
Synthesis, structure and applications of graphene-based 2D heterostructures
 b
and
Hiroki
Ago
*acd
b
and
Hiroki
Ago
*acd
Abstract
With the profuse amount of two-dimensional (2D) materials discovered and the improvements in their synthesis and handling, the field of 2D heterostructures has gained increased interest in recent years. Such heterostructures not only overcome the inherent limitations of each of the materials, but also allow the realization of novel properties by their proper combination. The physical and mechanical properties of graphene mean it has a prominent place in the area of 2D heterostructures. In this review, we will discuss the evolution and current state in the synthesis and applications of graphene-based 2D heterostructures. In addition to stacked and in-plane heterostructures with other 2D materials and their potential applications, we will also cover heterostructures realized with lower dimensionality materials, along with intercalation in few-layer graphene as a special case of a heterostructure. Finally, graphene heterostructures produced using liquid phase exfoliation techniques and their applications to energy storage will be reviewed.
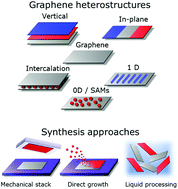
- This article is part of the themed collection: Graphene Chemistry
 Please wait while we load your content...
Please wait while we load your content...

