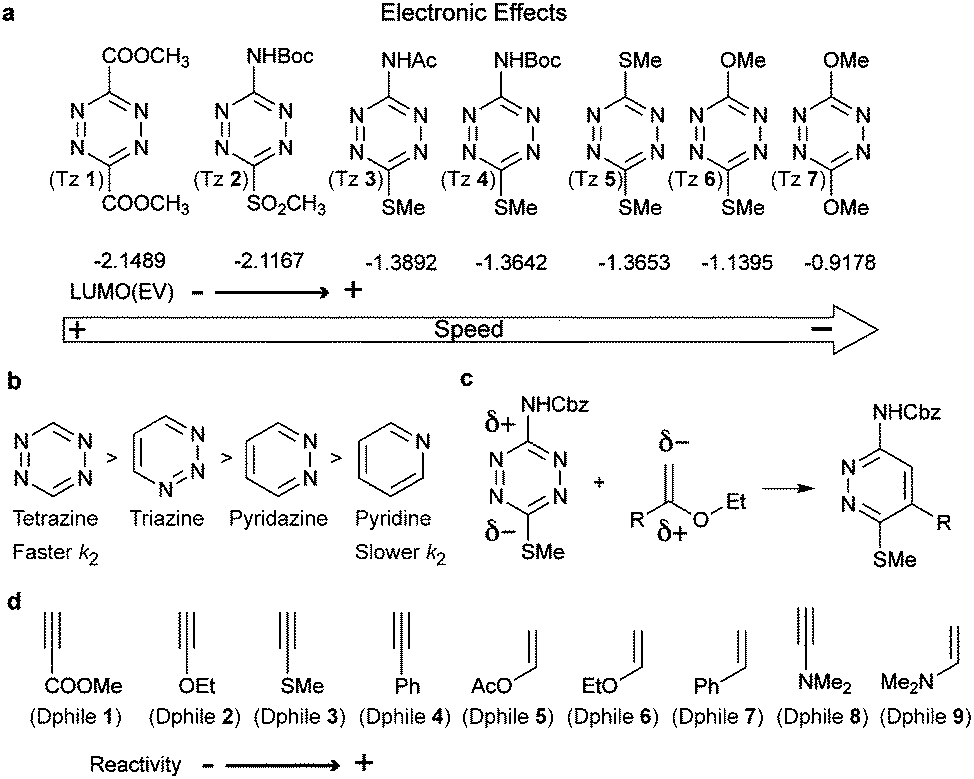 | ||
| Fig. 5 (a) Dependence of reactivity against LUMO energy of tetrazine. (b) Reactivity of heterocyclic ring bearing various numbers of nitrogen atoms. (c) Electronic effects on regioselectivity. (d) Electronic effects on olefinic and acetylenic dienophiles. | ||
Very recently, 1,2,4-triazines have been described as a new class of bioorthogonal reagent for IEDDA reactions, however, they are less reactive than 1,2,4,5-tetrazines.45,46 Boger et al. and Saez et al. also showed that the electronic properties of the substituent not only influence tetrazine's reactivity but also affect the regioselectivity of the cycloadditions (Fig. 5c). Accordingly, the authors demonstrated experimentally and theoretically that it is possible to predict the product formation by analysing the partial net charge on C3 and C6 positions (Fig. 5c).38,39,47
An even more pronounced impact on the IEDDA kinetics arises from the type of dienophile. In this case, dienophiles with electron-rich substituents are preferred to achieve fast kinetics. So far, a large number of papers, mostly by Sauer and co-workers,48 have been devoted to discussing the reactivity of different substituted unstrained dienophiles and several principal rules have been summarized based on the electronic effect.49–55 Briefly, olefinic dienophiles surpass acetylenic dienophiles in terms of reaction rates, (Dphile 1–4versus Dphile 5–7, Fig. 5d) due to the increased electron withdrawing character of the triple bond, therefore lowering the energy of the HOMO.49,55 Introduction of EDGs, such as dialkylamino, make enamines (Dphile 8) and ynamines (Dphile 9) highly reactive dienophiles. Similar dependence between electronic properties and kinetics are also observed with phenylacetylenes (Dphile 4) and styrene (Dphile 7) if EDGs are introduced in either the phenyl group or in the olefinic/acetylenic part.49,55
 | ||
| Fig. 6 Influence of the strain effect on the IEDDA reaction. (a) Degree of ring strain along a series of cyclic alkenes. Similar strain effects are also observed for cyclo-enamines and cyclic enol ethers.49 (b) The crown conformation of TCO makes it 7 orders of magnitude more reactive than cis-cyclooctene towards 3,6-bismethyoxycarbonyl-1,2,4,5-tetrazine and 3,6-bistrifluoromethyl-1,2,4,5-tetrazine.56 (c and d) Via strained ring fusion, the crown conformation was furthered locked, resulting in rate enhancement against TCO. | ||
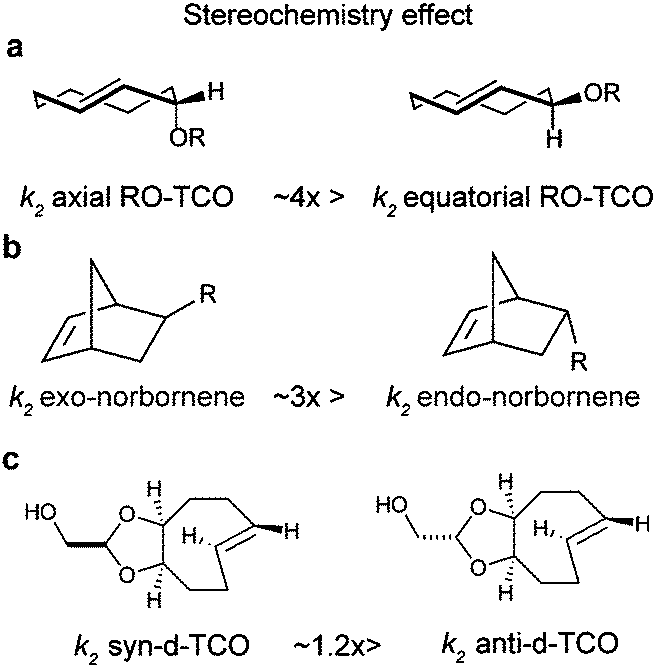 | ||
| Fig. 7 Effect of stereochemistry on stereoisomers of (a) TCO, (b) norbornene and (c) d-TCO. | ||
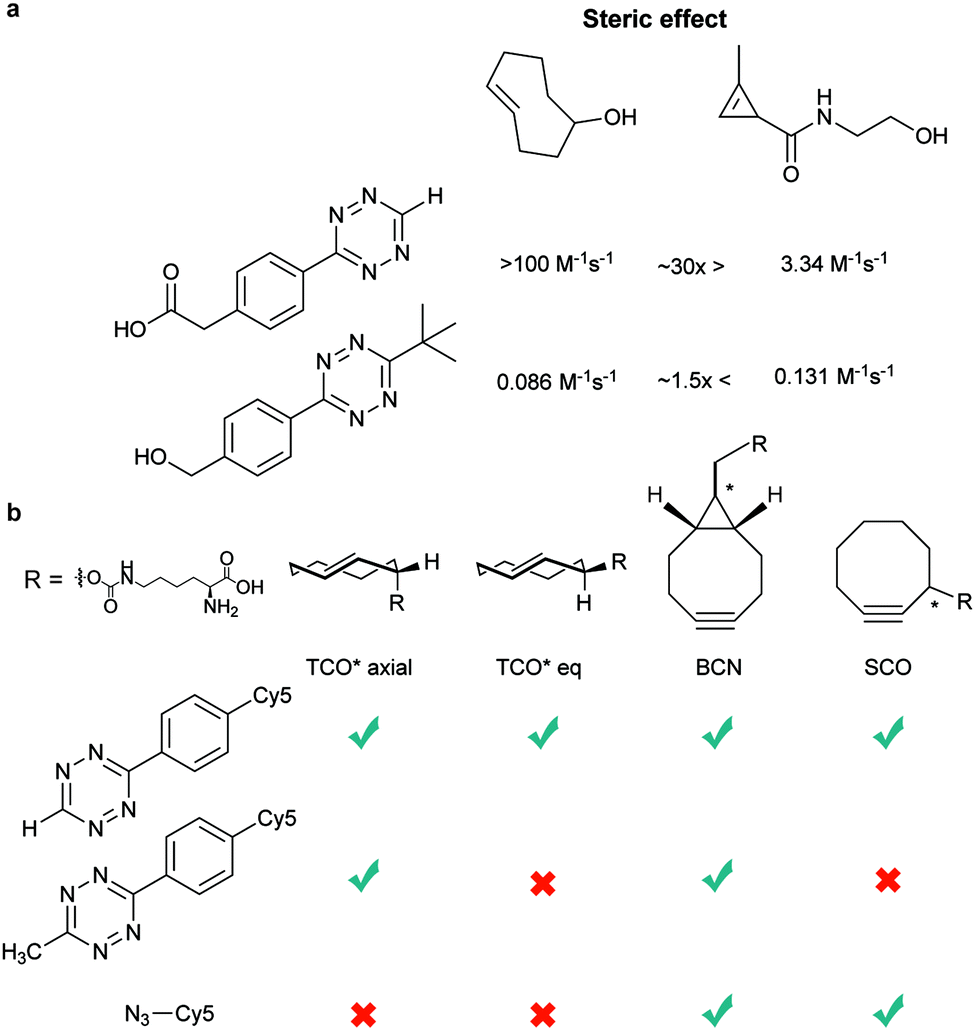 | ||
| Fig. 8 Influence of the steric effect on the IEDDA reaction (a) bulky tetrazine reverse the reactivity of TCO and cyclopropene compared with less bulky tetrazine. (b) Reactivities of TCO*, SCO, and BCN with azide, H-Tz, and Me-Tz. By fine-tuning the reaction partners mutual bioorthogonality could be obtained. | ||
2.3 Synthesis of tetrazine derivatives
Conventionally tetrazines are obtained via the synthesis of dihydrotetrazines by reacting the appropriate precursors (e.g. aromatic/aliphatic nitriles and imidoesters) with hydrazine, followed by oxidation to afford the reactive tetrazine cores. Since recently, new methodologies based on carbon–carbon bond formation have been explored in the generation of various tetrazines, especially tetrazines functionalized with fluorescent dyes. In the following sections these methods are presented and discussed in detail.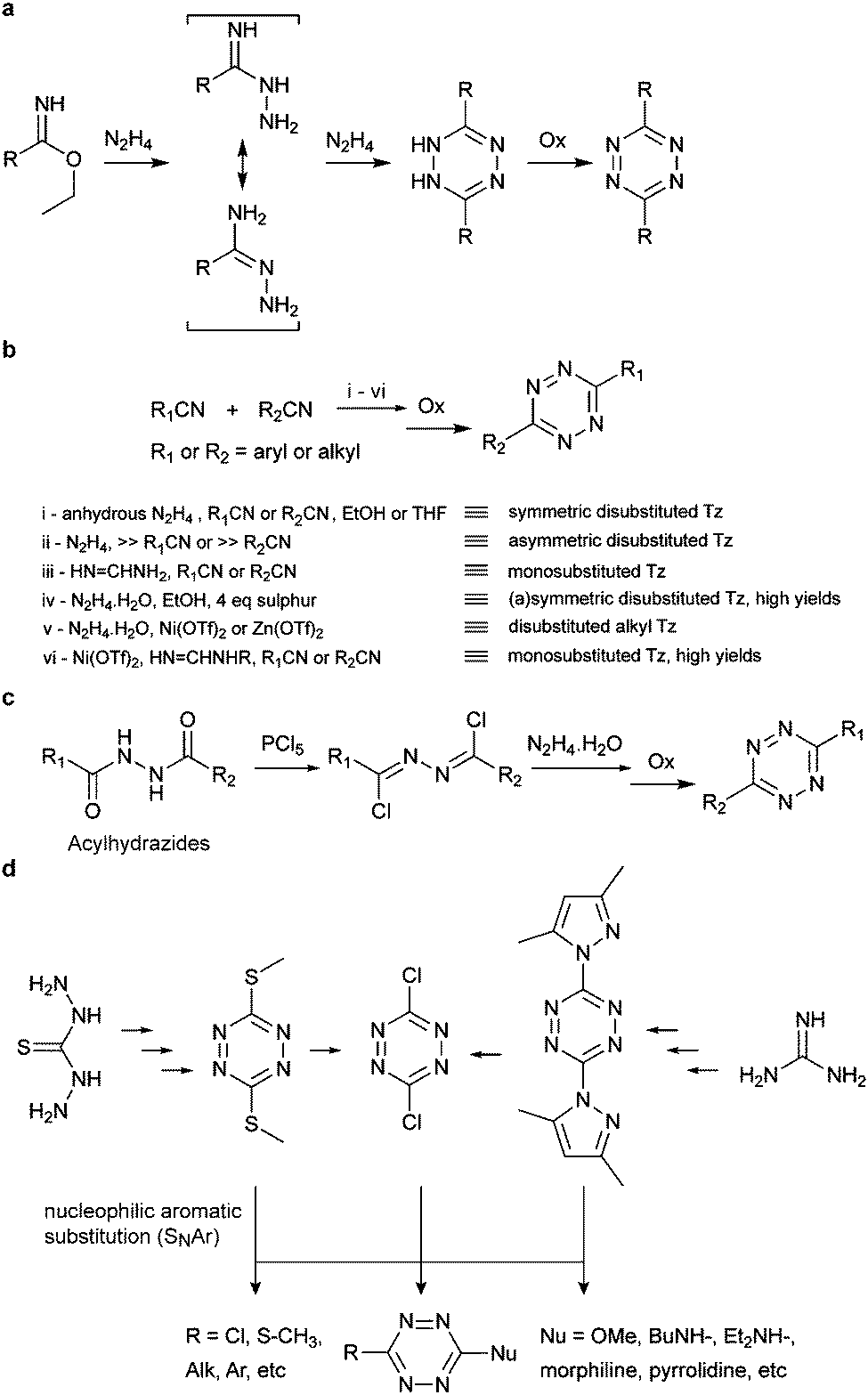 | ||
| Fig. 9 Classic synthesis routes for tetrazines using (a) imidoesters, (b) nitriles, (c) acylhydrazides and (d) nucleophilic aromatic substitution. | ||
Recently, new synthetic routes have been explored for the synthesis of tetrazines. Devaraj et al. reported the use of Lewis acids as catalysts for the activation of nitriles, including unreactive alkyl nitriles, by coordinating to the nitrile and promoting the nucleophilic addition of hydrazine. It was shown that nickel and zinc triflates allowed the one-pot synthesis of both symmetric and asymmetric dialkyl tetrazines in good yields (Fig. 9b(v)).79 Ni(OTf)2 catalyst was later used for the preparation of asymmetric monosubstituted tetrazines in high yield (∼75%) on gram-scale synthesis (Fig. 9b(vi)), whereas sulphur-promoted reaction only resulted in yields lower than 20%.80 Importantly, this method was applied in the synthesis of coumarin-tetrazine and BODIPY–tetrazine fluorescent probes.81,82 Although high yields were achieved, this one-step synthesis method has limitations of substrate scope. The hydrazine and heating conditions are not compatible with several functional groups that are susceptible to either nucleophilic addition or reduction, such as carbonyls and alkyl halides. To overcome these limitations acylhydrazides have been used for the synthesis of (a) symmetric substituted tetrazines, in a stepwise reaction first reported by Stolle et al., involving the synthesis of 1,2-dichloromethylene hydrazines by treatment with PCl5 and subsequent condensation with hydrazine and oxidation (Fig. 9c).83,84 With this general methodology, both symmetric and asymmetric tetrazines with EWGs and aliphatic substituents were synthesized with modest yields.85 Microwave irradiation has also been proved to assist the formation of tetrazine.86 A further optimization on the condensation of 1,2-dichloromethylene hydrazine with hydrazine monohydrate by the same group showed that microwave irradiation could reduce the reaction time from 5–16 h to 30 mins with an extra 10–20% yield increase, compared to refluxing conditions.42 Another alternative method commonly used for tetrazine synthesis is the nucleophilic aromatic substitution (SNAr) starting from the relevant precursors (e.g. methylthio-1,2,4,5-tetrazine, 3,6-dichloro-1,2,4,5-tetrazine, 3,6-(3,5-dimethylpyrazolyl)-1,2,4,5-tetrazine, Fig. 9d).87
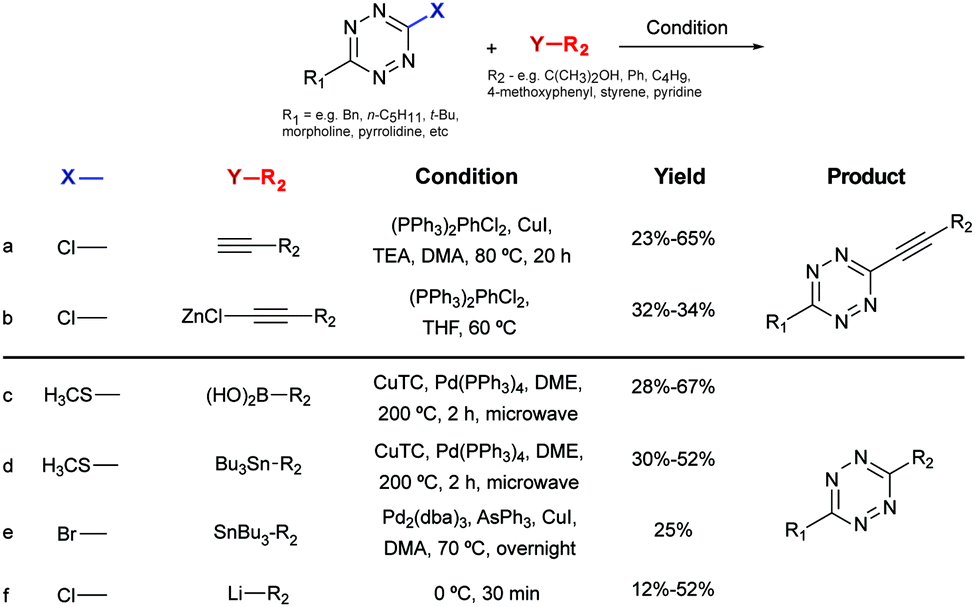 | ||
| Fig. 10 Tetrazine crosslinking reactions based on C–C bond formation. | ||
Negishi coupling of chlorotetrazines with hexynylzinc chloride also affords coupling products with 30% yield (Fig. 10b).88 Under Suzuki-like and Stille-like conditions Guillaumet et al. reported the reaction of 3-methylthio-1,2,4,5-tetrazine derivatives with boronic acids and organostannane precursors (Fig. 10c and d).89 Yields of 30–70% were achieved under microwave conditions for both reactions. This Pd-catalysed cross-coupling reaction represents the first of few methods to construct asymmetrical tetrazines using vinyl, aryl and heteroaryl substituents.89 Recently, Pd-catalysed Stille cross coupling reactions were developed for the derivatization of tetrazines at positions C3 and C6 with fluorescent probes (Fig. 10e). Wombacher et al. identified 3-bromo-6-methyl-1,2,4,5-tetrazine as a suitable building block for coupling with protected fluorophore-organotin derivatives, giving yields around 20%. Interestingly, this method is applicable for fluorescein and Oregon green derivatives but not for rhodamine or Si-rhodamine dyes.90 The replacement of the chlorine on tetrazines by nucleophilic aromatic substitution using alkyllithium is another alternative method developed by Tang et al. (Fig. 10f).91 A number of tetrazine derivatives have been also prepared by Audebert et al. by nucleophilic substitution of chlorotetrazines with ethynyl compounds in the presence of n-BuLi, although in low yields (∼12%).92
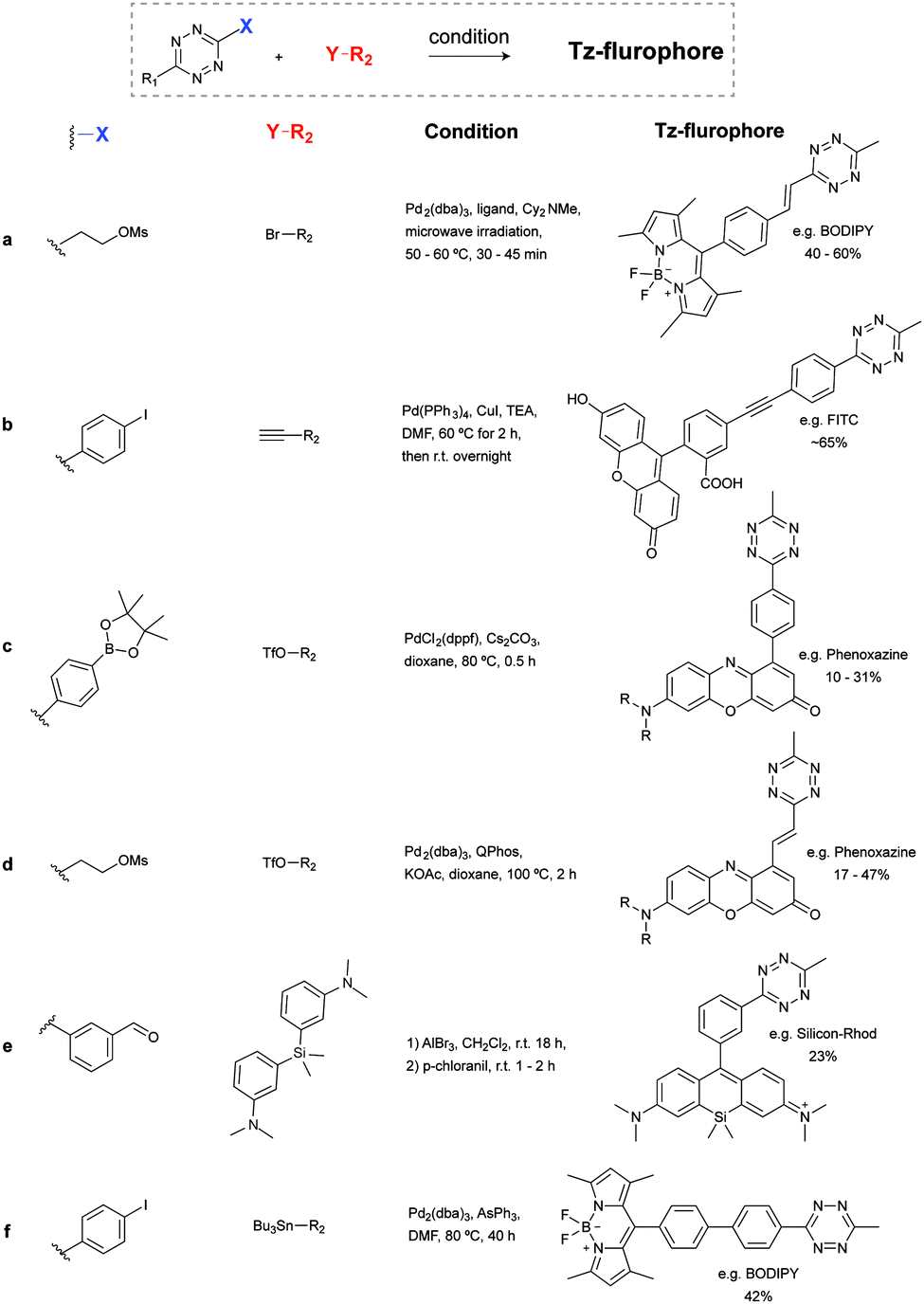 | ||
| Fig. 11 Synthesis of tetrazine conjugates by functionalization of tetrazine substituents. | ||
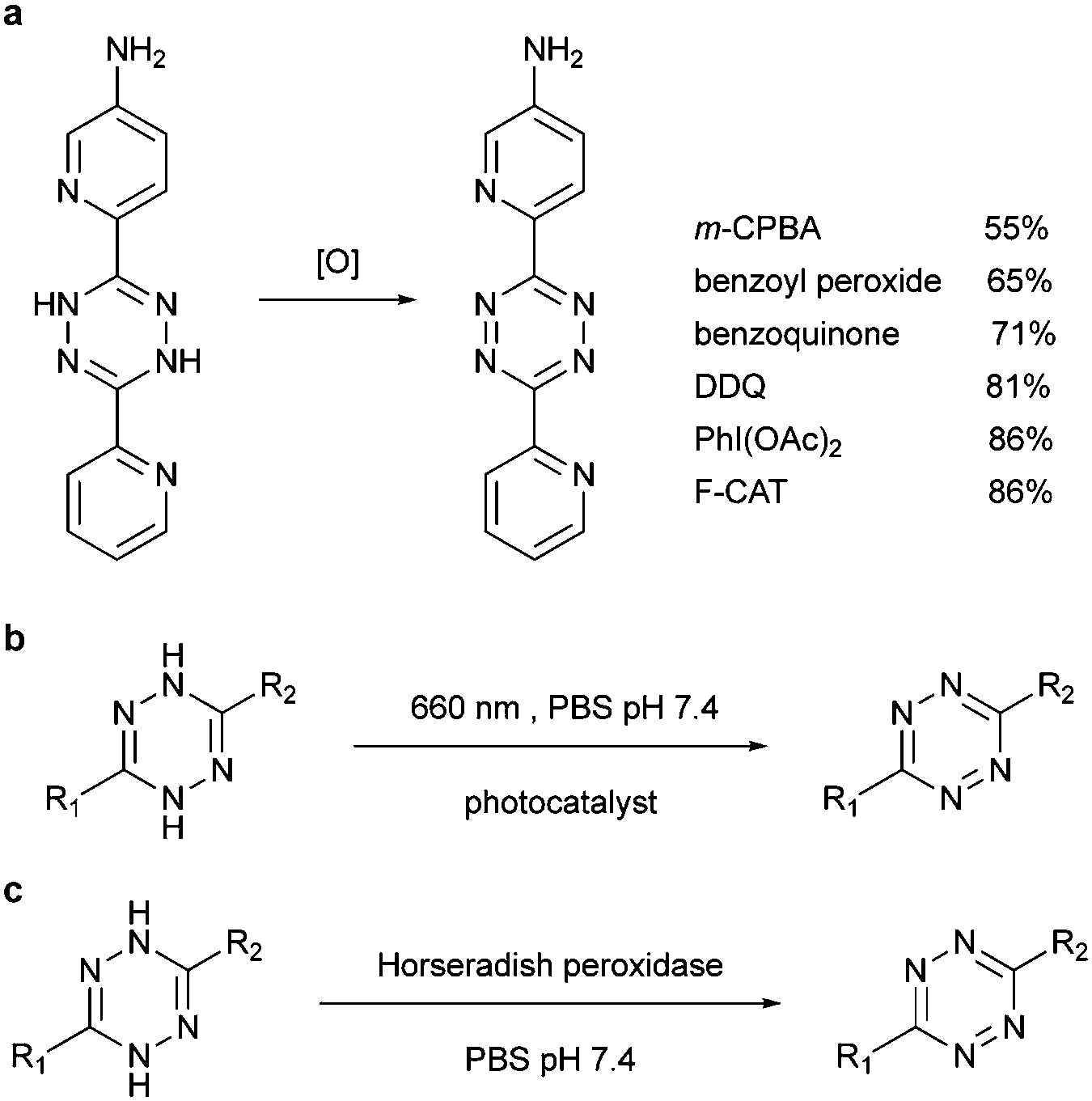 | ||
| Fig. 12 Methods to oxidize dihydrotetrazine by (a) organic oxidants, (b) photocatalyst and (c) enzyme. | ||
Alternatively, by using photocatalysts, the dihydrotetrazine could be oxidized to tetrazine in quantitative yields with 660 nm LED in the presence of methylene blue within 200 s. Even under ambient light, the photoredox reaction could be afforded in 47% conversion in 2 h (Fig. 12b). Similarly, horseradish peroxidase could also oxidize dihydrotetrazine with fast reaction rates (Km = 1.0 × 10−4 M, kcat = 27 s−1, and kcat/Km = 2.7 × 105 M−1 s−1, Fig. 12c).103
2.4 Synthesis of dienophiles
For metabolic oligosaccharide engineering and genetic encoding applications, the synthesis of dienophile-bearing sugars and amino acids is usually performed by using commercially available NHS- and p-nitrophenyl-activated dienophiles. Otherwise, multistep synthesis is required where unstrained dienophiles, such as terminal alkenes, are of advantage. Cyclopropenes are usually accessed from TMS-protected propynes via rhodium-catalysed cyclopropenation with diazo compounds (Fig. 13a).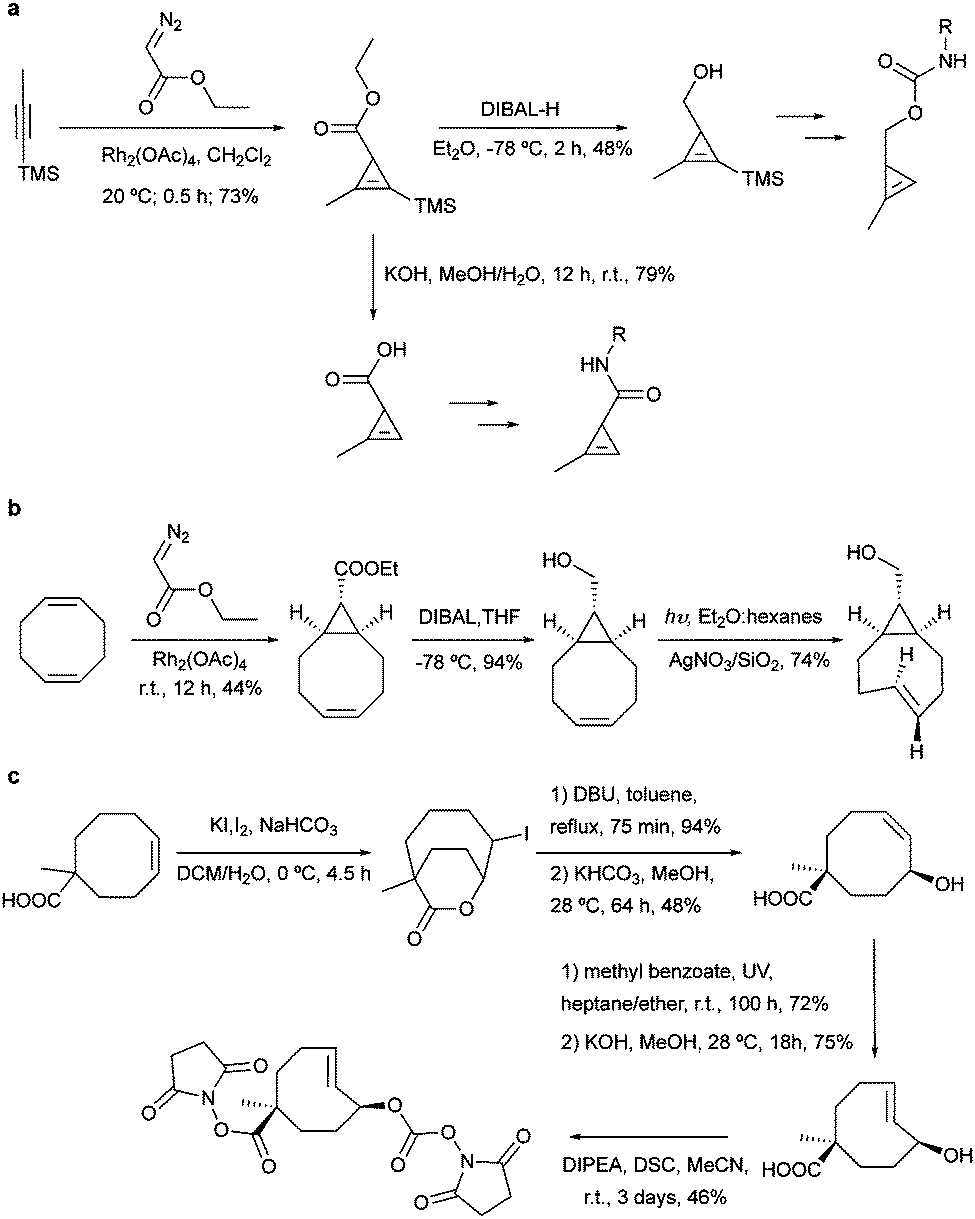 | ||
| Fig. 13 Common synthetic routes for the preparation of most common dienophiles. | ||
For the preparation of cyclopropene amide-linked derivatives, the TMS-protected ester subsequently undergoes base-catalysed hydrolysis (e.g. potassium hydroxide) to give the TMS-free acid, which is then subjected to the corresponding amine to afford the desired amide (Fig. 13a). For the synthesis of cyclopropenes containing a carbamate scaffold, DIBAL reduction agent is typically used to produce the appropriate alcohol, followed by carbamate formation and TMS removal.104–106 Importantly, methylcyclopropenyl methanol cannot be isolated as it is prone to rapid polymerization upon concentration, where a one-pot synthesis is required.106,107
The fast grow of IEDDA reaction in the bioorthogonal chemistry field is closely boosted with the advanced TCO synthesis methodology. A robust synthesis of this IEDDA partner was reported by Fox et al. in 2008 using a one-step reaction via flow photo isomerisation. Recently, in order to improve the trans/cis ratios and reduce the photo degradation of the active trans-cyclooctene, a new synthetic route was proposed in a closed-loop flow apparatus.108,109 In this synthesis, the reaction mixture is continuously photo irradiated at 254 nm and pumped through AgNO3 impregnated silica, which binds strongly with the desired trans-isomer, eluting the cis-isomer to be isomerised again until completely consumed. This direct method was further applied in the synthesis of most TCO derivatives published so far since then.108
Despite the extensively use of TCO in bioorthogonal chemistry, the commercial availability of TCO is still limited. All typical syntheses for TCO derivatives involve the construction of the corresponding cis-derivatives and then trans-isomerisation. Efforts have been made to introduce extra ring strain and functionality at defined positions. For example, rhodium-catalysed cyclopropenation on bicyclo[6.1.0]non-4-yne was also used to annihilate the cyclopropane ring to maintain the half chair conformation of bicyclo[6.1.0]non-4-ene (Fig. 13b).59 Robillard et al. designed an elegant synthesis route to construct a bis-NHS-activated TCO-bearing linker for antibody-targeted therapy. A methyl group near the carboxylic acid was introduced to prevent epimerization and to control regioselectivity15 (Fig. 13c).
3. The toolbox of bioorthogonal partners for IEDDA reactions
3.1 Dienophiles as IEDDA precursors
As abovementioned, the IEDDA reaction can be tuned to reach rate constants from 1 up to 106 M−1 s−1 by changing the electron deficiency of the 1,2,4,5-tetrazine precursors, or by manipulating the ring strain and electronic effects on the dienophiles. Significant efforts have focused on accelerating the reaction rate by expanding the scope of dienophiles. The first reported dienophile for protein bioconjugation via IEDDA reaction was the strained TCO introduced by Fox et al. that revealed an unusually fast second-order rate constant towards a dipyridyl-tetrazine precursor (Tz 8) of k2 1140 M−1 s−1 in MeOH and 2000 M−1 s−1 in 9:1 MeOH/water (Fig. 14a).100 In pure water at 25 °C, the equatorial-diastereomer of TCO reacts with a water-soluble dipyridyl-tetrazine derivative (Tz 9) with an increased rate constant of 22600 M−1 s−1 while the axial-diastereomer reacts faster with a k2 of 80200 M−1 s−1 (Fig. 14a).60 Robillard et al. also reported that the axial diastereomer of 5-hydroxy-TCO is more reactive than the equatorial diastereomer due to instability of the axially substituted ring induced by transannular interactions.110 Fox et al. subsequently optimized TCO in terms of reactivity by fusing a cis-cyclopropane onto the cyclooctene moiety to increase the ring strain (s-TCO, Fig. 14b).59 When Tz 8 is reacted in MeOH at 25 °C, this strained derivative is 19 times more reactive than the parent TCO (k2 = 22000 M−1 s−1).59 Later, it was demonstrated that a water soluble s-TCO derivative bearing a PEG linker reacts with Tz 9 in pure water at a rate constant of 3300000 M−1 s−1 (s-TCO, Fig. 14b), which makes this dienophile the fastest to date.60 There are some limitations, however, that emerge from the high reactivity of the “TCO dienophiles”. One is the deactivation of TCO by isomerisation in the presence of high thiol concentrations. As an example, Robillard et al. showed that in fresh mouse serum at 37 °C the TCO isomer converts into cis-cyclooctene with a half-life of 3.26 h.110 Secondly, the “TCO dienophiles” TCO/s-TCO are not stable enough for prolonged storage. Recently, Johnson et al. introduced a new variant of s-TCO complexed with AgNO3, which has improved stability and can be used directly in cells for rapid bioorthogonal reactions with tetrazines.111 Alternatively, to address the stability limitations of s-TCO Fox et al. designed a new conformationally-strained dioxolane-fused trans-cyclooctene (d-TCO), which maintains high reactivity towards tetrazines (k2 366000 M−1 s−1 for syn isomer and 318000 M−1 s−1 for anti-isomer at 25 °C in pure water with Tz 9) but displays enhanced stability and is more easily prepared (Fig. 14c).60 Stability studies of the syn isomer in human serum at room temperature showed no isomerisation or decomposition after 24 h neither after 4 days (>97% trans isomer).
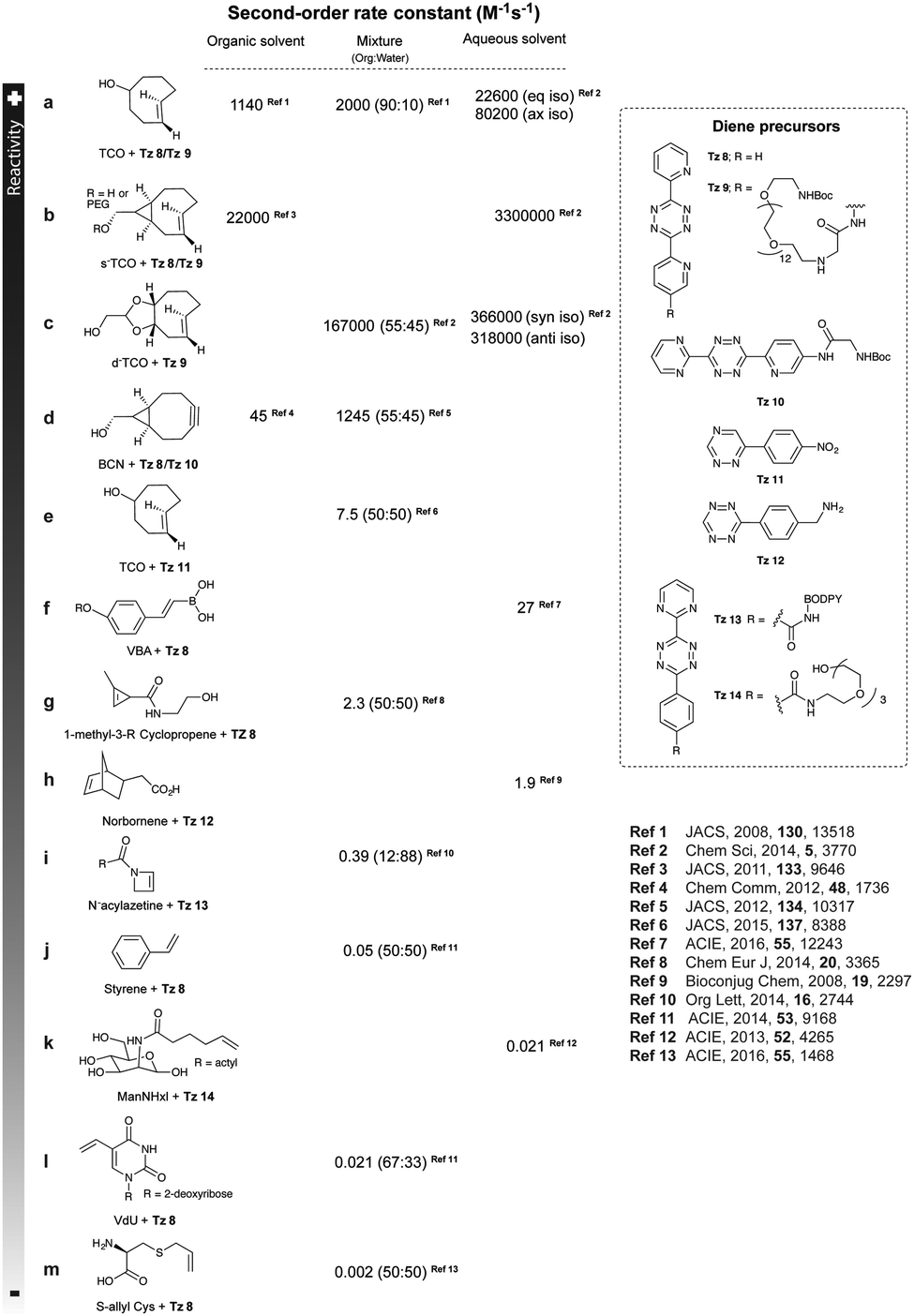 | ||
| Fig. 14 Examples of strained and unstrained dienophiles for IEDDA reactions and their corresponding reaction rates. | ||
Bicyclooctynes (bicyclo[6.1.0]nonyne, BCN) were found also to undergo fast IEDDA reactions with tetrazines (k2 = 45 M−1 s−1 in MeOH at 24 °C with Tz 8; k2 = 1245 M−1 s−1 in 55% MeOH/H2O at 25 °C with Tz 10; Fig. 14d).61,112 Recently, Prescher et al. showed that 1,2,4-triazines participate in IEDDA reactions with strained alkenes (Fig. 14e).45 In particular, it was found that 1,2,4-triazines react with TCO, but not with norbornene or cyclopropene.45 Considering the latter two undergo efficient cycloadditions with 1,2,4,5-tetrazines, this suggests an opportunity for sequential dual-labelling. However, this methodology remains unexplored most likely because the relatively slow kinetics of the 1,2,4-triazine: TCO reaction (k2 = 7.5 M−1 s−1 in 50% ACN/PBS at 25 °C with Tz 11, Fig. 14e). Importantly, this new class of IEDDA reagents showed enhanced stability enabling its direct use in recombinant protein production (fully stable in PBS and in excess of cysteine for 1 week at 37 °C).45 A contemporaneous study showed that 1,2,4-triazines also react with strained alkynes (BCN), however, with even with more sluggish kinetics (k2 = 0.38 × 10−3 M−1 s−1 with BCN in acetonitrile).46
Recently, vinylboronic acids (VBA) appeared as a new interesting class of compounds for fast and water-soluble reactions with tetrazines (27 M−1 s−1 with Tz 8; Fig. 14f).113 The use of cyclopropenes as tetrazine reactive partners was also proposed as an alternative to TCO dienophiles (Fig. 14g). The Devaraj group demonstrated that modulating the substituents of cyclopropenes has a dramatic effect on their stability and on the kinetics towards tetrazines. Indeed, it is well described that unsubstituted cyclopropenes are susceptible to both nucleophilic attack and polymerization reactions. To overcome these limitations, Devaraj et al. “protected” the double bond with a methyl group, and the resulting 1-methyl-3-substituted cyclopropene derivatives appeared as fast and in vivo stable dienophiles with one of these analogues having a second-order rate constant of 2.3 M−1 s−1 (in 50% aqueous solutions with Tz 8; Fig. 14g).69,104,106 Faster reaction rates for this dienophile are expected if measured under purely aqueous conditions. Norbornenes, another class of cyclic dienophiles, offer an excellent balance between facile strain-promoted reactivity with tetrazines and overall chemical stability (Fig. 14h). Tetrazine Tz 12 reacts rapidly with norbornene in aqueous buffer with a second-order rate constant of 1.9 M−1 s−1 (Fig. 14h).114 Recently, a new bioorthogonal N-acylazetine small tag showed equal efficiency as the norbornene for protein modification via IEDDA reaction with a rate constant of 0.39 M−1 s−1 against Tz 12 (Fig. 14i). Vinylbenzyl groups (styrene) also react efficiently in IEDDA reactions (0.05 M−1 s−1 against Tz 8 in 50% MeOH/water, Fig. 14j)115,116 Other minimal-tags include terminal unstrained alkene-decorated sugars117 (0.021 M−1 s−1 against Tz 14) and nucleosides (5-vinyl-2′-deoxyuridine, VdU, 0.021 M−1 s−1 against Tz 8, Fig. 14k).115 Recently, our group explored chemical installed S-allyl handles for “IEDDA labelling” with tetrazines (Fig. 14l). Although, this handle reacts significantly slower (0.002 M−1 s−1 against Tz 8 in 50% MeOH/PBS) compared to trans-cyclooctenes, it was suitable for pretargeting live cell imaging.118
While most studies of reaction kinetics are performed on small molecule models, it is important to study what is the actual kinetics of these reactions in the protein context. In one example, Chin et al. showed that the second-order rate constant for the labelling of sfGFP bearing a 1,3 disubstituted cyclopropene with a fluorescent analogue of Tz 8 was 27 ± 1.8 M−1 s−1 (Fig. 15). Importantly, it was shown that the rate constant for this reaction in the test tube and in cells is approximately 10-fold faster when compared to the cyclopropene model (2.3 M−1 s−1, Fig. 15). The differences in the reaction rates should be related with the reaction solvent (water for protein and 50% water/org for small molecule). Similarly, Lemke et al. demonstrated that the tetrazine ligation (Tz 12-TAMRA) with a BCN-GFP bearing protein occurs with a reaction rate constant of k2 = 29000 ± 7500M−1 s−1 (Fig. 15a).119 Progress of the labelling reaction was determined using Förster resonance energy transfer (FRET) to follow the decrease in GFP fluorescence and the increase of the TAMRA fluorescence upon GFP excitation. Just recently, the Lemke group developed a new hydrophilic TCO analogue (DOTCO; dioxo-TCO) for genetic encoding which showed fast reactions with a Cy5 (cyanine dye) tetrazine when incorporated into GFP (6370 M−1 s−1 in pure water with Tz 12-Cy5; Fig. 15a).120
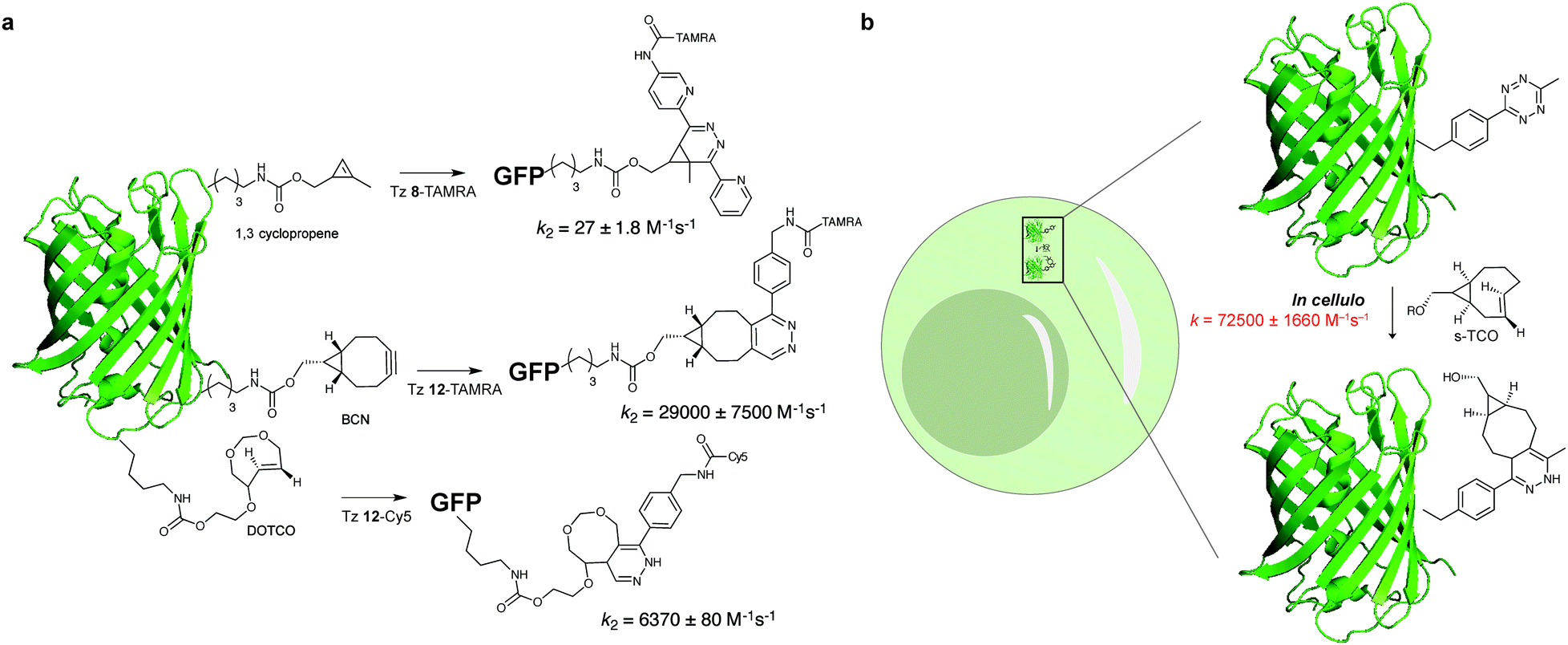 | ||
| Fig. 15 On protein and in cellular rate constant determination for reaction of GFP bearing dienophiles and tetrazine. | ||
An ideal bioorthogonal reaction requires a rate of >104 M−1 s−1 inside cells to reach completion in seconds to min at biological concentrations (μM to nM) of the bioorthogonal target.121 To meet these rates, Mehl et al. developed a tetrazine-modified GFP (GFP-Tet-v2.0) that reacts in cells with s-TCO with a rate constant of 72500 ± 1660 M−1 s−1 (Fig. 15).121 The authors predict that this reaction rate allows 95% labelling in <1 min at 1 μM concentrations of Tet-v2.0-protein and s-TCO, meeting the needs of the ideal bioorthogonal ligation. Compared to the “cuvette second-order rate” (87000 ± 1440 M−1 s−1) the rate constant reduction is justified by the in vivo cellular environment or hampered uptake of s-TCO.121
3.2 Dienes as IEDDA precursors – fast kinetics versus stability
Tetrazine partners are also a key factor in the IEDDA reaction rates. Hilderbrand et al. have shown that electron-withdrawing substituents enhanced IEDDA reactivity of tetrazines, however, these substituents may also induced their degradation.40 An optimal balance between reactivity and stability must be achieved for effective labelling in in vivo systems. The disubstituted tetrazines Tz 15–Tz 16 comprising electron-donating substituents show the highest stability (∼90% stable), but also suffer from slow kinetics (200–2000 M−1 s−1) (Fig. 16). Hydrogen substituted tetrazines Tz 22 and Tz 23 demonstrate a good balance of stability (∼70% stable for Tz 23) and fast reaction kinetics (30000 M−1 s−1). These compounds are not the most electronically favourable (no electron-withdrawing substituents) but the fastest suggesting these tetrazines may react faster due to less steric interference. The least stable tetrazine, but also one of the fastest, comprises electron-withdrawing groups (R2 = pyrimidine; Tz 21).
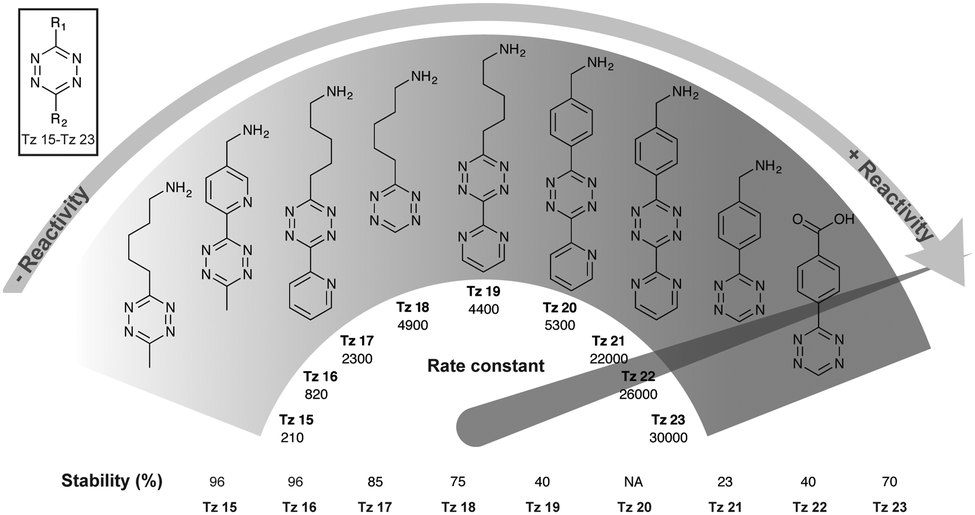 | ||
| Fig. 16 Second order rate constants of selected tetrazines with TCO in PBS at 37 °C and corresponding stability assessed in PBS at 37 °C for 10 h. NA, not assessed.40 | ||
3.3 Fluorogenic tetrazines for IEDDA ligations
In bioorthogonal labelling approaches a large amount of the bioorthogonal fluorescent dye is often required to increase reaction efficiency. In these examples the excess of the fluorescent probe has to be removed using several washing steps in order to reduce background fluorescence of the unreacted probes. One alternative approach to minimize background fluorescence is based on the use of fluorogenic probes, which become fluorescent only upon transformation. Such fluorogenic dyes are extremely advantageous for in vivo labelling applications, since they usually result in highly conspicuous images without requiring washing steps.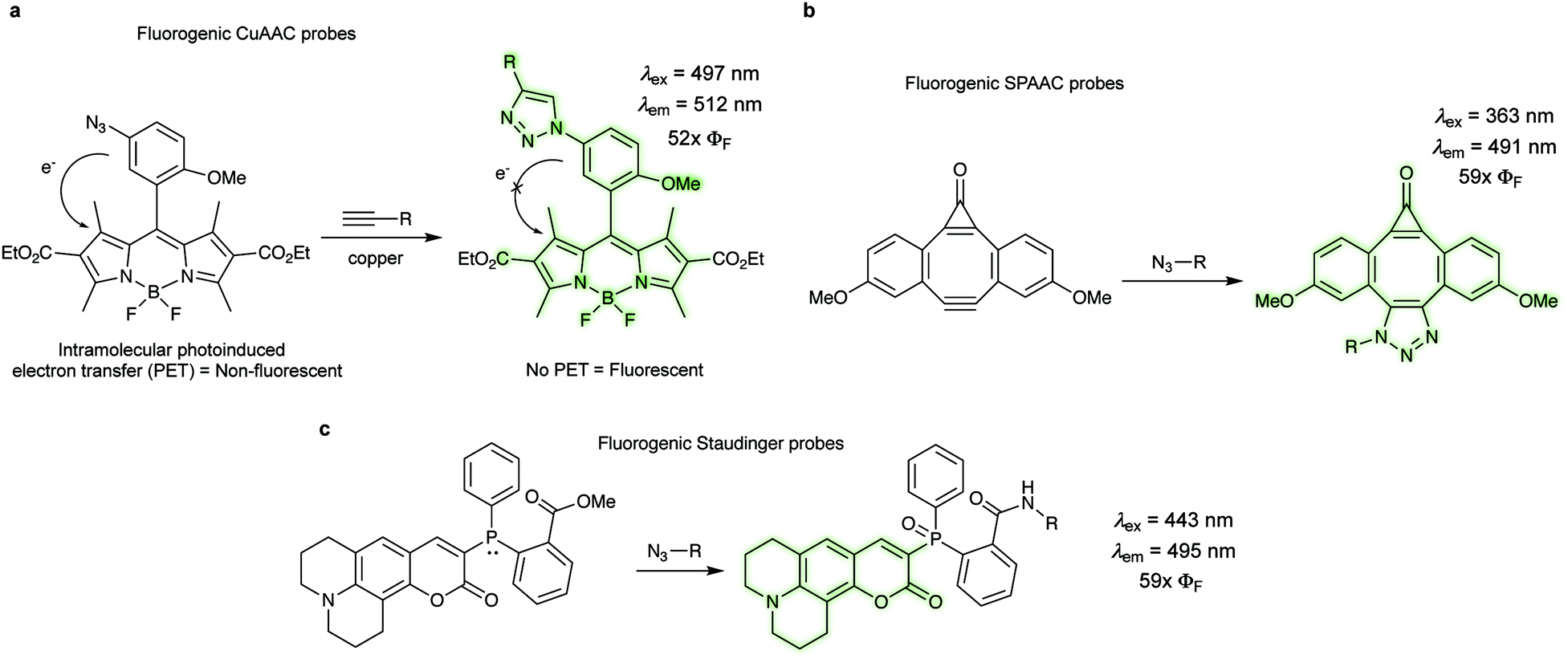 | ||
| Fig. 17 Bioorthogonal fluorogenic probes. (a) Fluorogenic CuAAC reaction of azido-BODIPY with alkynes. (b) Fluorogenic dyes activated by SPAAC. (c) Coumarin-phosphine fluorogenic dye activated by the Staudinger ligation. ΦF = fluorescent quantum yield. | ||
There are three major photophysical quenching mechanisms, which involve Förster resonance energy transfer (FRET), through-bond energy transfer (TBET), and photoinduced electron transfer (PET).125 In the CuAAC reaction the increase in fluorescence quantum yield (52×) upon triazole formation is due to the lowering of the HOMO energy level of the aryl moiety that reduce the PET to the BODIPY acceptor (Fig. 17a).124 The presence of an alkyne motif can also function as a quencher, although not so frequently. After the pioneering work on terminal alkynes by Farhni et al.126 and on cyclooctyne-fused coumarins by Bertozzi et al.,127 Boons et al. reported a dibenzocyclooctyne, which exhibited a ∼59-fold fluorescent increase after SPAAC reaction with alkynes (Fig. 17b).128 Fluorescence increase is due to large differences in oscillator strengths of the S0 ↔ S1 transitions in the planar C2v-symmetry of the starting material compared to the nonplanar cycloaddition product.128 Staudinger ligation has also been exploited for the development of fluorogenic probes (Fig. 17c). In one example, a phosphane-modified coumarin showed a 59-fold increase in fluorescence quantum yield after click reaction (λexc 443, λem 495nm).129 Prior to the Staudinger reaction the original phosphine is responsible for the quenching via internal charge transfer (ICT). Upon oxidation to the phosphine oxide, the ICT originating from the phosphorous' lone pair is no longer possible, therefore the fluorescence is restored.129 For more information about the applications of click fluorogenic probes the reader is directed to a recent review by Kele et al.130
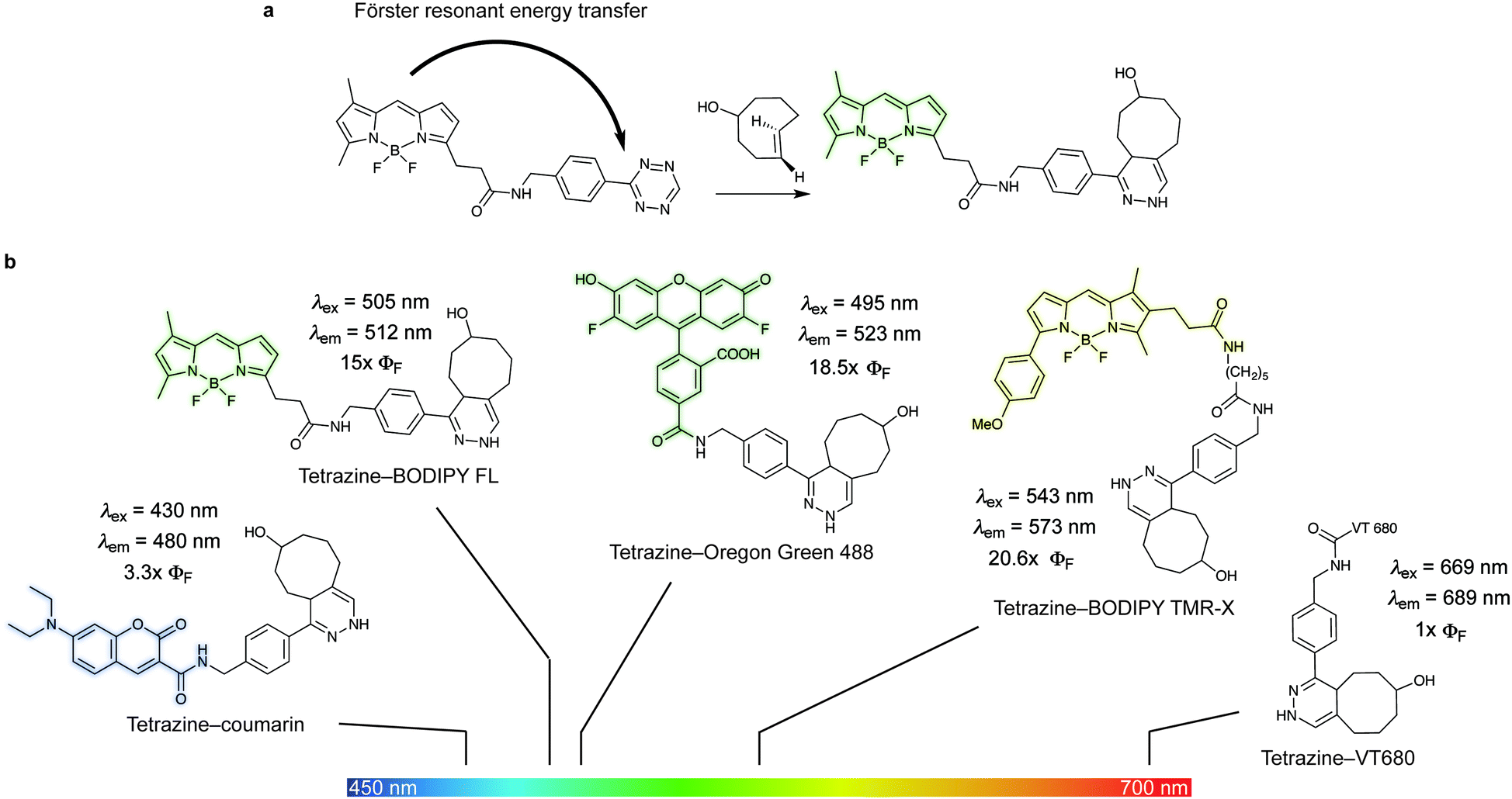 | ||
| Fig. 18 (a) Example of the mechanism of quenching of fluorophore–tetrazine conjugates by Förster Resonance Energy Transfer. As the tetrazine is consumed by cycloaddition the FRET quenching is forbidden and fluorescence is restored. (b) Photophysical properties of the depicted dyes before and after reaction with TCO.133 The extent of fluorescence quenching depends on the wavelength emission of the fluorophores and the absorption spectra of the tetrazine. ΦF = fluorescent quantum yield. | ||
Fluorogenic BODIPY probes. We and the Chin group also showed that tetrazines can quench tetramethylrhodamine (yellow-emitting, λex = 550 nm, λem = ∼585 nm, 4–5-fold increase) but the fluorescence enhancement is less pronounced.72,118 In a following work the group of Weissleder proposed an alternative way for designing more efficient turn-on probes, which consisted in the design of conformationally restricted tetrazine–BODIPY conjugates for fluorescence quenching through bond energy transfer (TBET).82 TBET systems require the energy donor and acceptor moieties to be connected through twisted, but otherwise conjugated, π-electron systems. In addition, unlike traditional FRET, TBET quenching does not require overlapping emission and absorption bands of fluorophores and tetrazines, respectively.134 This approach resulted in new fluorogenic probes with a turn-on up to 1600 times (Fig. 19a). It was shown that structures with enhanced spatial donor–acceptor proximity resulted in higher magnitudes of fluorescence turn-on. The exceptional fluorogenic turn-on ratios of the new probes compared to the flexible linked tetrazine–fluorophore conjugates (e.g. 80-fold greater than tetrazine–BODIPY TMR-X) suggest that TBET is the most likely quenching mechanism for these compounds. The suitability of this fluorophore for biological applications was demonstrated by imaging EGFR expression with TCO-functionalized antibodies on both fixed and live cells.82 Later in 2014 Wombacher et al. reported other rigid tetrazine–BODIPY conjugates, which showed less fluorogenicity upon reaction with TCO (Fig. 19b). This work further demonstrated that the close proximity between the tetrazine and the fluorophore as well as fixation of transition dipoles is crucial to obtain strong quenching.94 Indeed, the new probes Tz–BODIPY 4 and Tz–BODIPY 5 show little structural flexibility and identical geometrical layouts concerning their donor and acceptor transition dipoles. However, the BODIPY derivative with a shorter interchromophore (Tz–BODIPY 5) exhibits twice of the fluorescence of the one with a longer linker (Tz–BODIPY 4).94
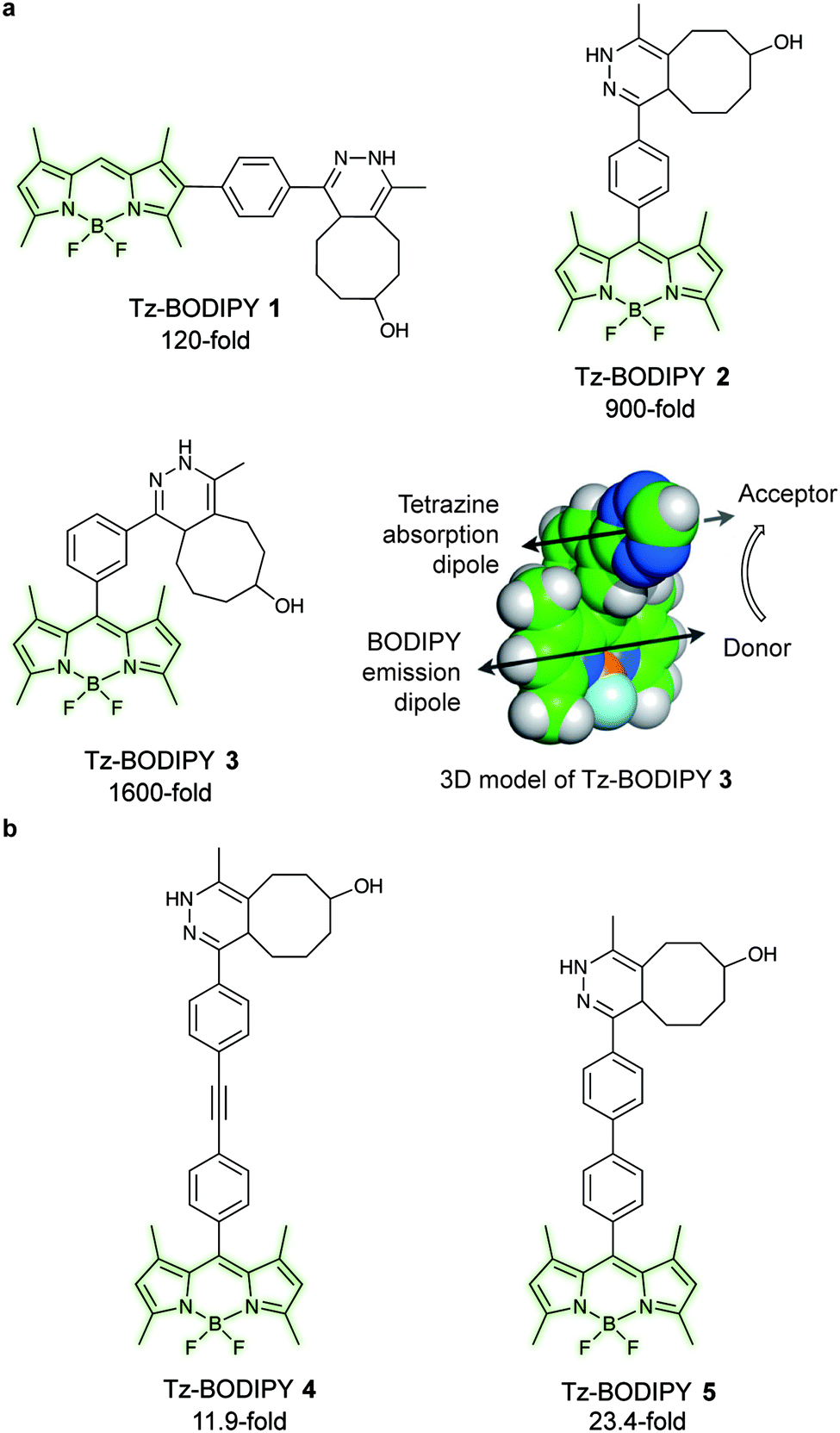 | ||
| Fig. 19 TBET-based tetrazine quenched fluorogenic probes. (a) Superbright fluorogenic tetrazine–BODIPY derivatives where the tetrazine was directly attached off the BODIPY core with a rigid phenyl linker for TBET quenching. 3D model of Tz–BODIPY 3 is depicted, illustrating a twisted phenyl linker between the BODIPY and the tetrazine chromophore and the orientation of the donor and acceptor transition dipoles.82 (b) New BODIPY derivatives with longer interchromophore linkers.94 Turn-on effect (e.g. 1600-fold for Tz–BODIPY 3) denotes the relative fluorescence quantum yield of the fluorophores before and after reaction with TCO. From ref. 82, Copyright © 2013 by John Wiley & Sons, Inc. Reprinted in part by permission of John Wiley & Sons, Inc. | ||
Fluorogenic coumarin probes. A series of coumarin–tetrazine probes were also prepared and their design included aligned transition dipoles of the coumarin fluorophore and the tetrazine in order to maximize contributions from FRET in addition to TBET (Fig. 20a). These probes, named HELIOS probes (HELIOS = hyperemissive ligation-initiated orthogonal sensing), exhibit the highest brightness enhancements reported for any bioorthogonal fluorogenic dyes with fluorescence enhancements up to 11
000-fold. The fluorescence quenching of the coumarin cores by tetrazines occurs mainly via a TBET mechanism.81 Imaging of epidermal growth factor receptor (EGFR) on the surface of cancer cells using a TCO labelled anti-EGFR antibody (Cetuximab) and HELIOS 370H was performed within seconds of dye addition and exhibited no nonspecific signal according to the control experiments (Fig. 20b).
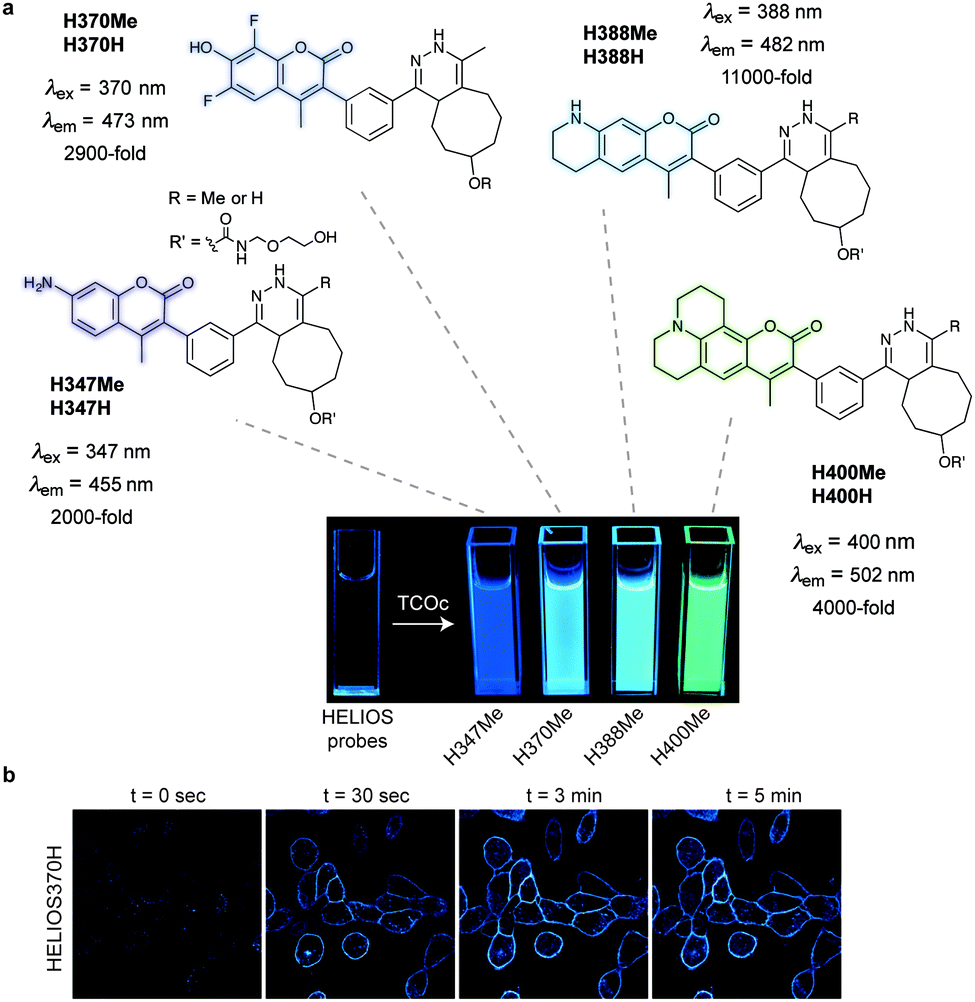 | ||
| Fig. 20 Ultra-bright fluorogenic coumarin–tetrazine probes. (a) Turn-on response of HELIOS probes using a handheld UV lamp. Fluorescence enhancement (e.g. 11000-fold for H388H) represents the relative fluorescence quantum yield of the fluorophores before and after reaction with TCO. (b) No-wash fluorogenic imaging of EGFR expression on A431 cells. Cells were incubated with an α-EGFR–TCO antibody, washed briefly, and then imaged sequentially after addition of HELIOS 370H. From ref. 81, Copyright © 2014 by John Wiley & Sons, Inc. Reprinted in part by permission of John Wiley & Sons, Inc. | ||
Fluorogenic xanthene probes. Xanthene dyes, such as fluorescein and rhodamine derivatives, are arguably the most popular class of fluorescent probes for cellular imaging and are typically highly soluble in aqueous solutions.93 In this regard, Devaraj et al. synthesized tetrazine-conjugated xanthene dyes such as Oregon-Green and tetramethylrhodamine (TAMRA) conjugated to tetrazines via a rigid styrenyl linker.93 The Oregon Green probe (Tz–OG 1) underwent a 400-fold fluorescence enhancement upon TCO reaction while the red-emitting TAMRA (Tz–Rh 1) showed a 75-fold enhancement in fluorescence (Fig. 21a).93
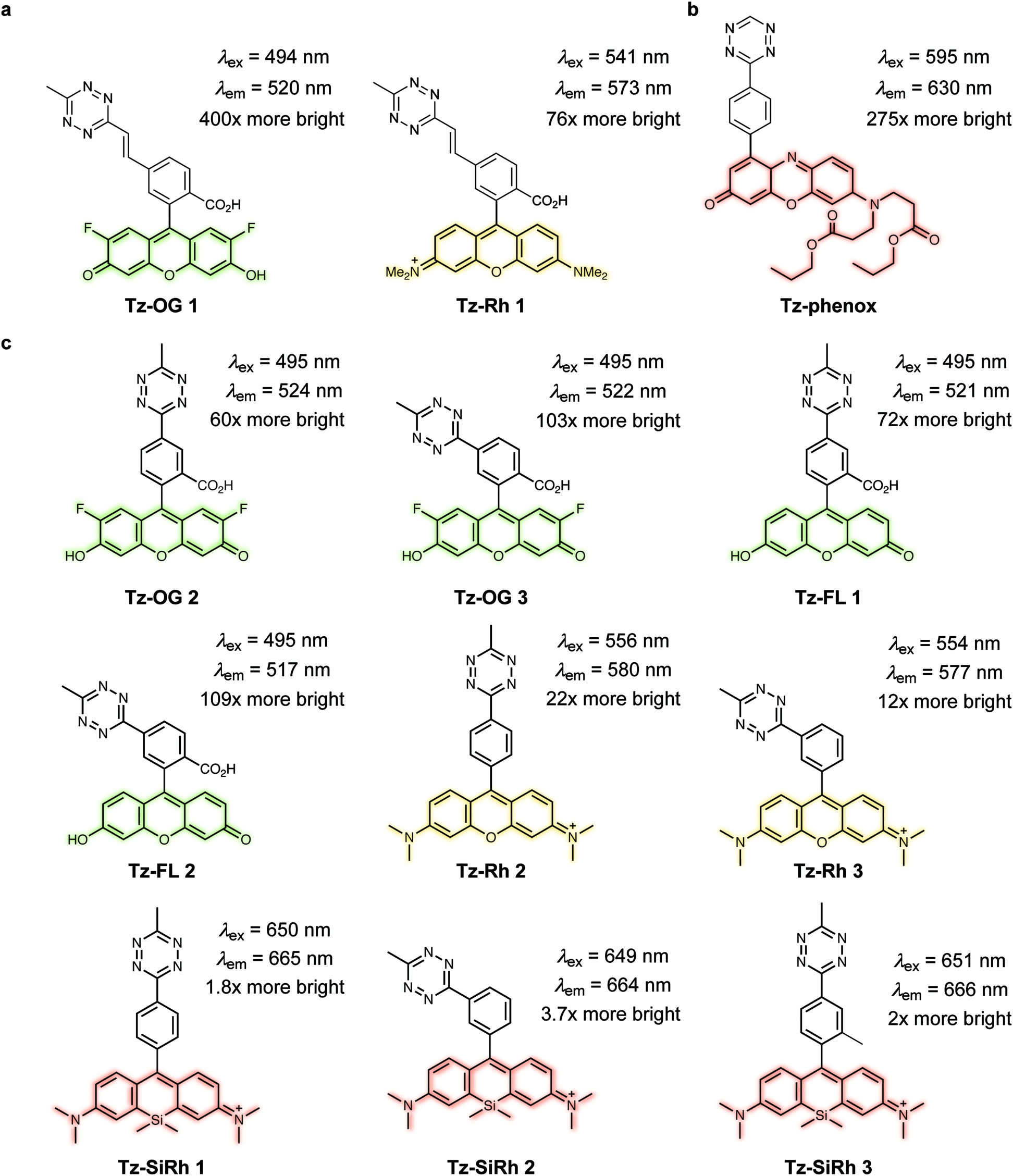 | ||
| Fig. 21 TBET-based tetrazine quenched fluorophores. (a) OG and Rh fluorophores conjugated to tetrazines via a rigid styrenyl linker.93 (b) Red-emitting fluorogenic phenoxazine probe.95 (c) Green- to far-red-emitting fluorogenic tetrazines-fluorophore conjugates.90 Turn-on effect values represent the relative fluorescence quantum yield of the fluorophores before and after IEDDA reaction. FL = fluorescein; OG = oregon Green; Phenox = phenoxazine; Rh = rhodamine; SiRh = silicon-rhodamine. | ||
Recently, Kele et al. reported new tetrazine-phenoxazine fluorogenic probes. The red excitability and emission properties of these probes ensure minimal autofluorescence, while TBET fluorogenicity reduces nonspecific background fluorescence. Indeed, due to its high lipophilicity phenoxazine is often compromised by high background fluorescence, however, the high fluorogenicity of the new tetrazine-phenoxazine probes (up to 275× turn-on) allowed efficient labelling of live cells (Fig. 21b).95 To expand on this approach in the context of multicolor imaging, Wombacher et al. synthesized a panel of rigidly-linked green- to far-red-emitting fluorogenic tetrazine probes (Fig. 21c).90 Once again the authors found that the distance between the tetrazine and the fluorophore, and the fluorophore itself, are crucial for efficient quenching and for the resulting fluorescence turn-on upon conversion by IEDDA reaction.
000 M−1 cm−1, Stokes shift = 24), PDHP (ex = 360 nm, em = 465 nm) has relatively low quantum yield (0.011 in MeOH and 0.251 in ACN) and extinction coefficient (4329 M−1 cm−1 in MeOH and 3769 M−1 cm−1 in ACN).135 On the other hand, PDHP has a large Stokes shift (∼100 nm), which could be beneficial (e.g. less self-quenching and/or auto-fluorescence background) in certain imaging applications. Finally, a lysine-derived non-canonical amino acid containing styrene moiety (KStyr) was genetically encoded into an intracellular stress response protein, HdeA, and labelled in living cells after tetrazine reaction. Recently, Vrabel et al. reported a fluorogenic reaction between axial-TCO with tetrazine (Fig. 22b). By screening a series of tetrazines, the photochemical properties of the resulting 1,4-dihydropyridazines could be tuned towards 91-fold fluorescence intensity increase and emissions ranging from 480 to 605 nm. By modifying tetrazines with peptides on resin, the addition of axial-TCO resulted in rapid fluorescent product within 1.5 min. Furthermore, taxol- and triphenylphosphonium-tetrazine derivatives were also synthesized for targeting subcellular compartments and efficient and fast fluorogenic labelling was observed on microtubules and mitochondria.136
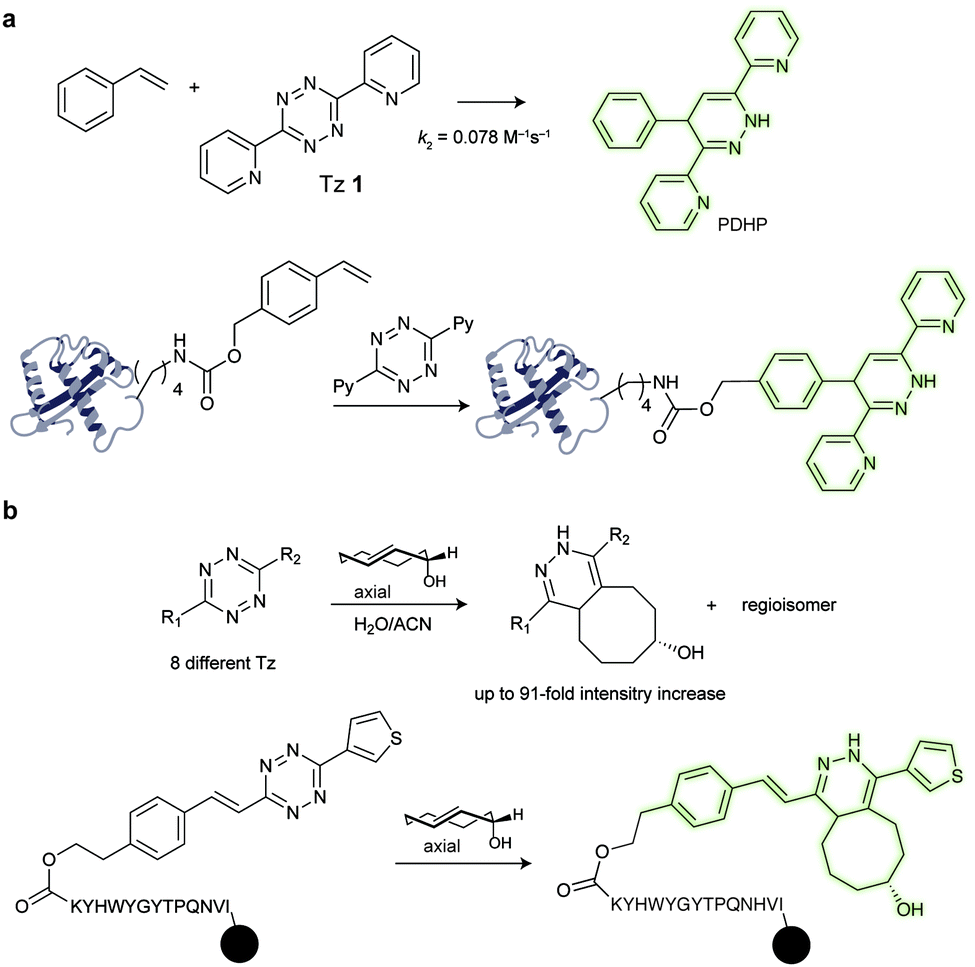 | ||
| Fig. 22 (a) Fluorogenic reaction between styrene and tetrazine, and selective labelling of HdeA. A Plasmid pHdeA was constructed to encode an HdeA mutant containing the lysine-styrene non-canonical amino acid (KStyr) at position 28 (HdeA-F28KStyr). E. coli cells expressing HdeA-F28KStyr were successfully labelled after tetrazine reaction. (b) Fluorogenic reaction between a-TCO and tetrazines, and fluorogenicity with a peptide-linked beads. | ||
4. Methods for installing IEDDA chemical reports on biomolecules
Selective and site-specific labelling of proteins is essential for studying protein structure, activity, localization and trafficking. During the last decade, a wide variety of IEDDA bioorthogonal reporters have been incorporated into biomolecules through genetic encoding of non-canonical amino acids (ncAAs) during protein synthesis, or through enzymatic and chemical modification of native residues on post-expressed proteins. These methods will be presented and discussed in the following sections.4.1 Genetic code expansion
The development of strategies to genetically encode ncAAs into proteins has had arguably the greatest impact upon the progress of bioorthogonal chemistry in recent years. In this approach, an aminoacyl-tRNA synthetase (aaRS)/transfer RNA (tRNA) pair is used to insert an ncAA into the growing protein chain in response to an amber stop codon (UAG) on the mRNA.137–139 A plethora of ncAAs bearing ketones, aldehydes, azides, alkynes, alkenes, tetrazines, or aryl halides reactive groups have been developed in recent years using this strategy and incorporated into E. coli, yeast, mammalian cells140 and even animals.141,142The vast majority of studies on the incorporation of ncAAs in IEDDA applications so far have used the pyrrolysine synthetase/tRNA pair (PylRS/tRNACUA) from methanosarcinia for three reasons. Firstly, this synthetase, in its native form, incorporates none of the 20 canonical amino acids. Secondly, the high promiscuity of PylRS and the evolutionary mutants allowed the incorporation of more than 300 hundreds kind of ncAAs.143 Finally, while the use of other synthetase–tRNA pairs for incorporating ncAAs is limited to cells from a particular organisms, orthogonal PylRS variants can be selected for E. coli and then used to incorporate ncAAs into yeast, mammalian cells and animals.143,144 The enormous progresses made in the field of genetic code expansion over the recent years has resulted in up to 26 ncAAs incorporated into proteins for IEDDA applications (Fig. 23). A summary of the most significant examples is described in the following sections.
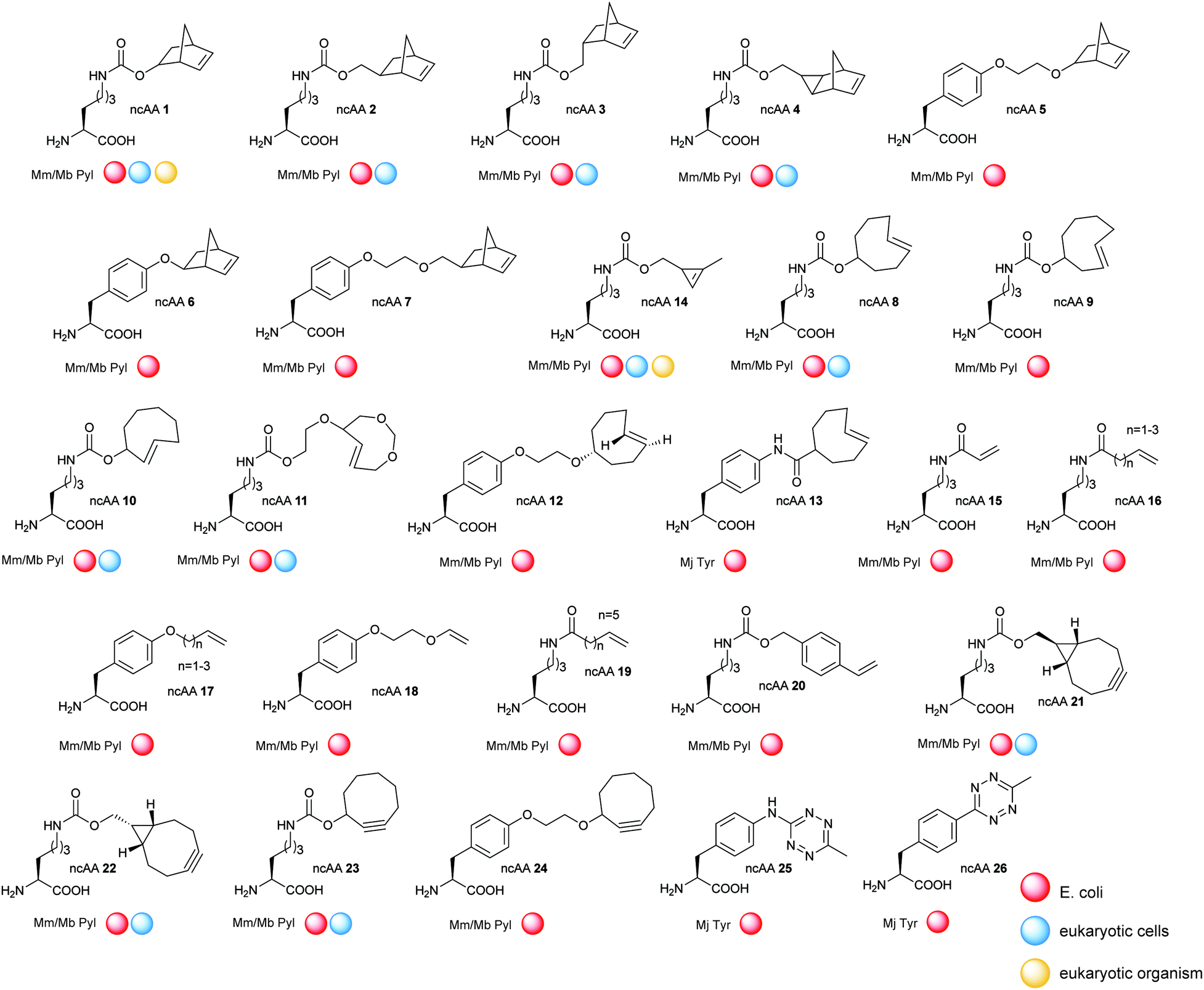 | ||
| Fig. 23 Summary of ncAAs that have been genetically encoded into proteins for IEDDA application. | ||
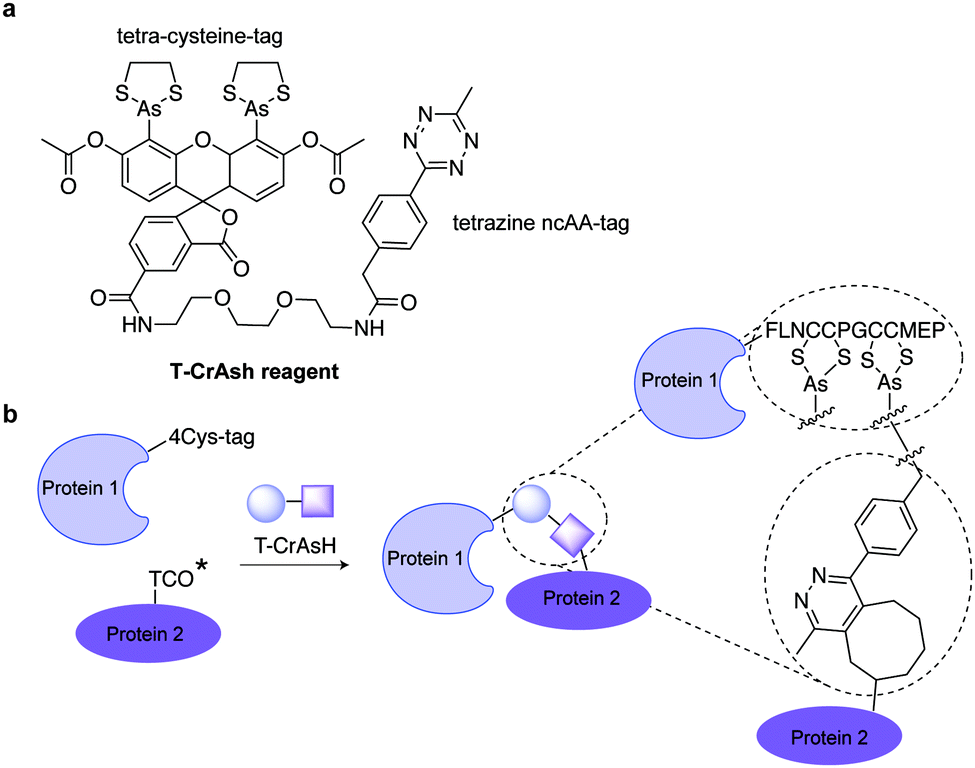 | ||
| Fig. 24 (a) Chemical structure of T-CrAsH reagent and the products of the two covalent reactions. (b) General scheme of the T-CrAsH method. | ||
Although TCO has been successfully used for labelling genetically encoded proteins, several reports have described that the ncAA TCO-Lys is relatively sticky resulting in high non-specific fluorescence background upon addition of the tetrazine dyes, even if extra washing steps are applied to remove excess of non-incorporated ncAA.8,112,152 With this in mind, an hydrophilic TCO-Lys derivative containing exo- and endo-cyclic heteroatoms introduced in the TCO ring was developed in order to increase hydrophilicity. The new hydrophilic ncAA DOTCO-Lys (ncAA 11) showed high incorporation efficiency in both prokaryotic and mammalian cells using a double-mutant pyrrolysyl-tRNA synthetase.120 Importantly, comparative washout studies revealed that only 5 min are required to remove DOTCO-Lys from cells whereas more than 6 h are needed to remove excess of BCN-Lys and TCO*Lys (Fig. 25). Thus, this hydrophilic DOTCO-Lys could be used to label and track intracellular proteins with rapid turn-over and expressed in low concentrations.120 Apart from Lys derivatives, TCO-Tyr ncAAs (ncAA 12, ncAA 13) have been also incorporated into proteins in E. coli.148,153
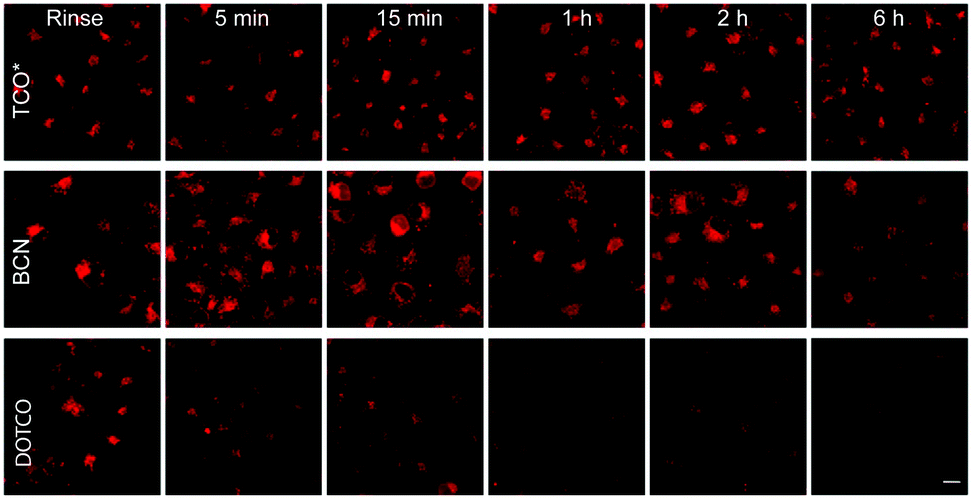 | ||
| Fig. 25 Comparative washout studies with DOTCO, BCN and TCO ncAAs. Confocal microscopy imaging of non-transfected cells washed for different times before labelling with a tetrazine dye. With DOTCO intracellular nonspecific background is easily removed from the cytoplasm after washing for 5 min. From ref. 120, Copyright © 2016 by John Wiley & Sons, Inc. Reprinted in part by permission of John Wiley & Sons, Inc. | ||
Bicyclo6.1.0nonyne (BCN). BCN-Lys (ncAA 21) was first encoded into sfGFP in E. coli and live mammalian cells by Chin et al. using a MbPylRS triple mutant (Y271M, L274G, and C313A).112 Contemporaneous with this work Lemke et al. reported the incorporation of BCN (ncAA 22) into several other proteins (EGFR, hepatitis D virus, actin, vimentin and FK506-binding protein), via MbPylRS and MmPylRS mutants.151,152,159,160 Recently Chin et al. developed a genetically directed bioorthogonal ligand tethering (BOLT) method and used it for selective inhibition (iBOLT) of protein function. Specifically, BCN-MEK variants (MEK = mitogen-activated protein kinase) and specific MEK inhibitors conjugated to a tetrazine through different linkers were used for rapid and covalent inhibition of MEK isozymes. By introducing an azobenzene linker between the tetrazine and the inhibitor (photo-BOLT), reversible and optical regulation of MEK kinase activity was achieved in live cells by cis/trans isomerisation of the azobenzene. This technique was shown useful for spatiotemporal control of protein function.161 In another example, BCN encoded sfGFP has been found to undergo IEDDA ligations with tetrazine glycan conjugates both in vitro and in vivo, providing another method for rapidly and homogenous engineering of glycoproteins.162
Strained cyclooctyne (SCO). First developed for site-specific protein labelling through SPAAC with fluorogenic azides,163 genetically encoded SCO handles were later used for IEDDA applications. SCO (ncAA 23) was incorporated into GFP via a double mutant synthetase from Mm and labelled with a Cy5 tetrazine dye. Detectable fluorescence was observed only after 2 h with excess of dye that led to significant background staining.119,149 Apart from Lys derivatives, SCO-Tyr ncAA (ncAA 24) has been also incorporated into proteins in E. coli and modified through IEDDA.148
500 M−1 s−1 and a half reaction time of 12–14 seconds against s-TCO. This new ncAA enabled the efficient sub-stoichiometric labelling of proteins in live cells, eliminating the need for washing out excess labelling agents.121
Dual genetic encoding has been also described using the promiscuity of tRNA synthetases. Indeed, Lemke et al. showed that a PylRS/tRNACUA double mutant from Mm could accept both SCO and TCO*. As a result, simultaneous incorporation of two different ncAAs into proteins was possible via a pulse-chase labelling strategy.8 By fine-tuning the diene partners, selective labelling of these ncAAs allowed dual colour labelling of biological relevant proteins (Fig. 26). Specifically, Lemke's lab found that, under certain reaction conditions and timescale of the experiment, TCO* reacts with both mono-substituted (H-Tz) and di-substituted (Me-Tz) tetrazines, while SCO reacts only with mono-substituted variants (Fig. 26). Of note, the reactivity of H-Tz and Me-Tz with other dienophiles was recently studied in detail and to proceed as follows: H-Tz:endoBCN > or ≈ exoBCN > TCO*mix isomers ≫ SCO ≫ norbornene; Me-Tz: TCO*mix isomers ⋙ endoBCN > or ≈ exoBCN ⋙ SCO > norbornene. This provides guidance for constructing orthogonal reactions and dual labelling systems.67
 | ||
| Fig. 26 Dual protein labelling on live cell surface for super-resolution microscopy. TCO-bearing and SCO-bearing amino acids were incorporated into insulin receptors. The labelled proteins were visualized via treatment with a fluorescent Me-Tz (cyan, Me-Tz-Cy5, to tag TCO), followed by a fluorescent mono-substituted tetrazine probe (magenta, H-Tz-Atto532 to tag SCO). From ref. 8, Copyright © 2014 by John Wiley & Sons, Inc. Reprinted in part by permission of John Wiley & Sons, Inc. | ||
4.2 Alternative enzymatic methods or enzyme tags for protein modification by IEDDA
An alternative to genetic encoding methods for protein bioconjugation uses engineered enzymes that covalently attach a labelled substrate to a tagged protein of interest by recognizing a peptide sequence or a small fused protein (e.g. trypsiligase, lipoic acid ligase, PFTase, sortase). Another approach involves the use of self-labelling enzymes that have been engineered to attach a fluorescent molecule to one of its own amino acid residues (SNAP/Clip tag, HaloTag).1,3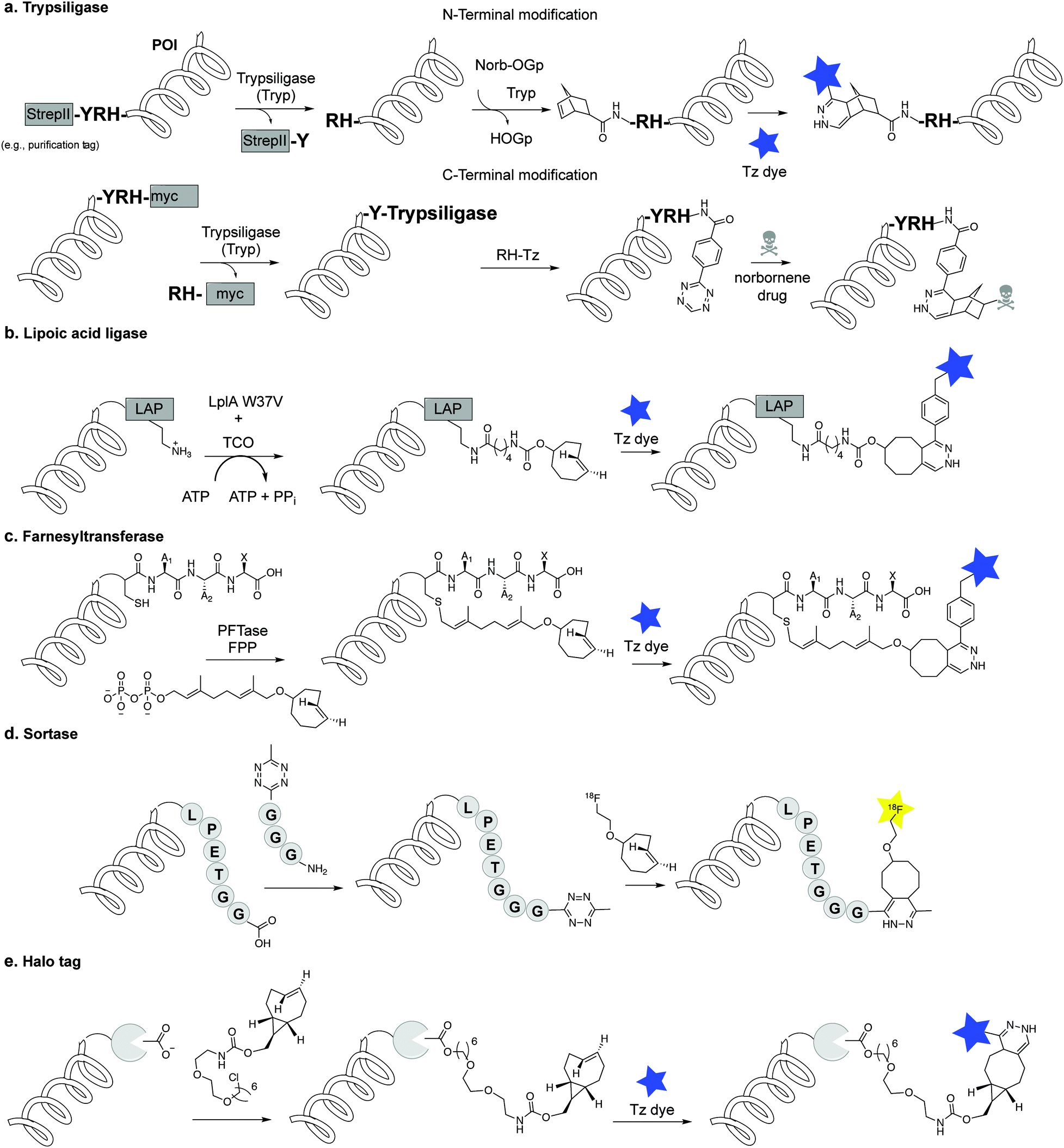 | ||
| Fig. 27 Enzymatic methods for protein labelling through IEDDA. (a) Protein modification via trypsiligase. (b) Site-specific fluorescence labelling of proteins using LplA and tetrazine reactions. (c) Labelling of CaaX peptide motif at the C-terminus with farnesyl isoprenoid analogs catalysed by PFTase. (d) Sortase bioconjugation. (e) HaloTag-based bioconjugation. | ||
4.3 Direct incorporation of unnatural nucleotides into RNA/DNA for IEDDA modification
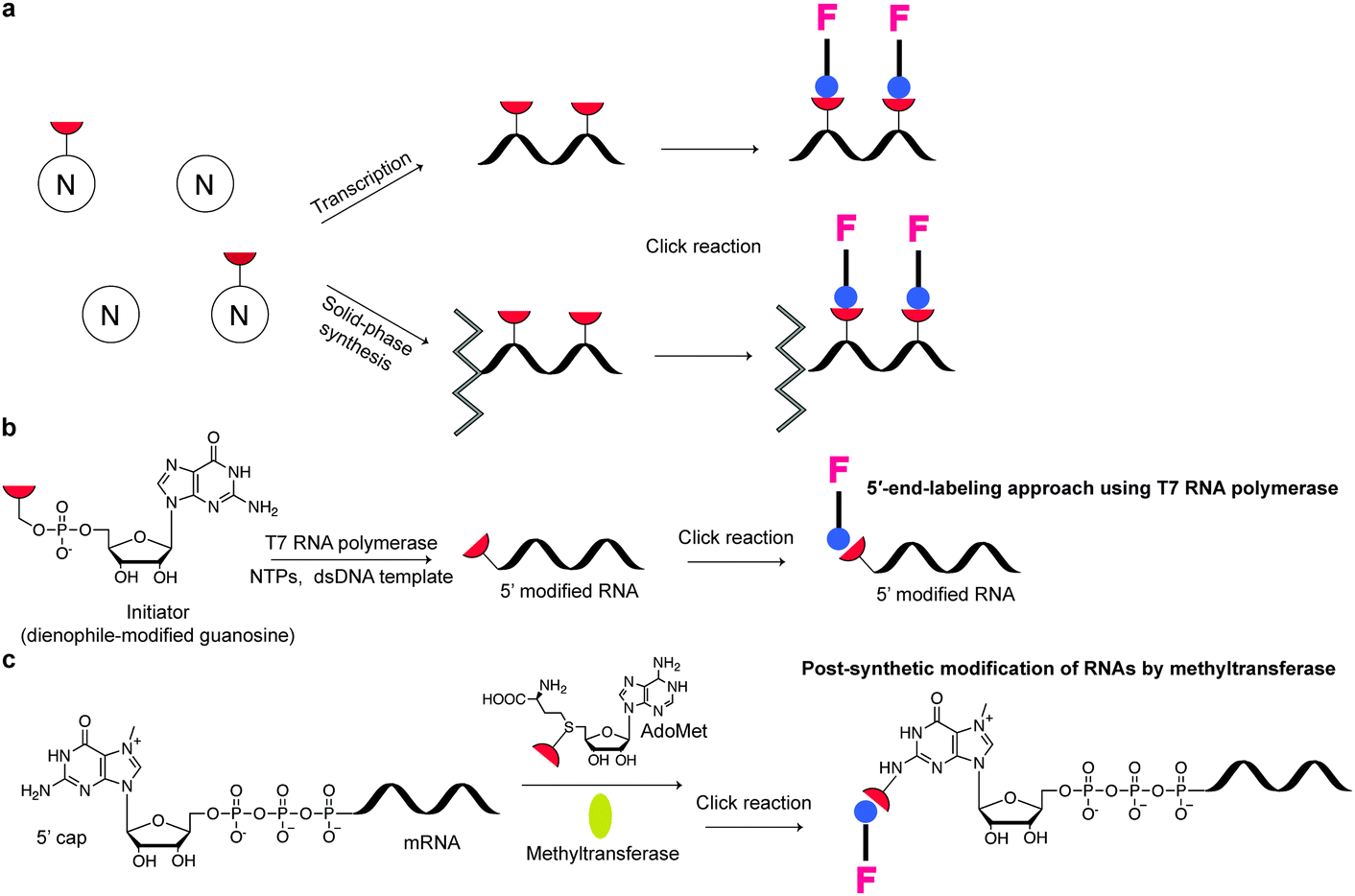 | ||
| Fig. 28 Methods used for incorporation of IEDDA bioorthogonal handles into RNA for subsequent labelling by click chemistry. (a) The promiscuity of various RNA polymerases tolerates the incorporation of unnatural nucleotides, producing modified RNAs. (b) Labelling of RNAs using the 5′-end-labelling approach employing the T7 RNA polymerase. (c) Post-synthetic modification of RNA by chemo-enzymatic labelling of 5′-capped RNAs. A methyltransferase (MTase) variant can be used to modify the mRNA cap with the reactive handle if the respective S-adenosylmethionine (AdoMet) analog is provided. NTP = Nucleoside triphosphate. | ||
Recently, Rentmeister et al. showed that a variant of trimethylguanosine synthase 2 from Giardia lamblia (GlaTgs2-Var1) recognises a novel AdoMet-analog bearing a vinylbenzyl group, allowing the modification of capped RNA structures at the N2 position of the 5′ cap (30% yield in 3 h with 10% of GlaTgs2-Var1).116 Similarly, an allyl modified RNA was prepared in 90% yield. The differences in yields were attributed to steric constraints in the substrate-binding pocket of the methyltransferase. Subsequent IEDDA labelling with a tetrazine-TAMRA derivative showed quantitative conversion of the vinylbenzyl modified 5′-capped RNA, but not on the allyl-modified RNA, possibly due to the relatively low reactivity of the allyl handle. This approach was proven to be efficient for a 106-long capped RNA.116 It has also been shown that a cap (guanine N7) methyltransferase from the microsporidian parasite Encephalitozoon cuniculi (Ecm1) could transfer bulky side chains from AdoMet analogues to 5′-capped mRNAs at position N7 with almost quantitative conversion, which allowed efficient labelling with a tetrazine dye.184 By using the above-mentioned methyltransferases and exploring their regioselectivity, dual 5′ Cap labelling of mRNAs was also achieved.185
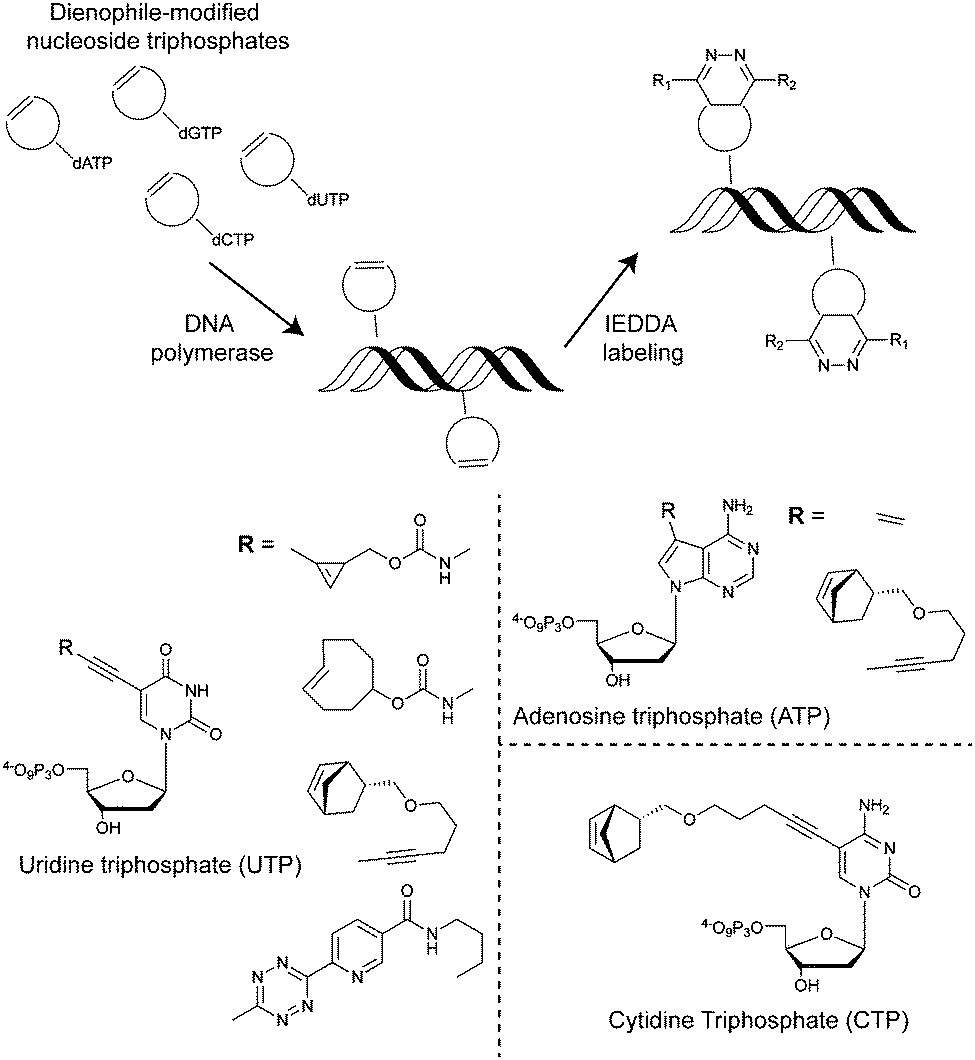 | ||
| Fig. 29 Examples of IEDDA nucleotides incorporated into DNA. | ||
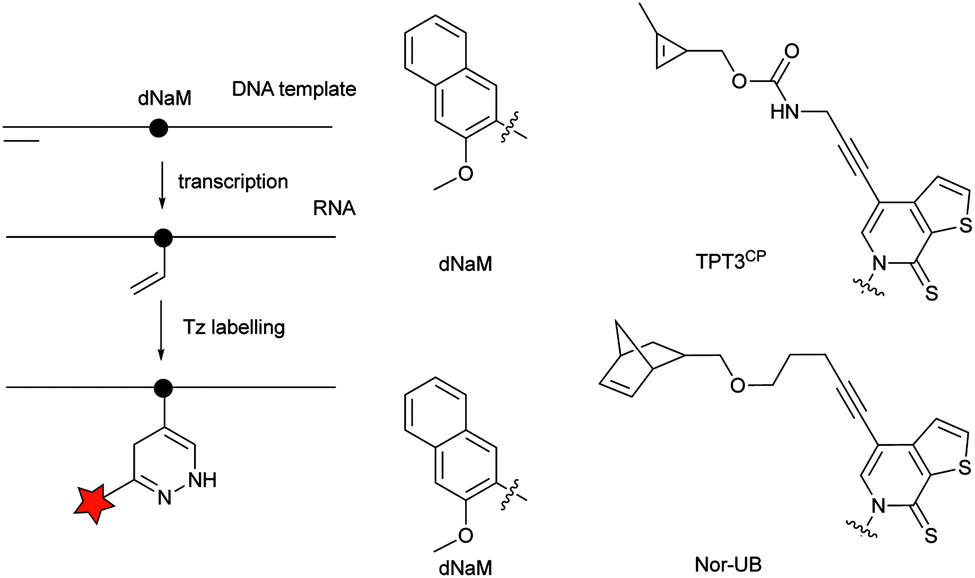 | ||
| Fig. 30 Unnatural nucleobase 2-methoxy-3-methylnaphthalene (dNaM) is placed in the DNA template to site-specifically incorporate the complementary cyclopropene (TPT3CP) or norbornene-bearing unnatural base (Nor-UB) during T7 in vitro transcription. | ||
4.4 Metabolically incorporation for glycan modification
The simplest method for chemically labelling relevant biomolecules in living systems is metabolic labelling, in which the endogenous machinery of living cells is used to incorporate reactive handles into the biomolecules. In general, this is accomplished by growing cells or organisms in media in which a specific natural substrate (e.g. amino acid, nucleotide and carbohydrate) is replaced with a close analogue bearing a chemical reporter for subsequent modification with affinity or biophysical tags. Consequently, cells use the chemical analogue instead of the natural substrate to synthesize or modify glycans, nucleic acids or proteins. Numerous bioorthogonal probes have been developed for metabolic labelling including azidohomoanaline for labelling newly synthesized proteins by replacement of natural Met residues, azido sugars for labelling glycoproteins and 5-ethynyl-2′-deoxyuridine (EdU) for labelling nucleic acids.195 To the best of our knowledge, IEDDA reactions have thus far been successfully applied for metabolic labelling of glycans, and nucleic acids, but the possibility of using the tetrazine click reaction to label proteins tagged with a dienophile-bearing ncAA (or vice versa), incorporated using the natural translational machinery, is still to be explored.Glycans participate in diverse biological processes such as extracellular matrix formation, protein folding, protein stability and function, regulation of the interaction of proteins with one another, etc.196 In addition, disordered glycosylation is associated with a number of diseases.197 The ability to monitor glycans could provide fundamental insights into their roles in cell biology. While azide and alkyne reactive handles have been the most popular for labelling tagged glycans using bioorthogonal chemistry,198 IEDDA-based approaches are now advancing.
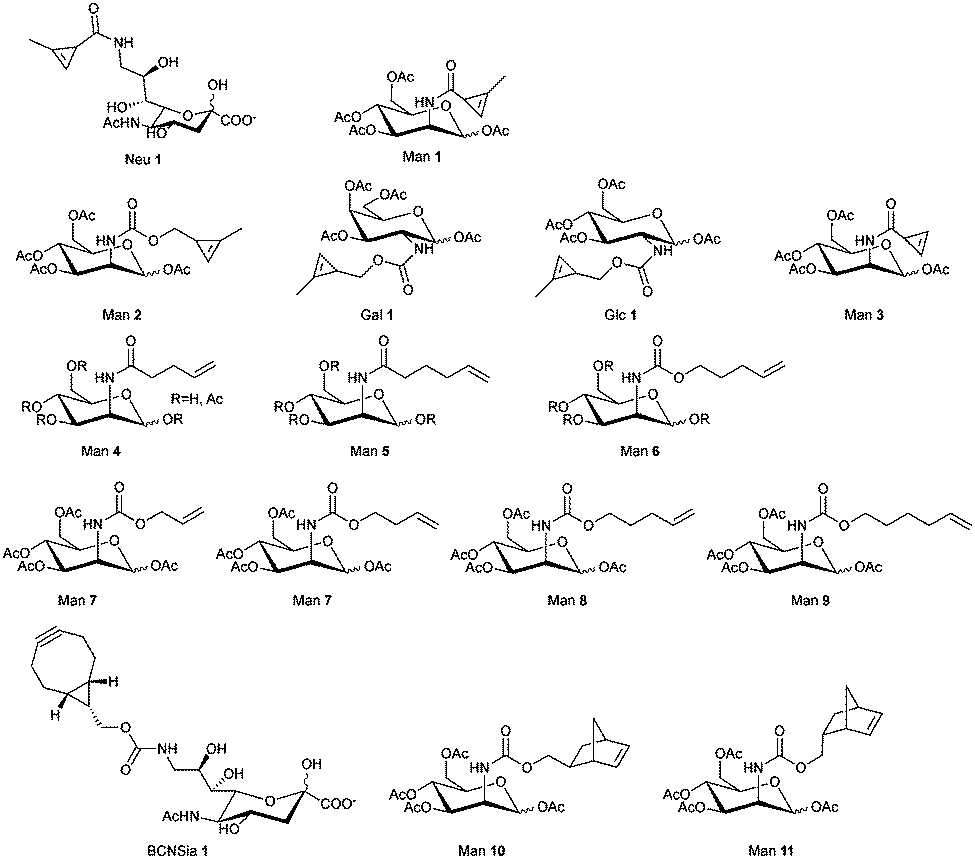 | ||
| Fig. 31 Carbohydrate derivatives used in glycan engineering to date for IEDDA labelling. | ||
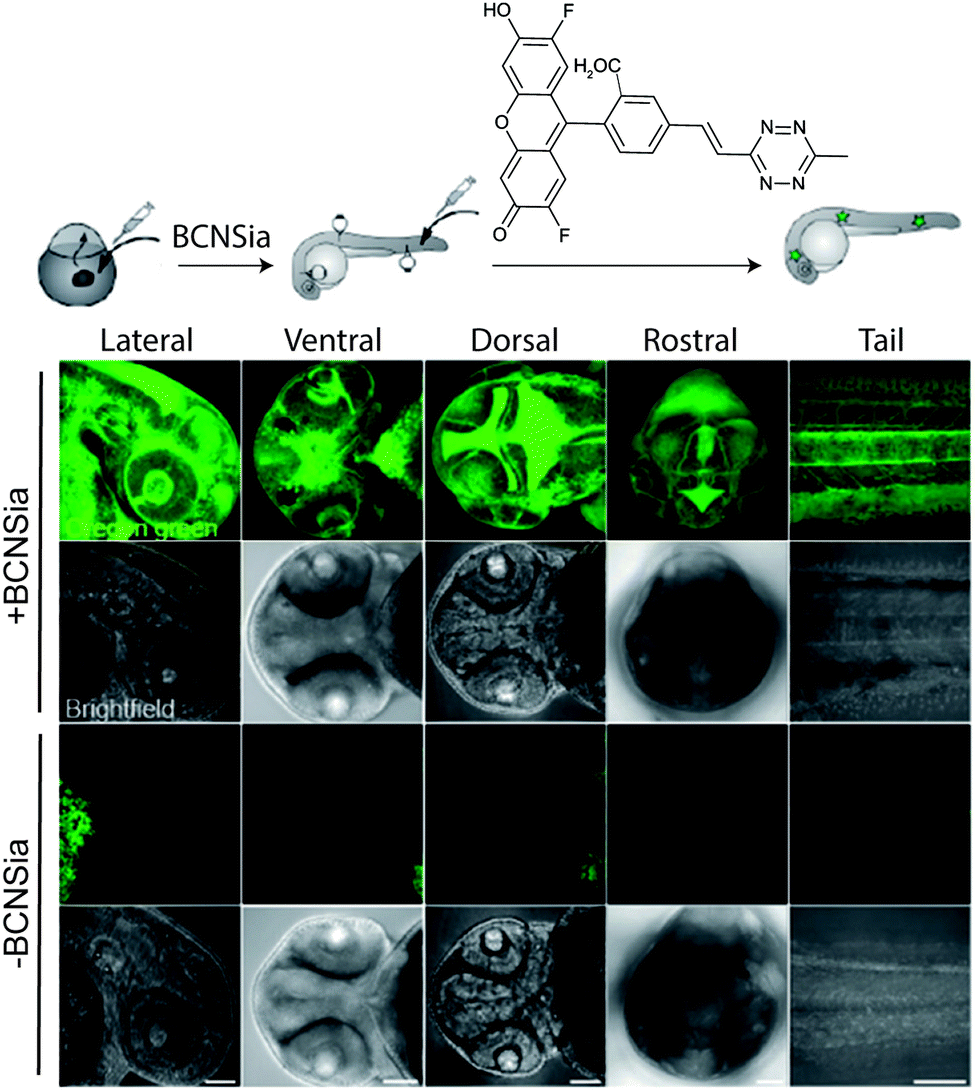 | ||
| Fig. 32 Zebrafish embryos treated with or without BCNSia and subsequently injected with a fluorescent tetrazine. From ref. 207, Copyright © 2014 by John Wiley & Sons, Inc. Reprinted in part by permission of John Wiley & Sons, Inc. | ||
4.5 Unnatural D-amino acids IEDDA reporters
Exogenous unnatural D-amino acids have been incorporated in place of the terminal D-alanine of bacterial peptidoglycans and are being used for remodelling and imaging of bacterial surfaces. Thanks to the promiscuity of transpeptidase, various alkene-displaying D amino acids (from terminal alkenes to norbornene) were incorporated onto the cell surface and labelled by reaction with fluorescent tetrazines.208 A D-Dap-norbornene (Dap = D-diaminopropionic acid) showed the best labelling efficiency with a balance between incorporation efficiency and IEDDA reactivity. A tetrazine bearing D-amino acid (D-Dap-Tet-NH2) was also tested for metabolic remodelling and subsequent TCO imaging. Despite the expected fast kinetics, labelling efficiency with D-Dap-Tet-NH2 was lower than with D-Dap-NB-NH2, presumably due to limited incorporation levels.2084.6 Bioorthogonal IEDDA lipid chemical reporters
Bioorthogonal chemical reporters for lipid labelling and imaging have also attracted widespread interest in recent years.209 In one example, a cyclopropene tagged phospholipid was used for visualizing phospholipid uptake and distribution in human cells (SKBR3 breast cancer cells). Upon treatment with a fluorogenic tetrazine, rapid membrane staining was observed.104 Following these studies, a TCO-tagged ceramide lipid and a reactive tetrazine were used for imaging Golgi structures and dynamics in live cells by 3D confocal and stimulated emission depletion (STED) microscopy.2104.7 Chemical introduction of IEDDA bioorthogonal handles
In addition to genetic encoding, the development of reactions that enable the selective installation of reactive handles on relevant biomolecules for subsequent modification/imaging would facilitate the study of the biological function of these targets. Within the last few years few examples were reported for the chemical installation of IEDDA reporters on proteins. Functionalization of proteins with IEDDA partners using NHS-chemistry has been widely developed for conjugation; however, this method is not site-selective resulting in heterogeneous mixtures of conjugates. As example, Weissleder et al. converted TCO into the reactive succinimidyl carbonate for conjugation to amine-containing biomolecules, including antibodies. Cetuximab–TCO conjugate was used for pre-targeted imaging of A549 cancer cells that overexpress EGFR (Fig. 33a). Recently, we demonstrated that S-allyl Cys could be easily chemically installed into apoptotic protein markers through a [2,3]-sigmatropic rearrangement with allyl selenocyanate. We further demonstrate the utility of this minimal handle for the efficient pre-targeted labelling of apoptosis in cells using fluorogenic tetrazine dyes (Fig. 33b).118 Recently, Smith et al. developed a protocol for the direct labelling of peptides/proteins using a crosslinking dichlorotetrazine reagent that reacts with two proximate cysteine sulfhydryl groups by nucleophilic displacement. Importantly, the authors could extend the strategy to label a protein (thioredoxin), which contains a single, solvent-exposed disulfide bond necessary for its activity. An attractive feature of the S,S-tetrazine crosslinking is the possibility to regenerate the starting disulfide by a UV-catalysed tetrazine decomposition release process and thus, in the case of thioredoxin, restore original activity (Fig. 33c).211 Another method for chemical modification uses a bifunctional linker that contains a “IEDDA clickable” handle and a maleimide to tag cysteine/selenocysteine residues on protein surfaces.212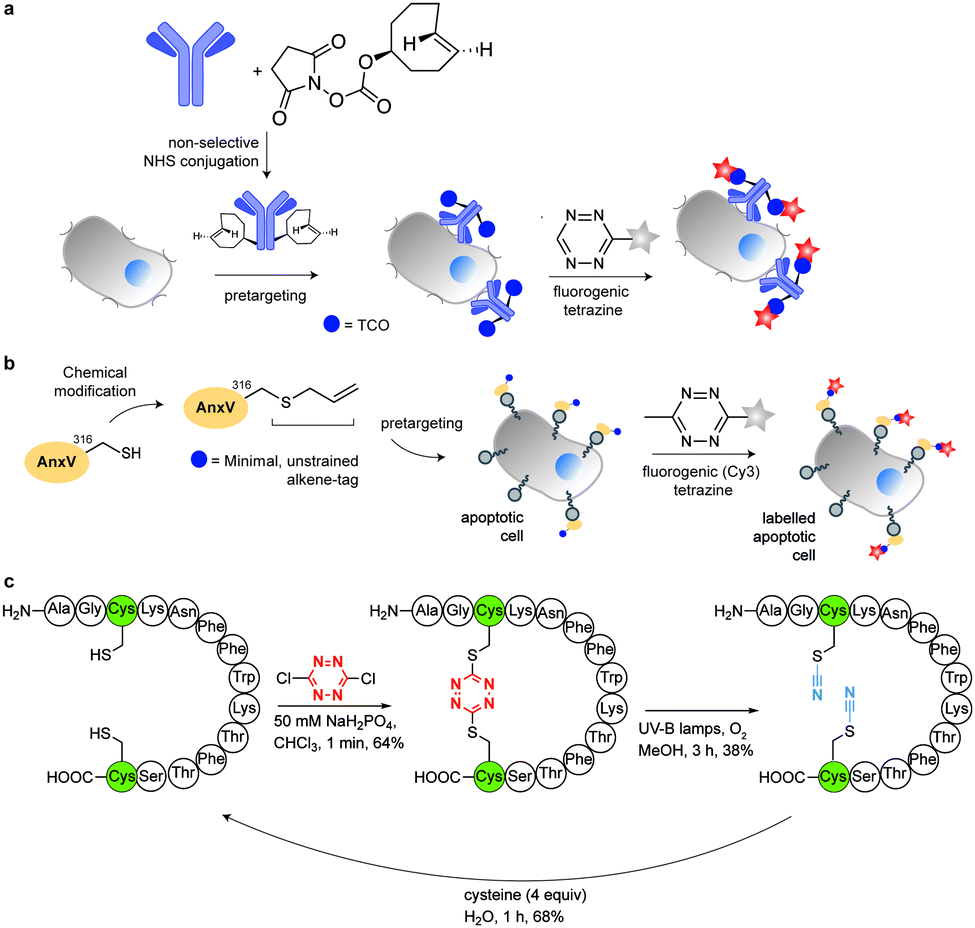 | ||
| Fig. 33 Chemical installation of IEDDA reporters for pretargeting imaging approaches. (a) TCO-antibody prepared by NHS-chemistry for pre-targeted antibody labelling. (b) Chemical installation of terminal alkene handles for imaging apoptotic cells. (c) S,S-tetrazine crosslinking mechanism that regenerates the starting disulfide by a UV-catalysed tetrazine decomposition release process. | ||
5. Selected applications of IEDDA reaction in biological systems: examples where tetrazine ligation is needed
IEDDA ligations have enormous potential for bioorthogonal approaches and thus have been used to get insights into the behaviour and function of a variety of biomolecules such as proteins, antibodies, nucleic acids, glycans, lipids or bioactive small molecules. As an example, the very fast kinetics in biological media at low concentrations make the tetrazine–TCO ligation particularly useful for tracking and imaging fast biological processes.167,213 Other applications of the “tetrazine ligation” include fluorescent labelling of low-abundance proteins within living cells for super-resolution imaging,152 identification of the targets of bioactive small molecules and proteins in living cells,214 protein profiling in living systems,155 and sequence-specific detection of DNA and mRNA.215 The most exciting and recent examples on these topics are summarized in the next sections.5.1 Super-resolution imaging of site-specifically labelled proteins
Until today, fluorescent proteins (e.g. green fluorescent protein (GFP)) have been widely used to image and track recombinant proteins in live cells. Although powerful, fluorescent protein tags are limited since their relatively large sizes may potentially interfere with protein function.216 In contrast, methods to site-specifically label proteins in their cellular context with small, bright and photostable fluorophores by means of bioorthogonal approaches would substantially advance super-resolution imaging.3,216 In fact, coupling fluorophores directly to a protein of interest has, comparing with fused fluorescent proteins, the tremendous advantage of minimizing the distance between probe and target, increasing spatial resolution. This becomes apparent when comparing the resolution achieved in live-cell stimulated emission depletion (STED) imaging experiments of microtubules labelled with SiRh fluorophore coupled via a SNAP-tag to a microtubule-binding protein217 or directly using a docetaxel-SiR derivative218,219 (SiR-tubulin).3 In addition, synthetic fluorophores can have more favourable spectroscopic properties (e.g. brightness and photostability) than fluorescent proteins.220 Recent advances in genetic code expansion and bioorthogonal chemistry have enabled the efficient labelling of proteins. However, super-resolution imaging has been accomplished only just recently.8,152 Two challenges have limited progress in this area: (i) the low efficiency of non-canonical amino acid incorporation that limits labelling density and therefore spatial resolution and (ii) the uncharacterized specificity of intracellular labelling that will define signal-to-noise, and ultimately resolution, in imaging.152,221 Optimization of incorporation efficiency, coupling chemistry, and labelling protocols recently opened the way to the application of IEDDA chemistry in super-resolution microscopy. In one example, Chin et al. reported the efficient production of cytoskeletal proteins (β-actin and vimentin) containing a bicyclo[6.1.0]nonyne-Lys at a specific site. These functionalized proteins could be efficiently labelled with SiRh tetrazines creating densely labelled cytoskeletal ultrastructures. Super-resolution imaging by stochastic optical reconstruction microscopy (STORM) revealed sub-diffraction features, including nuclear actin filaments (Fig. 34).152 Furthermore, actin with distinct morphologies at different cellular locations could be observed with nanometre-scale refined features (Fig. 34).152 Even though the super-resolution imaging was performed on fixed samples, which usually show much less background signal than live samples because it is easier to wash out unreacted probe, the experiments underline the potential of the combination of genetic encoding and bioorthogonal chemistry for super-resolution microscopy.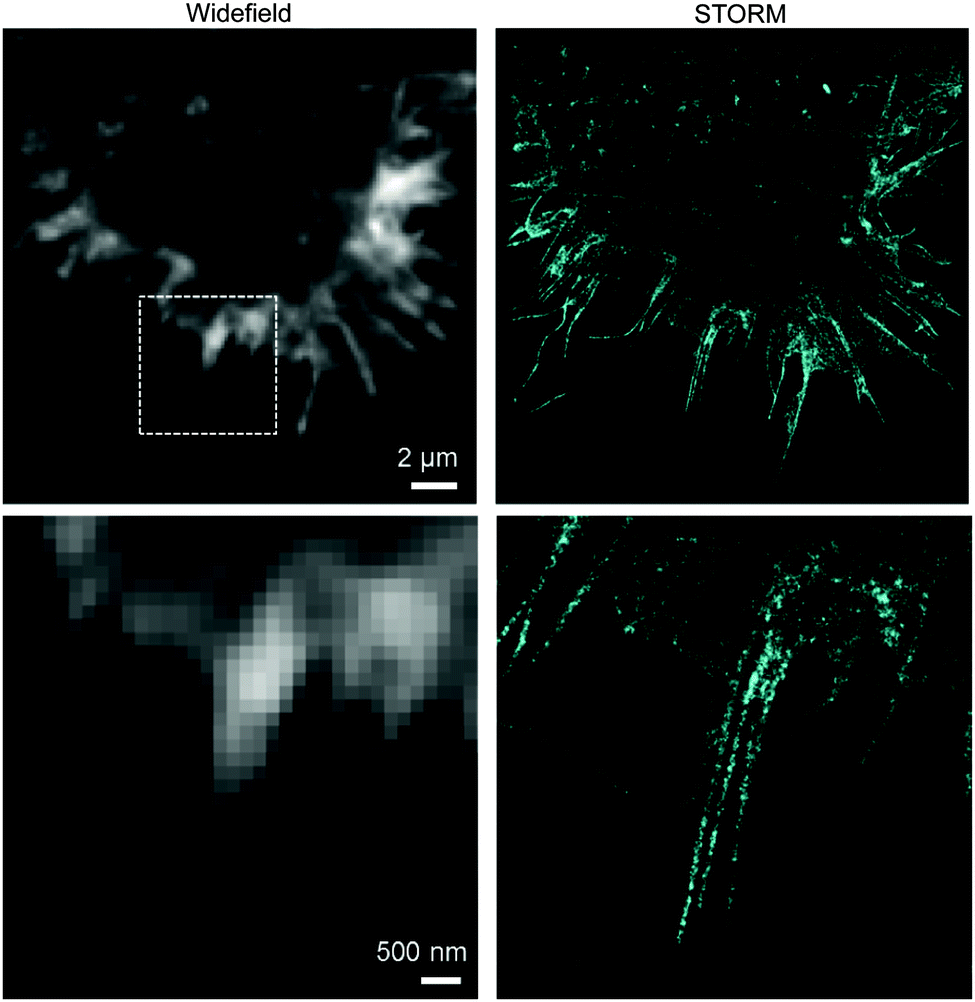 | ||
| Fig. 34 STORM imaging of actin in COS-7 cells. Upper images: wide field image of actin and corresponding STORM image. Bottom images are zoom-in images of the boxed region in the upper wide field image. Adapted in part with permission from ref. 152. Copyright 2015 American Chemical Society. | ||
Even though ncAA encoding technology and IEDDA reactions have been used for super-resolution imaging of highly abundant cytoskeletal proteins and surface proteins as in the previous example, applications to less abundant proteins are largely obscured by the limited efficiency of incorporation that requests the use of excess of the ncAA. This fact, results in high concentrations of non-incorporated ncAA that can also react with the dyes leading to nonspecific binding (sticking) of the fluorescent dyes. For intracellular labelling, new hydrophilic tetrazine dyes with higher “turn-on” fluorescence upon reaction, which may be washed out easily, would help to achieve higher signal to noise.120,221,222 Alternatively, Lemke's group developed a site-specific click-PAINT super-resolution microscopy method for imaging of low-abundant proteins inside mammalian cells, namely, the protein nucleoporin Nup153, a component of the nuclear pore complex (NPC) (Fig. 35).222 A schematic representation of this approach is outlined and the respective imaging data is presented in Fig. 35.
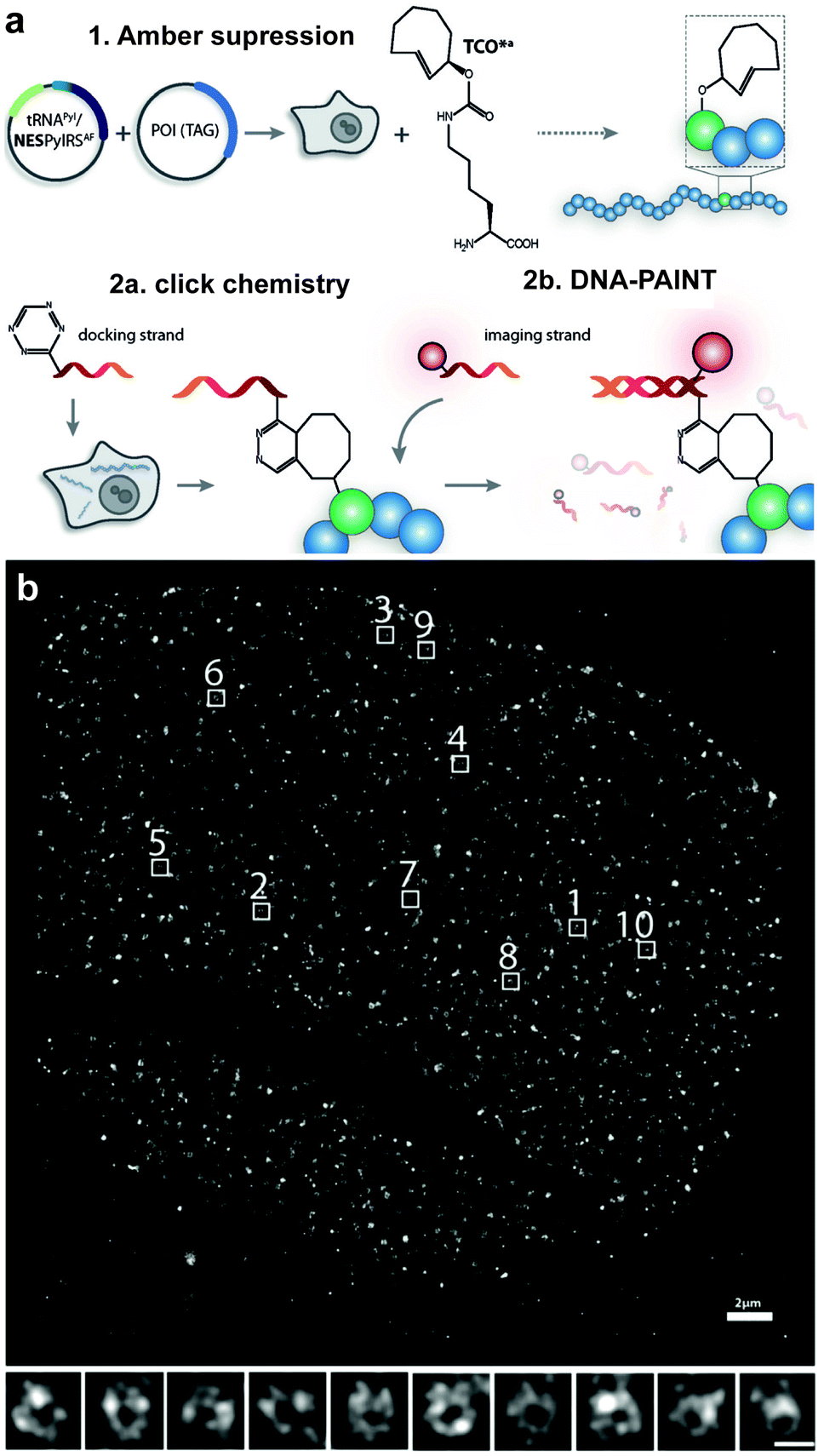 | ||
| Fig. 35 Super-resolution imaging of NPC. (a) Schematic representation of the Click-PAINT method. A protein of interest and a PyltRNA/NESPylRS pair (NES = nuclear export signal coupled to PylRS to reinforce cytoplasmic localization) are expressed in mammalian cells in the presence of TCO*. The expressed TCO*-tagged protein is first reacted with a tetrazine-functionalized DNA strand and then with a complementary imaging DNA strand conjugated to a dye. (b) Super-resolved images showing the typical circular appearance of NPCs. From ref. 222, Copyright © 2016 by John Wiley & Sons, Inc. Reprinted by permission of John Wiley & Sons, Inc. | ||
5.2 Study of fast dynamic process in living cells: where speed makes the difference
Although the strategy of combining site-specific incorporation of ncAAs with IEDDA bioorthogonal reactions may be advantageous for the labelling of intracellular proteins, there are several criteria that must be satisfied. One of these criteria is the careful selection of the tetrazine-conjugated fluorophores, as their cellular properties, such as membrane permeability, intracellular distribution, and retention, can affect protein labelling efficiency and specificity. Recently, Hang et al. performed a systematic evaluation of bioorthogonal IEDDA reactions between site-specifically incorporated ncAAs and various tetrazine fluorogenic probes for fluorescence labelling and imaging of low-abundance intracellular proteins in live cells. In particular, it was reported that the bioorthogonal fluorescence imaging of the intracellular protein interferon (IFN)-inducible transmembrane protein 3 (IFITM3), a small vesicle-associated membrane protein that is involved in host restriction of influenza virus and many other pathogenic viruses. Fluorescent labelling of the TCO-tagged HA-IFITM3 (HA, Human influenza hemagglutinin) with various tetrazine fluorophores resulted in fluorescence puncta that are characteristic to IFITM3-containing vesicles (Fig. 36a).213 Importantly, immunofluorescence microscopy performed in parallel with tetrazine labelling showed that tetrazine fluorophore signals overlapped with anti-HA fluorescence signals confirming labelling efficiency and specificity (Fig. 36a). Notably, when the imaging of IFITM3 in live cells was performed using N- or C-terminal fusions with fluorescent proteins (e.g. GFP or mCherry), both cellular localization and antiviral activity of IFITM3 were disrupted.213 To explore the trafficking of IFITM3 in live cells, the authors evaluated the dynamics of IFITM3 with exogenously added fluorescent cargoes that are internalized into endocytic vesicles. Interestingly, it was shown that red dextran particles were internalized into cells and then fused with BODIPY-labelled green IFITM3-residing puncta to yield yellow vesicular structures, demonstrating that IFITM3 traffics to the same location as exogenously acquired cargoes after their internalization in the endocytic pathway (Fig. 36b). Moreover, the time-lapse imaging showed that this fusion process is completed within min. These studies significantly expand the scope of site-specific bioorthogonal imaging of intracellular proteins in live cells, and demonstrated the utility of fast and fluorogenic IEDDA reactions for the localization and real-time dynamic trafficking of fast biological events.213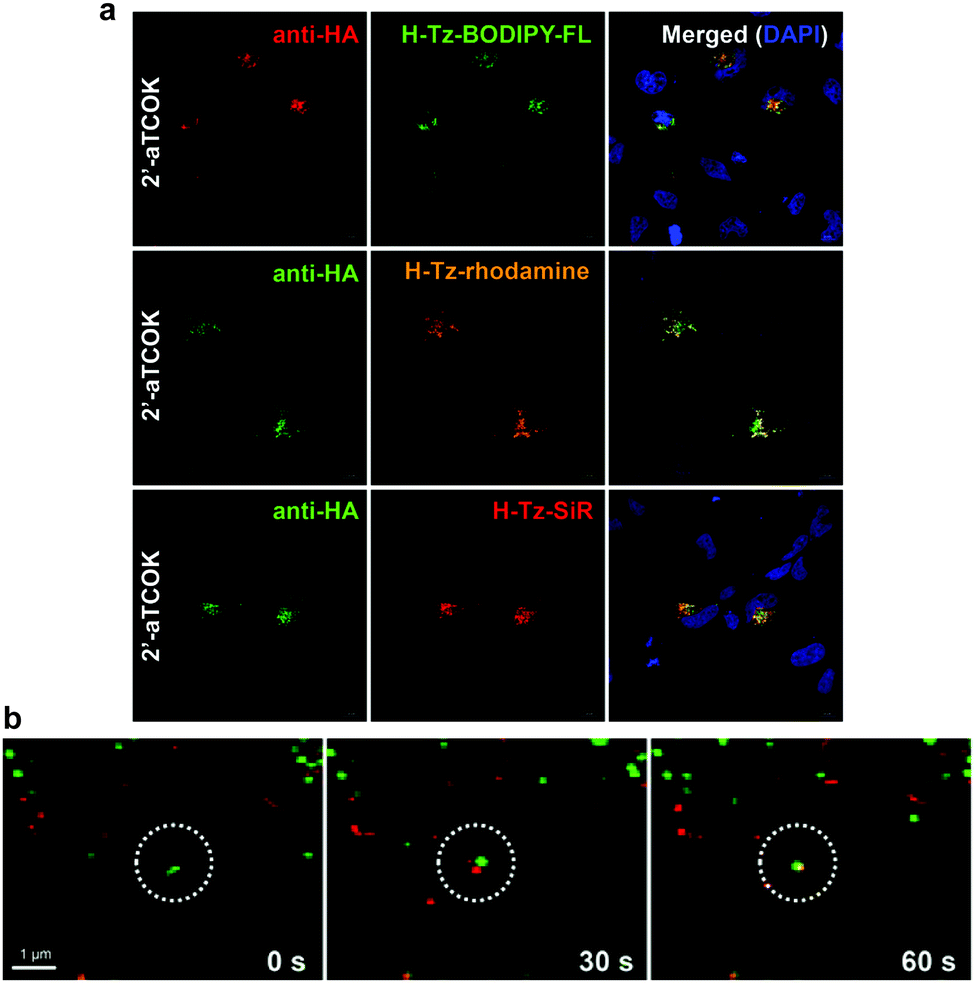 | ||
| Fig. 36 Bioorthogonal fluorescence imaging of HA-IFITM3-TCOK. (a) HeLa cells expressing HA-IFITM3-TCOK were labelled with H-Tz–BODIPY–FL, H-Tz–rhodamine, or H-Tz–SiR and then subjected to anti-HA immunofluorescence staining. DAPI (blue) was used to stain nuclei. (b) Time-lapse imaging of the fusion process of IFITM-containing vesicles with dextran particles. IFITM3-TCOK was labelled with H-Tz–BODIPY–FL and dextran particles were labelled with pHrodo Red. Images were acquired every 30 seconds. Reprinted in part with permission from ref. 213. Copyright 2016 American Chemical Society. | ||
5.3 Identification of bioactive targets of small molecules and proteins in living cells
Monitoring how, when, and where small molecules engage their targets intracellularly is a major challenge in chemical biology and pharmacological research, because it provides insights that can assist the design or more efficient binders and to understand their mode(s) of action.223 For this purpose, Bantscheff et al. developed a modular strategy that enabled small molecule localization, target identification and target occupancy measurements of unmodified drugs in cells using bioorthogonal ligation reactions.224 Generally, the strategy uses a click-probe derived from a drug of interest that can be functionalized either with tags for affinity purification of potential target proteins or with fluorophores for high-resolution imaging. The feasibility of the modular probe strategy was demonstrated with noncovalent PARP inhibitors. Using a proteomic assay, the authors showed that dose-dependent competitive titration with the unmodified drug (Olaparib) allowed the determination of target and off-target affinity by quantitative mass spectrometry of the click-probe targets that are also bound to the free drug (Fig. 37(1)). In addition, attachment of a fluorophore to the click-probe allows localization of the probe inside cells by confocal microscopy (Fig. 37(2)).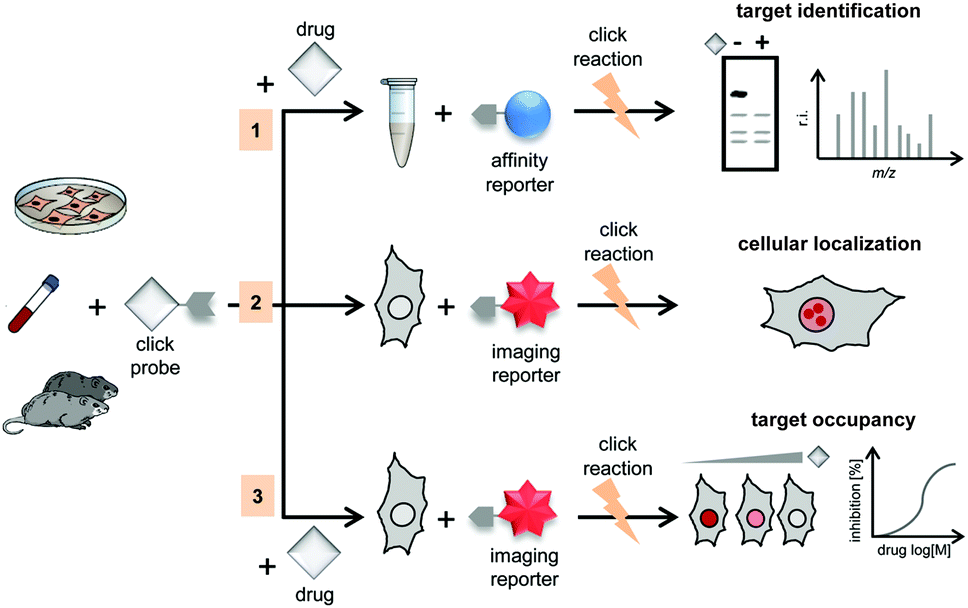 | ||
| Fig. 37 A single functionalized probe containing a bioorthogonal moiety (click-probe) for (1) target/off-target identification, (2) compound colocalization with the target protein, and (3) measurement of target occupancy of unmodified compounds. Reprinted with permission from ref. 224. Copyright 2016 American Chemical Society. | ||
This approach also allowed the determination of target occupancy by measuring the reduction of the localized fluorescence signal in the presence of unmodified drug (Fig. 37(3)).224 Of note, the authors systematically evaluated the compatibility and efficiency of a number of bioorthogonal reactions (IEDDA, SPAAC and CuAAC) and identified TCO labelled probes and tetrazine-tagged reporters to be the most efficient bioorthogonal coupling partners.224 Similarly, a dienophile-modified paclitaxel derivative with identical activity to the parent drug was used for imaging microtubular networks in cells upon IEDDA labelling with tetrazine fluorophore probes.133 Similarly, TCO-modified kinase inhibitors labelled with tetrazine-tagged fluorescent reporters could be used to track and image kinase activities in live cells.225
The detection of protein–protein interactions (PPI) within native cellular environments could provide key insights for drug design. To address this challenging question, minimalist linkers cyclopropene and diazirine were synthetically introduced within a small molecule bromodomain inhibitor. In this approach, the inhibitor is used for target identification, while the diazirine photo-cross-linker enables covalent linking. Lastly, the cyclopropene handle is used either for bioimaging or affinity-based protein labelling using biotin-tetrazine derivatives. The resulting probes were used for proteomic analysis and resulted in the identification of several hundreds of protein candidates targeted by the bromodomain BRD-4 inhibitor (+)-JQ1 (Fig. 38).
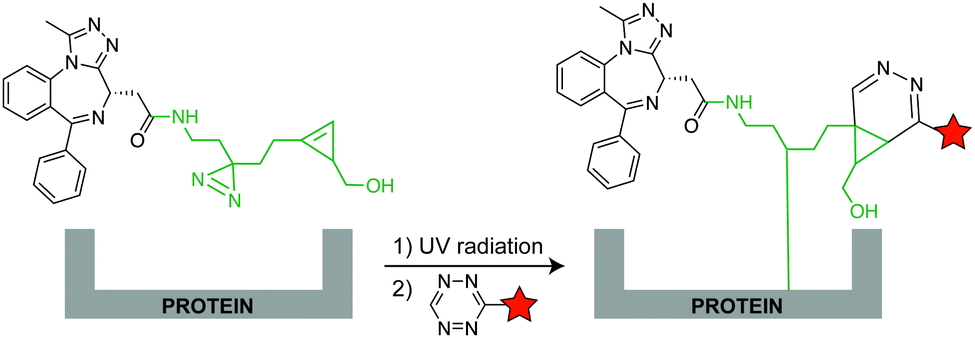 | ||
| Fig. 38 Minimalist linkers for proteome profiling. The diazirine moiety generates reactive species upon ultraviolet (UV) irradiation creating covalent bonds with targeted proteins. Covalent ligation allows for affinity pull-down assays of the target for further proteome profiling using the cyclopropene handle as chemically tractable tags. | ||
5.4 Protein profiling in organisms
The ability to identify newly synthesised proteins at specific times in cells of interest will facilitate the study of dynamic cellular processes. In this regard, several protein tagging strategies through genetic code expansion have been employed to interrogate several complex cellular processes.226 In one example, an azide-containing analogue (L-azidohomoalaine, AHA) of methionine (Met) was introduced at Met codons and subsequently labelled through cycloaddition with alkyne-bearing probes.227 Based on auxotrophic strains, incorporation of the amino acid analogues is often achieved in minimal media under starvation conditions for the amino acid to be replaced. This limits the availability of the natural amino acid and decreases labelling efficiency.A novel technology that addresses some of the above-mentioned issues was developed – stochastic orthogonal recoding of translation with chemoselective modification (SORT-M) (Fig. 39).155 SORT-M uses a set of reengineered orthogonal pyrrolysyl-tRNA synthetase/tRNA pairs that enable codon-selective incorporation of a cyclopropene Lys derivative, which can be chemoselectively modified with various tetrazine probes for fluorescence imaging and proteome profiling of engineered bacteria, mammalian cells and in specific cell types or tissues in animals (Fig. 39).155 Importantly, the method could be performed directly in flies for developmental stage-specific proteomic studies.155 More specifically, by confining PylRS expression to the ovaries of Drosophila melanogaster using a specific promoter, selective incorporation of a cyclopropene-bearing amino acid into proteins derived from these organs was achieved. This was confirmed by fluorescent labelling of proteins specifically synthesised in the ovary. Protein identification could be also performed combining 2-D in-gel fluorescence electrophoresis and MS analysis.155 More recently, an improved version of SORT-M called SORT-E (SORT with enrichment) was developed. In this version, SORT-tagged cellular proteins are reacted with a tetrazine diazobenzene biotin compound and then captured on streptavidin beads. The beads can be washed and enriched proteins may be detected using MS after reductive cleavage of the diazobenzene linker.156
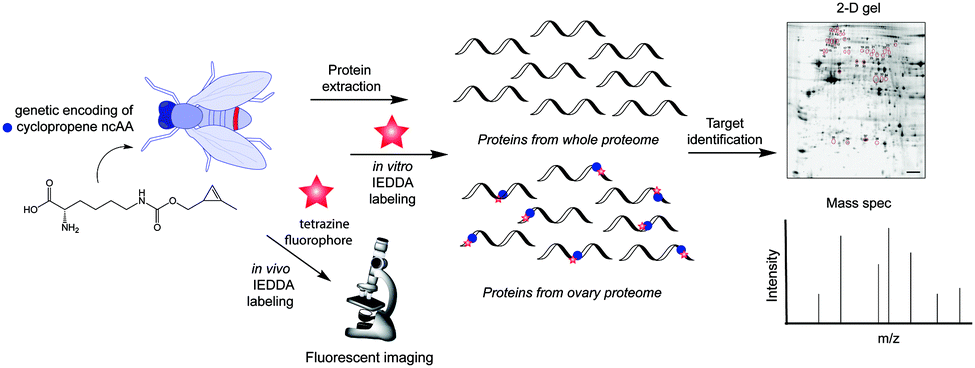 | ||
| Fig. 39 “SORT-M” allows the incorporation of diverse chemical groups into the proteome, at diverse codons by PylRS aminoacylation of the corresponding tRNAXXX (PyltRNAXXX, XXX indicates choice of anticodon). This system results in stochastic incorporation of the ncAA (yellow star, and yellow triangle) in the resulting protein. A GAL4 fly lines expressing PyltRNAXXX, PylRS (under GAL-4 control) is fed with an ncAA (red star, cyclopropene-Lys derivative). The ncAA is incorporated into the proteome of specific tissues where the GAL-4 reporter is active (red ovary). Combining fluorescent 2D-gel electrophoresis and mass spectrometry is possible to identify these proteins. Direct visualization of synthesized proteins by tetrazine labelling in tissue has been also by demonstrated fluorescent microscopy. | ||
5.5 Sequence-specific detection of DNA and mRNA in living systems
As mentioned a beneficial characteristic concerning the use of tetrazines during IEDDA is the “turn-on” effect in case a tetrazine–fluorophore conjugate is used. By using these probes, fluorescence is only observed when the bioorthogonal partners meet and, thus, where exactly the bioorthogonal reaction occurs. This feature has been used in the development of tetrazine smart probes for the sequence-specific detection of DNA and mRNA in living systems. Devaraj et al. first applied template-driven reactions with fluorogenic tetrazines for nucleic acid detection.215 Specifically, a quenched fluorophore-tetrazine and a methyl-cyclopropene group were conjugated to two different oligonucleotides and showed to react by IEDDA ligation only in the presence of the DNA template that can hybridize with both oligonucleotides. The use of the template increases the effective molarity (52–123 mM) by confining the tetrazine and cyclopropene partners in close proximity, and the bioorthogonal reaction rapidly takes place, leading to strongly fluorescent products. This approach allowed detection of template DNA inside living mammalian cells (Fig. 40a).215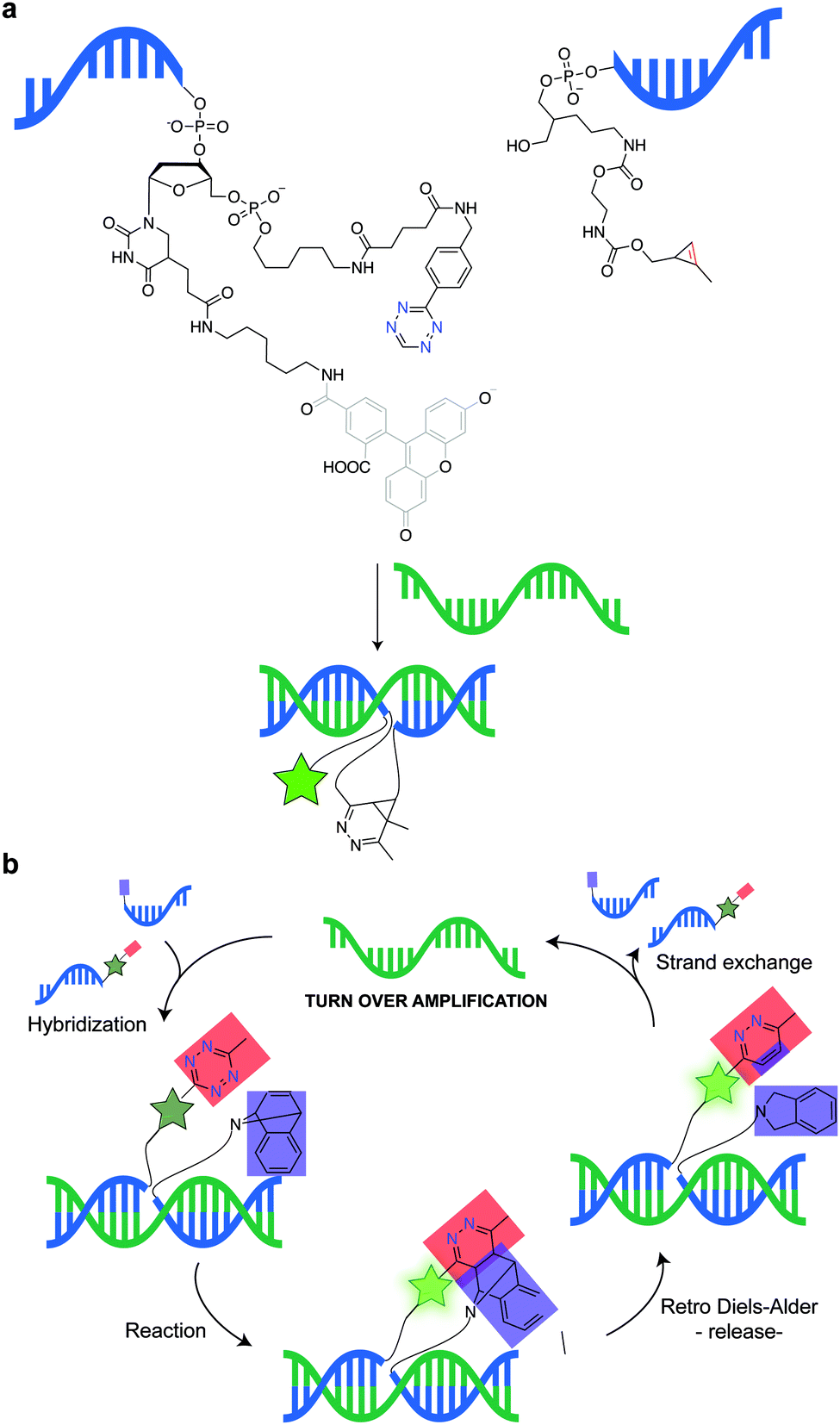 | ||
| Fig. 40 (a) Schematic of DNA-template fluorogenic tetrazine ligations. (b) Schematic of DNA-template tetrazine-mediated transfer reaction for live cell miRNA detection. | ||
One limitation of this approach is that the resulting product reacts with a similar affinity for the template as the reactive starting oligonucleotides, impeding reaction turnover and signal amplification. To address these issues, the same research group reported an optimized system using oligonucleotide templates derivatised with a norbornadiene and a highly fluorogenic tetrazine–BODIPY probe. The use of a highly quenched tetrazine probe enables a >100-fold increase in fluorescence in response to hybridization and subsequent proximity-driven reaction. In addition, the new antisense probes undergo dynamic strand exchange, which resulted in high turnover amplification allowing the detection of endogenous oncogenic miRNA target mir-21 (in vitro and cells) down to low-picomolar levels (Fig. 40b). Importantly, this system was able to distinguish single-base mismatches in miRNA templates.228
6. IEDDA applications in nuclear medicine
6.1 Overview of click radiochemistry
Nuclear imaging techniques are based on the radiolabelling of specific molecular probes using radioisotopes that are used as contrast agents for imaging of pathologic processes or cellular functions either by single-photon computed tomography (SPECT) or positron emission tomography (PET). A major advantage of SPECT and PET over other imaging techniques (e.g. optical and magnetic resonance imaging) is the high sensitivity that allows the detection of radiotracers in concentrations as low as 10−10 to 10−12 M.229,230 For SPECT, more than 80% of radiotracers used in nuclear medicine centers are with Technetium-99m,231,232 while for PET imaging the most clinically useful isotope is Fluorine-18.233 Other attractive isotopes for medical applications are Indium-111, Iodine-123, and Thalium-201 for SPECT and Carbon-11, Copper-64, Gallium-68 and Zirconium-89 for PET.234,235 Usually, radioisotopes are linked to targeting vectors (i.e., small bioactive molecules, peptides, proteins or antibodies) via a (i) bifunctional chelating agent for labelling radiometals or by (ii) direct or (iii) indirect methods for labelling with non-metallic isotopes (e.g.11C, 18F or 131I).233–239Over the past decade, bioorthogonal chemistry has gained momentum and has been increasingly applied in the field of radiopharmaceutical chemistry.240,241 The chemoselectivity and fast kinetics of such reactions in aqueous media makes them ideal for the radiosynthesis of molecular imaging agents. Marik and Sutcliffe et al. reported the first application of click chemistry for 18F-labelling of peptides through a CuAAC reaction between azido-decorated peptides and various [18F]fluoroalkynes groups or vice versa (Fig. 41a(i)).242,243 Since these initial studies a variety of 18F-prosthetic groups for CuAAC labelling have been prepared in radiochemical yields (RCY) ranging from 25% to 71% and reaction times of 5–10 min. Some examples of these 18F-building blocks are depicted in Fig. 41a(i) ([18F]PEG-alkyne,243 [18F]FDG-β-N3,244 [18F]Fluorobenzyl-N3245). The CuAAC click reaction has found applications in the development of PET probes for in vivo imaging, with one example (18F-RGD-K5)246 entering clinical trials in the US for diagnosis of integrin αvβ3 expressing tumours. The same click reaction was applied for the design and synthesis of radiometal-based radiotracers; Schibli et al. introduced the click-to-chelate concept to form novel ligand scaffolds containing a triazole group known to efficiently chelate the “99mTc(CO)3” core (Fig. 41a(ii)).247 Although a large number of radiotracers have been developed using this reaction some limitations have been reported. One obvious disadvantage is the use of a large excess of potential cytotoxic copper, which should be avoided for in vivo applications. For such applications, the SPAAC reaction was expanded for quick radiolabelling under physiological conditions in the absence of catalyst and widely used for the incorporation of radioisotopes into biomolecules for PET and SPECT imaging in animals (Fig. 41b(i)).248–255 Typical conditions for SPAAC labelling involves reaction times within 15–30 min, with isolated RCY of 19–98% and specific activities of ∼60 GBq μmol−1 (Fig. 41b(ii)).248–253 Another example of the click reaction in radiochemistry is the traceless Staudinger reaction.27,256 The first 18F variants for Staudinger radioligation were described by Steinbacha et al. and Gouverneur et al. (Fig. 41c).257–259 The Staudinger ligation has, however, one major limitation whereby the phosphine precursor is subject to oxidation, abolishing its reactivity. In addition to the classical click chemistry ligations, the reaction of unprotected aminooxy- or hydrazine-modified peptides with 18F-labelled aldehydes or ketones have also been explored for radiolabelling, resulting in the corresponding oxime or hydrazone conjugates (Fig. 41d(i)).260–262 These reactions are however limited due to the large excess of the components required, which results in very low specific activities. In addition, oximes and hydrazones are known to be prone to hydrolysis. The cyanobenzothiazole–cysteine (CBT-Cys) condensation reaction has also been recently established as an efficient, aqueous-compatible, and generally applicable method for labelling with 18F (Fig. 41d(ii)).263–265
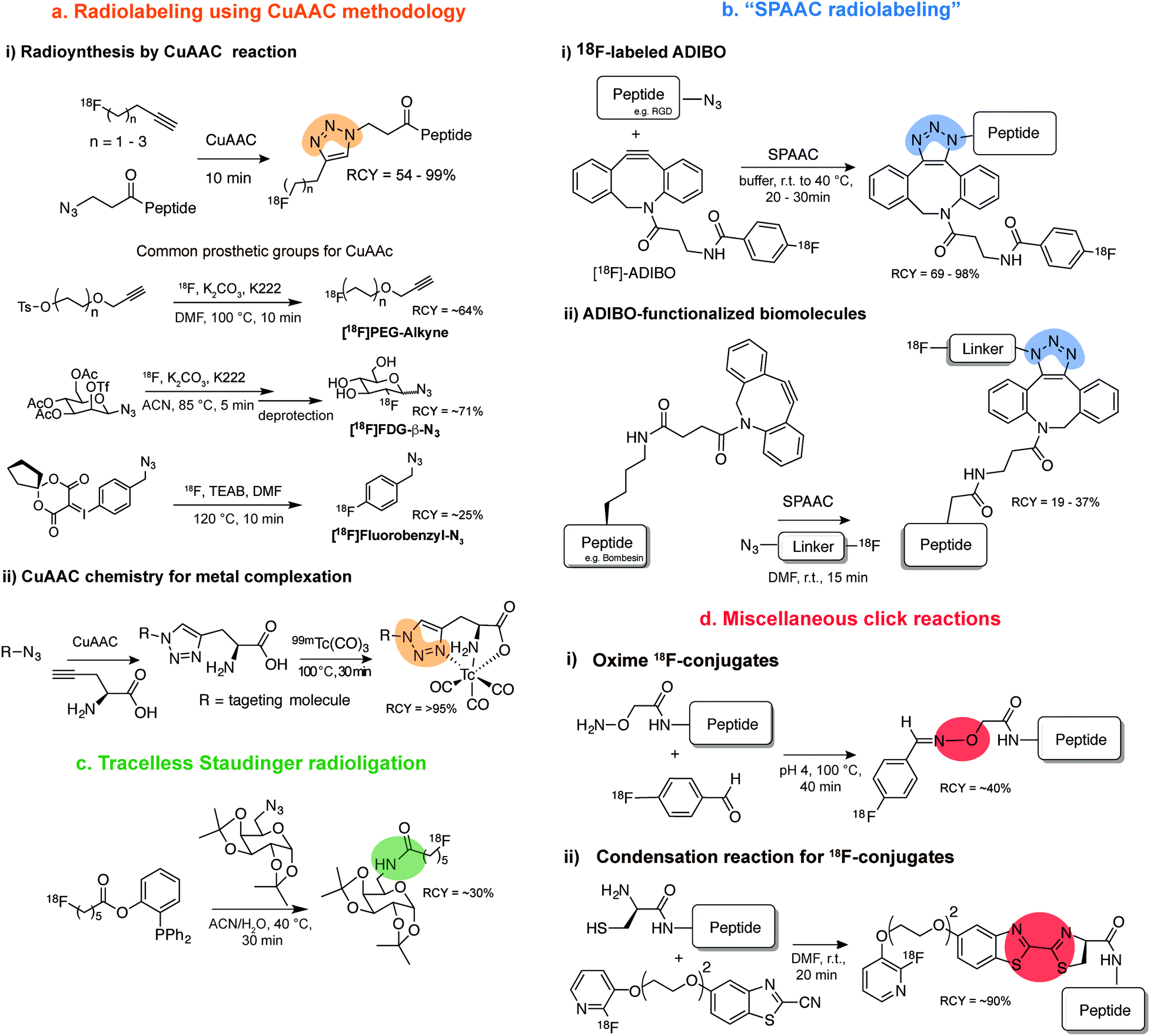 | ||
| Fig. 41 (a) Click methods for radiosynthesis. (i) CuAAC click reaction for preparation of radiotracers including the synthesis of the most common prosthetic groups. (ii) 99mTc(CO)3(H2O)3+ radiolabelling through complexation by triazole moieties formed by CuAAC cycloaddition. (b) Copper-free click radiolabelling by SPAAC reaction. (i) 18F introduced into the cyclooctyne prosthetic group (18F-labelled aza-dibenzocyclooctyne derivative, [18F]-ADIBO) and then subjected to strain-promoted cyclization with azide-functionalized biomolecules.248 (ii) Labelling of ADIBO-functionalized biomolecules with 18F-azides.249 (c) Staudinger radiolabelling.257 (d) Other examples of click radiolabelling. (i) Radiolabelling by oxime formation between an aminooxy-functionalized peptide and a 18F-fluorobenzaldehyde ([18F]FBA). (ii) Cyanobenzothiazole–cysteine (CBT–Cys) condensation reaction for 18F-radiolabelling. | ||
Although the mild conditions of the abovementioned click reactions are compatible with radiochemical approaches, they often require large amounts of purified peptide/protein precursor to obtain usable reaction rates for radiosynthesis. This leads to low specific activities (i.e., the ratio of labelled-to-unlabelled molecule), which is an important issue since the in vivo targets of PET/SPECT tracers are often receptors or enzymes that are present in low copy number. The slower reaction rates also exclude the use of these click reactions for in vivo pretargeting applications. These limitations have led to the search of alternative fast and catalyst-free methods applicable to radiochemistry, i.e., fast and high (radiochemical) yielding reactions that proceed under mild aqueous conditions using low concentrations of precursors. Notably, the IEDDA ligation has shown promising features, which make this click reaction suitable for both radiolabelling and “in vivo labelling”, i.e. pretargeting labelling. As mentioned, this reaction achieves unusually fast rates with no need for excess amount of reactants, or catalyst, resulting in radioactive preparations with high specific activities. The reaction is also not dependent on the solvent (tolerates water, organic solvents, cell media and blood plasma without reducing the reaction yields) and is highly selective in the presence of a broad range of functional groups. A pioneering work in this field was first reported by Robillard et al.266 In this work an 111In-DOTA-tetrazine derivative was used to label in vivo an antibody bearing TCO groups. Although first applied for radiolabelling with metals, over the past ∼8 years the tetrazine–TCO ligation (Tz/TCO ligation) has instead attracted attention for the construction of 18F- and 11C-based imaging probes, primarily due to the short half-life of these isotopes that required fast labelling reactions.
6.2 IEDDA applications with 18F
The application of Tz/TCO ligation for 18F labelling was first published by Fox et al. in 2010, where it was demonstrated that Tz 8 reacts with [18F]TCO in an excellent RCY of 98% at room temperature within seconds, using micromolar concentrations of the precursors (Fig. 42a).267 The synthesis of the [18F]TCO precursor was accomplished also in good yields (RCY of 71% within 15min) by reacting the corresponding nosylate derivative with [18F]-fluoride/tetrabutylammonium bicarbonate (TBAB) in acetonitrile (Fig. 42a). Further studies on the stability of this precursor have found that it is quickly metabolized in vivo (plasma metabolites observed within 5 min). At later time points (240 min post-injection, p.i.) PET imaging and ex vivo biodistribution studies showed a significant bone uptake indicating defluorination.268 The first in vivo imaging approach using a 18F-labelled peptide prepared via IEDDA chemistry consisted of the labelling of a tetrazine-RGD derivative with the [18F]TCO precursor (Fig. 42b). The resulting 18F-labelled tracer was prepared with a RCY of 90% within 5min at room temperature showing a high tumour accumulation, but also a high liver uptake associated to the hydrophobicity of this first-generation system (Fig. 42c).269
 | ||
| Fig. 42 (a) IEDDA conjugation between 18F-TCO and Tz 8. (b) Synthesis of [18F]TCO. (c) PET imaging and ex vivo biodistribution of Tz-18F-TCO-RGD. Reprinted in part (panel c) with permission from ref. 269. Copyright (2011) Elsevier B.V. | ||
In the previous example the tetrazine functionality is introduced through Lys acylation, which results in non-specific labelling of Lys residues that may be important for biologic activity. In this context, Conti et al. developed a bifunctional tetrazine–maleimide partner for efficient 18F-labelling of free Cys residues on peptides (cRGDyC) and proteins (VEGF-SH).270 As an example, labelling of Tz-18F-cRGDyC peptide was obtained in >95% RCY with a specific activity of 111–222 GBq μmol−1.270 More recently exendin-4, a glucagon like peptide-1 receptor (GLP-1R), was also modified via tetrazine–maleimide conjugation, and labelled with 18F-TCO for PET imaging of intraportally transplanted islet cells.271 Importantly, the 18F-labelled prosthetic groups N-[2-(4-18F-fluorobenzamido)ethyl]maleimide (18F-FBEM) and [18F]fluorobenzaldehyde (18F-FBA) have also been used for radiolabelling exendin. In this case the 18F-labelled exendin tracers were obtained in low specific activity, which resulted in low binding to the GLP-1R in tissues in comparison with the tetrazine-labelled exendin.271,272 The disadvantage of the tetrazine–maleimide approach relates with the occurrence of known thiol-exchange reactions that the maleimide products undergo in plasma.273,274
[18F]TCO has also been used for labelling poly(ADP-ribose) polymerase 1 (PARP1) inhibitors. In vivo studies have shown that 18F-AZD2281, one of the labelled molecules, accumulates in tumours overexpressing PARP1. Interestingly, the authors have used magnetic TCO-decorated beads to “pull out” the excess of unlabelled tetrazine-conjugated AZD2281 from the reaction mixture resulting in high specific activities without lengthy HPLC purifications.275
Toward the development of faster IEDDA 18F-labelling reactions Fox et al. developed a new radiotracer based on s-TCO by treating the corresponding tosylate precursor with 18F-tetrabutylammonium fluoride (18F-TBAF) in acetonitrile at 85 °C for 10 min (∼29% isolated yield) (Fig. 43a).27618F-sTCO is stable in PBS pH 7.4 up to 2 h at 37 °C. In fetal bovine serum 18F-sTCO retains 74% of radiochemical purity, after 1 h. This degree of degradation is not an issue, as the IEDDA reaction takes place within few minutes. In fact, it was shown that 18F-sTCO completely reacts with a RGD-tetrazine in less than 5 min (26–40 GBq μmol−1) and can be used to rapidly assemble probes for PET imaging in human U87MG tumour-bearing mice.277 More recently, a 18F version of d-TCO was prepared by nucleophilic substitution of the corresponding tosyl analogue with 12% radiochemical yield (18F-d-TCO, Fig. 43b).278 In PBS pH 7.4, 18F-d-TCO is stable, with 94% of the compound being intact after 2 h incubation at 37 °C. In rat plasma, 18F-d-TCO slowly isomerises into the corresponding cis-derivative, with 52% and 34% of the intact trans-compound being present after 1 and 2 hours incubation at 37 °C, respectively.
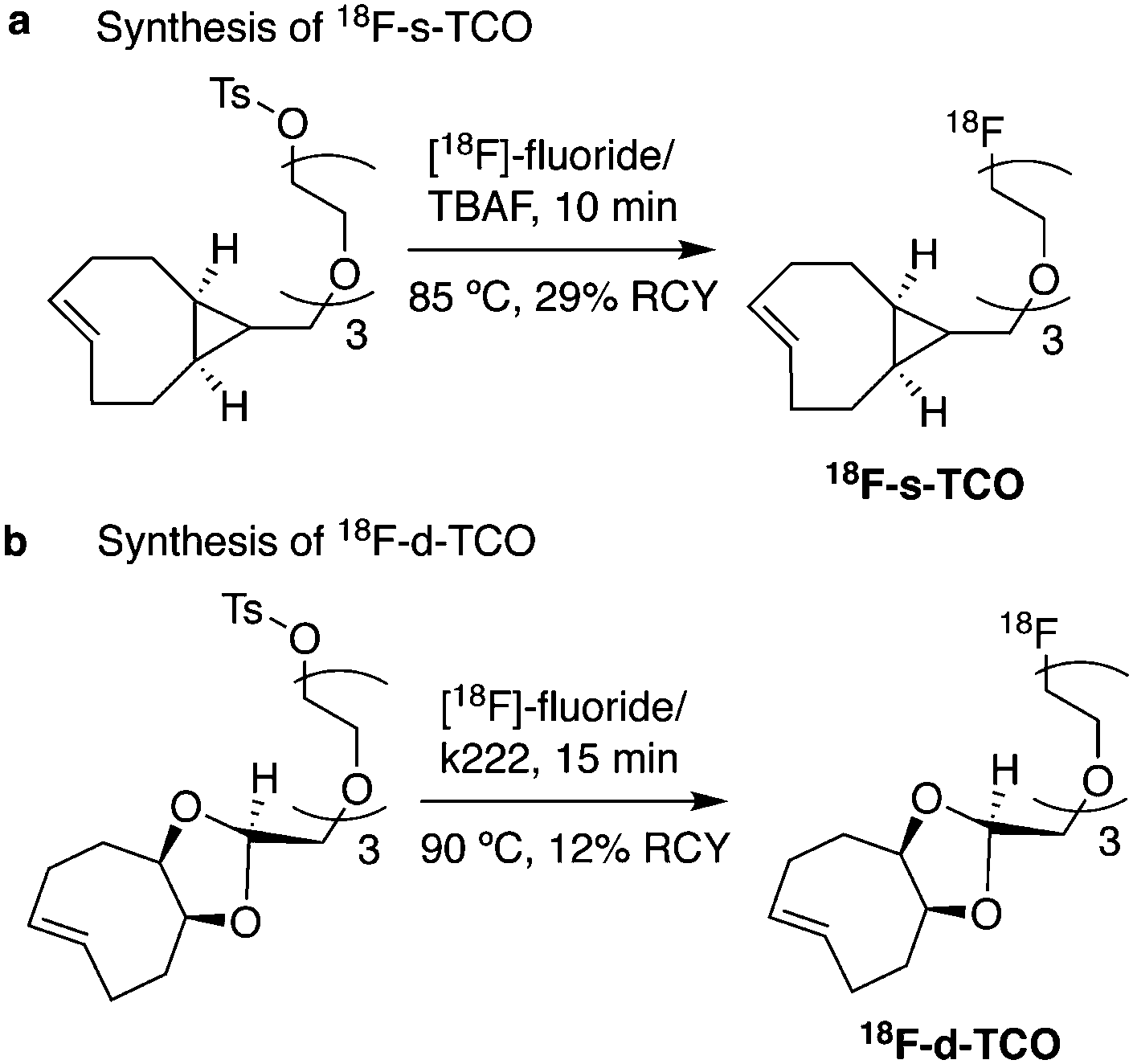 | ||
| Fig. 43 Synthesis of (a) 18F-s-TCO and (b) 18F-d-TCO. | ||
“Hot IEDDA” ligations are not only restricted to the 18F-TCO moiety. For example, Knight et al. used a 18F-norbornene derivative (18F-Norb) for labelling of a tetrazine-bombesin peptide. The authors demonstrated that 18F-Norb is capable of undergoing IEDDA click reactions with tetrazine species with promising reaction rates and high specific activities.279
| Radiolabelled tetrazine | Yield conditions | |
|---|---|---|
| 1 |
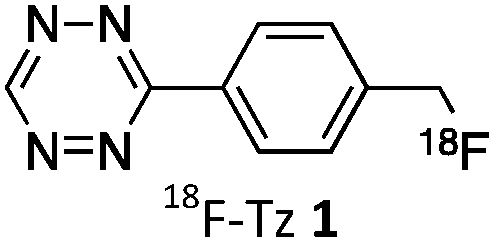
|
<1% MeCN, 85 °C, 15 min |
| 2 |
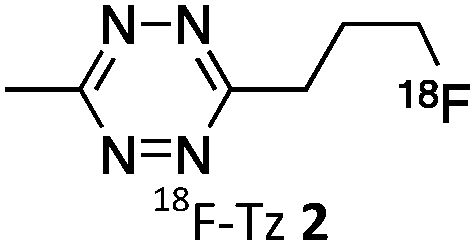
|
<18% MeCN, 90 °C, 5 min |
| 3 |
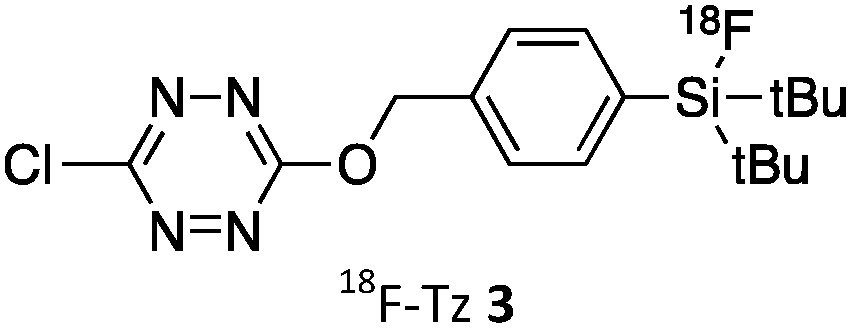
|
∼78% MeCN/DCM, r.t., 20–25 min |
| 4 |
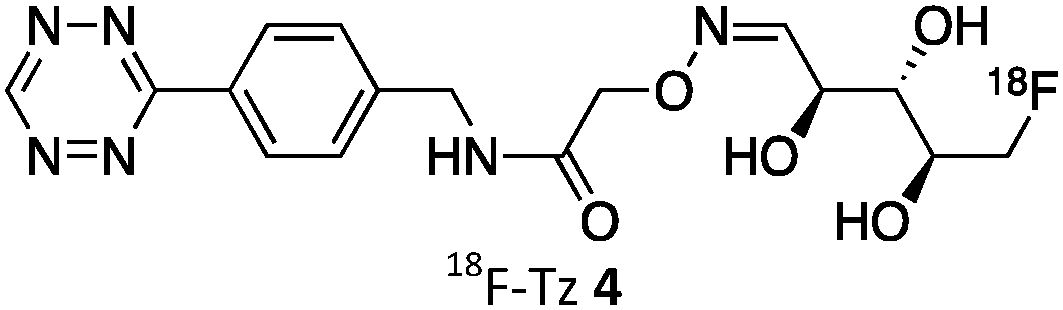
|
96% anilinium acetate buffer pH 4.6, r.t., 10 min |
| 5 |
|
10% DMF, Et3N, 60 °C, 7 min |
Despite this progress, the radiolabelling of small tetrazine derivatives was still limited by low RCYs due to the instability of the tetrazine moiety under conditions typically used in direct 18F-fluorination. A more efficient synthesis of a 18F-labelled tetrazine derivative used a [18F]SiFA–OH building block (RCY = ∼78%) that effectively reacts with 1,4-dichlorotetrazine to generate [18F]SiFA-OTz (18F-Tz 3). Using a specific activity of 7.0–8.5 GBq μmol−1 18F-Tz 3 reacts quantitatively with TCO in 1 minute (Table 1, entry 3).281 Recently, a new 18F-labelled tetrazine derivative was developed also in high yields and with specific activities of 12–16 GBq μmol−1 under mild reaction conditions via conjugation of 5-[18F]fluoro-5-deoxyribose with an aminooxy functionalized tetrazine to give 18F-Tz 4 (Table 1, entry 4). The 18F-tetrazine exhibited sufficient blood circulation time and favourable elimination characteristics.282 More recently Liang et al. labelled a tetrazine with [18F]SFB (RCY = 10%) to give 18F-Tz 5 and successfully constructed PET probes with TCO tagged antibody fragments for tumour imaging (Table 1, entry 5).283
6.3 “Tetrazine radiolabelling” with other relevant isotopes
Few examples of the application of the IEDDA reaction for direct labelling of bioactive molecules with other relevant isotopes have been described. In one example, a modular strategy for antibody labelling was developed. Specifically, norbornene was conjugated to trastuzumab – an anti-HER2 targeting antibody – and reacted with a tetrazine bearing different chelators (DOTA and desferrioxamine, DFO) for labelling with radiometals (64Cu and 89Zr) (Fig. 44a). In vivo PET imaging and ex vivo biodistribution experiments revealed significant and specific uptake of the 64Cu-DOTA and 89Zr–DFO–trastuzumab bioconjugates in HER2-positive BT-474 xenografts (Fig. 44a). The authors showed that a dienophile modified antibody can be reacted selectively with various radiometal chelators facilitating cross-comparisons in the development of antibody-based radiopharmaceuticals since the modified antibodies are synthesized using identical ligation conditions.284 Additionally the use of mild IEDDA conditions avoids the use of high temperatures that are usually required for labelling antibodies bearing coordinating agents. Recently, Weissleder et al. developed a tetrazine conjugate that incorporated the DFO ligand for installation of 89Zr and a BODIPY fluorophore for multimodal imaging. 89Zr–DFO–BODIPY–trastuzumab was prepared within minutes by Tz/TCO ligation and PET imaging showed high tumour conspicuity. In addition, biodistribution studies confirmed high specific probe uptake in BT474 xenografts (HER2+) (237.3 ± 14.5% ID per g in BT474) compared to low, nonspecific probe uptake in BT20 xenografts (HER2−) (16.4 ± 5.6% ID per g) 96 h p.i. (Fig. 44b). Ex vivo fluorescence of selected tissues confirmed target localization and persistence of the fluorescence of 89Zr–DFO–BODIPY–trastuzumab. Similar systems may find utility to identify targets intraoperatively through fluorescence.285 Similarly, Spivey et al. used a tetrazine modified 68Ga-DOTA to construct probes for labelling norbornene-containing biomolecules.286 In another example, Reiner et al. used tetrazine bearing amino acids as building blocks for the synthesis of peptides and performed labelling studies with these peptides applying a 89Zr–DFO TCO derivative.287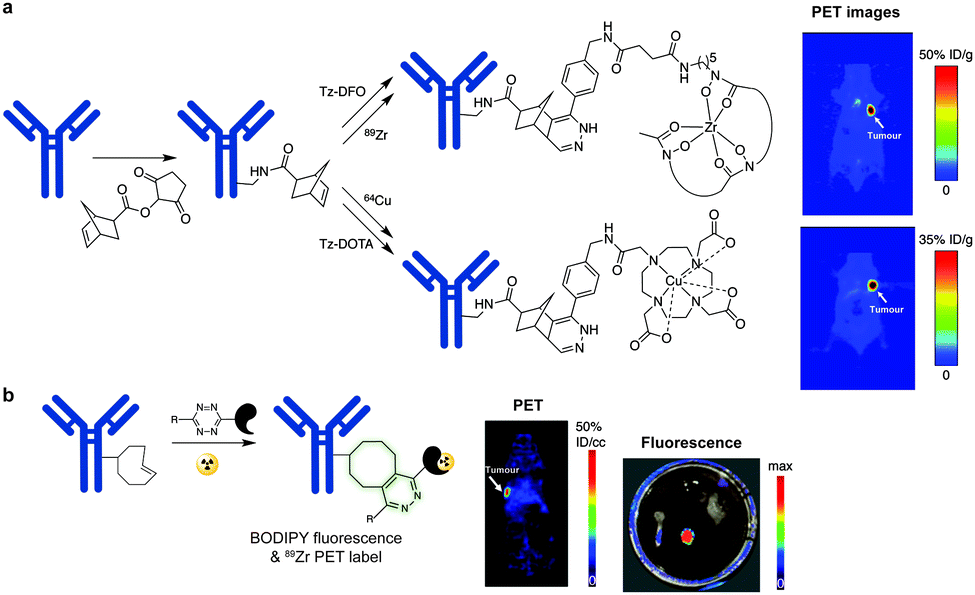 | ||
| Fig. 44 (a) Schematic of the modular radiolabelling strategy based on the ligation of norbornene-modified antibody and radiometal-labelled tetrazines. (b) Tz/TCO chemistry for multimodal imaging by using trastuzumab–TCO and a tetrazine incorporating 89Zr–DFO for PET imaging and BODIPY for fluorescence. Reprinted in part with permission from ref. 284 (panel a) and ref. 285 (panel b). Copyright (2011 and 2016) American Chemical Society. | ||
6.4 Pretargeting based on IEDDA
The IEDDA reaction between tetrazines and TCOs has been employed in a variety of settings from fluorescence imaging of targeted biomolecules to magnetic resonance imaging with nanoparticles,72,133,288,289 however, the most significant impact of this ligation may lie in in vivo pretargeting imaging applications. For such challenging approaches the bioorthogonal reactant pairs should have high stability towards the amino and thiol nucleophiles (among others) present in biological systems as well as high selectivity to ensure that only the target biomolecules are labelled in vivo. In addition, the bioorthogonal reaction should proceed with fast kinetics even in the case of low concentration of the target protein, as is the case with in vivo pretargeting, where the reacting partners are diluted in the blood pool.290 Among all the existing bioorthogonal reactions the IEDDA ligation is the one with most potential for pretargeting applications.291 In these systems, the radionuclide is administered separately from the targeting protein, most often a tumour-targeting antibody. In the first step, the targeting antibody bearing a strained alkene (or tetrazine) is administered and allowed to accumulate in the specific tissue. Lastly, when the excess antibody has cleared from the circulation, a radiolabelled tetrazine (or dienophile) is administered (Fig. 45). In a general way pretargeting can be regarded as in vivo labelling, since the antibody is radiolabelled at the target tissue after in vivo administration.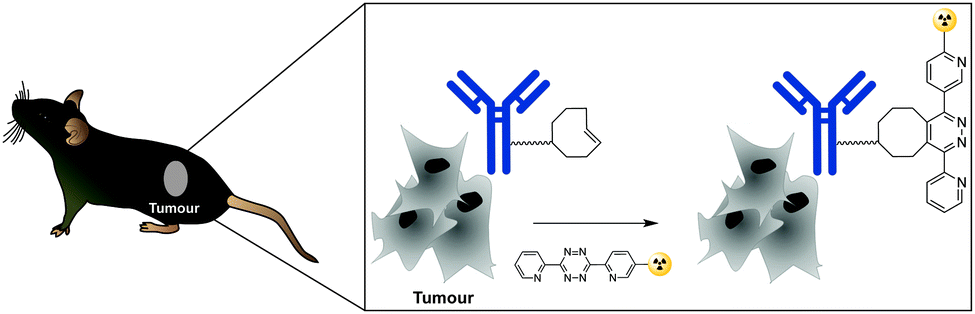 | ||
| Fig. 45 Schematic representation of tumour pretargeting with IEDDA reaction in mice. | ||
Until recently, most pretargeting approaches for radioimmunoimaging and radioimmunotherapy were based on the ligation of an antibody and a radionuclide using the avidin–biotin system, exploiting the high specificity and strong affinity (kd = 10−15 M) of avidin (or streptavidin) for biotin. Indeed, this pretargeting approach has been shown to be unaffected by extremes of pH, high temperatures, organic solvents and biological systems.292 Despite these features, the avidin–biotin system does have limitations, in particular, the high immunogenicity of the (strept)avidin, which cannot be humanized precluding its repeated use. The antibody-based pretargeting systems based on bioorthogonal click chemistry address such limitations. The use of IEDDA in pretargeting was pioneered largely by Robillard et al. In this work TCO was conjugated to anti-TAG72 monoclonal antibody [CC49] via an oligoethyleneglycol linker and used as a tagging compound while a DOTA modified tetrazine moiety was used for labelling with 111In. The 111In–DOTA–tetrazine conjugate was injected one day after administration of the TCO–antibody conjugate (CC49–TCO) in mice bearing colon cancer xenografts. Impressively, the chemically tagged tumours reacted rapidly with 111In–DOTA–tetrazine using only a small excess (3.4 equivalents) of the tetrazine radioactive tracer, resulting in pronounced accumulation in the tumour, as demonstrated by SPECT (Fig. 46).266Ex vivo biodistribution studies confirmed a high tumour uptake (∼3.0 ID per g) but a lower tumour-to-blood (T/B) ratio, which was assigned to the reaction of 111In–tetrazine with freely circulating CC49–TCO. In addition, although this pretargeting approach enabled straightforward antibody modification the highly strained antibody-conjugated TCO tag was prone to slow in vivo deactivation (25% in 24 h).266
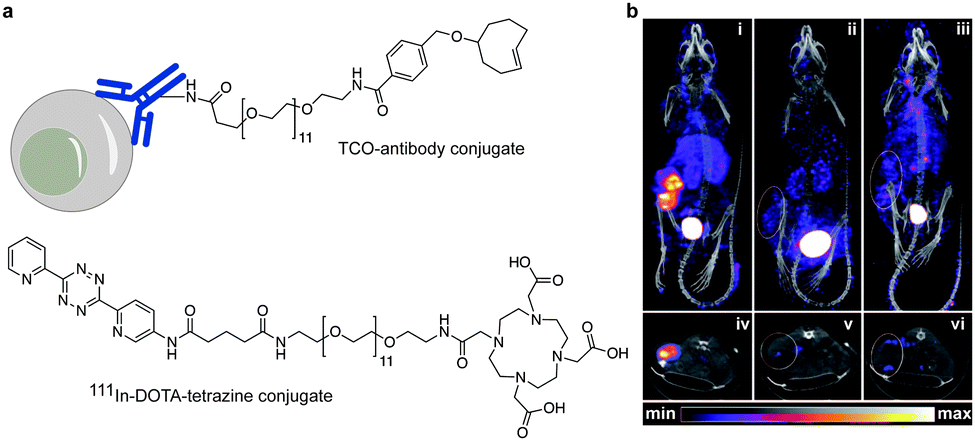 | ||
| Fig. 46 (a) Pretargeting components: TCO–antibody conjugate and 111In–DOTA–tetrazine conjugate. (b) SPECT/CT imaging of mice bearing colon carcinoma xenografts: SPECT images of mice preinjected with (i) TCO–antibody conjugate followed one day later by 111In–DOTA–tetrazine conjugate. (ii) Mice treated with unmodified antibody (no TCO tag) followed one day later by 111In–DOTA–tetrazine conjugate. (iii) Mice treated with TCO-modified rituximab, which lacks specificity for TAG72, followed by 111In–DOTA–tetrazine conjugate. (iv)–(vi) Single transverse slices passing through the tumours in (i)–(iii). From ref. 266, Copyright © 2013 by John Wiley & Sons, Inc. Reprinted in part (panel b) by permission of John Wiley & Sons, Inc. | ||
In a follow up study, the same researchers showed that the TCO tag is potentially deactivated in vivo through isomerisation to the unreactive cis-cyclooctene isomer by interactions with copper-containing proteins.293 By attaching the TCO through a shorter linker to hamper interaction with the copper-binding site in albumin, the deactivation half-life of TCO in circulation in mice was enhanced to 4 days.293 In another study, a pretargeting approach was developed for radioimmunotherapy using a 177Lu-labelled tetrazine probe and the CC49–TCO antibody.294 It was demonstrated that the use of a less hydrophobic tag (TCO–oxymethylbenzamide) afforded a CC49–TCO conjugate with a longer clearance half-life, improved tumour accumulation and markedly increased in vivo tag stability.295 Very recently, the authors also successfully employed a TCO–tetrazine pretargeting approach with antibody fragments and affibodies.296,297 In addition, a number of other groups, have reported the development of a pretargeted imaging strategy based on the IEDDA reaction. Lewis et al. described an A33 TCO–antibody for in vivo pretargeting with a 64Cu-labelled tetrazine in SW1222 tumour-bearing mice.298 Relatively low T/B ratios were observed caused by the reaction between the labelled tetrazine and the freely circulating A33-TCO.
To circumvent the high blood uptake, tetrazine-functionalized clearing agents were developed (e.g. galactose–albumin–tetrazine and polystyrene beads coated with tetrazine-conjugated albumin). These enable rapid reaction with and removal of the TCO-tagged antibody from blood resulting in a significant improvement of the tumour-to-blood (125-fold) ratios (Fig. 47a).294 The utility of the IEDDA reaction for pretargeting extends beyond 111In, 64Cu and 89Zr metals. Recently, a bisphosphonate-modified variant of TCO was used to pretarget bone and for subsequent bone imaging after in vivo labelling with a 99mTc–tetrazine (Fig. 47b). This approach explores the high avidity of bisphosphonates to osteoclasts. In the same study a therapeutic radioisotope was also used (177Lu conjugated to a tetrazine).299
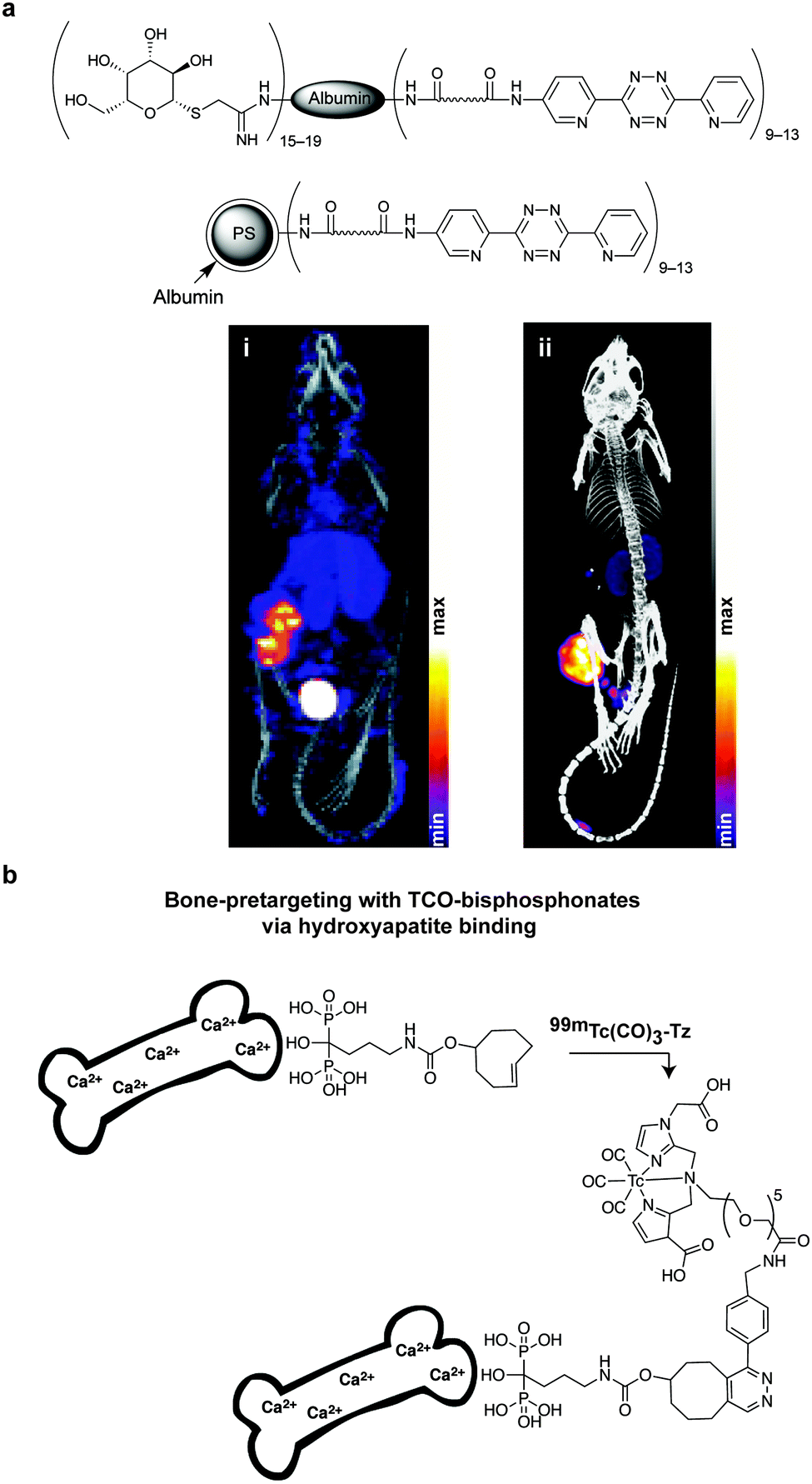 | ||
| Fig. 47 Clearing agents and TCO–bisphophonates for pretargeting applications. (a) Schematic representations of galactose–albumin–tetrazine and polystyrene (PS) beads coated with tetrazine-conjugated albumin for rapid removal of freely circulating antibody–TCO. (i) SPECT/CT image of a CC49–TCO pretargeted mouse bearing LS174T xenograft, after 3 h injection of the 111In–DOTA–tetrazine conjugate (24 h post-antibody injection).266 (ii) SPECT/CT imaging using same animal model pretreated with CC49–TCO after two doses of the clearing agent galactose–albumin–tetrazine followed by 111In–tetrazine injection.294 (b) Bisphosphonate-modified TCO for bone pretargeting. Reprinted in part (panel a) with permission from ref. 266 and 294. Copyright (2010) by John Wiley & Sons, Inc. and Society of Nuclear Medicine and Molecular Imaging, Inc. | ||
A robust and modular chemoenzymatic strategy was employed for the preparation of site-specifically modified antibody–TCO immunoconjugates on the glycans present in the heavy chain.300 This approach relies on the enzymatic activity of β-1,4-galactosidase and galactosyltransferase [Gal-T(Y289L)] for the incorporation of azide-modified monosaccharides (GalNAz) into the oligosaccharide chain. The resulting azide-modified antibody was then conjugated to a dibenzocyclooctyne-bearing variant of TCO (DIBO–PEG12–TCO) via SPAAC ligation. The site-specifically labelled huA33–TCO immunoconjugate was used for pretargeting PET imaging of colorectal cancer with a 64Cu-labelled Tz radioligand (64Cu–Tz–SarAr).300 The same chemoenzymatic strategy was used to construct a TCO- and fluorophore-bearing immunoconjugate of the huA33 antibody (huA33–Dye800–TCO) for multimodal PET/NIRF imaging of colorectal cancer using a 64Cu-labelled tetrazine (64Cu–Tz–SarAr). Importantly, simulated tumour resections using a near-infrared fluorescence (NIRF) camera showed its potential for real-time delineation of malignant tissue during image-guided surgery.301
IEDDA ligation has also been successfully used in the synthesis of radioactive iodine labelled constructs.302,303 For example, Valliant et al. developed a convenient method to prepare radioiodinated tetrazines that could be used for labelling a TCO–anti-VEGFR2 conjugate with iodine-125 for in vivo studies. The 125I–tetrazine was prepared by employing an oxidative halo destannylation reaction. Under these conditions the tetrazine showed high stability resulting in high iodination yields (80% RCY, 15 min reaction).303 Importantly, the 125I–tetrazine labelled antibody was shown to be 10 times more stable to deiodination than the same antibody labelled through direct iodination of for example tyrosines using iodogen.
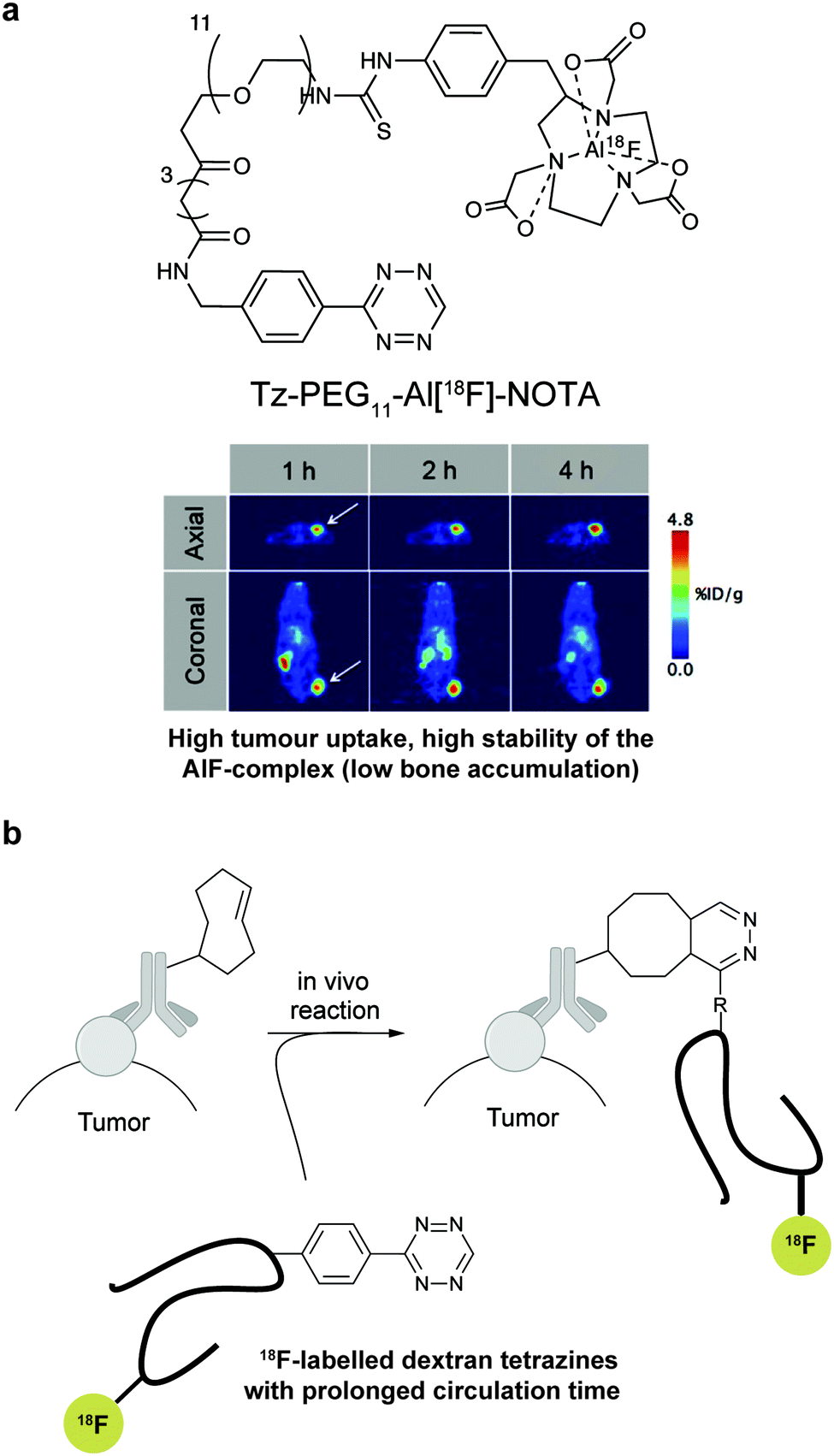 | ||
| Fig. 48 IEDDA pretargeting with short-lived isotopes. (a) PET images of pretargeted subcutaneous xenograft tumour-bearing mice with Tz–PEG11–Al[18F]–NOTA/5B1–TCO. (b) 18F-labelled dextran tetrazines with optimized pharmacokinetics. Reprinted in part (panel a) with permission from ref. 305. Copyright (2016) American Chemical Society. | ||
The fast rates of the IEDDA ligation has made this reaction so attractive that even labelling with 11C (half-life of 20.3 min) was explored in pretargeting approaches. The first radiosynthesis of a 11C-tetrazine was reported in 2013 by Kristensen et al. in moderate yields (33% RCY) in 2 min but with a total preparation time of 50–60 min that included HPLC purification. The final click ligation with TCO is completed within 20 seconds, however, imaging, biodistribution, and stability data were not reported using this probe.308 More recently a new 11C-tetrazine precursor was prepared in 50% RCY and an overall synthesis time of 30 min including purification. The in vivo profile of this tetrazine was investigated by dynamic PET/MR scanning and ex vivo biodistribution studies. These results confirmed this 11C-radiotetrazine as a promising tool for pretargeted PET imaging. To investigate the applicability of the 11C-tetrazine for rapid in vivo pretargeting and PET imaging, mesoporous silica was functionalized with TCO. The TCO-modified silica was shown be localized in the lung by PET after in vivo click reaction with 11C-tetrazine, demonstrating the potential of 11C-labelled tetrazine for pretargeting applications.309
7. Tetrazines – an emerging trigger for bioorthogonal cleavage reactions
Together with the explosion of bioorthogonal ligation reactions, using biocompatible reagents to trigger bond cleavage under physiological conditions has recently gained much attention.4,5 Instead of attaching additional functionality on biomolecules of interest, bioorthogonal cleavage reactions offer an alternative way to restore function of protected motifs, such as caged-fluorophores, prodrugs or masked biomolecules. In order to induce such a gain-of-function event, not only the caging groups, but the trigger, should be bioorthogonal, non-toxic, stable and reactive in cellular environment. In this section, we report recent progress in bioorthogonal cleavage reactions drawing particular attention to tetrazine-mediated IEDDA decaging reactions and its application in vitro and in vivo.7.1 Bioorthogonal bond cleavage chemistry
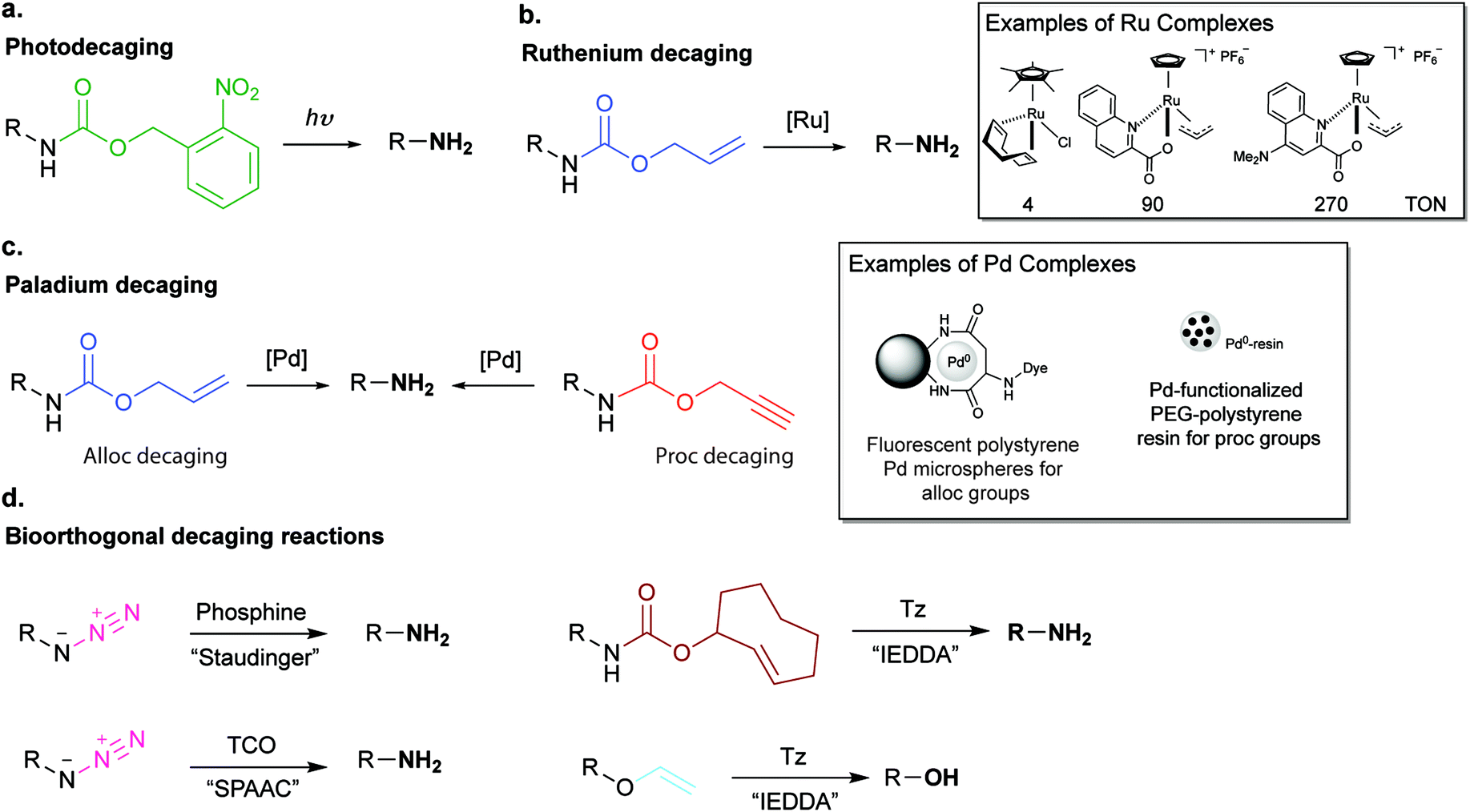 | ||
| Fig. 49 Examples of bioorthogonal decaging methods. | ||
Apart from organoruthenium, Pd species have also been shown to be a useful catalyst for O-deallylation (Fig. 49c).323,324 Using fluorescent polystyrene microspheres, Bradley et al. showed that Pd nanoparticles could cross cell membrane and cleave allylcarbamate linkages intracellularly on mammalian cells (Fig. 49c).325 Additionally, propargyl groups (proc) could also be cleaved by Pd catalysts and used to activate anticancer drugs in cells and, most impressively, in zebrafish.326–328 In a recent work, Chen et al. managed to restore the function of propargyl-carbamates (proc)-caged proteins by Pd catalysis in vitro and in vivo.329 They also managed to manipulate cell-surface sugars by depropargylation of caged glycans using in situ-generated Pd nanoparticles.330 Recently, novel cleavable groups such as an allenyl ether on Tyr and non-terminal allylcarbamate linkages were also developed to undergo decaging reactions catalysed by Pd.331,332 Notably, other organometallic complexes, such as mercury,333 copper,334,335 iron318,336 and cobalt species337 were also found reactive for bond-cleavage reaction on allyl, propargyl and azide groups under physiological conditions, in living human cells and even in nematodes and zebrafish.
It is noteworthy that even though reaction proceeds efficiently at the small molecule level with 10% catalyst loading, when applied for protein and drug activation in cell studies, large amounts of catalyst is always required due to nonspecific binding of metal to proteins, especially for Pd catalysed reactions.329 Despite the moderate to low toxicity of these metal catalysts, their use is still limited for in vivo applications. In addition, further studies are needed to understand the cellular uptake of these catalysts and their influence on normal cellular processes.
7.2 IEDDA triggered “click-to-release” of TCO bearing molecules.
TCO pro-drug activation. Robillard et al. first demonstrated in 2013 that the superfast IEDDA reaction could serve as a cleavage reaction for drug activation (Fig. 50a).65 By reacting a doxorubicin carbamate-linked TCO derivative with tetrazine, the resulting 1,4-dihydropyridazine product could undergo elimination of doxorubicin and CO2 if TCO is modified at the allylic position (Fig. 50b). Notably, it was shown that the axially substituted TCO presents 156-fold higher reactivity than equatorial corresponding isomer (Fig. 50c), which presumably derives from the steric interaction between tetrazine and equatorial benzyl carbamate moiety.65 In a recent example, this tetrazine-triggered reaction was used for pretargeting antibody–drug conjugate (ADC) therapy. Here, the masked drug is conjugated to a tumour-homing antibody via a TCO linker (CC49-TCO-Dox), which could be readily released in antibody pretargeted tumour cells upon tetrazine decaging reaction (Fig. 50d). Indeed, in vivo studies in mice bearing colon carcinoma xenografts showed efficacious drug release and high tumour retention of doxorubicin as confirmed by ex vivo fluorescence RP-HPLC analysis of tumour homogenates. Of note the TCO linker displays high in vivo stability with a half-life of 5 days, comparable to conventional cleavable ADC linkers. Although some isomerisation of TCO is expected, this only results in loss of reactivity and not in non-specific payload release, and thus potential side-toxicity.15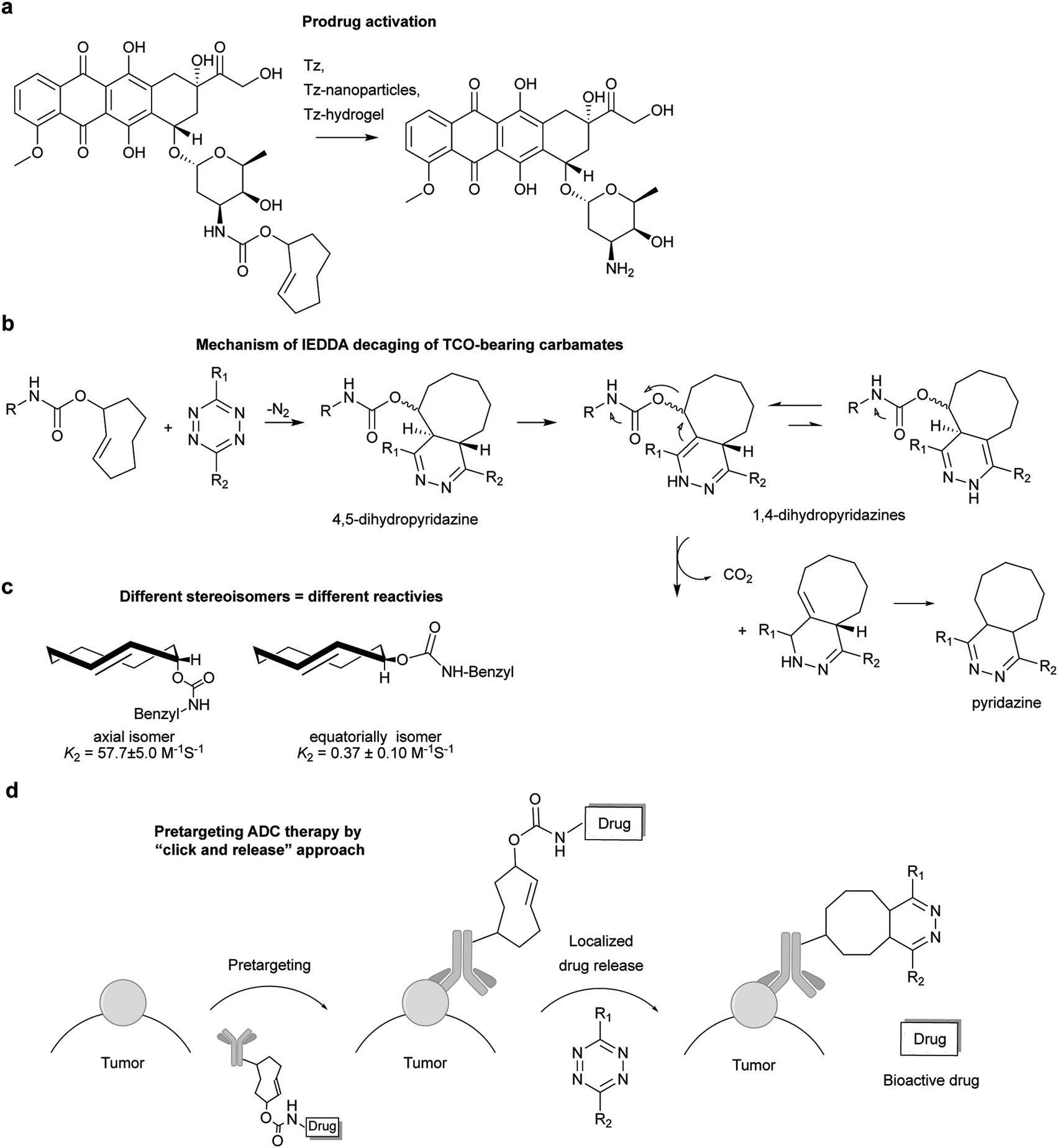 | ||
| Fig. 50 (a) Non-metal cleavage reactions, mediated by bio-friendly chemicals. (b) Doxorubicin release upon tetrazine ligation. (c) Mechanism of decaging by IEDDA tetrazine reaction. (d) Equatorial and axial isomers of TCO carbamates and the respective reaction rates. (e) Triggered drug release from an ADC by tetrazine reaction. | ||
Multivalence of nanoparticles and hydrogels enables the introduction of a high copy number of tetrazine motifs, enhancing activation efficiency and local drug concentration, and thus endow these materials as potential vectors for drug activation. In conventional chemotherapy, high doses of a drug need to be used because of the lack of specificity to the target diseased tissue. This leads to side toxicity and limits the general efficacy of the treatment. Royzen et al. envisioned that by systemic administration of non-toxic tetrazine-bearing magnetic nanoparticles (MNP), enhanced permeability and retention (EPR) effect would guide MNP accumulation in cancerous tissues, minimizing side effects by localized drug release. As a proof of concept, MNPs decorated with tetrazines and fluorescent probes could be used to image in situ prodrug activation of TCO-modified doxorubicin in cancer cells (Fig. 51a). Indeed, the pro-drug was activated completely after 1 hour incubation of fluorescent tetrazine bearing MNP. This multi-model platform has potential for in vivo image-guided drug release either by fluorescence or magnetic resonance imaging.344
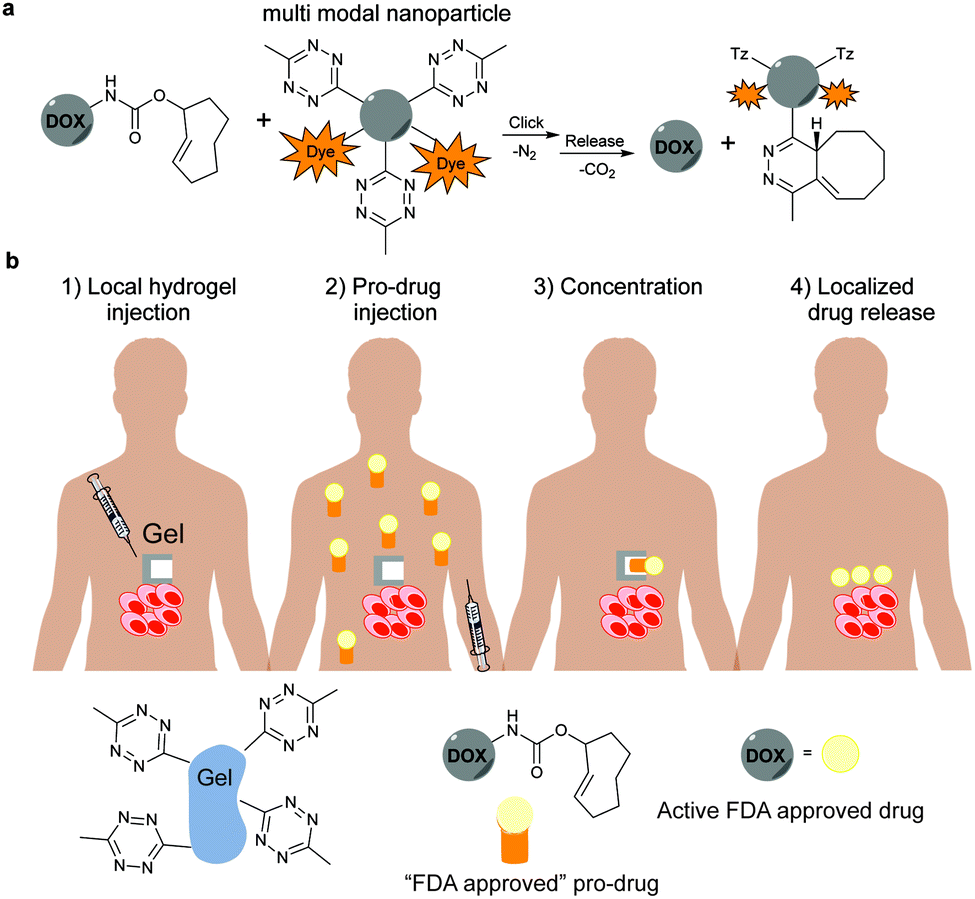 | ||
| Fig. 51 (a) NP functionalization with tetrazines and fluorescent probes for image-guided prodrug activation. (b) “Click-to-release” for concentration and activation of pro-drugs at a diseased site. | ||
Similarly, a tetrazine-based hydrogel was synthesized for local drug activation. In contrast to systemic administration, the hydrogel could be injected directly into the tumour site, which allowed release of drug upon subsequent intravenously injection of the TCO modified drug (Fig. 51b).345 Animal studies revealed an increase in the efficacy of doxorubicin, minimizing systemic toxicity (no signs of toxicity, including weight loss, changes in coat texture and concentration of reticulocytes).
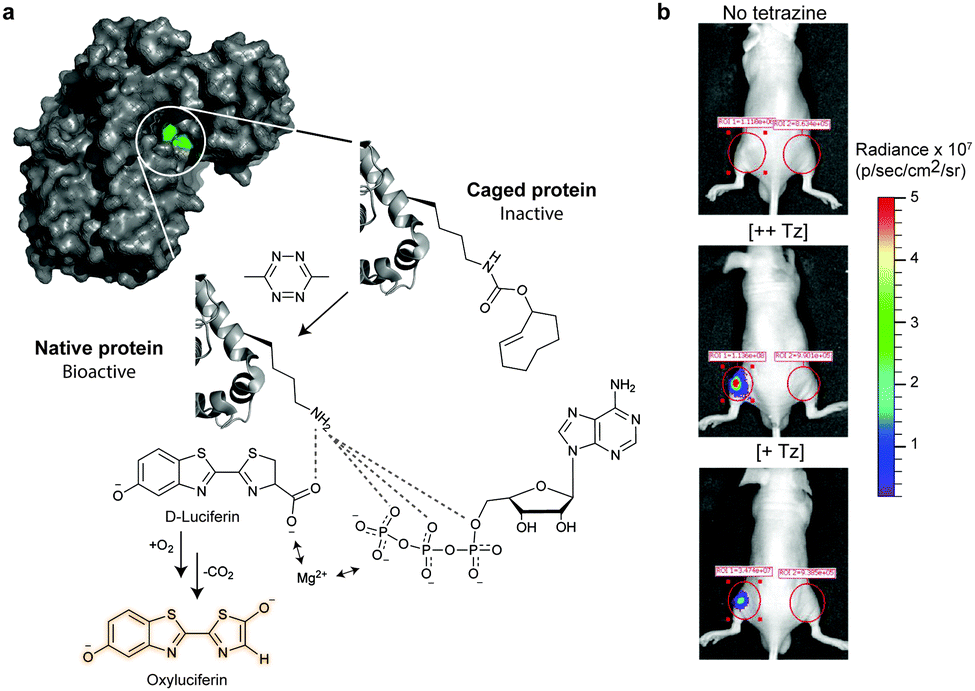 | ||
| Fig. 52 (a) Schematic of the mechanism of protein gain-of-function. Caging the critical Lys residue of fLuc (TCOK-529) blocked protein function that could be restored by tetrazine cleavage and reformation of hydrogen bonding network between ε-amine of K529 and ATP. (b) Bioorthogonal chemical activation strategy in mice by using chemically caged fLuc as a model. HEK293T cells expressing the TCOK-a caged fLuc were injected into living mice subcutaneously (left legs), followed by the treatment of tetrazine (66 or 6.6 mg kg−1 body weight; [++Tz] and [+Tz], respectively) via tail vein injection. After in vivo decaging recover of luciferase bioluminescence was observed. Adapted in part with permission from ref. 311. Copyright 2016 American Chemical Society. | ||
TCO decaging has not been limited only to drug release and protein gain-of-function studies. In another example, a tetrazine triggered approach has been described for the HPLC-free solid phase synthesis of RNA.347 Additionally, a new strategy for bioorthogonal carbonyl sulphide and hydrogen sulphide (COS/H2S) donation initiated by tetrazines has also been achieved (Fig. 53).348
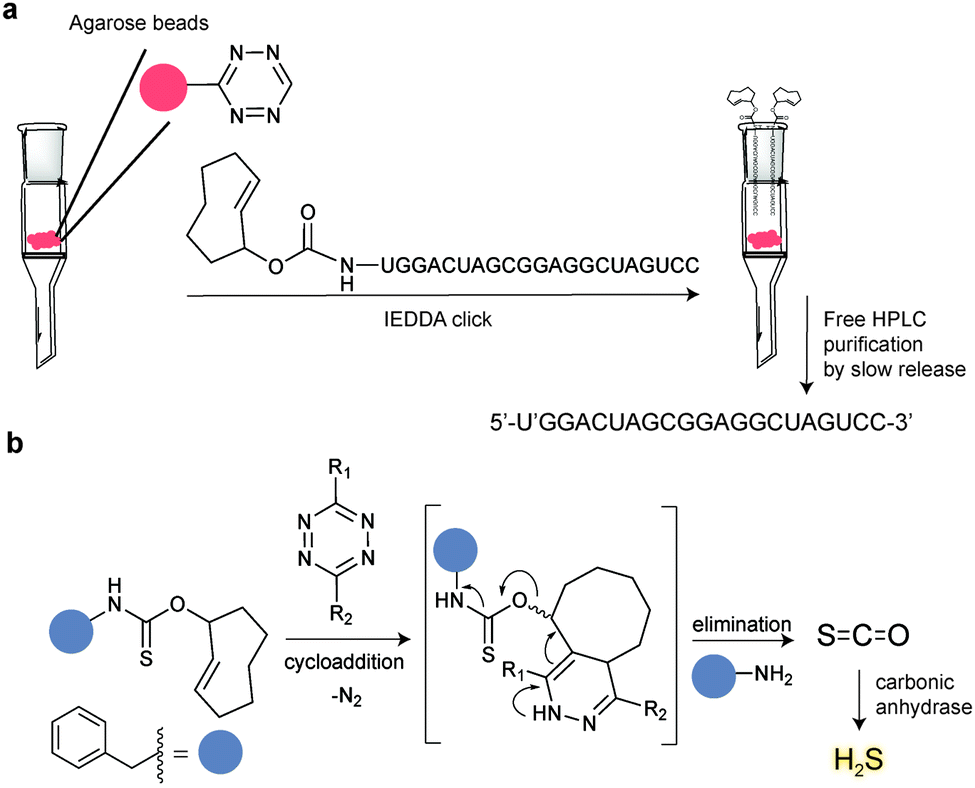 | ||
| Fig. 53 (a) Click-to-release system for HPLC-free purification of RNAs bearing a TCO group. Prior generated failure capped sequences are collected in the early-eluted fractions before the slow release of TCO. (b) Self-immolative thiocarbamates triggered by tetrazines decompose and release SCO, which is rapidly converted to H2S by the ubiquitous enzyme carbonic anhydrase. | ||
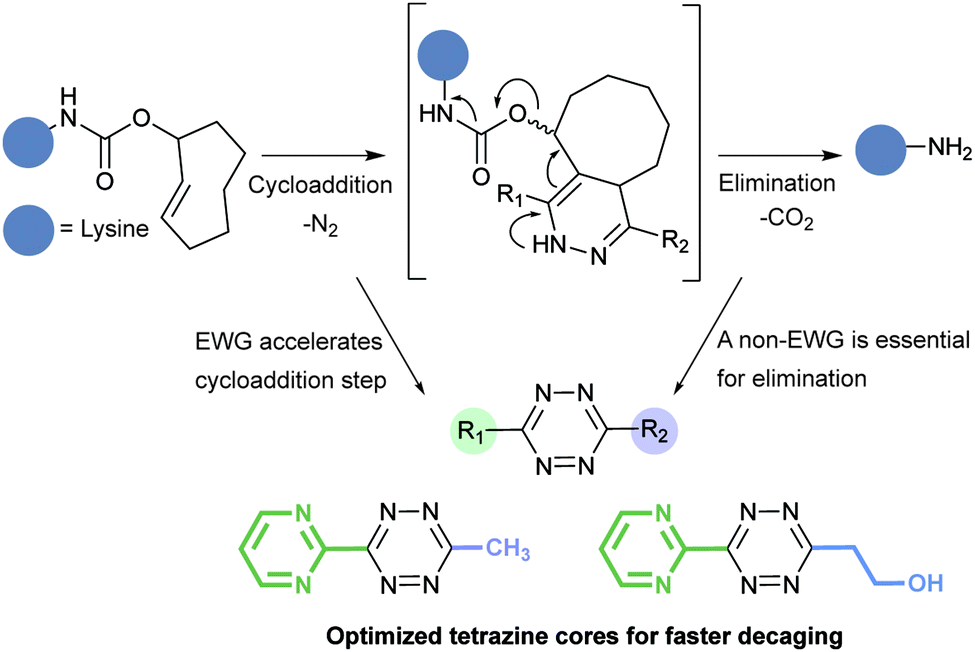 | ||
| Fig. 54 Structure parameters determining the kinetics of TCO decaging. | ||
7.3 Vinyl-ether for function reactivation by IEDDA ligation
Apart from TCO derivatives, vinyl ethers could also serve as a masking group for alcohols.350,351 Our group recently reported a vinyl ether decaging reaction on a cytotoxic drug for activation of a halogen-bearing duocarmycin derivative via a Winstein spirocyclization (Fig. 55). In cells the double prodrug showed similar cytotoxicity to the active drug. The broad applicability of this reaction on other chemotypes, including the protected amino acids serine and tyrosine, an 1,6-anhydro sugar, and a fluorophore, expand the scope and potential of this reaction for applications on proteins and glycans.352 | ||
| Fig. 55 The double pro-drug reacts with tetrazine leading to the formation of an intermediate that undergoes a Winstein spirocyclization to afford the bioactive cyclopropanyl duocarmycin analogue. | ||
Furthermore, based on the vinyl ether decaging chemistry, a tetrazine responsive self-immolative cleavable linker was developed by Bradley et al. to achieve molecular triggered drug delivery (Fig. 56a). Accordingly, the vinyl ether cleavable moiety was embedded on a PEG polymer and upon addition of tetrazine, the decaged prodrug could trigger 80% cell death with 48 h incubation.353 Also recently, Devaraj et al. explored the vinyl ether decaging for mRNA detection. In particular, a caged near-infrared (NIR)-emitting cyanine dye was shown to restore the fluorescence by up to 70-fold after tetrazine reaction (Fig. 56b). Further this NIR fluorogenic probe was applied for mRNA detection in vitro and in cellular context (Fig. 56c).354
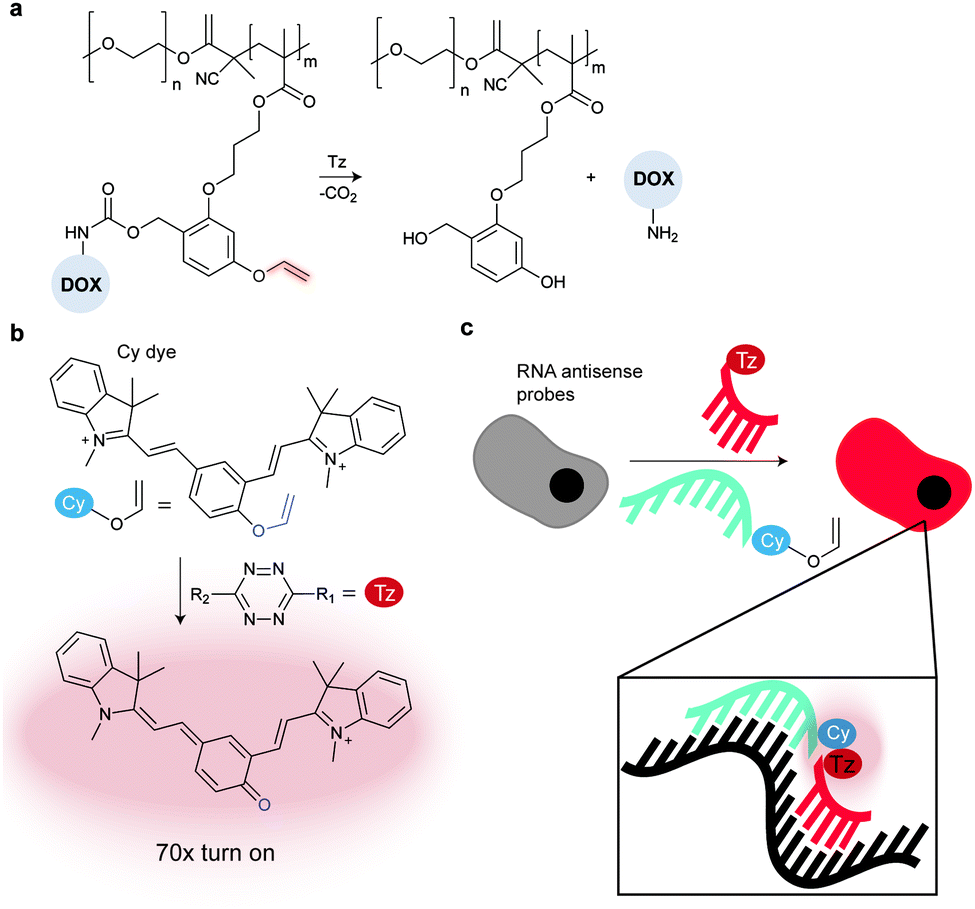 | ||
| Fig. 56 (a) PEG polymer conjugated to a doxorubicin pro-drug through a self-immolative, which is eliminated upon reaction with tetrazine liberating the bioactive drug. (b) NIR-emitting vinyl ether cyanine dye activated by tetrazine reaction. (c) A fluorogenic NIR RNA-templated tetrazine uncaging probe could be used for cell imaging of mRNA. | ||
8. IEDDA reactions in materials and nanomaterials with biological applications
The aqueous compatibility of IEDDA reactions without involving any toxic catalyst or solvent makes it an optimal reaction for bio- and nanomaterial applications. Most importantly, due to its modular nature, IEDDA reactions allow the facile cross-linking of building blocks and thus enable an alternative way for the construction and modification of molecular structures. Another major advantage of IEDDA reactions is the facilitated purification since none of the conjugation partners has to be used in excess to reach fast reactions. Based on these favourable characteristics, the IEDDA reaction has been explored for biomaterials and nanomaterials preparation for applications in biology and medicine. Some examples of these applications are presented in the following sections.8.1 Polymer hydrogel formation via IEDDA bioorthogonal reaction
Due to their high-water content, excellent mass transport properties, and tissue-like elasticity, ionically crosslinked hydrogels have been used extensively to encapsulate cells and bioactive molecules (e.g. 3D cell culture and drug delivery in vivo) as well as for tissue engineering but so far, these polymers have been largely limited by low crosslinking efficiency and poor integrity of the resulting materials. To overcome these limitations, IEDDA ligation, along with other bioorthogonal reactions,355 have been proposed as an alternative method for the synthesis of biocompatible hydrogels. In one example, Joshi et al. reported the synthesis of alginate hydrogels via tetrazine–norbornene bioorthogonal chemistry. By using tetrazine/norbornene alginate derivatives, the hydrogel could be prepared in 1 hour displaying tunable mechanical and swelling properties depending on the use of different tetrazine/norbornene alginate ratios (Fig. 57). Compared with the ionically crosslinked hydrogel, IEDDA-clicked gels showed higher cell viability and cell metabolic activity. Minimal inflammatory response and high stability was also observed after in vivo injections. | ||
| Fig. 57 Schematic of the click synthesis of alginate polymer. Coupling chemistry is used to modify alginate backbone carboxylic acids with tetrazine or norbornene, resulting in Alg–Tz or Alg–N polymers respectively. Alg–Tz and Alg–N polymers are reacted together to create a covalently crosslinked click alginate hydrogel network. | ||
Furthermore, IEDDA combined with thiol–ene chemistry could serve as a robust reaction for further functionalization. Indeed, post-gelation modification could be introduced by functionalizing excess norbonene units with Cys-containing RGD peptide (RGDS) by thiol–ene chemistry. This construct showed 3-fold increase in cell adherence compared with unmodified gels.356 In another example, Anseth et al. demonstrated that hydrogels formed by multi-functional PEG–tetrazine units could be functionalized with norbornene-containing peptides (e.g. an ECM mimetic cell degradable peptide, a RGDS peptide or a short peptide bearing a thiol group) for 3D cell culture. This hydrogel network formulated to have pendant thiols could be subsequently used for functionalization with fluorescein and rhodamine fluorescent norbornene-BSA proteins by photoinitiated thiol–ene click reaction with excellent pattern fidelity (Fig. 58).357
 | ||
| Fig. 58 Sequential biochemical patterning in tetrazine click gels. Tetrazine click hydrogels were formed to leave pendant thiols in the network for functionalization with fluorescent norbornene–BSA proteins by thiol–ene chemistry. Scalebars = 200 μm. Adapted with permission from ref. 357. Copyright 2013 American Chemical Society. | ||
In order to develop a dual marking hydrogel system, Weissleder et al. encapsulated an X-ray radiography insoluble contrast agent and a pigment into a gelatin hydrogel, which was prepared by IEDDA reaction between tetrazine- and norbornene-modified gelatines. In vivo studies showed good performance for in vivo CT imaging and ex vivo visual localization of the radiopaque tinted hydrogel.358 Double network (DN) hydrogels have also received much attention because of its high-water percentage (ca. 90 wt%) and high strength. In another work, Dove et al. reported the synthesis of a DN hydrogel in one-step under physiological conditions and within 3 min by using tetrazine–norbornene click reaction for formation of the loose network (norbornene-functionalized chitosan and a linear PEG ditetrazine) and thiol–ene chemistry for formation of the dense network (PEG–tetraalkyne and a linear PEG–dithiol). The resulting hydrogels exhibited excellent mechanical properties and display high cytocompatibility (>99% cell viability after 48 h).359
Due to its ability to coordinate with metal ions, PEG polymers terminally functionalized with bipyridyltetrazines could form multi-functional supermolecular gels in the presence of metal ions (Fe2+ and Ni2+). This platform was explored to prepare metallo-hydrogel scaffolds for photo-triggered drug release (Fig. 59). This approach is, however, limited by slow photo release rates due to the competitive absorption from the dark-coloured gel, and thus, the development of new systems with alternative metals or ligands with desirable optical properties is demanded.360
 | ||
| Fig. 59 Synthesis of metallo-hydrogels. (a) Diels–Alder reaction and metal coordination of bpTz. (b) Schematic representation of the drug release approach triggered by photo decaging from the metallo-hydrogel. | ||
The IEDDA reaction has also emerged as an important tool for producing microsphere hydrogels by interfacial crosslinking using a tetrazine-hyaluronic derivative (HA-Tz) and bis-TCO cross-linker (Fig. 60a and b). Covalent tagging with diffusible Alexa-TCO yielded shell- or core-labelled microspheres. Alternating the presence and absence of TCO-dye afford onion-like structures with 3, 5, and 7 layers (Fig. 60c). These 3D microspheres were tested for 3D culture of prostate cancer cells with good cell proliferation and viability (98% viable at 5 days). Notably, cells aggregated to form clusters instead of homogeneous disperse.361
 | ||
| Fig. 60 Synthesis of microsphere hydrogels. (a) Instantaneous cross-linking via tetrazine–TCO ligation. (b) Gel interface forms when a droplet of tetrazine-modified hyaluronic (HA-Tz) contacts a solution of bis-TCO. Cross-linking at the gel/liquid interface is faster than the rate of diffusion through the gel interface. (c) Onion-like structures obtained by alternating the presence and absence of Alexa-TCO during the cross-linking procedure. Adapted with permission from ref. 361. Copyright 2014 American Chemical Society. | ||
8.2 Application of IEDDA reaction in polymer formation and modification
Tetrazine bioorthogonal reaction is increasingly being applied for polymer modification or polymer-polymer coupling. In 2011, O’Reilly demonstrated firstly the suitability of the IEDDA conjugation method for efficient synthesis of block copolymers.362 Importantly, the norbornene–tetrazine combination was found to be compatible with the trithiocarbonate group in RAFT (reversible addition–fragmentation chain transfer polymerisation) chain transfer agents (Fig. 61).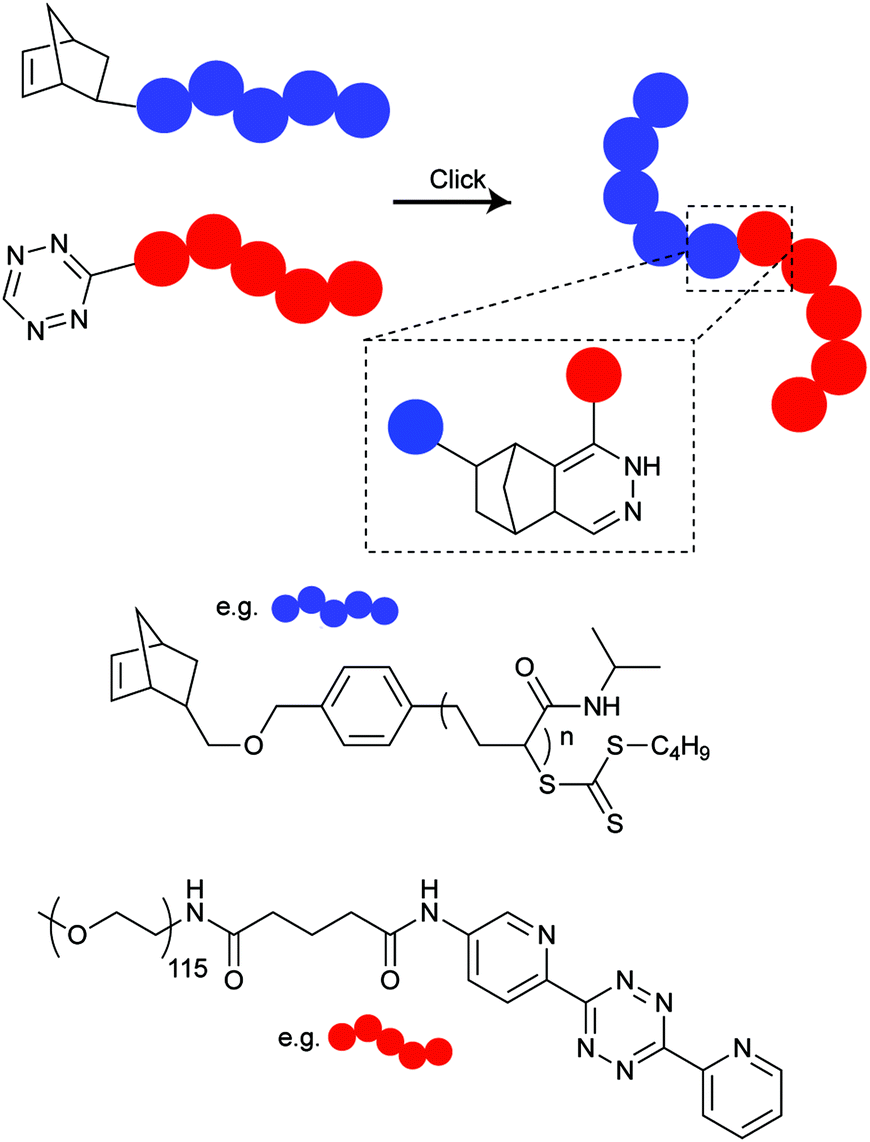 | ||
| Fig. 61 Polymer–polymer coupling by norbornene–tetrazine reaction between norbornenyl-functionalized poly(styrene) trithiocarbonate and a tetrazine-functionalized PEG. | ||
Another application by Jia et al. describes the synthesis of multiblock copolymers through rapid tetrazine–TCO bioorthogonal polymerization (Fig. 62a). Further modification of the copolymer fibres with a fibronectin-derived cell adhesive peptide promotes the attachment and alignment of fibroblasts with various shapes and orientations, whereas when incubated with peptide-free fibres, cells remained round and unbound.363 In a related paper, Fox et al. discovered that dihydrotetrazine could undergo photocatalytic oxidation to tetrazine thereby turning on its reactivity towards s-TCO (Fig. 62b). Interfacial polymerization between tetrazine-bearing monomers and bis-s-TCO monomers conjugated to dihydrotetrazine moieties (DHTz) produced dihydrotetrazine fibres (Fig. 62b), which could be further reacted with TCO bearing proteins, peptides and dyes (Fig. 62c) under irradiation with sensitizers. Notably, the fibres conjugated to a cell adhesive RGD peptide exhibited a dramatically increased ability to mediate contact guidance of cells (Fig. 62d).103
 | ||
| Fig. 62 Synthesis of copolymer fibres. (a) Interfacial polymerization between PEG tetrazine-derived monomer bearing peptides and bis-s-TCO. (b) Schematic representation of fibre photoactivation and subsequent conjugation. DHTz-containing fibres were irradiated with visible light and allowed to react with an s-TCO conjugate bearing fluorophores or cell adhesive peptides. (c) Confocal images (10×, scale bar 100 μm) of activated fibres that were treated with Alexa-s-TCO. (d) Confocal images (10×) of activated fibres that were treated with RGD-s-TCO. Cell culture with NIH 3T3 fibroblasts showed that the cells selectively adhered and spread on the fibres. Cell attachment and spreading on the fibres was not observed in control experiments. Adapted with permission from ref. 103. Copyright 2016 American Chemical Society. | ||
8.3 Application of IEDDA reaction in the synthesis of nanoparticles for biomedicine
Due to a large number of interesting properties (e.g. multivalent targeting capability, high payload, etc.), imaging and therapeutic nanoparticles have been widely used in nanomedicine research.364 Quantum dots (QDs) are highly fluorescent nanoscale crystals with size-dependent emission spectra and excellent photophysical properties (e.g. high quantum yield, low photobleaching) that have been demonstrated to have several advantages over fluorescent organic dyes for cell imaging.365 The first proof of concept study using “bioorthogonal QD” was reported in 2010 by Bawendi et al. In this work norbornene-coated QDs were used to target cancer cells previously decorated with tetrazine-modified epidermal growth factor antibody (Fig. 63).366 Similarly, Duda et al. conjugated tetrazine-modified antibodies to quantum dots modified with norbornene. Using these QD/antibody conjugates, the authors target a rare cell population in bone marrow at the single cell level in live animals using multiphoton microscopy.367 | ||
| Fig. 63 Pre-targeting of EGFRs on live cells with a tetrazine-modified EGF antibody and labelling with norbornene-coated QDs. | ||
Beyond QDs Weissleder et al. described the use of magneto-fluorescent nanoparticles (MFNPs; fluorescent iron oxide nanoparticles) in different configurations to improve binding efficiency and cell detection for clinical diagnostics. This technique was referred to as ‘bioorthogonal nanoparticle detection’ (BOND). The rational for the development of this new methodology was that magnetic based sensing schemes benefit from high sensitivity, low cost, and the ability to perform measurements in samples that are not optically transparent. Their study compared two BOND assays to determine the ability to specifically target cancer cells. Cellular labelling was performed by exposing cells directly to antibody–MFNP conjugates (one step BOND-1 strategy) or by incubating cells first with TCO decorated antibodies, followed by tetrazine–MFNPs (two steps strategy: BOND-2) (Fig. 64).288 The authors demonstrated that BOND-2 consistently yielded higher nanoparticle binding to cells by a factor of 10 to 15 compared to direct immuno-conjugates obtained from BOND-1 method.
 | ||
| Fig. 64 “BOND” strategy. Antibodies against biomarkers are modified with TCO and used as scaffolds to deliver magnetic fluorescent nanoparticles (MFNPs) onto live cells through tetrazine reaction. | ||
Importantly, a similar two-step targeting strategy using avidin/biotin, although exceeds the direct “BOND-1 conjugate”, falls behind BOND-2, which was attributed to the large size of avidin proteins blocking neighbouring biotin binding sites and potential constraining binding configurations. Using this system, different biomarkers of human circulating tumour cells were detected and evaluated via μ-nuclear magnetic resonance on clinical samples.368–371 Alternatively, the Hall effect of MNP enables rapid and sensitive high-throughput screening on mammalian cells and bacteria.372,373 Other medical applications of BOND-2 technique are on-chip detection of microvesicles liberated into the blood circulation from glioblastomas,374 detection of microvesicle urinary markers,375,376 and detection of mammalian and bacteria cells for diagnosis of infectious disease.377–379 BOND-2 was also evaluated for cell tracking by spatiotemporally labelling with photoactivatable fluorophores,380 and for targeting intracellular biomarkers in permeabilized and fixed cells by confocal fluorescence imaging and diagnostic magnetic resonance.381 In one attempt to improve BOND-2 assay sensitivity and robustness, multiple consecutive conjugation steps of fluorescent nanoparticles containing TCO and tetrazine groups were used to amplify the corresponding signals.382 Detection sensitivity could be increased up to 2 orders of magnitude over current method.
There have also been many advances in the development of nanoparticle systems for in vivo radionuclide imaging. Liposomes are attractive nanoparticle vesicles considered promising systems for controlled drug delivery. Reiner et al. introduced in 2013 a dual-delivery pretargeting approach using a pH low insertion peptide conjugated to a tetrazine (pHLIP–Tz) to tether “TCO-liposomes” to tumour tissue in live animals (Fig. 65). The long circulating radiolabelled liposomes showed a significant accumulation in tumour sites mainly via enhanced permeability and retention (EPR) effect, after in vivo IEDDA click reaction between pHLIP–tetrazine and the 18F-labelled liposomes bearing TCO lipids.383 The described liposomes might be particularly useful for imaging the delivery of drugs immobilized either on the liposomal surface or in the endoliposomal space. Similarly, mesoporous silicon (PSi) is a very attractive material for targeted drug delivery in nanomedicine due to their biodegradability, low toxicity, and high drug loading capacity. Recently, Airaksinen et al. reported the first successful pretargeted PET imaging of TCO-modified mesoporous silicon nanoparticles (PSi-NPs), using 18F-labelled tetrazine as a tracer. In vivo IEDDA reaction between the 18F-Tz and TCOs on the surface of the PSi-NPs was fast and occurred within the first 10 min. Unfortunately, administration of the 18F-Tz tracer 24 h after the injection of the nanoparticles, resulted in no reaction, suggesting cis–trans isomerisation in vivo of the TCO groups. Further optimization is still needed to maintain the high reactivity of TCO groups to permit pretargeted imaging over the course of days, which would be relevant for longer-circulating nanotheranostic agents.309
 | ||
| Fig. 65 Nanoparticle systems for radionuclide imaging. (a) In vivo pretargeting with liposomes. At low pH levels, the polar C-terminus of the peptide pHLIP–Tz changes its conformation to a helical structure, resulting in the insertion of the peptide into the cellular membrane. The tetrazine on the tumour surface can then be targeted by 18F-labelled bioorthogonal liposomes bearing TCO groups (left shoulder, pre-injected intra-tumourally with pHLIP–Tz, SKOV3; right shoulder, untreated; bioorthogonally TCO liposomes encapsulating 18F PET isotope were administered intravenously). Reprinted in part with permission from ref. 383. Copyright 2013 American Chemical Society. (b) Pretargeted PET–CT tracing of mesoporous silicon nanoparticles. (i) TCO-modified PSi-NPs were administered 15 min before the intravenous injection of the 18F-tetrazine probe. (ii) Controls were injected only with 18F-tetrazine. The arrows indicate the location of spleen, the expected target organ. Reprinted in part with permission from ref. 309. Copyright 2017 American Chemical Society. | ||
Carbon nanotubes (CNT) and graphene are another class of nanoparticles that have potential for biomedical applications. Functionalization of these nanocomposites using tetrazine-chemistry is described as a promising approach to control and manipulate their characteristics.384–386 To the best of our knowledge, application of these constructs in a biomedical context is still to be explored.
Conclusions
Owing to its high selectivity, unparalleled fast kinetics, and free catalyst nature, the IEDDA reactions have emerged during the last decade as the state-of-the-art approach for selective modification of proteins in live cells and notably in animals. Herein we reviewed recent advances in this field with a focus on examples that highlight the utility of IEDDA ligations over other bioorthogonal reactions. The superfast kinetics and unique fluorogenicity makes IEDDA particularly suitable for super resolution imaging and tracking fast dynamics process in cells with minimal background. In addition, with the deepening understanding of IEDDA reactivity, researchers have realized that by choosing a fine-tuned tetrazine-dienophile pair, two parallel IEDDA reactions can be orthogonal to each other, which allows fast simultaneous multicolour labelling. Although recent developments are very promising a number of key challenges can be envisaged that must be addressed in the future. The fastest IEDDA ligations usually involve relatively large reactive groups, leading to higher chances of influencing biological processes. In addition, the hydrophobicity of large reporting groups could also be disadvantageous in certain circumstances, affording high nonspecific labelling. To circumvent this, a challenge lies in the development of small, reactive and hydrophilic dienophiles that possess at the same time high stability and permeability to achieve fast and selective labelling with minimal background. In this regard, the troublesome synthesis of the tetrazine and dienophiles partners, combined with their limited commercial availability, is still restraining IEDDA as a widely used tool.In terms of pretargeting, the development of clearing agents introduced before administration of the radiolabelled probe results in improved tissue to non-tissue ratios with pronounced localization of radioactivity in the tumour. However, the next challenges lie in the modulation of the pharmacokinetics of the Tz/TCO components to reduce in vivo nonspecific accumulation of the IEDDA radio ligands. More attention should be also given to the therapeutic potential of pretargeted radio immunotherapy. It is envisaging that the use of radioligands with optimum pharmacokinetic profiles combined with clearing agents would substantially reduce accumulation in healthy tissues, maximizing the therapeutic efficacy and minimizing side effects. Regarding future perspectives, we anticipate the use of IEDDA reactions will go beyond the straightforward attachment of a given functional moiety to a protein of interest. There are already published work supporting that tetrazine decaging reactions will emerge as efficient tools for study for example the effect of PTMs on protein function and bioorthogonal drug activation. In vivo examples of ADCs containing a dienophile chemically linked to a prodrug could be already used for direct release of the drug after tumour binding triggered by the IEDDA reaction. Future studies will aim to optimize the therapeutic effect of this class of ADCs for pre-targeting tumour treatment. In resume, the potential of IEDDA reactions are enormous and suggest a rich future of this bioorthogonal reaction in the field of chemical biology.
Acknowledgements
We thank the European Commission (Marie Skłodowska-Curie ITN Protein Conjugates; Marie Skłodowska-Curie IEF to B. L. O.), China Scholarship Council (PhD studentship to Z. G.), FCT Portugal (FCT Investigator to G. J. L. B.), and the EPSRC for financial support. We also thank Lavinia Dunsmore, Sarah Davies and Benjamin Stenton for helpful carefully reading the manuscript. G. J. L. B. is a Royal Society University Research Fellow and the recipient of a European Research Council Starting Grant (TagIt).Notes and references
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS PubMed.
- O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed.
- L. Xue, I. A. Karpenko, J. Hiblot and K. Johnsson, Nat. Chem. Biol., 2015, 11, 917–923 CrossRef CAS PubMed.
- J. Li and P. R. Chen, Nat. Chem. Biol., 2016, 12, 129–137 CrossRef CAS PubMed.
- N. Krall, F. P. da Cruz, O. Boutureira and G. J. L. Bernardes, Nat. Chem., 2016, 8, 103–113 CAS.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed.
- O. S. Walker, S. J. Elsasser, M. Mahesh, M. Bachman, S. Balasubramanian and J. W. Chin, J. Am. Chem. Soc., 2016, 138, 718–721 CrossRef CAS PubMed.
- I. Nikic, T. Plass, O. Schraidt, J. Szymanski, J. A. Briggs, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2014, 53, 2245–2249 CrossRef CAS PubMed.
- T. H. Wright, B. J. Bower, J. M. Chalker, G. J. L. Bernardes, R. Wiewiora, W. L. Ng, R. Raj, S. Faulkner, M. R. Vallee, A. Phanumartwiwath, O. D. Coleman, M. L. Thezenas, M. Khan, S. R. Galan, L. Lercher, M. W. Schombs, S. Gerstberger, M. E. Palm-Espling, A. J. Baldwin, B. M. Kessler, T. D. Claridge, S. Mohammed and B. G. Davis, Science, 2016, 354, 597 CrossRef CAS PubMed.
- P. V. Robinson, C. T. Tsai, A. E. de Groot, J. L. McKechnie and C. R. Bertozzi, J. Am. Chem. Soc., 2016, 138, 10722–10725 CrossRef CAS PubMed.
- M. H. Wright, B. Clough, M. D. Rackham, K. Rangachari, J. A. Brannigan, M. Grainger, D. K. Moss, A. R. Bottrill, W. P. Heal, M. Broncel, R. A. Serwa, D. Brady, D. J. Mann, R. J. Leatherbarrow, R. Tewari, A. J. Wilkinson, A. A. Holder and E. W. Tate, Nat. Chem., 2014, 6, 112–121 CrossRef CAS PubMed.
- D. Rosner, T. Schneider, D. Schneider, M. Scheffner and A. Marx, Nat. Protoc., 2015, 10, 1594–1611 CrossRef PubMed.
- N. Stefan, M. Zimmermann, M. Simon, U. Zangemeister-Wittke and A. Pluckthun, Bioconjugate Chem., 2014, 25, 2144–2156 CrossRef CAS PubMed.
- P. C. Trippier, ChemMedChem, 2013, 8, 190–203 CrossRef CAS PubMed.
- R. Rossin, S. M. van Duijnhoven, W. Ten Hoeve, H. M. Janssen, L. H. Kleijn, F. J. Hoeben, R. M. Versteegen and M. S. Robillard, Bioconjugate Chem., 2016, 27, 1697–1706 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- K. E. Beatty, J. D. Fisk, B. P. Smart, Y. Y. Lu, J. Szychowski, M. J. Hangauer, J. M. Baskin, C. R. Bertozzi and D. A. Tirrell, Chembiochem, 2010, 11, 2092–2095 CrossRef CAS PubMed.
- B. Amgarten, R. Rajan, N. Martinez-Saez, B. L. Oliveira, I. S. Albuquerque, R. A. Brooks, D. G. Reid, M. J. Duer and G. J. L. Bernardes, Chem. Commun., 2015, 51, 5250–5252 RSC.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- S. I. Presolski, V. Hong, S. H. Cho and M. G. Finn, J. Am. Chem. Soc., 2010, 132, 14570–14576 CrossRef CAS PubMed.
- D. Soriano Del Amo, W. Wang, H. Jiang, C. Besanceney, A. C. Yan, M. Levy, Y. Liu, F. L. Marlow and P. Wu, J. Am. Chem. Soc., 2010, 132, 16893–16899 CrossRef PubMed.
- J. Dommerholt, F. P. Rutjes and F. L. van Delft, Top. Curr. Chem., 2016, 374, 16 CrossRef PubMed.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS PubMed.
- B. L. Nilsson, L. L. Kiessling and R. T. Raines, Org. Lett., 2000, 2, 1939–1941 CrossRef CAS PubMed.
- E. Saxon, J. I. Armstrong and C. R. Bertozzi, Org. Lett., 2000, 2, 2141–2143 CrossRef CAS PubMed.
- S. S. van Berkel, M. B. van Eldijk and J. C. van Hest, Angew. Chem., Int. Ed., 2011, 50, 8806–8827 CrossRef CAS PubMed.
- J. Y. Axup, K. M. Bajjuri, M. Ritland, B. M. Hutchins, C. H. Kim, S. A. Kazane, R. Halder, J. S. Forsyth, A. F. Santidrian, K. Stafin, Y. Lu, H. Tran, A. J. Seller, S. L. Biroc, A. Szydlik, J. K. Pinkstaff, F. Tian, S. C. Sinha, B. Felding-Habermann, V. V. Smider and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16101–16106 CrossRef CAS PubMed.
- W. Song, Y. Wang, J. Qu, M. M. Madden and Q. Lin, Angew. Chem., Int. Ed., 2008, 47, 2832–2835 CrossRef CAS PubMed.
- A. Herner and Q. Lin, Top. Curr. Chem., 2016, 374, 1 CrossRef PubMed.
- D. A. MacKenzie, A. R. Sherratt, M. Chigrinova, L. L. Cheung and J. P. Pezacki, Curr. Opin. Chem. Biol., 2014, 21, 81–88 CrossRef CAS PubMed.
- J. M. Chalker, C. S. Wood and B. G. Davis, J. Am. Chem. Soc., 2009, 131, 16346–16347 CrossRef CAS PubMed.
- N. Li, R. K. Lim, S. Edwardraja and Q. Lin, J. Am. Chem. Soc., 2011, 133, 15316–15319 CrossRef CAS PubMed.
- Y. A. Lin, J. M. Chalker, N. Floyd, G. J. L. Bernardes and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 9642–9643 CrossRef CAS PubMed.
- M. Yang, J. Li and P. R. Chen, Chem. Soc. Rev., 2014, 43, 6511–6526 RSC.
- R. A. Carboni and R. V. Lindsey, J. Am. Chem. Soc., 1959, 81, 4342–4346 CrossRef CAS.
- D. L. Boger, R. P. Schaum and R. M. Garbaccio, J. Org. Chem., 1998, 63, 6329–6337 CrossRef CAS PubMed.
- A. Hamasaki, R. Ducray and D. L. Boger, J. Org. Chem., 2006, 71, 185–193 CrossRef CAS PubMed.
- M. R. Karver, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2011, 22, 2263–2270 CrossRef CAS PubMed.
- S. Jain, K. Neumann, Y. Zhang, J. Geng and M. Bradley, Macromolecules, 2016, 49, 5438–5443 CrossRef CAS.
- D. Wang, W. Chen, Y. Zheng, C. Dai, K. Wang, B. Ke and B. Wang, Org. Biomol. Chem., 2014, 12, 3950–3955 CAS.
- C. Li, H. X. Ge, B. Yin, M. Y. She, P. Liu, X. D. Li and J. L. Li, RSC Adv., 2015, 5, 12277–12286 RSC.
- J. Balcar, G. Chrisam, F. X. Huber and J. Sauer, Tetrahedron Lett., 1983, 24, 1481–1484 CrossRef CAS.
- D. N. Kamber, Y. Liang, R. J. Blizzard, F. Liu, R. A. Mehl, K. N. Houk and J. A. Prescher, J. Am. Chem. Soc., 2015, 137, 8388–8391 CrossRef CAS PubMed.
- K. A. Horner, N. M. Valette and M. E. Webb, Chem. – Eur. J., 2015, 21, 14376–14381 CrossRef CAS PubMed.
- L. R. Domingo, M. T. Picher and J. A. Saez, J. Org. Chem., 2009, 74, 2726–2735 CrossRef CAS PubMed.
- J. Sauer and D. Lang, Angew. Chem., Int. Ed. Engl., 1964, 76, 603 Search PubMed.
- J. Sauer, D. K. Heldmann, J. Hetzenegger, J. Krauthan, H. Sichert and J. Schuster, Eur. J. Org. Chem., 1998, 2885–2896 CrossRef CAS.
- J. Sauer, D. Lang and H. Wiest, Chem. Ber., 1964, 97, 3208–3218 CrossRef CAS.
- J. Sauer and G. Heinrichs, Tetrahedron Lett., 1966, 7, 4979 CrossRef.
- D. K. Heldmann and J. Sauer, Tetrahedron Lett., 1997, 38, 5791–5794 CrossRef CAS.
- A. Meier and J. Sauer, Tetrahedron Lett., 1990, 31, 6855–6858 CrossRef CAS.
- K. Müller and J. Sauer, Tetrahedron Lett., 1984, 25, 2541–2544 CrossRef.
- D. R. Soenen, J. M. Zimpleman and D. L. Boger, J. Org. Chem., 2003, 68, 3593–3598 CrossRef CAS PubMed.
- F. Thalhammer, U. Wallfahrer and J. Sauer, Tetrahedron Lett., 1990, 31, 6851–6854 CrossRef CAS.
- F. Liu, Y. Liang and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 11483–11493 CrossRef CAS PubMed.
- F. Liu, R. S. Paton, S. Kim, Y. Liang and K. N. Houk, J. Am. Chem. Soc., 2013, 135, 15642–15649 CrossRef CAS PubMed.
- M. T. Taylor, M. L. Blackman, O. Dmitrenko and J. M. Fox, J. Am. Chem. Soc., 2011, 133, 9646–9649 CrossRef CAS PubMed.
- A. Darko, S. Wallace, O. Dmitrenko, M. M. Machovina, R. A. Mehl, J. W. Chin and J. M. Fox, Chem. Sci., 2014, 5, 3770–3776 RSC.
- W. Chen, D. Wang, C. Dai, D. Hamelberg and B. Wang, Chem. Commun., 2012, 48, 1736–1738 RSC.
- J. A. Wagner, D. Mercadante, I. Nikic, E. A. Lemke and F. Grater, Chem. – Eur. J., 2015, 21, 12431–12435 CrossRef CAS PubMed.
- J. E. Hoffmann, T. Plass, I. Nikic, I. V. Aramburu, C. Koehler, H. Gillandt, E. A. Lemke and C. Schultz, Chem. – Eur. J., 2015, 21, 12266–12270 CrossRef CAS PubMed.
- J. Li, S. Jia and P. R. Chen, Nat. Chem. Biol., 2014, 10, 1003–1005 CrossRef CAS PubMed.
- R. M. Versteegen, R. Rossin, W. ten Hoeve, H. M. Janssen and M. S. Robillard, Angew. Chem., Int. Ed., 2013, 52, 14112–14116 CrossRef CAS PubMed.
- M. Vrabel, P. Kolle, K. M. Brunner, M. J. Gattner, V. Lopez-Carrillo, R. de Vivie-Riedle and T. Carell, Chem. – Eur. J., 2013, 19, 13309–13312 CrossRef CAS PubMed.
- I. Nikic, J. H. Kang, G. E. Girona, I. V. Aramburu and E. A. Lemke, Nat. Protoc., 2015, 10, 780–791 CrossRef CAS PubMed.
- G. B. Cserep, O. Demeter, E. Batzner, M. Kallay, H. A. Wagenknecht and P. Kele, Synthesis, 2015, 2738–2744 CAS.
- J. Yang, Y. Liang, J. Seckute, K. N. Houk and N. K. Devaraj, Chem. – Eur. J., 2014, 20, 3365–3375 CrossRef CAS PubMed.
- A. Meijer, S. Otto and J. B. F. N. Engberts, J. Org. Chem., 1998, 63, 8989–8994 CrossRef CAS.
- J. W. Wijnen, S. Zavarise, J. B. F. N. Engberts and M. Charton, J. Org. Chem., 1996, 61, 2001–2005 CrossRef CAS.
- K. Lang, L. Davis, J. Torres-Kolbus, C. Chou, A. Deiters and J. W. Chin, Nat. Chem., 2012, 4, 298–304 CrossRef CAS PubMed.
- M. A. Grishina, V. A. Potemkin, E. V. Bartashevich, A. N. Sinyaev, G. L. Rusinov, N. I. Latosh, I. N. Ganebnykh, O. V. Koryakova and R. I. Ishmetova, J. Struct. Chem., 2006, 47, 1155–1160 CrossRef CAS.
- A. Pinner, Ber. Dtsch. Chem. Ges., 1893, 26, 2126–2135 CrossRef.
- P. Audebert, S. Sadki, F. Miomandre, G. Clavier, M. C. Vernieres, M. Saoud and P. Hapiot, New J. Chem., 2004, 28, 387–392 RSC.
- N. O. Abdel, M. A. Kira and M. N. Tolba, Tetrahedron Lett., 1968, 9, 3871–3872 CrossRef.
- J. F. Ding, Z. Li, Z. Cui, G. P. Robertson, N. H. Song, X. M. Du and L. Scoles, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3374–3386 CrossRef CAS.
- M. Savastano, C. Bazzicalupi, C. Giorgi, C. Garcia-Gallarin, M. D. Lopez de la Torre, F. Pichierri, A. Bianchi and M. Melguizo, Inorg. Chem., 2016, 55, 8013–8024 CrossRef CAS PubMed.
- J. Yang, M. R. Karver, W. Li, S. Sahu and N. K. Devaraj, Angew. Chem., Int. Ed., 2012, 51, 5222–5225 CrossRef CAS PubMed.
- D. L. Alge, D. F. Donohue and K. S. Anseth, Tetrahedron Lett., 2013, 54, 5639–5641 CrossRef CAS PubMed.
- L. G. Meimetis, J. C. Carlson, R. J. Giedt, R. H. Kohler and R. Weissleder, Angew. Chem., Int. Ed., 2014, 53, 7531–7534 CrossRef CAS PubMed.
- J. C. Carlson, L. G. Meimetis, S. A. Hilderbrand and R. Weissleder, Angew. Chem., Int. Ed., 2013, 52, 6917–6920 CrossRef CAS PubMed.
- D. S. Liu, A. Tangpeerachaikul, R. Selvaraj, M. T. Taylor, J. M. Fox and A. Y. Ting, J. Am. Chem. Soc., 2012, 134, 792–795 CrossRef CAS PubMed.
- R. Stollé, J. Prakt. Chem., 1906, 73, 277–287 CrossRef.
- D. Z. Wang, W. X. Chena, Y. Q. Zheng, C. F. Dai, L. F. Wang and B. H. Wang, Heterocycl. Commun., 2013, 19, 171–177 CAS.
- E. Sagot, A. Le Roux, C. Soulivet, E. Pasquinet, D. Poullain, E. Girard and P. Palmas, Tetrahedron, 2007, 63, 11189–11194 CrossRef CAS.
- S. G. Tolshchina, G. L. Rusinov and V. N. Charushin, Chem. Heterocycl. Compd., 2013, 49, 66–91 CrossRef CAS.
- Z. Novak and A. Kotschy, Org. Lett., 2003, 5, 3495–3497 CrossRef CAS PubMed.
- F. Suzenet, N. Leconte, A. Keromnes-Wuillaume and G. Guillaumet, Synlett, 2007, 0204–0210 CrossRef.
- A. Wieczorek, P. Werther, J. Euchner and R. Wombacher, Chem. Sci., 2017, 8, 1506–1510 RSC.
- Q. Zhou, P. Audebert, G. Clavier, F. Miomandre and J. Tang, RSC Adv., 2014, 4, 7193–7195 RSC.
- C. Quinton, V. Alain-Rizzo, C. Dumas-Verdes, G. Clavier, L. Vignau and P. Audebert, New J. Chem., 2015, 39, 9700–9713 RSC.
- H. Wu, J. Yang, J. Seckute and N. K. Devaraj, Angew. Chem., Int. Ed., 2014, 53, 5805–5809 CrossRef CAS PubMed.
- A. Wieczorek, T. Buckup and R. Wombacher, Org. Biomol. Chem., 2014, 12, 4177–4185 CAS.
- G. Knorr, E. Kozma, A. Herner, E. A. Lemke and P. Kele, Chem. – Eur. J., 2016, 22, 8972–8979 CrossRef CAS PubMed.
- D. Nhu, S. Duffy, V. M. Avery, A. Hughes and J. B. Baell, Bioorg. Med. Chem. Lett., 2010, 20, 4496–4498 CrossRef CAS PubMed.
- J. Sauer, G. R. Pabst, U. Holland, H. S. Kim and S. Loebbecke, Eur. J. Org. Chem., 2001, 697–706 CrossRef CAS.
- S. Pican, V. Lapinte, J. F. Pilard, E. Pasquinet, L. Beller, L. Fontaine and D. Poullain, Synlett, 2009, 731–734 CAS.
- R. Selvaraj and J. M. Fox, Tetrahedron Lett., 2014, 55, 4795–4797 CrossRef CAS PubMed.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- S. A. Albu, S. A. Al-Karmi, A. Vito, J. P. Dzandzi, A. Zlitni, D. Beckford-Vera, M. Blacker, N. Janzen, R. M. Patel, A. Capretta and J. F. Valliant, Bioconjugate Chem., 2016, 27, 207–216 CrossRef CAS PubMed.
- J. P. Dzandzi, D. R. Beckford Vera, A. R. Genady, S. A. Albu, L. J. Eltringham-Smith, A. Capretta, W. P. Sheffield and J. F. Valliant, J. Org. Chem., 2015, 80, 7117–7125 CrossRef CAS PubMed.
- H. Zhang, W. S. Trout, S. Liu, G. A. Andrade, D. A. Hudson, S. L. Scinto, K. T. Dicker, Y. Li, N. Lazouski, J. Rosenthal, C. Thorpe, X. Jia and J. M. Fox, J. Am. Chem. Soc., 2016, 138, 5978–5983 CrossRef CAS PubMed.
- J. Yang, J. Seckute, C. M. Cole and N. K. Devaraj, Angew. Chem., Int. Ed., 2012, 51, 7476–7479 CrossRef CAS PubMed.
- D. N. Kamber, L. A. Nazarova, Y. Liang, S. A. Lopez, D. M. Patterson, H. W. Shih, K. N. Houk and J. A. Prescher, J. Am. Chem. Soc., 2013, 135, 13680–13683 CrossRef CAS PubMed.
- D. M. Patterson, L. A. Nazarova, B. Xie, D. N. Kamber and J. A. Prescher, J. Am. Chem. Soc., 2012, 134, 18638–18643 CrossRef CAS PubMed.
- A. K. Spate, H. Busskamp, A. Niederwieser, V. F. Schart, A. Marx and V. Wittmann, Bioconjugate Chem., 2014, 25, 147–154 CrossRef PubMed.
- M. Royzen, G. P. Yap and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 3760–3761 CrossRef CAS PubMed.
- D. Svatunek, C. Denk, V. Rosecker, B. Sohr, C. Hametner, G. Allmaier, J. Fröhlich and H. Mikula, Monatsh. Chem., 2016, 147, 579–585 CrossRef CAS PubMed.
- R. Rossin, S. M. van den Bosch, W. Ten Hoeve, M. Carvelli, R. M. Versteegen, J. Lub and M. S. Robillard, Bioconjugate Chem., 2013, 24, 1210–1217 CrossRef CAS PubMed.
- H. E. Murrey, J. C. Judkins, C. W. Am Ende, T. E. Ballard, Y. Fang, K. Riccardi, L. Di, E. R. Guilmette, J. W. Schwartz, J. M. Fox and D. S. Johnson, J. Am. Chem. Soc., 2015, 137, 11461–11475 CrossRef CAS PubMed.
- K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 10317–10320 CrossRef CAS PubMed.
- S. Eising, F. Lelivelt and K. M. Bonger, Angew. Chem., Int. Ed., 2016, 55, 12243–12247 CrossRef CAS PubMed.
- N. K. Devaraj, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2008, 19, 2297–2299 CrossRef CAS PubMed.
- U. Rieder and N. W. Luedtke, Angew. Chem., Int. Ed., 2014, 53, 9168–9172 CrossRef CAS PubMed.
- J. M. Holstein, D. Stummer and A. Rentmeister, Chem. Sci., 2015, 6, 1362–1369 RSC.
- A. Niederwieser, A. K. Spate, L. D. Nguyen, C. Jungst, W. Reutter and V. Wittmann, Angew. Chem., Int. Ed., 2013, 52, 4265–4268 CrossRef CAS PubMed.
- B. L. Oliveira, Z. Guo, O. Boutureira, A. Guerreiro, G. Jimenez-Oses and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2016, 55, 14683–14687 CrossRef CAS PubMed.
- A. Borrmann, S. Milles, T. Plass, J. Dommerholt, J. M. Verkade, M. Wiessler, C. Schultz, J. C. van Hest, F. L. van Delft and E. A. Lemke, ChemBioChem, 2012, 13, 2094–2099 CrossRef CAS PubMed.
- E. Kozma, I. Nikic, B. R. Varga, I. V. Aramburu, J. H. Kang, O. T. Fackler, E. A. Lemke and P. Kele, ChemBioChem, 2016, 17, 1518–1524 CrossRef CAS PubMed.
- R. J. Blizzard, D. R. Backus, W. Brown, C. G. Bazewicz, Y. Li and R. A. Mehl, J. Am. Chem. Soc., 2015, 137, 10044–10047 CrossRef CAS PubMed.
- X. Li, X. Gao, W. Shi and H. Ma, Chem. Rev., 2014, 114, 590–659 CrossRef CAS PubMed.
- K. Sivakumar, F. Xie, B. M. Cash, S. Long, H. N. Barnhill and Q. Wang, Org. Lett., 2004, 6, 4603–4606 CrossRef CAS PubMed.
- J. J. Shie, Y. C. Liu, Y. M. Lee, C. Lim, J. M. Fang and C. H. Wong, J. Am. Chem. Soc., 2014, 136, 9953–9961 CrossRef CAS PubMed.
- Y. C. Chen, K. Tsao and J. W. Keillor, Can. J. Chem., 2015, 93, 389–398 CrossRef CAS.
- Z. Zhou and C. J. Fahrni, J. Am. Chem. Soc., 2004, 126, 8862–8863 CrossRef CAS PubMed.
- J. C. Jewett and C. R. Bertozzi, Org. Lett., 2011, 13, 5937–5939 CrossRef CAS PubMed.
- F. Friscourt, C. J. Fahrni and G. J. Boons, J. Am. Chem. Soc., 2012, 134, 18809–18815 CrossRef CAS PubMed.
- G. A. Lemieux, C. L. De Graffenried and C. R. Bertozzi, J. Am. Chem. Soc., 2003, 125, 4708–4709 CrossRef CAS PubMed.
- B. C. Gergely, H. András and K. Péter, Methods Appl. Fluoresc., 2015, 3, 042001 CrossRef.
- P. Audebert, F. Miomandre, G. Clavier, M. C. Vernieres, S. Badre and R. Meallet-Renault, Chem. – Eur. J., 2005, 11, 5667–5673 CrossRef CAS PubMed.
- Yong-Hua Gong, Fabien Miomandre, R. Méallet-Renault, S. Badré, L. Galmiche, J. Tang, P. Audebert and G. Clavier, Eur. J. Org. Chem., 2009, 6121–6128 CrossRef CAS.
- N. K. Devaraj, S. Hilderbrand, R. Upadhyay, R. Mazitschek and R. Weissleder, Angew. Chem., Int. Ed., 2010, 49, 2869–2872 CrossRef CAS PubMed.
- T. G. Kim, J. C. Castro, A. Loudet, J. G. Jiao, R. M. Hochstrasser, K. Burgess and M. R. Topp, J. Phys. Chem. A, 2006, 110, 20–27 CrossRef CAS PubMed.
- X. Shang, X. Song, C. Faller, R. Lai, H. Li, R. Cerny, W. Niu and J. Guo, Chem. Sci., 2017, 8, 1141–1145 RSC.
- A. Vazquez, R. Dzijak, M. Dracinsky, R. Rampmaier, S. J. Siegl and M. Vrabel, Angew. Chem., Int. Ed., 2017, 56, 1334–1337 CrossRef CAS PubMed.
- C. C. Liu and P. G. Schultz, Annu. Rev. Biochem., 2010, 79, 413–444 CrossRef CAS PubMed.
- J. Xie and P. G. Schultz, Nat. Rev. Mol. Cell Biol., 2006, 7, 775–782 CrossRef CAS PubMed.
- L. Davis and J. W. Chin, Nat. Rev. Mol. Cell Biol., 2012, 13, 168–182 CAS.
- A. Gautier, D. P. Nguyen, H. Lusic, W. An, A. Deiters and J. W. Chin, J. Am. Chem. Soc., 2010, 132, 4086–4088 CrossRef CAS PubMed.
- A. Bianco, F. M. Townsley, S. Greiss, K. Lang and J. W. Chin, Nat. Chem. Biol., 2012, 8, 748–750 CrossRef CAS PubMed.
- S. Greiss and J. W. Chin, J. Am. Chem. Soc., 2011, 133, 14196–14199 CrossRef CAS PubMed.
- W. Wan, J. M. Tharp and W. R. Liu, Biochim. Biophys. Acta, 2014, 1844, 1059–1070 CrossRef CAS PubMed.
- A. Dumas, L. Lercher, C. D. Spicer and B. G. Davis, Chem. Sci., 2015, 6, 50–69 RSC.
- E. Kaya, M. Vrabel, C. Deiml, S. Prill, V. S. Fluxa and T. Carell, Angew. Chem., Int. Ed., 2012, 51, 4466–4469 CrossRef CAS PubMed.
- S. Schneider, M. J. Gattner, M. Vrabel, V. Flugel, V. Lopez-Carrillo, S. Prill and T. Carell, ChemBioChem, 2013, 14, 2114–2118 CrossRef CAS PubMed.
- J. R. Kramer, B. Onoa, C. Bustamante and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12574–12579 CrossRef CAS PubMed.
- Y. Kurra, K. A. Odoi, Y. J. Lee, Y. Yang, T. Lu, S. E. Wheeler, J. Torres-Kolbus, A. Deiters and W. R. Liu, Bioconjugate Chem., 2014, 25, 1730–1738 CrossRef CAS PubMed.
- T. Plass, S. Milles, C. Koehler, J. Szymanski, R. Mueller, M. Wiessler, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2012, 51, 4166–4170 CrossRef CAS PubMed.
- M. Baumdick, Y. Bruggemann, M. Schmick, G. Xouri, O. Sabet, L. Davis, J. W. Chin and P. I. Bastiaens, eLife, 2015, 4, e12223 Search PubMed.
- A. Rutkowska, T. Plass, J. E. Hoffmann, D. A. Yushchenko, S. Feng and C. Schultz, ChemBioChem, 2014, 15, 1765–1768 CrossRef CAS PubMed.
- C. Uttamapinant, J. D. Howe, K. Lang, V. Beranek, L. Davis, M. Mahesh, N. P. Barry and J. W. Chin, J. Am. Chem. Soc., 2015, 137, 4602–4605 CrossRef CAS PubMed.
- R. B. Cooley, P. A. Karplus and R. A. Mehl, ChemBioChem, 2014, 15, 1810–1819 CrossRef CAS PubMed.
- Z. Yu, Y. Pan, Z. Wang, J. Wang and Q. Lin, Angew. Chem., Int. Ed., 2012, 51, 10600–10604 CrossRef CAS PubMed.
- T. S. Elliott, F. M. Townsley, A. Bianco, R. J. Ernst, A. Sachdeva, S. J. Elsasser, L. Davis, K. Lang, R. Pisa, S. Greiss, K. S. Lilley and J. W. Chin, Nat. Biotechnol., 2014, 32, 465–472 CrossRef CAS PubMed.
- T. S. Elliott, A. Bianco, F. M. Townsley, S. D. Fried and J. W. Chin, Cell Chem. Biol., 2016, 23, 805–815 CrossRef CAS PubMed.
- Y. J. Lee, Y. Kurra, Y. Yang, J. Torres-Kolbus, A. Deiters and W. R. Liu, Chem. Commun., 2014, 50, 13085–13088 RSC.
- W. W. Wang, Y. Zeng, B. Wu, A. Deiters and W. R. Liu, ACS Chem. Biol., 2016, 11, 1973–1981 CrossRef CAS PubMed.
- Y. Yang, S. Lin, W. Lin and P. R. Chen, ChemBioChem, 2014, 15, 1738–1743 CrossRef CAS PubMed.
- S. Lin, H. Yan, L. Li, M. Yang, B. Peng, S. Chen, W. Li and P. R. Chen, Angew. Chem., Int. Ed., 2013, 52, 13970–13974 CrossRef CAS PubMed.
- Y. H. Tsai, S. Essig, J. R. James, K. Lang and J. W. Chin, Nat. Chem., 2015, 7, 554–561 CrossRef CAS PubMed.
- T. Machida, K. Lang, L. Xue, J. W. Chin and N. Winssinger, Bioconjugate Chem., 2015, 26, 802–806 CrossRef CAS PubMed.
- T. Plass, S. Milles, C. Koehler, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2011, 50, 3878–3881 CrossRef CAS PubMed.
- J. L. Seitchik, J. C. Peeler, M. T. Taylor, M. L. Blackman, T. W. Rhoads, R. B. Cooley, C. Refakis, J. M. Fox and R. A. Mehl, J. Am. Chem. Soc., 2012, 134, 2898–2901 CrossRef CAS PubMed.
- H. Neumann, K. Wang, L. Davis, M. Garcia-Alai and J. W. Chin, Nature, 2010, 464, 441–444 CrossRef CAS PubMed.
- A. Sachdeva, K. Wang, T. Elliott and J. W. Chin, J. Am. Chem. Soc., 2014, 136, 7785–7788 CrossRef CAS PubMed.
- K. Wang, A. Sachdeva, D. J. Cox, N. M. Wilf, K. Lang, S. Wallace, R. A. Mehl and J. W. Chin, Nat. Chem., 2014, 6, 393–403 CrossRef CAS PubMed.
- C. Meyer, S. Liebscher and F. Bordusa, Bioconjugate Chem., 2016, 27, 47–53 CrossRef CAS PubMed.
- M. Best, A. Degen, M. Baalmann, T. T. Schmidt and R. Wombacher, ChemBioChem, 2015, 16, 1158–1162 CrossRef CAS PubMed.
- J. K. Dozier, S. L. Khatwani, J. W. Wollack, Y. C. Wang, C. Schmidt-Dannert and M. D. Distefano, Bioconjugate Chem., 2014, 25, 1203–1212 CrossRef CAS PubMed.
- M. Rashidian, E. Keliher, M. Dougan, P. K. Juras, M. Cavallari, G. R. Wojtkiewicz, J. Jacobsen, J. G. Edens, J. M. Tas, G. Victora, R. Weissleder and H. Ploegh, ACS Cent. Sci., 2015, 1, 142–147 CrossRef CAS PubMed.
- M. Rashidian, E. J. Keliher, A. M. Bilate, J. N. Duarte, G. R. Wojtkiewicz, J. T. Jacobsen, J. Cragnolini, L. K. Swee, G. D. Victora, R. Weissleder and H. L. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6146–6151 CrossRef CAS PubMed.
- J. E. Glasgow, M. L. Salit and J. R. Cochran, J. Am. Chem. Soc., 2016, 138, 7496–7499 CrossRef CAS PubMed.
- J. J. Gruskos, G. Zhang and D. Buccella, J. Am. Chem. Soc., 2016, 138, 14639–14649 CrossRef CAS PubMed.
- D. Schulz and A. Rentmeister, Chembiochem, 2014, 15, 2342–2347 CrossRef CAS PubMed.
- S. Kath-Schorr, Top. Curr. Chem., 2016, 374, 4 CrossRef PubMed.
- K. Gutsmiedl, C. T. Wirges, V. Ehmke and T. Carell, Org. Lett., 2009, 11, 2405–2408 CrossRef CAS PubMed.
- J. Schoch, S. Ameta and A. Jaschke, Chem. Commun., 2011, 47, 12536–12537 RSC.
- A. M. Pyka, C. Domnick, F. Braun and S. Kath-Schorr, Bioconjugate Chem., 2014, 25, 1438–1443 CrossRef CAS PubMed.
- J. Schoch, M. Staudt, A. Samanta, M. Wiessler and A. Jaschke, Bioconjugate Chem., 2012, 23, 1382–1386 CrossRef CAS PubMed.
- J. M. Holstein and A. Rentmeister, Methods, 2016, 98, 18–25 CrossRef CAS PubMed.
- S. Ameta, J. Becker and A. Jaschke, Org. Biomol. Chem., 2014, 12, 4701–4707 CAS.
- P. N. Asare-Okai, E. Agustin, D. Fabris and M. Royzen, Chem. Commun., 2014, 50, 7844–7847 RSC.
- J. M. Holstein, L. Anhauser and A. Rentmeister, Angew. Chem., Int. Ed., 2016, 55, 10899–10903 CrossRef CAS PubMed.
- J. M. Holstein, F. Muttach, S. H. H. Schiefelbein and A. Rentmeister, Chem. – Eur. J., 2017, 23, 6165–6173 CrossRef CAS PubMed.
- H. Busskamp, E. Batroff, A. Niederwieser, O. S. Abdel-Rahman, R. F. Winter, V. Wittmann and A. Marx, Chem. Commun., 2014, 50, 10827–10829 RSC.
- M. Merkel, S. Arndt, D. Ploschik, G. B. Cserep, U. Wenge, P. Kele and H. A. Wagenknecht, J. Org. Chem., 2016, 81, 7527–7538 CrossRef CAS PubMed.
- J. Schoch and A. Jaschke, RSC Adv., 2013, 3, 4181–4183 RSC.
- K. Wang, D. Wang, K. Ji, W. Chen, Y. Zheng, C. Dai and B. Wang, Org. Biomol. Chem., 2015, 13, 909–915 CAS.
- X. Ren, A. H. El-Sagheer and T. Brown, Analyst, 2015, 140, 2671–2678 RSC.
- J. Schoch, M. Wiessler and A. Jaschke, J. Am. Chem. Soc., 2010, 132, 8846–8847 CrossRef CAS PubMed.
- M. L. Winz, E. C. Linder, T. Andre, J. Becker and A. Jaschke, Nucleic Acids Res., 2015, 43, e110 CrossRef PubMed.
- C. Domnick, F. Eggert and S. Kath-Schorr, Chem. Commun., 2015, 51, 8253–8256 RSC.
- F. Eggert and S. Kath-Schorr, Chem. Commun., 2016, 52, 7284–7287 RSC.
- J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13–21 CrossRef CAS PubMed.
- A. Varki, Glycobiology, 2017, 27, 3–49 CrossRef PubMed.
- K. Ohtsubo and J. D. Marth, Cell, 2006, 126, 855–867 CrossRef CAS PubMed.
- T. J. Sminia, H. Zuilhof and T. Wennekes, Carbohydr. Res., 2016, 435, 121–141 CrossRef CAS PubMed.
- C. M. Cole, J. Yang, J. Seckute and N. K. Devaraj, ChemBioChem, 2013, 14, 205–208 CrossRef CAS PubMed.
- D. M. Patterson, K. A. Jones and J. A. Prescher, Mol. BioSyst., 2014, 10, 1693–1697 RSC.
- D. C. Xiong, J. Zhu, M. J. Han, H. X. Luo, C. Wang, Y. Yu, Y. Ye, G. Tai and X. S. Ye, Org. Biomol. Chem., 2015, 13, 3911–3917 CAS.
- A. K. Spate, V. F. Schart, J. Hafner, A. Niederwieser, T. U. Mayer and V. Wittmann, Beilstein J. Org. Chem., 2014, 10, 2235–2242 CrossRef PubMed.
- F. Doll, A. Buntz, A. K. Spate, V. F. Schart, A. Timper, W. Schrimpf, C. R. Hauck, A. Zumbusch and V. Wittmann, Angew. Chem., Int. Ed., 2016, 55, 2262–2266 CrossRef CAS PubMed.
- A. K. Spate, V. F. Schart, S. Schollkopf, A. Niederwieser and V. Wittmann, Chem. – Eur. J., 2014, 20, 16502–16508 CrossRef PubMed.
- A. K. Spate, J. E. Dold, E. Batroff, V. F. Schart, D. E. Wieland, O. R. Baudendistel and V. Wittmann, ChemBioChem, 2016, 17, 1374–1383 CrossRef PubMed.
- A. A. Neves, H. Stöckmann, Y. A. Wainman, J. C. H. Kuo, S. Fawcett, F. J. Leeper and K. M. Brindle, Bioconjugate Chem., 2013, 24, 934–941 CrossRef CAS PubMed.
- P. Agarwal, B. J. Beahm, P. Shieh and C. R. Bertozzi, Angew. Chem., Int. Ed., 2015, 54, 11504–11510 CrossRef CAS PubMed.
- S. E. Pidgeon and M. M. Pires, Chem. Commun., 2015, 51, 10330–10333 RSC.
- H. C. Hang, J. P. Wilson and G. Charron, Acc. Chem. Res., 2011, 44, 699–708 CrossRef CAS PubMed.
- R. S. Erdmann, H. Takakura, A. D. Thompson, F. Rivera-Molina, E. S. Allgeyer, J. Bewersdorf, D. Toomre and A. Schepartz, Angew. Chem., Int. Ed., 2014, 53, 10242–10246 CrossRef CAS PubMed.
- S. P. Brown and A. B. Smith, 3rd, J. Am. Chem. Soc., 2015, 137, 4034–4037 CrossRef CAS PubMed.
- J. D. Thomas, H. Cui, P. J. North, T. Hofer, C. Rader and T. R. Burke, Bioconjugate Chem., 2012, 23, 2007–2013 CrossRef CAS PubMed.
- T. Peng and H. C. Hang, J. Am. Chem. Soc., 2016, 138, 14423–14433 CrossRef CAS PubMed.
- Z. Li, D. Wang, L. Li, S. Pan, Z. Na, C. Y. Tan and S. Q. Yao, J. Am. Chem. Soc., 2014, 136, 9990–9998 CrossRef CAS PubMed.
- J. Seckute, J. Yang and N. K. Devaraj, Nucleic Acids Res., 2013, 41, e148 CrossRef CAS PubMed.
- K. M. Dean and A. E. Palmer, Nat. Chem. Biol., 2014, 10, 512–523 CrossRef CAS PubMed.
- G. Lukinavicius, K. Umezawa, N. Olivier, A. Honigmann, G. Yang, T. Plass, V. Mueller, L. Reymond, I. R. Correa, Jr., Z. G. Luo, C. Schultz, E. A. Lemke, P. Heppenstall, C. Eggeling, S. Manley and K. Johnsson, Nat. Chem., 2013, 5, 132–139 CrossRef CAS PubMed.
- G. Lukinavicius, L. Reymond, E. D'Este, A. Masharina, F. Gottfert, H. Ta, A. Guther, M. Fournier, S. Rizzo, H. Waldmann, C. Blaukopf, C. Sommer, D. W. Gerlich, H. D. Arndt, S. W. Hell and K. Johnsson, Nat. Methods, 2014, 11, 731–733 CrossRef CAS PubMed.
- T. S. Elliott, A. Bianco and J. W. Chin, Curr. Opin. Chem. Biol., 2014, 21, 154–160 CrossRef CAS PubMed.
- J. B. Grimm, B. P. English, J. Chen, J. P. Slaughter, Z. Zhang, A. Revyakin, R. Patel, J. J. Macklin, D. Normanno, R. H. Singer, T. Lionnet and L. D. Lavis, Nat. Methods, 2015, 12, 244–250 CrossRef CAS PubMed.
- I. Nikic and E. A. Lemke, Curr. Opin. Chem. Biol., 2015, 28, 164–173 CrossRef CAS PubMed.
- I. Nikic, G. Estrada Girona, J. H. Kang, G. Paci, S. Mikhaleva, C. Koehler, N. V. Shymanska, C. Ventura Santos, D. Spitz and E. A. Lemke, Angew. Chem., Int. Ed., 2016, 55, 16172–16176 CrossRef CAS PubMed.
- M. Schurmann, P. Janning, S. Ziegler and H. Waldmann, Cell Chem. Biol., 2016, 23, 435–441 CrossRef PubMed.
- A. Rutkowska, D. W. Thomson, J. Vappiani, T. Werner, K. M. Mueller, L. Dittus, J. Krause, M. Muelbaier, G. Bergamini and M. Bantscheff, ACS Chem. Biol., 2016, 11, 2541–2550 CrossRef CAS PubMed.
- K. S. Yang, G. Budin, T. Reiner, C. Vinegoni and R. Weissleder, Angew. Chem., Int. Ed., 2012, 51, 6598–6603 CrossRef CAS PubMed.
- J. T. Ngo and D. A. Tirrell, Acc. Chem. Res., 2011, 44, 677–685 CrossRef CAS PubMed.
- D. C. Dieterich, A. J. Link, J. Graumann, D. A. Tirrell and E. M. Schuman, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9482–9487 CrossRef CAS PubMed.
- H. Wu, B. T. Cisneros, C. M. Cole and N. K. Devaraj, J. Am. Chem. Soc., 2014, 136, 17942–17945 CrossRef CAS PubMed.
- R. Weissleder and M. J. Pittet, Nature, 2008, 452, 580–589 CrossRef CAS PubMed.
- M. L. James and S. S. Gambhir, Physiol. Rev., 2012, 92, 897–965 CrossRef CAS PubMed.
- J. R. Ballinger, Semin. Nucl. Med., 2015, 45, 470–478 CrossRef PubMed.
- M. M. Khalil, J. L. Tremoleda, T. B. Bayomy and W. Gsell, Int. J. Mol. Imaging, 2011, 2011, 796025 Search PubMed.
- O. Jacobson, D. O. Kiesewetter and X. Chen, Bioconjugate Chem., 2015, 26, 1–18 CrossRef CAS PubMed.
- E. W. Price and C. Orvig, Chem. Soc. Rev., 2014, 43, 260–290 RSC.
- J. P. Holland, M. J. Williamson and J. S. Lewis, Mol. Imaging, 2010, 9, 1–20 CAS.
- C. Kai and C. Xiaoyuan, Curr. Top. Med. Chem., 2010, 10, 1227–1236 CrossRef.
- S. Richter and F. Wuest, Molecules, 2014, 19, 20536 CrossRef PubMed.
- S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719–766 CrossRef CAS PubMed.
- M. J. Adam and D. S. Wilbur, Chem. Soc. Rev., 2005, 34, 153–163 RSC.
- D. Zeng, B. M. Zeglis, J. S. Lewis and C. J. Anderson, J. Nucl. Med., 2013, 54, 829–832 CrossRef CAS PubMed.
- J. Y. Choi and B. C. Lee, Nucl. Med. Mol. Imaging, 2015, 49, 258–267 CrossRef CAS PubMed.
- J. Marik and J. L. Sutcliffe, Tetrahedron Lett., 2006, 47, 6681–6684 CrossRef CAS.
- H. S. Gill and J. Marik, Nat. Protoc., 2011, 6, 1718–1725 CrossRef CAS PubMed.
- S. Maschauer and O. Prante, Carbohydr. Res., 2009, 344, 753–761 CrossRef CAS PubMed.
- B. H. Rotstein, N. A. Stephenson, N. Vasdev and S. H. Liang, Nat. Commun., 2014, 5, 4365 CAS.
- L. Mirfeizi, J. Walsh, H. Kolb, L. Campbell-Verduyn, R. A. Dierckx, B. L. Feringa, P. H. Elsinga, T. de Groot, I. Sannen, G. Bormans and S. Celen, Nucl. Med. Biol., 2013, 40, 710–716 CrossRef CAS PubMed.
- T. L. Mindt, H. Struthers, L. Brans, T. Anguelov, C. Schweinsberg, V. Maes, D. Tourwé and R. Schibli, J. Am. Chem. Soc., 2006, 128, 15096–15097 CrossRef CAS PubMed.
- V. Bouvet, M. Wuest and F. Wuest, Org. Biomol. Chem., 2011, 9, 7393–7399 CAS.
- L. S. Campbell-Verduyn, L. Mirfeizi, A. K. Schoonen, R. A. Dierckx, P. H. Elsinga and B. L. Feringa, Angew. Chem., Int. Ed., 2011, 50, 11117–11120 CrossRef CAS PubMed.
- M. E. Martin, S. G. Parameswarappa, M. S. O’Dorisio, F. C. Pigge and M. K. Schultz, Bioorg. Med. Chem. Lett., 2010, 20, 4805–4807 CrossRef CAS PubMed.
- N. J. Baumhover, M. E. Martin, S. G. Parameswarappa, K. C. Kloepping, M. S. O’Dorisio, F. C. Pigge and M. K. Schultz, Bioorg. Med. Chem. Lett., 2011, 21, 5757–5761 CrossRef CAS PubMed.
- R. D. Carpenter, S. H. Hausner and J. L. Sutcliffe, ACS Med. Chem. Lett., 2011, 2, 885–889 CrossRef CAS PubMed.
- K. Sachin, V. H. Jadhav, E.-M. Kim, H. L. Kim, S. B. Lee, H.-J. Jeong, S. T. Lim, M.-H. Sohn and D. W. Kim, Bioconjugate Chem., 2012, 23, 1680–1686 CrossRef CAS PubMed.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS PubMed.
- J. A. Codelli, J. M. Baskin, N. J. Agard and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 11486–11493 CrossRef CAS PubMed.
- B. L. Nilsson, L. L. Kiessling and R. T. Raines, Org. Lett., 2000, 2, 1939–1941 CrossRef CAS PubMed.
- M. Pretze, F. Wuest, T. Peppel, M. Köckerling and C. Mamat, Tetrahedron Lett., 2010, 51, 6410–6414 CrossRef CAS.
- L. Carroll, S. Boldon, R. Bejot, J. E. Moore, J. Declerck and V. Gouverneur, Org. Biomol. Chem., 2011, 9, 136–140 CAS.
- M. Pretze, D. Pietzsch and C. Mamat, Molecules, 2013, 18, 8618–8665 CrossRef CAS PubMed.
- M. Namavari, Z. Cheng, R. Zhang, A. De, J. Levi, J. K. Hoerner, S. S. Yaghoubi, F. A. Syud and S. S. Gambhir, Bioconjugate Chem., 2009, 20, 432–436 CrossRef CAS PubMed.
- R. R. Flavell, P. Kothari, M. Bar-Dagan, M. Synan, S. Vallabhajosula, J. M. Friedman, T. W. Muir and G. Ceccarini, J. Am. Chem. Soc., 2008, 130, 9106–9112 CrossRef CAS PubMed.
- M. Glaser, P. Iveson, S. Hoppmann, B. Indrevoll, A. Wilson, J. Arukwe, A. Danikas, R. Bhalla and D. Hiscock, J. Nucl. Med., 2013, 54, 1981–1988 CrossRef CAS PubMed.
- J. Jeon, B. Shen, L. Xiong, Z. Miao, K. H. Lee, J. Rao and F. T. Chin, Bioconjugate Chem., 2012, 23, 1902–1908 CrossRef CAS PubMed.
- J. A. H. Inkster, D. J. Colin and Y. Seimbille, Org. Biomol. Chem., 2015, 13, 3667–3676 CAS.
- X. Su, K. Cheng, J. Jeon, B. Shen, G. T. Venturin, X. Hu, J. Rao, F. T. Chin, H. Wu and Z. Cheng, Mol. Pharmaceutics, 2014, 11, 3947–3956 CrossRef CAS PubMed.
- R. Rossin, P. Renart Verkerk, S. M. van den Bosch, R. C. M. Vulders, I. Verel, J. Lub and M. S. Robillard, Angew. Chem., Int. Ed., 2010, 49, 3375–3378 CrossRef CAS PubMed.
- Z. Li, H. Cai, M. Hassink, M. L. Blackman, R. C. D. Brown, P. S. Conti and J. M. Fox, Chem. Commun., 2010, 46, 8043–8045 RSC.
- L. Wyffels, D. Thomae, A. M. Waldron, J. Fissers, S. Dedeurwaerdere, P. Van der Veken, J. Joossens, S. Stroobants, K. Augustyns and S. Staelens, Nucl. Med. Biol., 2014, 41, 513–523 CrossRef CAS PubMed.
- R. Selvaraj, S. Liu, M. Hassink, C.-w. Huang, L.-p. Yap, R. Park, J. M. Fox, Z. Li and P. S. Conti, Bioorg. Med. Chem. Lett., 2011, 21, 5011–5014 CrossRef CAS PubMed.
- S. Liu, M. Hassink, R. Selvaraj, L.-P. Yap, R. Park, H. Wang, X. Chen, J. M. Fox, Z. Li and P. S. Conti, Mol. Imaging, 2013, 12, 121–128 CAS.
- Z. Wu, S. Liu, M. Hassink, I. Nair, R. Park, L. Li, I. Todorov, J. M. Fox, Z. Li, J. E. Shively, P. S. Conti and F. Kandeel, J. Nucl. Med., 2013, 54, 244–251 CrossRef CAS PubMed.
- Y. Wang, K. Lim, M. Normandin, X. Zhao, G. W. Cline and Y.-S. Ding, Nucl. Med. Biol., 2012, 39, 167–176 CrossRef CAS PubMed.
- B. Bernardim, P. M. S. D. Cal, M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, I. S. Albuquerque, E. Perkins, F. Corzana, A. C. B. Burtoloso, G. Jiménez-Osés and G. J. L. Bernardes, Nat. Commun., 2016, 7, 13128 CrossRef CAS PubMed.
- P. M. S. D. Cal, G. J. L. Bernardes and P. M. P. Gois, Angew. Chem., Int. Ed., 2014, 53, 10585–10587 CrossRef CAS PubMed.
- T. Reiner, E. J. Keliher, S. Earley, B. Marinelli and R. Weissleder, Angew. Chem., Int. Ed., 2011, 50, 1922–1925 CrossRef CAS PubMed.
- M. T. Taylor, M. L. Blackman, O. Dmitrenko and J. M. Fox, J. Am. Chem. Soc., 2011, 133, 9646–9649 CrossRef CAS PubMed.
- M. Wang, D. Svatunek, K. Rohlfing, Y. Liu, H. Wang, B. Giglio, H. Yuan, Z. Wu, Z. Li and J. Fox, Theranostics, 2016, 6, 887–895 CrossRef CAS PubMed.
- E. M. F. Billaud, E. Shahbazali, M. Ahamed, F. Cleeren, T. Noel, M. Koole, A. Verbruggen, V. Hessel and G. Bormans, Chem. Sci., 2017, 8, 1251–1258 RSC.
- J. C. Knight, S. Richter, M. Wuest, J. D. Way and F. Wuest, Org. Biomol. Chem., 2013, 11, 3817–3825 CAS.
- C. Denk, D. Svatunek, T. Filip, T. Wanek, D. Lumpi, J. Fröhlich, C. Kuntner and H. Mikula, Angew. Chem., Int. Ed., 2014, 53, 9655–9659 CrossRef CAS PubMed.
- J. Zhu, S. Li, C. Wangler, B. Wangler, R. Bruce Lennox and R. Schirrmacher, Chem. Commun., 2015, 51, 12415–12418 RSC.
- O. Keinänen, X.-G. Li, N. K. Chenna, D. Lumen, J. Ott, C. F. M. Molthoff, M. Sarparanta, K. Helariutta, T. Vuorinen, A. D. Windhorst and A. J. Airaksinen, ACS Med. Chem. Lett., 2016, 7, 62–66 CrossRef PubMed.
- M. Rashidian, L. Wang, J. G. Edens, J. T. Jacobsen, I. Hossain, Q. Wang, G. D. Victora, N. Vasdev, H. Ploegh and S. H. Liang, Angew. Chem., Int. Ed., 2016, 55, 528–533 CrossRef CAS PubMed.
- B. M. Zeglis, P. Mohindra, G. I. Weissmann, V. Divilov, S. A. Hilderbrand, R. Weissleder and J. S. Lewis, Bioconjugate Chem., 2011, 22, 2048–2059 CrossRef CAS PubMed.
- L. G. Meimetis, E. Boros, J. C. Carlson, C. Ran, P. Caravan and R. Weissleder, Bioconjugate Chem., 2016, 27, 257–263 CrossRef CAS PubMed.
- H. L. Evans, L. Carroll, E. O. Aboagye and A. C. Spivey, J. Labelled Compd. Radiopharm., 2014, 57, 291–297 CrossRef CAS PubMed.
- B. M. Zeglis, F. Emmetiere, N. Pillarsetty, R. Weissleder, J. S. Lewis and T. Reiner, ChemistryOpen, 2014, 3, 48–53 CrossRef CAS PubMed.
- J. B. Haun, N. K. Devaraj, S. A. Hilderbrand, H. Lee and R. Weissleder, Nat. Nanotechnol., 2010, 5, 660–665 CrossRef CAS PubMed.
- T. Plass, S. Milles, C. Koehler, J. Szymański, R. Mueller, M. Wießler, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2012, 51, 4166–4170 CrossRef CAS PubMed.
- J. C. Knight and B. Cornelissen, Am. J. Nucl. Med. Mol. Imaging, 2014, 4, 96–113 CAS.
- R. Rossin and M. S. Robillard, Curr. Opin. Chem. Biol., 2014, 21, 161–169 CrossRef CAS PubMed.
- D. B. Axworthy, J. M. Reno, M. D. Hylarides, R. W. Mallett, L. J. Theodore, L. M. Gustavson, F.-M. Su, L. J. Hobson, P. L. Beaumier and A. R. Fritzberg, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 1802–1807 CrossRef CAS.
- R. Rossin, S. M. van den Bosch, W. ten Hoeve, M. Carvelli, R. M. Versteegen, J. Lub and M. S. Robillard, Bioconjugate Chem., 2013, 24, 1210–1217 CrossRef CAS PubMed.
- R. Rossin, T. Läppchen, S. M. van den Bosch, R. Laforest and M. S. Robillard, J. Nucl. Med., 2013, 54, 1989–1995 CrossRef CAS PubMed.
- R. Rossin, S. M. J. van Duijnhoven, T. Läppchen, S. M. van den Bosch and M. S. Robillard, Mol. Pharmaceutics, 2014, 11, 3090–3096 CrossRef CAS PubMed.
- S. M. J. van Duijnhoven, R. Rossin, S. M. van den Bosch, M. P. Wheatcroft, P. J. Hudson and M. S. Robillard, J. Nucl. Med., 2015, 56, 1422–1428 CrossRef CAS PubMed.
- M. Altai, A. Perols, M. Tsourma, B. Mitran, H. Honarvar, M. Robillard, R. Rossin, W. ten Hoeve, M. Lubberink, A. Orlova, A. E. Karlström and V. Tolmachev, J. Nucl. Med., 2016, 57, 431–436 CrossRef PubMed.
- B. M. Zeglis, K. K. Sevak, T. Reiner, P. Mohindra, S. D. Carlin, P. Zanzonico, R. Weissleder and J. S. Lewis, J. Nucl. Med., 2013, 54, 1389–1396 CrossRef CAS PubMed.
- A. Yazdani, H. Bilton, A. Vito, A. R. Genady, S. M. Rathmann, Z. Ahmad, N. Janzen, S. Czorny, B. M. Zeglis, L. C. Francesconi and J. F. Valliant, J. Med. Chem., 2016, 59, 9381–9389 CrossRef CAS PubMed.
- B. E. Cook, P. Adumeau, R. Membreno, K. E. Carnazza, C. Brand, T. Reiner, B. J. Agnew, J. S. Lewis and B. M. Zeglis, Bioconjugate Chem., 2016, 27, 1789–1795 CrossRef CAS PubMed.
- P. Adumeau, K. E. Carnazza, C. Brand, S. D. Carlin, T. Reiner, B. J. Agnew, J. S. Lewis and B. M. Zeglis, Theranostics, 2016, 6, 2267–2277 CrossRef PubMed.
- M. H. Choi, H. E. Shim, S.-J. Yun, H. R. Kim, S. Mushtaq, C. H. Lee, S. H. Park, D. S. Choi, D.-E. Lee, E.-B. Byun, B.-S. Jang and J. Jeon, Bioorg. Med. Chem., 2016, 24, 2589–2594 CrossRef CAS PubMed.
- S. A. Albu, S. A. Al-Karmi, A. Vito, J. P. K. Dzandzi, A. Zlitni, D. Beckford-Vera, M. Blacker, N. Janzen, R. M. Patel, A. Capretta and J. F. Valliant, Bioconjugate Chem., 2016, 27, 207–216 CrossRef CAS PubMed.
- H. L. Evans, Q.-D. Nguyen, L. S. Carroll, M. Kaliszczak, F. J. Twyman, A. C. Spivey and E. O. Aboagye, Chem. Commun., 2014, 50, 9557–9560 RSC.
- J. P. Meyer, J. L. Houghton, P. Kozlowski, D. Abdel-Atti, T. Reiner, N. V. Pillarsetty, W. W. Scholz, B. M. Zeglis and J. S. Lewis, Bioconjugate Chem., 2016, 27, 298–301 CrossRef CAS PubMed.
- N. K. Devaraj, G. M. Thurber, E. J. Keliher, B. Marinelli and R. Weissleder, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4762–4767 CrossRef CAS PubMed.
- B. Nichols, Z. Qin, J. Yang, D. R. Vera and N. K. Devaraj, Chem. Commun., 2014, 50, 5215–5217 RSC.
- M. M. Herth, V. L. Andersen, S. Lehel, J. Madsen, G. M. Knudsen and J. L. Kristensen, Chem. Commun., 2013, 49, 3805–3807 RSC.
- O. Keinänen, E. M. Mäkilä, R. Lindgren, H. Virtanen, H. Liljenbäck, V. Oikonen, M. Sarparanta, C. Molthoff, A. D. Windhorst, A. Roivainen, J. J. Salonen and A. J. Airaksinen, ACS Omega, 2017, 2, 62–69 CrossRef PubMed.
- W. Ren, A. Ji, M. X. Wang and H. W. Ai, ChemBioChem, 2015, 16, 2007–2010 CrossRef CAS PubMed.
- G. Zhang, J. Li, R. Xie, X. Fan, Y. Liu, S. Zheng, Y. Ge and P. R. Chen, ACS Cent. Sci., 2016, 2, 325–331 CrossRef CAS PubMed.
- E. Arbely, J. Torres-Kolbus, A. Deiters and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 11912–11915 CrossRef CAS PubMed.
- D. P. Nguyen, M. Mahesh, S. J. Elsässer, S. M. Hancock, C. Uttamapinant and J. W. Chin, J. Am. Chem. Soc., 2014, 136, 2240–2243 CrossRef CAS PubMed.
- C. Streu and E. Meggers, Angew. Chem., Int. Ed., 2006, 45, 5645–5648 CrossRef CAS PubMed.
- T. Volker, F. Dempwolff, P. L. Graumann and E. Meggers, Angew. Chem., Int. Ed., 2014, 53, 10536–10540 CrossRef PubMed.
- H. W. Ai, J. W. Lee and P. G. Schultz, Chem. Commun., 2010, 46, 5506–5508 RSC.
- M. I. Sanchez, C. Penas, M. E. Vazquez and J. L. Mascarenas, Chem. Sci., 2014, 5, 1901–1907 RSC.
- G. Y. Tonga, Y. Jeong, B. Duncan, T. Mizuhara, R. Mout, R. Das, S. T. Kim, Y. C. Yeh, B. Yan, S. Hou and V. M. Rotello, Nat. Chem., 2015, 7, 597–603 CrossRef CAS PubMed.
- H. T. Hsu, B. M. Trantow, R. M. Waymouth and P. A. Wender, Bioconjugate Chem., 2016, 27, 376–382 CrossRef CAS PubMed.
- K. K. Sadhu, E. Lindberg and N. Winssinger, Chem. Commun., 2015, 51, 16664–16666 RSC.
- P. K. Sasmal, S. Carregal-Romero, W. J. Parak and E. Meggers, Organometallics, 2012, 31, 5968–5970 CrossRef CAS.
- Y. Chen, A. S. Kamlet, J. B. Steinman and D. R. Liu, Nat. Chem., 2011, 3, 146–153 CrossRef CAS PubMed.
- F. Song, A. L. Garner and K. Koide, J. Am. Chem. Soc., 2007, 129, 12354–12355 CrossRef CAS PubMed.
- A. L. Garner, F. Song and K. Koide, J. Am. Chem. Soc., 2009, 131, 5163–5171 CrossRef CAS PubMed.
- R. M. Yusop, A. Unciti-Broceta, E. M. Johansson, R. M. Sanchez-Martin and M. Bradley, Nat. Chem., 2011, 3, 239–243 CrossRef CAS PubMed.
- M. Santra, S. K. Ko, I. Shin and K. H. Ahn, Chem. Commun., 2010, 46, 3964–3966 RSC.
- J. T. Weiss, J. C. Dawson, K. G. Macleod, W. Rybski, C. Fraser, C. Torres-Sanchez, E. E. Patton, M. Bradley, N. O. Carragher and A. Unciti-Broceta, Nat. Commun., 2014, 5, 3277 Search PubMed.
- J. T. Weiss, J. C. Dawson, C. Fraser, W. Rybski, C. Torres-Sanchez, M. Bradley, E. E. Patton, N. O. Carragher and A. Unciti-Broceta, J. Med. Chem., 2014, 57, 5395–5404 CrossRef CAS PubMed.
- J. Li, J. Yu, J. Zhao, J. Wang, S. Zheng, S. Lin, L. Chen, M. Yang, S. Jia, X. Zhang and P. R. Chen, Nat. Chem., 2014, 6, 352–361 CrossRef CAS PubMed.
- J. Wang, B. Cheng, J. Li, Z. Zhang, W. Hong, X. Chen and P. R. Chen, Angew. Chem., Int. Ed., 2015, 54, 5364–5368 CrossRef CAS PubMed.
- J. Wang, S. Zheng, Y. Liu, Z. Zhang, Z. Lin, J. Li, G. Zhang, X. Wang, J. Li and P. R. Chen, J. Am. Chem. Soc., 2016, 138, 15118–15121 CrossRef CAS PubMed.
- R. Friedman Ohana, S. Levin, M. G. Wood, K. Zimmerman, M. L. Dart, M. K. Schwinn, T. A. Kirkland, R. Hurst, H. T. Uyeda, L. P. Encell and K. V. Wood, ACS Chem. Biol., 2016, 11, 2608–2617 CrossRef CAS PubMed.
- M. Santra, D. Ryu, A. Chatterjee, S. K. Ko, I. Shin and K. H. Ahn, Chem. Commun., 2009, 2115–2117, 10.1039/b900380k.
- A. A. Kislukhin, V. P. Hong, K. E. Breitenkamp and M. G. Finn, Bioconjugate Chem., 2013, 24, 684–689 CrossRef CAS PubMed.
- M. Taki, S. Iyoshi, A. Ojida, I. Hamachi and Y. Yamamoto, J. Am. Chem. Soc., 2010, 132, 5938–5939 CrossRef CAS PubMed.
- P. K. Sasmal, S. Carregal-Romero, A. A. Han, C. N. Streu, Z. Lin, K. Namikawa, S. L. Elliott, R. W. Koster, W. J. Parak and E. Meggers, Chembiochem, 2012, 13, 1116–1120 CrossRef CAS PubMed.
- H. Y. Au-Yeung, E. J. New and C. J. Chang, Chem. Commun., 2012, 48, 5268–5270 RSC.
- J. Kim and C. R. Bertozzi, Angew. Chem., Int. Ed., 2015, 54, 15777–15781 CrossRef CAS PubMed.
- R. van Brakel, R. C. Vulders, R. J. Bokdam, H. Grull and M. S. Robillard, Bioconjugate Chem., 2008, 19, 714–718 CrossRef CAS PubMed.
- K. Gorska, A. Manicardi, S. Barluenga and N. Winssinger, Chem. Commun., 2011, 47, 4364–4366 RSC.
- J. B. Pawlak, G. P. Gential, T. J. Ruckwardt, J. S. Bremmers, N. J. Meeuwenoord, F. A. Ossendorp, H. S. Overkleeft, D. V. Filippov and S. I. van Kasteren, Angew. Chem., Int. Ed., 2015, 54, 5628–5631 CrossRef CAS PubMed.
- S. S. Matikonda, D. L. Orsi, V. Staudacher, I. A. Jenkins, F. Fiedler, J. Y. Chen and A. B. Gamble, Chem. Sci., 2015, 6, 1212–1218 RSC.
- Y. Ge, X. Fan and P. R. Chen, Chem. Sci., 2016, 7, 7055–7060 RSC.
- I. Khan, P. F. Agris, M. V. Yigit and M. Royzen, Chem. Commun., 2016, 52, 6174–6177 RSC.
- J. M. Mejia Oneto, I. Khan, L. Seebald and M. Royzen, ACS Cent. Sci., 2016, 2, 476–482 CrossRef CAS PubMed.
- J. Li, S. Jia and P. R. Chen, Nat. Chem. Biol., 2014, 10, 1003–1005 CrossRef CAS PubMed.
- E. Agustin, P. N. Asare Okai, I. Khan, M. R. Miller, R. Wang, J. Sheng and M. Royzen, Chem. Commun., 2016, 52, 1405–1408 RSC.
- A. K. Steiger, Y. Yang, M. Royzen and M. D. Pluth, Chem. Commun., 2017, 53, 1378–1380 RSC.
- X. Fan, Y. Ge, F. Lin, Y. Yang, G. Zhang, W. S. Ngai, Z. Lin, S. Zheng, J. Wang, J. Zhao, J. Li and P. R. Chen, Angew. Chem., Int. Ed., 2016, 55, 14046–14050 CrossRef CAS PubMed.
- A. Hamasaki, R. Ducray and D. L. Boger, J. Org. Chem., 2006, 71, 185–193 CrossRef CAS PubMed.
- D. L. Boger and S. M. Sakya, J. Org. Chem., 1988, 53, 1415–1423 CrossRef CAS.
- E. Jimenez-Moreno, Z. Guo, B. L. Oliveira, I. S. Albuquerque, A. Kitowski, A. Guerreiro, O. Boutureira, T. Rodrigues, G. Jimenez-Oses and G. J. Bernardes, Angew. Chem., Int. Ed., 2017, 56, 243–247 CrossRef CAS PubMed.
- K. Neumann, S. Jain, A. Gambardella, S. Walker, E. Valero, A. Lilienkampf and M. Bradley, Chembiochem, 2016, 18, 91–95 CrossRef PubMed.
- H. Wu, S. C. Alexander, S. Jin and N. K. Devaraj, J. Am. Chem. Soc., 2016, 138, 11429–11432 CrossRef CAS PubMed.
- C. A. DeForest and K. S. Anseth, Nat. Chem., 2011, 3, 925–931 CrossRef CAS PubMed.
- R. M. Desai, S. T. Koshy, S. A. Hilderbrand, D. J. Mooney and N. S. Joshi, Biomaterials, 2015, 50, 30–37 CrossRef CAS PubMed.
- D. L. Alge, M. A. Azagarsamy, D. F. Donohue and K. S. Anseth, Biomacromolecules, 2013, 14, 949–953 CrossRef CAS PubMed.
- S. Hong, J. Carlson, H. Lee and R. Weissleder, Adv. Healthcare Mater., 2016, 5, 421–426 CrossRef CAS PubMed.
- V. X. Truong, M. P. Ablett, S. M. Richardson, J. A. Hoyland and A. P. Dove, J. Am. Chem. Soc., 2015, 137, 1618–1622 CrossRef CAS PubMed.
- K. Kawamoto, S. C. Grindy, J. Liu, N. Holten-Andersen and J. A. Johnson, ACS Macro Lett., 2015, 4, 458–461 CrossRef CAS.
- H. Zhang, K. T. Dicker, X. Xu, X. Jia and J. M. Fox, ACS Macro Lett., 2014, 3, 727–731 CrossRef CAS PubMed.
- C. F. Hansell, P. Espeel, M. M. Stamenovic, I. A. Barker, A. P. Dove, F. E. Du Prez and R. K. O'Reilly, J. Am. Chem. Soc., 2011, 133, 13828–13831 CrossRef CAS PubMed.
- S. Liu, H. Zhang, R. A. Remy, F. Deng, M. E. Mackay, J. M. Fox and X. Jia, Adv. Mater., 2015, 27, 2783–2790 CrossRef CAS PubMed.
- O. Salata, J. Nanobiotechnol., 2004, 2, 3 CrossRef PubMed.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763–775 CrossRef CAS PubMed.
- H. S. Han, N. K. Devaraj, J. Lee, S. A. Hilderbrand, R. Weissleder and M. G. Bawendi, J. Am. Chem. Soc., 2010, 132, 7838–7839 CrossRef CAS PubMed.
- H. S. Han, E. Niemeyer, Y. Huang, W. S. Kamoun, J. D. Martin, J. Bhaumik, Y. Chen, S. Roberge, J. Cui, M. R. Martin, D. Fukumura, R. K. Jain, M. G. Bawendi and D. G. Duda, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1350–1355 CrossRef CAS PubMed.
- A. A. Ghazani, C. M. Castro, R. Gorbatov, H. Lee and R. Weissleder, Neoplasia, 2012, 14, 388–395 CrossRef CAS PubMed.
- A. A. Ghazani, S. McDermott, M. Pectasides, M. Sebas, M. Mino-Kenudson, H. Lee, R. Weissleder and C. M. Castro, Nanomedicine, 2013, 9, 1009–1017 CrossRef CAS PubMed.
- A. A. Ghazani, M. Pectasides, A. Sharma, C. M. Castro, M. Mino-Kenudson, H. Lee, J. A. Shepard and R. Weissleder, Nanomedicine, 2014, 10, 661–668 CrossRef CAS PubMed.
- J. B. Haun, C. M. Castro, R. Wang, V. M. Peterson, B. S. Marinelli, H. Lee and R. Weissleder, Sci. Transl. Med., 2011, 3, 71ra16 Search PubMed.
- D. Issadore, J. Chung, H. Shao, M. Liong, A. A. Ghazani, C. M. Castro, R. Weissleder and H. Lee, Sci. Transl. Med., 2012, 4, 141ra192 Search PubMed.
- D. Issadore, H. J. Chung, J. Chung, G. Budin, R. Weissleder and H. Lee, Adv. Healthcare Mater., 2013, 2, 1224–1228 CrossRef CAS PubMed.
- H. Shao, J. Chung, L. Balaj, A. Charest, D. D. Bigner, B. S. Carter, F. H. Hochberg, X. O. Breakefield, R. Weissleder and H. Lee, Nat. Med., 2012, 18, 1835–1840 CrossRef CAS PubMed.
- J. Rho, J. Chung, H. Im, M. Liong, H. Shao, C. M. Castro, R. Weissleder and H. Lee, ACS Nano, 2013, 7, 11227–11233 CrossRef CAS PubMed.
- H. J. Chung, K. L. Pellegrini, J. Chung, K. Wanigasuriya, I. Jayawardene, K. Lee, H. Lee, V. S. Vaidya and R. Weissleder, PLoS One, 2015, 10, e0133417 Search PubMed.
- M. Liong, M. Fernandez-Suarez, D. Issadore, C. Min, C. Tassa, T. Reiner, S. M. Fortune, M. Toner, H. Lee and R. Weissleder, Bioconjugate Chem., 2011, 22, 2390–2394 CrossRef CAS PubMed.
- H. J. Chung, T. Reiner, G. Budin, C. Min, M. Liong, D. Issadore, H. Lee and R. Weissleder, ACS Nano, 2011, 5, 8834–8841 CrossRef CAS PubMed.
- G. Budin, H. J. Chung, H. Lee and R. Weissleder, Angew. Chem., Int. Ed., 2012, 51, 7752–7755 CrossRef CAS PubMed.
- S. S. Agasti, R. H. Kohler, M. Liong, V. M. Peterson, H. Lee and R. Weissleder, Small, 2013, 9, 222–227 CrossRef CAS PubMed.
- J. B. Haun, N. K. Devaraj, B. S. Marinelli, H. Lee and R. Weissleder, ACS Nano, 2011, 5, 3204–3213 CrossRef CAS PubMed.
- V. M. Peterson, C. M. Castro, H. Lee and R. Weissleder, ACS Nano, 2012, 6, 3506–3513 CrossRef CAS PubMed.
- F. Emmetiere, C. Irwin, N. T. Viola-Villegas, V. Longo, S. M. Cheal, P. Zanzonico, N. Pillarsetty, W. A. Weber, J. S. Lewis and T. Reiner, Bioconjugate Chem., 2013, 24, 1784–1789 CrossRef CAS PubMed.
- J. Li, M. Li, L. L. Zhou, S. Y. Lang, H. Y. Lu, D. Wang, C. F. Chen and L. J. Wan, J. Am. Chem. Soc., 2016, 138, 7448–7451 CrossRef CAS PubMed.
- J. Zhu, J. Hiltz, M. A. Mezour, V. Bernard-Gauthier, R. B. Lennox and R. Schirrmacher, Chem. Mater., 2014, 26, 5058–5062 CrossRef CAS.
- J. Zhu, J. Hiltz, R. B. Lennox and R. Schirrmacher, Chem. Commun., 2013, 49, 10275–10277 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |