
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA review of flexible lithium–sulfur and analogous alkali metal–chalcogen rechargeable batteries
Hong-Jie
Peng
 a,
Jia-Qi
Huang
b and
Qiang
Zhang
*a
a,
Jia-Qi
Huang
b and
Qiang
Zhang
*a
aBeijing Key Laboratory of Green Chemical Reaction Engineering and Technology, Department of Chemical Engineering, Tsinghua University, Beijing 100084, China. E-mail: zhang-qiang@mails.tsinghua.edu.cn
bAdvanced Research Institute for Multidisciplinary Science, Beijing Institute of Technology, Beijing 100081, China
First published on 7th August 2017
Abstract
Flexible energy storage systems are imperative for emerging flexible devices that are revolutionizing our life. Lithium-ion batteries, the current main power sources, are gradually approaching their theoretical limitation in terms of energy density. Therefore, alternative battery chemistries are urgently required for next-generation flexible power sources with high energy densities, low cost, and inherent safety. Flexible lithium–sulfur (Li–S) batteries and analogous flexible alkali metal–chalcogen batteries are of paramount interest owing to their high energy densities endowed by multielectron chemistry. In this review, we summarized the recent progress of flexible Li–S and analogous batteries. A brief introduction to flexible energy storage systems and general Li–S batteries has been provided first. Progress in flexible materials for flexible Li–S batteries are reviewed subsequently, with a detailed classification of flexible sulfur cathodes as those based on carbonaceous (e.g., carbon nanotubes, graphene, and carbonized polymers) and composite (polymers and inorganics) materials and an overview of flexible lithium anodes and flexible solid-state electrolytes. Advancements in other flexible alkali metal–chalcogen batteries are then introduced. In the next part, we emphasize the importance of cell packaging and flexibility evaluation, and two special flexible battery prototypes of foldable and cable-type Li–S batteries are highlighted. In the end, existing challenges and future development of flexible Li–S and analogous alkali metal–chalcogen batteries are summarized and prospected.
 Hong-Jie Peng | Hong-Jie Peng completed his BE from the Department of Chemical Engineering, Tsinghua University, in 2013. He is currently a PhD candidate at Tsinghua University. His research interests include nanocarbon technology, advanced energy storage/conversion, lithium–sulfur batteries, and catalysis. |
 Jia-Qi Huang | Jia-Qi Huang is a professor in Advanced Research Institute for Multidisciplinary Science at Beijing Institute of Technology. He completed his PhD from the Department of Chemical Engineering, Tsinghua University, in 2012. His research interests include synthetic approaches and application of nanomaterials for rechargeable batteries. |
 Qiang Zhang | Qiang Zhang completed his bachelor degree and PhD from Tsinghua University in 2004 and 2009, respectively. After a stint in Case Western Reserve University, USA, and Fritz Haber Institute of the Max Planck Society, Germany, he joined Tsinghua University in 2011. He won the Newton Advanced Fellowship Scheme from Royal Society, UK, and NSFC for Young Scholars in China. His interests focus on energy materials, including Li–S batteries, Li metal anode, 3D graphene, and electrocatalysts. His research has been cited over 11 |
1. Introduction
The development of advanced and reliable technology for electrochemical energy storage is driven by rapidly growing markets of lighter, thinner, and smaller portable devices.1–3 Flexible electronics are of particular interest to realize exceptional portability and applicability for all-condition and all-environment usages. A wide range of applications, including rolled-up displays, implantable devices, touch screens, conformable active radio-frequency identification tags, epidermal sensors, and artificial skins, have been demonstrated.4–9 With the development of flexible electronics at an unrelenting pace, they are truly revolutionizing human society and life. Such a revolution, however, will become stagnant unless the development of corresponding power accessories keep pace with that of flexible electronics.10 On one hand, existing power sources are too heavy, thick, bulky, and rigid to meet the ubiquitous demand for flexible energy storage. Therefore, considerable efforts have to be devoted to the facile transition from these energy storage systems to their light, thin, small, and flexible counterparts. On the other hand, lithium-ion battery (LIB) technology, historically and presently the dominant element in portable devices, is gradually approaching theoretical limitations in terms of energy density, thus hindering device performance despite recent progress in flexible LIBs.10–13 Thus, the exploration of alternative, green, and high energy density chemistries has become an urgent research target for next-generation flexible batteries at present.14–16For a specific battery reaction, the cathode material (Cox) is reduced to accept both electrons and positive charge carriers (Mx+), whereas the anode material (Are–M) is oxidized via the loss of charge carriers during discharge (eqn (1)); the process should be reversible during charge (eqn (2)).
| Cox + xe− + Mx+ ↔ Cre–M (cathode reaction) | (1) |
| Are–M ↔ Aox + xe− + Mx+ (anode reaction) | (2) |
The specific capacity can be obtained based on Faraday's laws of electrolysis, where Q is the net charge capacity transferred during the reaction; Mcat and Mano, Wcat and Wano, Gcat and Gano, and Vcat and Vano refer to the weights, volumes, and gravimetric and volumetric specific capacities of the cathode and anode materials (Cox and Are–M or equivalently Cre–M and Aox), respectively (eqn (3) and (4)).
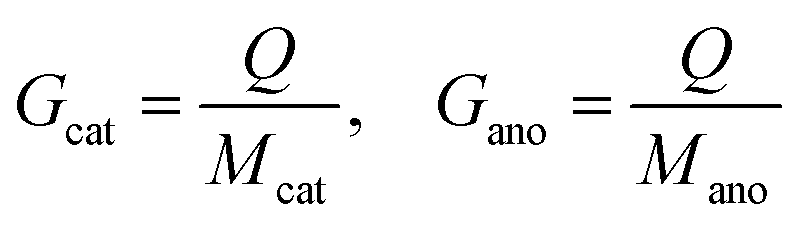 | (3) |
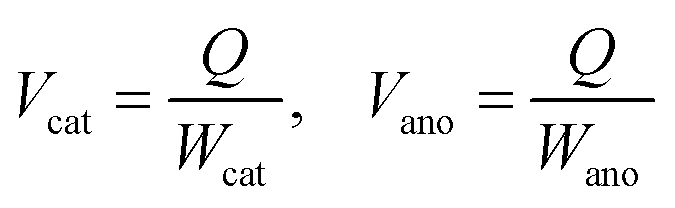 | (4) |
According to eqn (5) and (6), it can be clearly seen that energy density (EG and EV) of a specific battery is determined by the redox potential (Pcat and Pano) and specific capacity per unit weight or volume of each electrode (Gcat and Gano or Vcat and Vano). More positive potential of the cathode (Pcat), more negative potential of the anode (Pano), and higher capacities of the two electrodes (equivalent to lighter weights or smaller volume) contribute to higher energy densities.
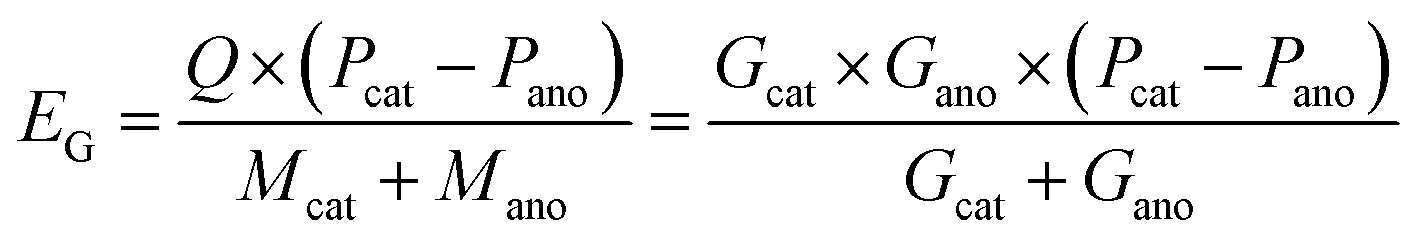 | (5) |
 | (6) |
To evaluate the potential of various electrode materials for next-generation flexible batteries, their redox potentials vs. Li/Li+ and gravimetric/volumetric specific capacities are shown in Fig. 1a and b, respectively. Cathode materials can be classified into (1) intercalated compounds such as lithium transition metal oxides/oxysalts, (2) halogens such as bromine (Br2) and iodine (I2), (3) oxygen (either in aqueous or non-aqueous conditions), and (4) other chalcogen elements such as sulfur (S), selenium (Se), and tellurium (Te); anode materials are also categorized into (1) intercalated compounds such as graphite (C) and lithium titanate, (2) alloys based on silicon (Si), germanium (Ge), tin (Sn), and phosphorous (P), (3) multivalent metals such as magnesium (Mg), aluminum (Al), and zinc (Zn), and (4) monovalent alkali metals such as lithium (Li), sodium (Na), and potassium (K).16–18
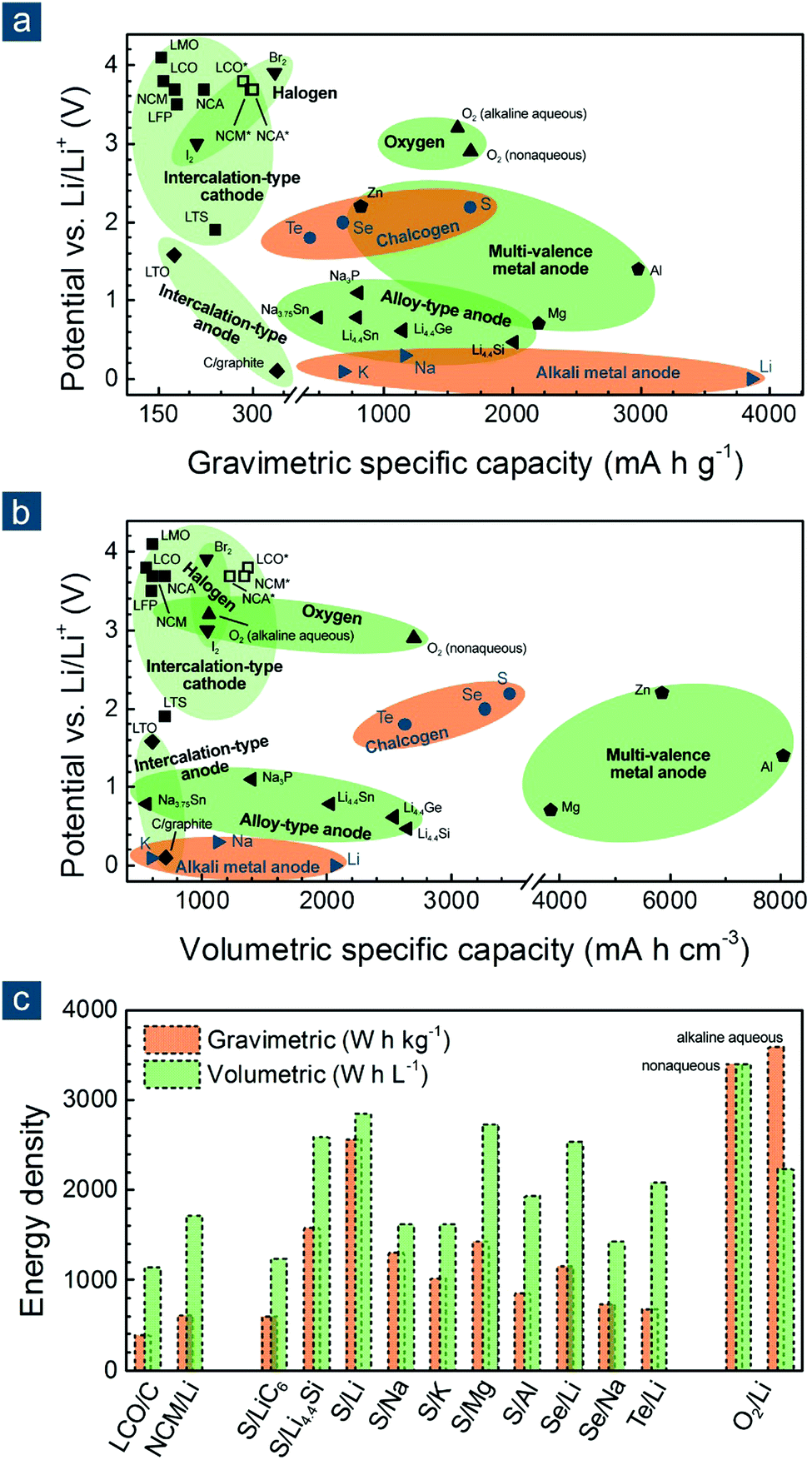 | ||
| Fig. 1 Potential vs. specific capacities of various electrode materials: (a) gravimetric and (b) volumetric. For a fair comparison, capacities of cathode and anode materials are calculated based on their weights in oxidized and reduced forms, namely being combined without and with cations (e.g., Li+, Na+, K+, Mg2+, Zn2+, and Al3+), respectively. (c) Energy density of various battery systems. [Some abbreviations: LTS: Li0TiS2; LCO and LCO*: Li0.45CoO2 and Li0CoO2; NCM and NCM*: Li0.4Ni0.33Co0.33Mn0.33O2 and Li0Ni0.33Co0.33Mn0.33O2; NCA and NCA*: Li0.25Ni0.8Co0.15Al0.05O2 and Li0.05Ni0.8Co0.15Al0.05O2; LMO: Li0Mn2O4; LFP: Li0FePO4; LTO: Li4Ti5O12; C/graphite: LiC6. Some definitions: * indicates the theoretical maximum capacity of LCO, NCM, and NCA by assuming that all redox-active metal cations (Ni2+/Ni3+ and Co3+) are fully oxidized to tetravalent ones (Ni4+ and Co4+) after charging; capacities of alkaline aqueous and non-aqueous oxygen electrodes are calculated based on reactions: 0.5O2 + H2O + 2Li+ + 2e− = 2LiOH (anhydrous) and O2 + 2Li+ + 2e− = Li2O2, using the weights of (0.5O2 + H2O) and O2 for gravimetric ones and the volumes of (LiOH + 0.5H2O) and Li2O2 for volumetric ones. Data are reproduced with permission.16 Copyright 2014, Elsevier.] | ||
Among these materials, most intercalated cathodes, also conventional LIB electrode materials, enable high output voltages (>3.5 V, vs. Li/Li+ and similarly hereinafter), which are endowed by redox-active transition metal cations (e.g., Ni2+/3+/4+, Co3+/4+, and Fe2+/3+), but deliver low gravimetric specific capacities (<200–300 mA h g−1) owing to large molecular weight of crystal hosts that accommodate charge carriers (normally cations like Li+, Na+, K+, Mg2+, Zn2+, and Al3+). The halogen electrodes offer high redox potentials (>3.0 V) and higher gravimetric specific capacities (221 and 335 mA h g−1 for I2 and Br2, respectively) than intercalated compounds, though they are volatile and hazardous elements and may be detrimental to the environment and human health. The oxygen electrodes are featured with a promising voltage (2.9/3.2 V) and a very high gravimetric capacity (1675/1576 mA h g−1) when working in a non-aqueous/alkaline aqueous condition, but are unfortunately restricted by several insurmountable obstacles like short cycling life, poor energy efficiency, electrode/electrolyte instability, and atmospheric contamination by carbon dioxide and nitrogen.19,20 The intercalated anodes mainly suffer from low capacity (<370 mA h g−1). The alloy anodes have enhanced gravimetric specific capacities (800–2000 mA h g−1) with large uptake of charge carriers, but compromise on battery voltage due to relatively high dealloying potential (>0.4 V). The multivalent metal anodes exhibit very attractive gravimetric and volumetric specific capacities (800–3000 mA h g−1 and 3800–8000 mA h cm−3, respectively), which are unfortunately offset to a considerable extent by their greatly upshifted redox potential (0.7–2.2 V). Therefore, taking the redox potential, gravimetric/volumetric specific capacity, and research status into considerations, the chemistry between alkali metals and chalcogen elements other than oxygen, representatively the Li–S battery, have been recognized as a promising candidate for next-generation batteries capable of offering balanced battery performance (Fig. 1c).18,19,21–23
Despite several instructive reviews on flexible LIBs,10–13,24–26 recent progress of flexible Li–S batteries and alkali metal–chalcogen analogues has not received specific attention; nevertheless, this is worthwhile to be the focus of future explorations. In this review, we attempt to summarize the recent progress on flexible Li–S and analogous alkali metal–chalcogen batteries (Fig. 2). The major content will focus on flexible materials for flexible Li–S batteries, including flexible sulfur cathodes based on carbon nanotubes (CNTs), graphene, commercial carbon cloths/papers, carbonized polymers, hybridized carbon, and their composites with polymers or inorganics, as well as flexible lithium metal anodes, and flexible solid-state electrolytes. Then, other flexible alkali metal–chalcogen batteries will be introduced, followed by discussions on device integration, proper evaluation of flexibility, and two unique flexible Li–S battery prototypes, i.e., foldable planar Li–S batteries and cable-type Li–S batteries. Hopefully, through this review, the understanding of alkali metal–chalcogen chemistry, especially under unusual conditions, will become clearer, the understanding and design considerations can be fed back to their conventional battery forms, and the development of other flexible energy storage/conversion systems such as lithium–air batteries and miniaturized fuel cells will be also benefited.
 | ||
| Fig. 2 An overview of flexible alkali metal–chalcogen batteries. | ||
2. Flexible materials for flexible Li–S batteries
Li–S batteries possess a high theoretical energy density of 2597 W h kg−1. Sulfur, as the cathode material, exhibits advantages such as abundance, low cost, and environmental benignity.23,27,28 A typical Li–S battery operates on reversible electrochemical reactions between elemental lithium and sulfur in a liquid organic electrolyte. During discharge, the lithium metal anode is oxidized to lithium ions, which further migrate to the cathode; the sulfur cathode undergoes typical two-stage electrochemical reactions, with solid–liquid–solid phase transitions concurrently taking place. Long-chain polysulfides (Li2Sx, x = 4–8) are produced at the first stage, and they dissolve in the electrolyte and then precipitate in the form of insoluble lithium disulfides and/or sulfides (Li2S2 or Li2S) at the second stage.| (x/8)S8 + 2e− + 2Li+ = Li2Sx (x = 4–8) the first stage of discharge (∼2.2–2.4 V) | (7) |
| Li2Sx + (2x − 2)e− + (2x − 2)Li+ = xLi2S (x = 4–8) the second stage of discharge (<∼2.2 V) | (8) |
The charge processes are basically the inverse of each other but probably follow different reaction pathways.29 The unique multielectron, multiphase, and multistage reaction nature induces the Li–S battery with not only outstanding energy densities but also some intrinsic issues: (1) the dissolution of both sulfur and lithium electrodes results in severe phase migrations and drastic structural changes in electrodes of a working battery; (2) the re-deposition of Li2S, which has a lower density (1.66 g cm−3) than that of sulfur (α-phase, 2.07 g cm−3), leads to a significant volume fluctuation (∼80%); (3) the electrodeposition of lithium is easily accompanied with its dendritic growth; and (4) the migration of soluble polysulfides, driven by concentration gradients and electric fields, causes side reactions between polysulfides and lithium metal, reducing battery efficiency.23,27,28,30–36 Besides, low electrical conductivity of the solid products involved in the reactions inhibit efficient utilization of active materials. As a result of these issues, Li–S batteries nowadays encounter low capacity, rapid capacity fading, short cycling life, and serious self-discharge. To address these problems, many efforts have been made for the development of cathodes,37–50 current collectors,51–53 electrolytes,54–60 anodes,61–64 and separators.65–74
For flexible Li–S batteries, flexible materials play a pivotal role. Compared to conventional batteries, the flexible Li–S battery must afford stable electrochemical performance when being repeatedly bended, folded, or stretched. As a consequence, all materials in this cell, including the cathode, anode, separator, electrolyte, and current collector, have to be flexible enough to withstand mechanical deformations. Moreover, each component should be connected steadily in their original sequence to maintain continuous electron/ion pathways and prevent cell failure. In typical Li–S batteries, the separator is usually a porous polymer membrane that is naturally flexible, while the current collector is a metal foil such as Al foil for the cathode and copper (Cu) foil for the anode, which offer very limited flexibility due to their plastic and ductile deformations. Therefore, electrodes designed for flexible batteries are commonly metal-current-collector-free. To improve battery safety by preventing leakage of flammable liquid electrolytes, solid-state electrolytes are preferred for flexible batteries. Generally, in the design of flexible Li–S batteries, several aspects need to be considered to differentiate them from the design of conventional Li–S batteries: (1) mechanical properties of each cell component, such as rupture strength, maximum allowed strain, Young's modulus, and dynamic response to certain mechanical deformations; (2) electrochemical performance under deformation; and (3) stricter safety standards to prevent potential risks induced and exaggerated by massive mechanical strains. To rigidly meet these demands, a holistic design principle is necessary to manage all cell components together with tight connections, be adapted to their different physicochemical properties and mechanics, and realize highly compliant electron/ion transportation networks. Before working on such a holistic design, deep understanding and advanced engineering of key individual components are prerequisite. Hence, in this section, the contents are divided into three categories: flexible sulfur-based cathodes, flexible lithium-based anodes, and flexible solid-state electrolytes.
2.1. Flexible sulfur-based cathodes
A conventional cathode for Li–S batteries consists of active sulfur materials, binders, and conductive agents, all of which are coated on a metal foil, which acts as the current collector. Such a configuration can hardly endure deformations due to cracks in the electrode layer and electrode delamination from the current collector. Flexible electrodes, however, have to withstand bending, twisting, stretching and folding while properly maintaining power supply, thereby demanding for a more delicate design. To fabricate flexible sulfur-based cathodes, there are two main synthetic approaches: post-sulfur loading and pre-sulfur loading. The former involves prior formation of a flexible skeleton, which is then loaded with sulfur using vapor infusion, melt diffusion, or reprecipitation of sulfur from a solution (usually in carbon disulfide (CS2) or toluene); while the latter is realized through assembly of pre-synthesized sulfur composites into a flexible cathode.For Li–S batteries, a flexible cathode manifests several desirable features as compared to conventional cathodes: (1) high content of active materials with respect to total mass of the electrode if the cathode is binder- and current-collector-free; (2) mechanically robust skeleton to accommodate volumetric and structural changes; (3) long-range interpenetrated conductive network; (4) porous structure for electrolyte infiltration and polysulfide preservation; and (5) three-dimensional (3D) scaffold to improve areal sulfur loading.75
2.1.1.1. CNT-based flexible cathodes. Since 1991, when brought under the global spotlight by Iijima's work,76 CNTs have emerged as the most highly regarded and studied nanomaterials owing to their unique structure and properties. CNTs are truly the key pioneer and continuous driving force in nanomaterials and nanotechnology.77 After decades of developments, CNTs can be mass produced at a relatively low cost of $100 per kg and an annual capacity of 103–104 tons through scalable and continuous chemical vapor deposition (CVD) in industrial reactors.78,79 The success in developing reliable technologies for CNT mass production consequently promotes its bulk applications for many target markets, such as electrochemical energy storage.2,80
On one hand, CNTs can be regarded as coaxially rolled graphene sheets, with wall numbers ranging from one to several tens. The covalently bonded sp2-carbon characteristic renders CNTs with extraordinary electrical conductivity and superb mechanical, chemical, and thermal stability, making them especially suitable for serving as alternative conductive agents to conventional ones such as carbon blacks and graphite sheets. An electrical percolation network composed of CNTs in an electrode allows higher fault tolerance than that composed of carbon blacks, even at a smaller additive amount.
On the other hand, CNTs are typical one-dimensional (1D) nanomaterials, thereby possessing ubiquitous superiorities in structure and properties, such as large aspect ratio, high surface area, and structural/chemical anisotropies. As a result, it is quite easy and facile to assemble CNTs to fabricate various macroscopic forms, such as films,81 arrays,82 sponges,83 and fibers,84 through various methods such as vacuum filtration,85 self-assembly,86 printing,87 drawing,88 and spinning.89 These macroscopic forms normally have a tailorable porous structure, 3D conductive networks, and excellent mechanical robustness and are therefore regarded as very promising material platforms for flexible electrodes.
In fact, CNT-based flexible electrode materials have widely been explored as electrode prototypes for flexible LIBs. For Li–S batteries, vertically aligned CNTs (VACNTs) grown on metal substrates were first employed as sulfur cathode architectures in two separate but contemporary works by Dörfler et al.90 and Barchasz et al.91 In the former, a binder-free, high-capacity VACNT/S composite cathode was demonstrated. Though inflexible, such an electrode sheds light on the potential of CNTs as building blocks of binder-free, flexible sulfur-based cathodes. Aiming at improving the utilization of active materials and stability, the main challenges are (1) to rationally assemble CNTs into mechanically stable and electrically conductive architectures and (2) to simultaneously achieve uniform dispersion of sulfur in the matrix. Effective and efficient strategies for fabricating macroscopic CNT architectures and incorporating sulfur into them are thus of great importance.
To obtain mechanically robust CNT films from solution, the use of surfactants or surface functionalization is normally adopted to improve dispersion of CNTs in solvents and enhance the interfacial adhesion between each CNT. For example, Manthiram and coworkers prepared a self-weaving S–CNT composite cathode via a one-pot, solution-based approach.92 Briefly, CNTs were dispersed in sodium thiosulfate (Na2S2O3) solution with the help of isopropyl alcohol and Triton X-100 surfactants; addition of hydrochloride acid triggered controlled precipitation of sulfur to form S–CNT composites, which then self-wove into binder-, current-collector-free, pliable composite cathodes after filtering and washing. The flexible cathodes (40 wt% sulfur) exhibited a high initial capacity of 1352(541) mA h gsul(ele)−1 and retained 68% of initial capacity after 100 cycles at a relatively high rate of 1.0C (1.0C = 1672 mA gsul−1, similarly for sulfur hereinafter).
Surface functionalization such as oxidation is another route. Liu and colleagues first obtained oxidized CNTs via refluxing raw, hydrophobic CNTs in concentrated sulfuric acid/nitric acid solution, and the as-obtained hydrophilic CNTs were well dispersed in distilled water and filtrated to form CNT films.93 Sulfur was further infused into CNT films through immersing CNT films in molten sulfur, and the excess sulfur had to be removed by an additional heating procedure in vacuum. In the S–CNT composite film, sulfur (65 wt%) was uniformly coated on the external surface of CNTs, giving rise to an initial capacity of 1100(715) mA h gsul(ele)−1 and 67% capacity retention after 100 cycles at 0.1C.
Employing surfactants or functionalized CNTs to fabricated CNT-based flexible films is straightforward and has been well engineered; however, it also inevitably introduces detrimental side effects with respect to electrical conductivity. For example, strong oxidation, normally during oxidative acid refluxing, renders a large quantity of sp3 carbon, structural defects, and non-conjugated regions, all of which reduce electrical conductivity. On the other hand, most surfactants are electrically insulating. They form micelles surrounding CNTs, impeding direct electron transfer to the active phase and between CNTs. Therefore, it is critical to enable good dispersion of CNTs in solution and simultaneously maintain its extraordinary electrical conductivity for superior flexible electrode materials.
In this regard, Zhou et al. described template-directed synthesis of S–CNT nanostructures and their use as a binder-free, highly conductive, and flexible film for high-rate Li–S batteries (Fig. 3).94 This is actually the first report on flexible sulfur electrodes with unambiguous and quantitative demonstration of flexibility. This concept is based on two key aspects: (1) elemental sulfur was incorporated on CNTs via in situ carbothermal reduction of sulfate during CNT growth in anodic aluminum oxide (AAO) templates, achieving favorable confinement of sulfur and avoiding any post-loading procedure (Fig. 3a), and (2) as-obtained S–CNTs could be easily integrated into a pliable film with an interwoven nanotube network through ethanol evaporation-induced assembly (Fig. 3b and c), the ease of which was probably due to the large diameter (>30 nm) and long length (30–50 μm) of AAO-templated CNTs that reduced nanoscale agglomeration (Fig. 3d). The S–CNT film could sustain a 10 MPa stress with a strain of 9% and exhibited a high electrical conductivity of 8.0 S cm−1, which remained unaltered during 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 bending cycles (Fig. 3e and f). These desirable features rendered the S–CNT nanostructured film an outstanding flexible sulfur cathode with remarkable electrochemical performance at high rates. At a current density of 6 A gsul−1 (∼3.6C), reversible capacities were 712(163) and 520(260) mA h gsul(ele)−1 for S–CNT flexible cathodes with 23 and 50 wt% sulfur, respectively.
000 bending cycles (Fig. 3e and f). These desirable features rendered the S–CNT nanostructured film an outstanding flexible sulfur cathode with remarkable electrochemical performance at high rates. At a current density of 6 A gsul−1 (∼3.6C), reversible capacities were 712(163) and 520(260) mA h gsul(ele)−1 for S–CNT flexible cathodes with 23 and 50 wt% sulfur, respectively.
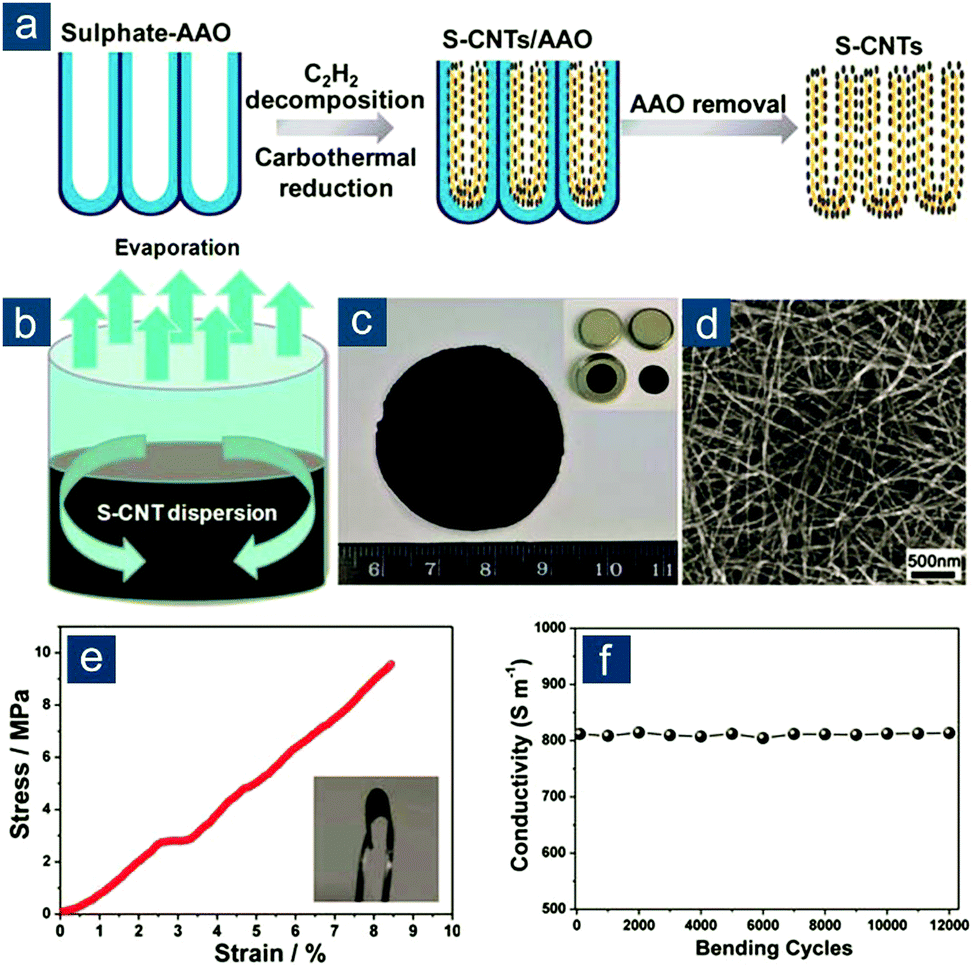 | ||
| Fig. 3 Non-functionalized self-assembled CNT-based flexible sulfur cathodes. Schematic for (a) AAO-template-directed synthesis of S–CNT nanostructures and (b) evaporation-induced assembly of a flexible membrane from S–CNT ethanol dispersion. (c) Image of S–CNT films. (d) Scanning electron microscopy (SEM) image of interwoven S–CNT. (e) Stress–strain curve and (f) electrical conductivity (measured upon repeated bending for 12000 cycles) of flexible S–CNT membrane. Reproduced with permission.94 Copyright 2012, Royal Society of Chemistry. | ||
Another promising route is to functionalize CNTs with sulfur before solution processing of flexible films. As an example, Yuan et al. developed a bottom-up strategy to fabricate hierarchical CNT–S cathodes (Fig. 4).95 “Bottom-up” herein corresponded to a pre-sulfur-loading approach, in contrast to a “top-down” one involving post-sulfur loading. In the fabrication, high-surface-area short multi-walled CNTs (MWCNTs, with an average diameter of 15 nm and a length of 10–50 μm) were first functionalized with sulfur to simultaneously (1) provide intimate short-range electrical contact and (2) reduce strong intertube forces. Ultralong CNTs (∼50 nm in diameter and 1000–2000 μm in length), obtained through vigorous shearing of VACNTs in solution, were employed to serve as (3) a long-range electrically conductive network and (4) robust mechanical support by fully exerting merits of a perfect sp2-carbon structure and an ultrahigh aspect ratio (a magnitude of ∼104) (Fig. 4a). No surfactant or extra surface modification of CNTs was required but facile co-dispersion of MWCNT/S composites and ultralong CNTs in ethanol was required, rendering as-obtained hierarchical CNT–S papers with a mixed short-/long-range conductive skeleton, interconnected porous structure as ion channels, and desirable flexibility (Fig. 4b). These characteristics resulted in significantly enhanced electrochemical performance of bottom-up CNT–S cathodes in comparison with those fabricated through conventional methods (Fig. 4c). With a moderate sulfur content of 54 wt% and a high sulfur loading of 6.3 mgsul cm−2, the bottom-up CNT–S cathodes maintained a reversible capacity of ∼700(380) mA h gsul(ele)−1 after 150 cycles at 0.05C (0.38 mA cm−2); in contrast, cathodes consisting of CNT papers with post-loaded sulfur showed rapid capacity decay to ∼200 mA h gsul−1 after only 80 cycles. More interestingly, flexible bottom-up CNT–S papers could be further stacked layer by layer into lasagna-like, high-sulfur-loading cathodes with ultrahigh areal capacities of up to 15.1 mA h cm−2. Such a rational integration of CNT dispersion with pre-impregnation of actual active materials (e.g., sulfur) holds great promise to fabricate flexible composite electrodes consisting of materials that are distinctive from each other.
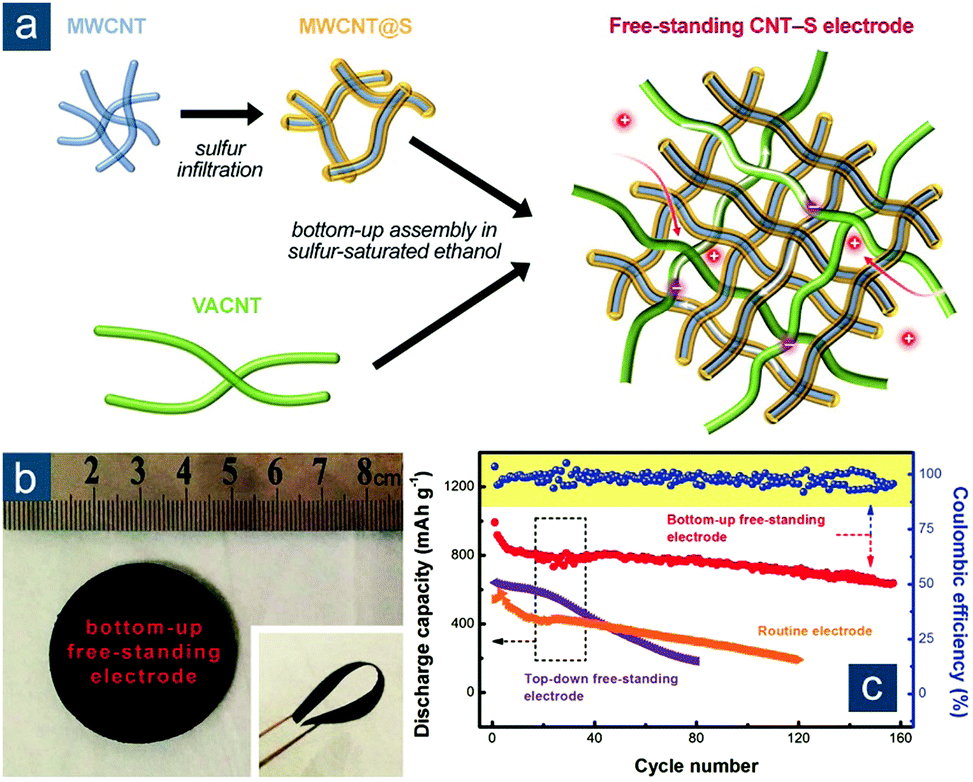 | ||
| Fig. 4 Elemental-sulfur-functionalized CNT-based flexible sulfur cathodes. (a) Schematic for the bottom-up fabrication of hierarchical freestanding CNT–S paper electrode with ultrahigh sulfur loadings. (b) Image of CNT–S papers in flat and bent (inset) states. (c) Cycling performance of various CNT–S cathodes prepared using conventional slurry coating, top-down (post-sulfur loaded) and bottom-up (pre-sulfur loaded) methods. Reproduced with permission.95 Copyright 2014, Wiley-VCH. | ||
Besides typical CNTs grown on metal catalysts or designed templates (e.g., AAO), special CNTs have been utilized to build CNT-based flexible sulfur electrodes. For example, Sun et al. explored the use of superaligned CNTs (SACNTs) in Li–S batteries (Fig. 5).96,97 SACNT arrays, first synthesized on silicon wafers by Fan and colleagues, were of high purity, high quality, high surface density, and quasi-unidirectional alignment, which differentiated them remarkably from conventional VACNT arrays.98 Through dispersion in a solvent under ultrasonication, SACNT bundles could facilely expand into a continuous 3D network that was conductive and highly porous. This unique feature enabled uniform distribution of sulfur nanocrystals within the SACNT matrix upon sulfur precipitation from SACNT/S ethanol solution, which was triggered by dropwise addition of water (Fig. 5a).96 Continuous SACNT network, as a physical barrier, prevented self-aggregation of sulfur, and as a result, nanosized sulfur (50 wt%) that was strongly anchored could rapidly access electrons and shorten lithium ion diffusion path, displaying impressive capacities of 1006(503), 960(480), and 879(440) mA h gsul(ele)−1 at high rates of 2.0, 5.0, and 10C, respectively. To achieve higher sulfur content and promote cycling performance, the same group combined the advantages of mesoporous structure and SACNTs through controlled oxidation of SACNTs in air, thus fabricating mesoporous SACNTs (PCNTs) (Fig. 5b).97 The controllable oxidation not only introduced abundant mesoporosity to PCNTs, but also altered surface properties of PCNTs, rendering flexible PCNT films with a more condensed structure, better mechanical strength (3.65 MPa), and higher electrical conductivity (58.0 S cm−1) than pristine films (0.46 MPa; 35.0 S cm−1) (Fig. 5c). Following a similar method to their aforementioned work, tunable sulfur contents of 50, 60, and 70 wt% were obtained to achieve flexible S–PCNT cathodes with capacities of 451, 511, 524 mA h gele−1, respectively, obtained at 0.1C after 100 cycles.
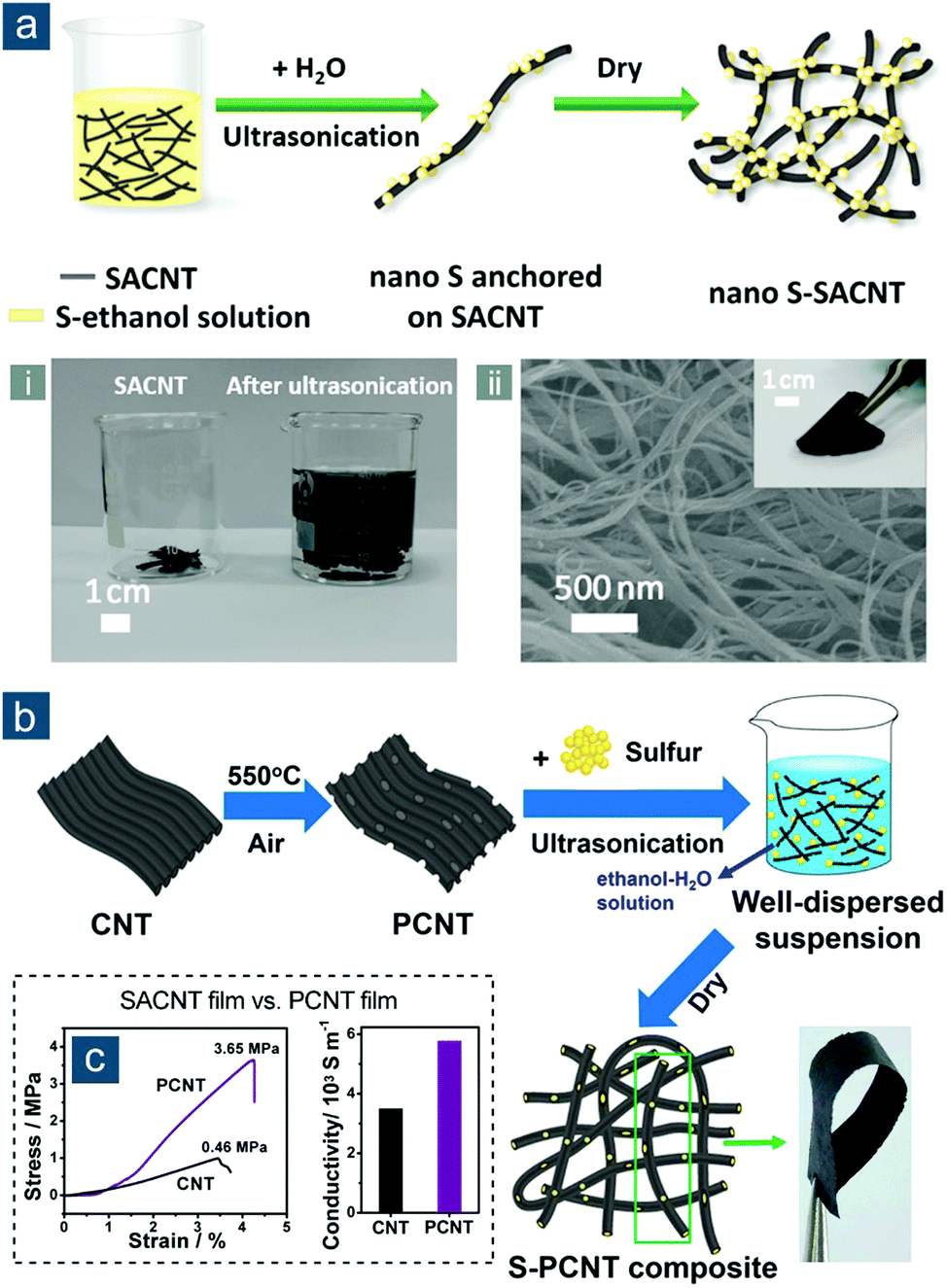 | ||
| Fig. 5 SACNT-based flexible sulfur cathodes. (a) Schematic for the synthesis of nano S–SACNT, along with (i) a photograph showing the volume expansion of SACNTs in solution after ultrasonication, (ii) SEM image of SACNT, and (ii, inset) photograph of flexible nano S–SACNT film. Reproduced with permission.96 Copyright 2014, American Chemical Society. (b) Schematic for the synthesis of S–PCNT and photograph showing a bent S–PCNT film. (c) Stress–strain curves (left) and electrical conductivities (right) of SACNT and PCNT films. Reproduced with permission.97 Copyright 2015, American Chemical Society. | ||
Qie and Manthiram employed commercial carbon nanofibers, known as stacked-cup CNTs, as raw materials to prepare activated carbon nanofiber (ACNF) sheets through carbon dioxide activation.99 Sulfur powders were laminated alternately between several ACNF sheets to obtain freestanding, high-sulfur-loading cathodes. With a sulfur content of 56 wt% and an ultrahigh sulfur loading of 11.4 mgsul cm−2, the layer-by-layer ACNF–S cathode delivered high capacities of 1002(564) and 762(430) mA h gsul(ele)−1 at 0.1 and 0.4C (1.92 and 7.68 mA cm−2, respectively), corresponding to areal capacities of 11.4 and 8.69 mA h cm−2, respectively.
Other than structural modulation, chemical modulation such as heteroatom doping is able to regulate the properties of CNTs significantly. For example, Zhao et al. adopted nitrogen-doped CNTs (N–CNTs) to fabricate CNT-based flexible sulfur cathodes, since N–CNTs were proven to exhibit stronger adsorption of polysulfides and stabilize sulfur electrochemical reactions.100 The freestanding S/N–CNT composite film exhibited a reversible capacity of 807(492) mA h gsul(ele)−1 after 100 cycles at 0.2C. Based on the success of dual-heteroatom-doped carbon in Li–S batteries,101–103 codoped CNTs may provide additional benefits for developing CNT-based flexible sulfur cathodes. The only concern is that the amounts, position, and varieties of dopants profoundly influence electrical conductivity of doped CNTs,104 which could offset the advantage of polysulfide immobilization.
Besides elemental sulfur, other sulfur-based active materials can be integrated with CNTs for fabricating flexible cathodes. For example, Fu and coworkers fabricated freestanding nano-Li2S/CNT paper via a simple Li2S solution filtration method. The 3D structure of CNT paper impeded Li2S particles from agglomeration into microsized particles during solvent evaporation, and nano-Li2S with good dispersion in CNT networks enabled high capacities of 264 and 359 mA h gele−1 at 0.1C when Li2S contents were 30 and 47 wt%, respectively.105 This method was further developed by exerting the principles of phase extraction, resulting in flexible electrodes based on an organodisulfide, phenyl disulfide (PDS, C6H5SSC6H5), and CNTs.106 This method allowed easy scaling up of active material loading to 19.3 mg cm−2. By combining the advantages of covalently bonded atomic sulfur and pliable CNT substrates, Bakenov and colleagues fabricated freestanding S/polyacrylonitrile (PAN)/CNT composite films, which exhibited an impressive initial capacity of 1457(585) mA h gsul(ele)−1 and 85% capacity retention after 260 cycles at 0.2C.107 To achieve dual-confinement of polysulfides and optimize electron/ion transport within the electrode, Li and Liu's group partially filled AAO-templated CNTs with a sulfur-rich copolymer in the hollow tube and then obtained a film consisting of interwoven sulfur-rich copolymer@CNT hybrid networks.108 The hybrid film served as a binder-free cathode, offering high capacities of 1300(826) and 700(445) mA h gsul(ele)−1 at 0.1 and 2.0C, respectively, and an excellent cycling stability with 98% capacity retention after 100 cycles at 1.0C. Flexible CNT papers or other macroscopic forms have also been widely employed in liquid-type Li–polysulfide batteries as Manthiram and others demonstrated,109–112 but liquid catholytes might not fit very well in flexible Li–S batteries because of the risk of electrolyte leakage and their flammable nature.
It should be noted that the above examples of CNT-based flexible sulfur cathodes mainly involve solution-processing procedures such as dispersion and vacuum filtration, which can barely be scaled up and require additional purifying/drying steps. To prevent the use of solvents, Giebeler and coworkers explored a facile fabrication of freestanding 3D interconnected carbon nanotube foams (CNTFs) through one-step spray pyrolysis and further employed such a CNTF as the sulfur host to build a mechanically flexible and binder-free sulfur cathode.113 With a moderate sulfur loading of 2 mgsul cm−2 (corresponding to a sulfur content of 40 wt%), a high sulfur utilization of 83% (a capacity of 1391/556 mA h gsul/ele−1) was obtained at 0.2C, while with a high sulfur loading of 7.1 mgsul cm−2 (corresponding to a sulfur content of 66 wt%), the flexible CNTF/S cathode exhibited an exceptional areal capacity of ∼9 mA h cm−2, equivalent to a gravimetric capacity of 1268(837) mA h gsul(ele)−1 at 0.1C. The investigation of solvent-free fabrication of CNT-based flexible sulfur cathodes paves the way for realizing high-energy-density and high-sulfur-loading Li–S batteries.
In summary, CNTs are especially suitable for constructing high-performance flexible sulfur-based cathodes owing to the following merits: (1) extraordinary electrical conductivity to wire up insulating sulfur-based active materials, (2) excellent chemical stability to withstand oxidative electrochemical corrosion that conventional Al foil current collectors normally undergo,53 (3) interconnected porous structure in CNT films to facilitate electrolyte permeation and ion transport, (4) interlinked structure to ensure good mechanical robustness and flexibility of CNT films, and (5) desirable and tailorable confinement of sulfur species through either internal hollow spaces or external physical/chemical anchoring sites. Further improvement in CNT-based flexible sulfur cathodes lies in the exploration of exotic CNT nanostructures and unique confinement strategies.
2.1.1.2. Graphene-based flexible cathodes. Graphene, since 2004, when it was first identified experimentally by Novoselov et al.,114 has been a rapidly rising star of materials science, and it is being developed at a truly relentless pace. Graphene is a strictly two-dimensional (2D), single-atom thin carbon film, exhibiting a plenty of new physics and chemistry.115 More specifically, aiming at its applications in electrochemical energy storage, its large specific surface area (theoretically being 2630 m2 g−1), stability within a wide range of potentials and temperatures, and high electron mobility/conductivity make graphene a wonder.116,117 Large-scale, low-cost, and environmentally friendly preparation of high-quality graphene is an urgent need and under booming development, mostly relying on efficient and scalable exfoliation methods.118,119 CVD-grown graphene has higher quality but, unlike CNTs, the yield of graphene through CVD is much lower.
Graphene is also an ideal flexible substrate. On one hand, perfect graphene consists of honeycomb-like sp2 carbon lattice and is the strongest existing material. On the other hand, its atomic thinness, in against lateral size of several to several tens micrometers, renders a large aspect ratio of 103–104, which is only enabled by quite a few cases of CNTs (e.g., VACNTs or SACNTs). Such in-plane toughness and sheet flexibility, in combination with ease of functionalization, dispersion, and assembly, render graphene an attractive and essential component in building flexible electrode materials. In forms of porous films,120,121 hydrogels/aerogels,122 and fibers,123 macroscopic graphene plays a vital role in flexible energy storage devices.
Graphene-based flexible sulfur cathodes were initiated by Wen and colleagues’ work.124 In this work, graphene sheets (GS) were dispersed in deionized water under ultrasonication, sulfur was precipitated from Na2S2O3, and GS/S paper was obtained through subsequent vacuum filtration (Fig. 6). The GS/S paper electrode was binder- and current-collector-free and flexible. With 67 wt% sulfur fraction, the GS/S paper electrode retained a specific capacity of 600(402) mA h gsul(ele)−1 after 100 cycles at 0.1C, corresponding to capacity retention of 83%.
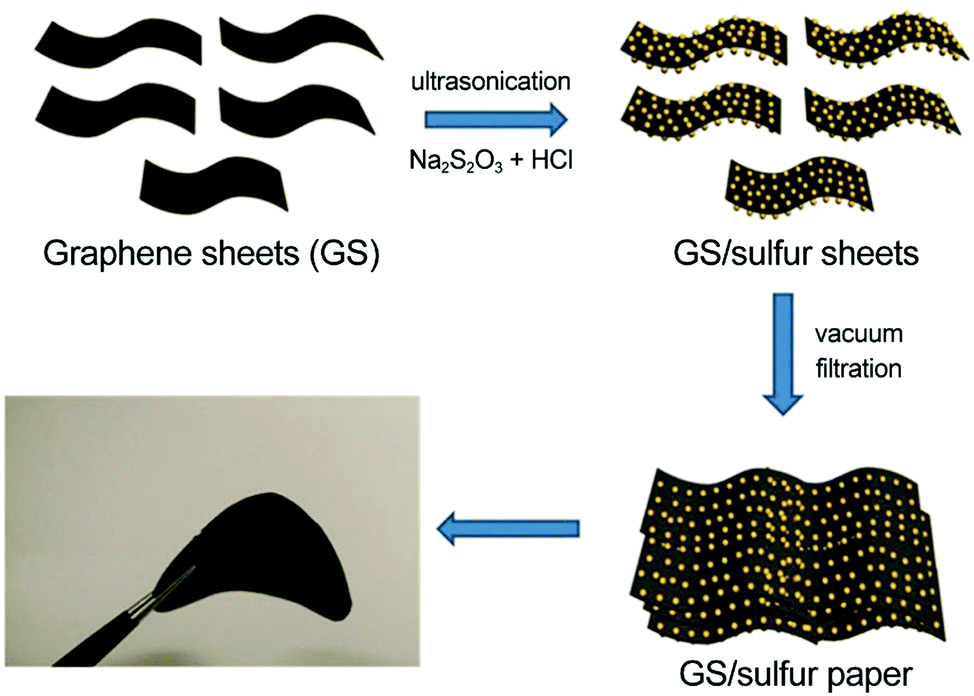 | ||
| Fig. 6 Vacuum-filtrated graphene-based flexible sulfur cathodes. Schematic and image of flexible GS/sulfur paper. Reproduced with permission.124 Copyright 2013, Royal Society of Chemistry. | ||
After that, various graphene-based flexible sulfur cathodes have been explored. One prototype is flexible graphene/sulfur composite film obtained through vacuum filtration or solvent evaporation. Graphene oxide (GO) or its reduced form (rGO) is preferred due to its abundant oxygen functionalities that stabilize colloidal interaction between GO or rGO sheets. Niu and coworkers reported a multistep fabrication of freestanding rGO–S composite films (Fig. 7a).125 Sulfur nanoparticles with a size of 9–22 nm were first deposited on GO via Na2S2O3 decomposition in acid solution (Fig. 7b). Subsequent immersion of a Zn foil in GO–S solution triggered synchronous reduction and assembly of GO–S into a compact thin film (Fig. 7c). Electrons were transferred from Zn to GO, resulting in Zn2+ cations and an rGO layer tightly attached to the Zn foil. On one hand, Zn2+ cations attracted negatively charged GO sheets in solution for further reduction and assembly. On the other hand, the electrostatic repulsion between rGO sheets was weakened to facilitate their layer-by-layer assembly. This synthetic method was smart and highly efficient in preparing ultraflexible rGO-based composite thin films. The nanostructured rGO–S paper could be arbitrarily rolled, bent, or folded (Fig. 7d), because of its extraordinary tensile strength of 68 MPa at a strain of 7.3%, corresponding to Young's modulus of 965 MPa. The electrochemical performance was also attractive as the rGO–S paper electrodes (56 wt%) exhibited high capacities of 1302(729) and 1003(562) mA h gsul(ele)−1 at 0.1 and 2.0C, respectively, as well as capacity retention of 66% after 500 cycles at 1.0C.
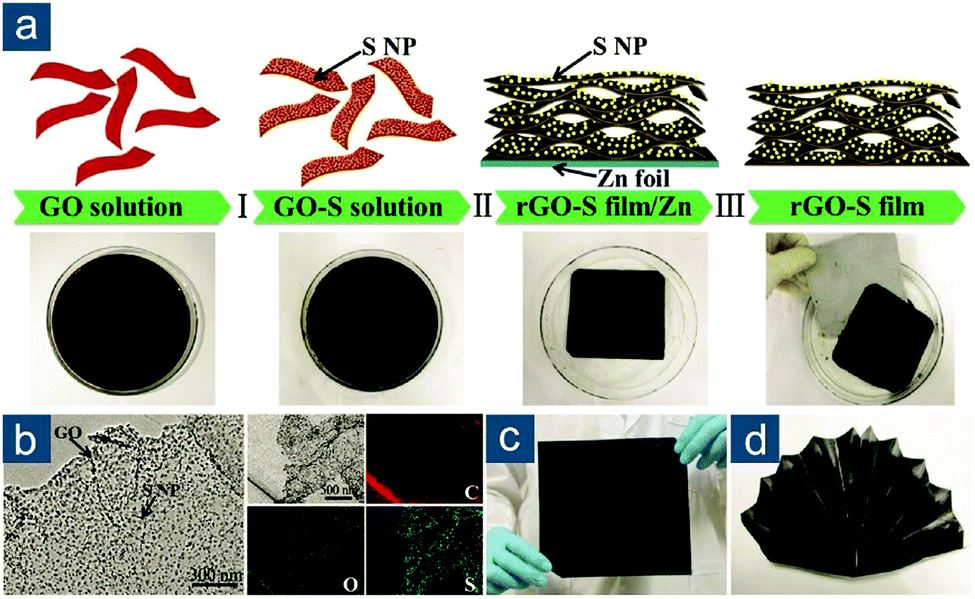 | ||
| Fig. 7 Self-assembled graphene-based flexible sulfur cathodes. (a) Schematic for the multistep fabrication of freestanding rGO–S films. (b) Transmission electron microscopy (TEM) images of GO sheet with sulfur nanoparticles (S NPs) and corresponding elemental mappings. Photographs of rGO–S film: (c) flat and (d) folded into a “folding fan”. Reproduced with permission.125 Copyright 2016, Wiley VCH. | ||
Such a multistep fabrication was further explored with rGO as the starting material.126 rGO sheets were first assembled into water-solvated rGO films by vacuum filtration, and the rGO films were immersed in ethanol to replace water, prior to adding S/CS2 mixture for sulfur impregnation. Solvation of the rGO films by water was crucial as it created abundant internal spaces to serve as continuous ion channels and allow facile sulfur loading via solution filtration. The rGO–S composite films resulting from freeze drying could be directly used as flexible nanostructured cathodes, displaying a desirable cycling stability as capacity retention of 65% after 300 cycles at 0.1C.
Unlike graphene sheets, graphene nanoribbons are regarded as the bridge between CNTs and graphene, possessing many intriguing physicochemical properties due to the dimensional confinement effects at the edge.127,128 Therefore, rGO nanoribbons (rGONRs), obtained from longitudinal unzipping of CNTs, have also been employed for flexible sulfur-based cathodes as Liu et al. demonstrated.129In situ generated sulfur anions were introduced to reduce GONRs and load sulfur simultaneously. Evaporation-induced self-assembly further guided the formation of S/rGONR paper, in which sulfur was chemically bonded with rGONR through C–S bonds and physically trapped by compact assembly. As a result, the flexible S/rGONR cathode exhibited good cycling performance at 3.0C, with a capacity of 455(248) mA h gsul(ele)−1 and flexibility retained after 300 cycles.
To prevent tedious vacuum filtration or solvent evaporation, another synthetic strategy toward graphene-based flexible sulfur cathodes was proposed: cutting electrode pieces from graphene/sulfur hydrogels/aerogels. Such a synthetic scheme was first explored by Zhou et al.130 A facile one-pot approach using S/CS2/alcohol mixed solution was proposed to reduce GO and anchor sulfur nanocrystals on interconnected fibrous graphene during solvothermal process. CS2 was employed to dissolve sulfur, while alcohol improved the miscibility of S/CS2 and GO aqueous solutions, synergistically facilitating the formation of homogenous graphene–S (G–S) hybrids. The oxygen functional groups in GO had profound influences on (1) gelation of G–S hybrids and (2) polysulfide immobilization via S–O/C–S bonds. As a result, the G–S hybrids possessed excellent electrical conductivity of ∼6.3–7.5 S cm−1 as a function of sulfur fraction from 71 to 55 wt%, shortened ion diffusion paths within sulfur nanocrystals, and strong anchoring of polysulfide intermediates. These favorable attributes rendered G–S hybrid electrodes (63 wt% sulfur) with high capacities of 1160(731) and 670(422) mA h gsul(ele)−1 at 0.3 and 1.5 A gsul−1 (∼0.18 and 0.9C), respectively, as well as excellent cycling performance.
To prepare graphene-based flexible sulfur cathodes from hydrogels/aerogels, elaborate multistep synthesis were developed, as demonstrated by Mai and Xu's group.131 After a series of synthetic steps, 3D graphene sponges with sulfur nanoparticles (3DGSs) were obtained mainly through reduction-induced self-assembly, which was facile and scalable. 3DGSs can be cut and pressed, rendering highly integral and flexible cathodes with superior cycling performance. At 1.5 A gsul−1 (∼0.9C), a reversible capacity of 580(316) mA h gsul(ele)−1 was maintained after 600 cycles, corresponding to capacity retention of 78%.
Multistep process allows precise control over each step but complicates fabrication. Thus, simple and scalable synthesis of graphene/sulfur composite monoliths is appreciated toward practical applications. To meet this requirement, Wang et al. have exploited a simple but efficient synthesis process for fabricating S–rGO paper materials that can serve directly as binder-free and freestanding electrodes.132 In the synthesis, raw materials, GO and commercial sulfur nanoparticles were mixed to form an aqueous suspension. After freeze drying, the suspension was converted into a foam-like monolithic S–GO precursor. Subsequent heat treatment, coupled with compression, resulted in a piece of S–rGO paper, which was highly pliable. Freeze drying induced highly porous structure in the S–rGO paper, and heat treatment simultaneously restored π-conjugated domains of graphene and made sulfur aggregates spread on conductive graphene sheets. Owing to these structural advantages, S–rGO paper electrodes exhibited high capacities of 1317(830) and 889(560) mA h gsul(ele)−1 at the 2nd and 200th cycle, respectively, when a low current density of 0.1 A gsul−1 (∼0.06C) was applied. The outstanding performance was retained at other current densities.
This synthetic methodology was also extended to fabricate flexible nano-Li2S/rGO paper electrodes, significantly overcoming the obstacles that conventional slurry-based approaches usually encounter for practical applications of Li2S-based cathodes.133 Similarly, rGO paper was first prepared via freeze drying and heat treatment. Li2S was loaded on the rGO paper by drop coating Li2S/anhydrous ethanol solution and annealing at 200 °C under argon protection (Fig. 8). rGO had abundant adhesive sites for Li2S nucleation and served as physical barriers to prevent Li2S agglomeration. Nano-Li2S confined in the rGO matrix, with a size of 25–50 nm, was proven to exhibit remarkable electrochemical activity (Li2S utilization of 96.0 and 51.2% at 0.1 and 7.0C, respectively) and good cycling performance. This was attributed to extraordinary electrical conductivity (307 S cm−1), structural flexibility to withstand volume change, and strong solvent adsorptivity of rGO paper. Compared to conventional slurry-based cathodes, such a binder- and current-collector-free electrode also substantially reduced unnecessary weight with an average capacity of ∼615 mA h gele−1.
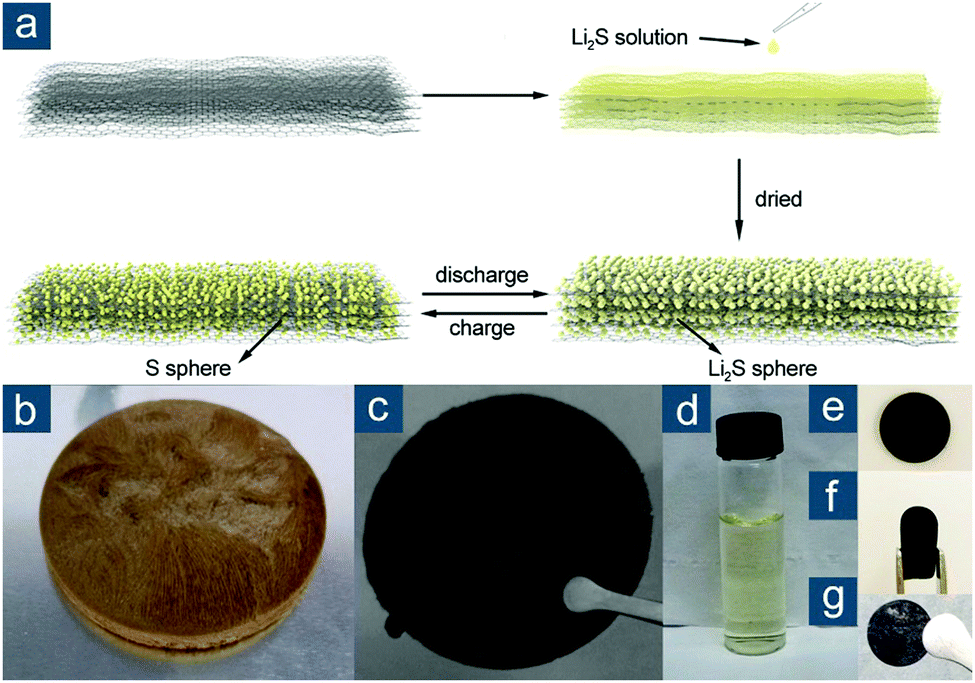 | ||
| Fig. 8 Monolith-derived graphene-based flexible sulfur cathodes. (a) Schematic for the preparation of nano-Li2S/rGO paper and its structural changes during cycling. Photographs showing (b) GO gel, (c) rGO paper, (d) Li2S/anhydrous ethanol solution, and flexible rGO disks in (e) flat and (f) bent states, as well as (g) being drop coated with nano-Li2S. Reproduced with permission.133 Copyright 2015, American Chemical Society. | ||
Analogous to heteroatom-doped CNTs, doped graphene also improves the entrapment of polysulfides and therefore is very promising for constructing flexible sulfur-based cathodes. For example, Han et al. demonstrated that flexible nitrogen-doped graphene (NG) paper promoted the performance of Li-dissolved polysulfide batteries.134 Zhou et al. prepared a series of 3D graphene aerogels through hydrothermal reduction of GO with or without elemental dopants.135 Li2S was further coated on freestanding rGO or doped graphene slices through drop coating. The as-obtained 3D Li2S/doped graphene aerogel-based electrodes possessed several preferable attributes including porous architecture, crosslinked network, uniform coating of Li2S, and strong binding affinities of heteroatoms toward Li2S/polysulfides. Li2S/NG showed the most promising electrochemical performance due to the high doping level of nitrogen (9.8 at%), while boron-doped graphene, with a small amount of boron (1.1 at%), also provided considerable enhancement since boron and its derivative dopants were theoretically predicted to be stronger anchoring sites than nitrogen in the presented work.
Besides the direct synthesis of flexible graphene/sulfur composite materials, an alternative concept to make sulfur cathodes flexible was proposed by Li and Cheng's group (Fig. 9a).136 By simply laminating conventional sulfur/carbon slurry between two pieces of flexible, highly conductive graphene films (with an electrical conductivity of 830 S cm−1) with one self-supported (GCC, Fig. 9b) and the other one coated on a polymer separator (G-separator, Fig. 9c), an integrated graphene/sulfur/graphene sandwich electrode was fabricated. Note that the sulfur/carbon slurry still consisted of conductive agents and binders, so the electrode was not “binder-free” but truly flexible. Such a concept circumvented the difficulty in preparing graphene/sulfur composite monoliths. More importantly, the fabrication was fully compatible with conventional processes in battery industry, and both graphene foils and the G-separator could be continuously produced. All these attributes rendered the design applicable for low-cost and large-scale practical applications. The electrochemical performance of sandwich electrodes was also impressive. At 1.5 A gsul−1 (∼0.9C), an initial capacity of ∼970(∼390) mA h gsul(ele)−1 was obtained and retained 70% of initial capacity after 300 cycles.
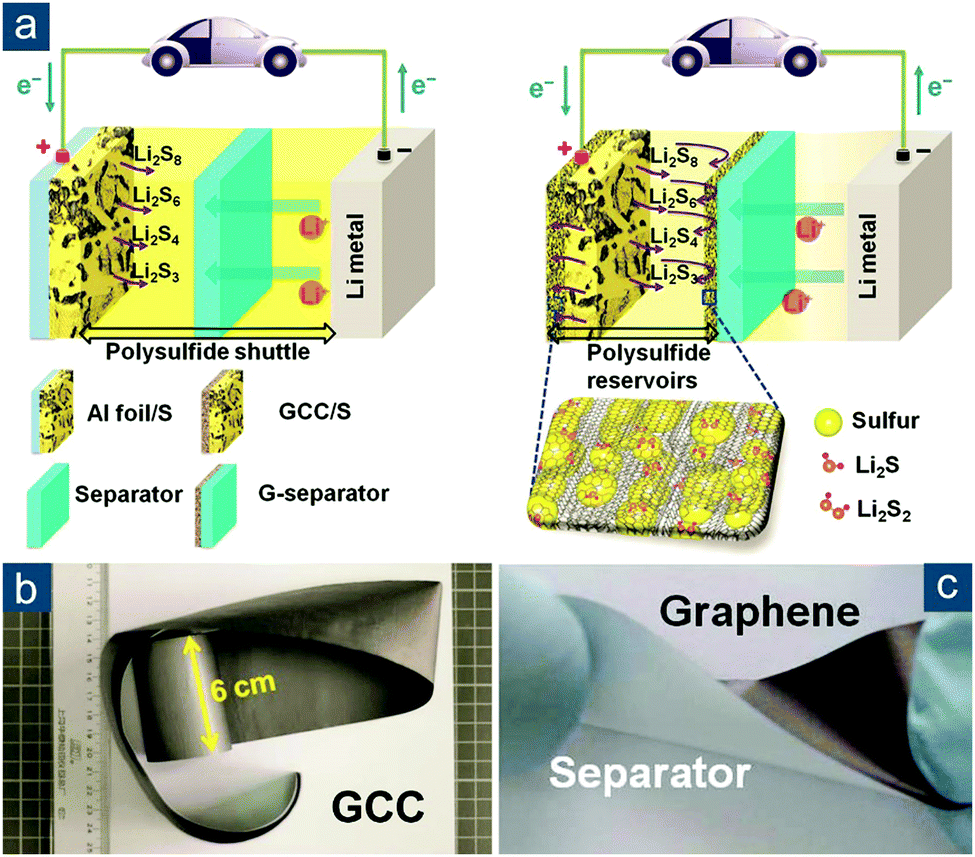 | ||
| Fig. 9 Sandwiched graphene-based flexible sulfur cathodes. (a) Schematic of Li–S batteries (left) with a conventional electrode configuration (Al foil + polymer separator) and (right) a sandwiched flexible electrode configuration (graphene current collector (GCC) + graphene-coated separator (G-separator)). Images of large pieces of flexible (b) GCC and (c) G-separator. Reproduced with permission.136 Copyright 2013, Wiley-VCH. | ||
In summary, as an intrinsic conductive and flexible material, graphene is of both scientific and technological interests and impacts for flexible sulfur-based electrodes. The superiority of graphene includes (1) large exposed external surface area (practically 400–1200 m2 g−1) to freely access electrons/ions, which is superior to most that of available MWCNTs (100–400 m2 g−1), (2) residual oxygen functional groups, especially in rGO, to ease manipulation and assembly in solution and provide chemical affinity to polar sulfur species, and (3) highly porous and robust graphene network to sustain electrode integrity. In comparison with CNTs, graphene has additional unique traits because of its inherent 2D-material feature: (4) more conformal coating on active particles to allow “plane-to-point” electrical contact, (5) sheet flexibility to accommodate volume fluctuation, and (6) large extended 2D plane to provide steric barrier or structural direction for sulfur materials.
However, it should be noted that the rich functionality on graphene not only brings advantages in dispersing graphene and strengthening macroscopic graphene, but also compromise on electrical conductivity. In many cases, graphene possesses inferior conductivity to CNTs. The reduction method for GO or other precursors should be carefully engineered to balance electrical and mechanical properties of graphene-based flexible electrodes.137 For example, increase in annealing time of GO films is obviously beneficial for restoring graphitic structure, but it unfortunately renders as-obtained rGO films inflexible and brittle. Therefore, choosing whether GO or rGO as the starting material, arranging the sequence of reduction and assembly, and selecting suitable reducing agents will have profound influence on both mechanical and electrochemical performance. Optimal structure, assembly, and functionality of graphene, as well as the mechanism of how graphene interacts with each other and various sulfur species, are still elusive, thus demanding for future progress on fundamental understanding and advanced synthetic strategies toward high-performance graphene-based flexible sulfur electrodes.
2.1.1.3. Commercial carbon cloth/paper-based flexible cathodes. Commercial carbon cloth or paper is a well-known flexible, conductive substrate, which is usually interwoven by carbon fibers. Although the history of carbon fibers can be traced to the late nineteenth century when Thomas Edison used carbon filaments in light bulbs, research on modern carbon fibers only started in 1959.138 After decades of development, carbon fibers now include a variety of highly standardized carbon products that have achieved global commercial success. Compared to low-dimensional carbon nanomaterials such as CNTs and graphene, the most outstanding advantage of commercial carbon cloth/paper is its much lower cost, wider availability, and stable quality. Besides, carbon cloth/paper has considerable electrical conductivity and extraordinary strength. Therefore, flexible electrodes based on commercial carbon cloth/paper have been widely explored for electrochemical energy storage.139,140
In Li–S battery research, Aurbach and colleagues demonstrated the first binder-free cathode using an activated carbon fiber cloth (ACFC) with sulfur impregnated in 2011.141 Stable capacity obtained was over 800 mA h gsul−1 after 80 cycles at 0.15 A gsul−1 (∼0.09C), but the relatively low sulfur content (∼33 wt%) limited overall capacity (∼260 mA h gele−1). Soon after that, Zhang et al. also employed commercial ACFC to enable electrochemical utilization of sulfur with an areal loading of 13 mgsul cm−2 but a moderate mass fraction of ∼48 wt%.142 Despite these successful demonstrations of commercial carbon cloth/paper in Li–S batteries, they have merely proven to be “binder-free” or “freestanding” sulfur cathodes, instead of truly “flexible” ones. A high sulfur content can barely be achieved on a sole carbon cloth/paper host. The main reason is the limited specific surface area of commercial carbon cloth, in which the diameter of a single carbon fiber is over 10 μm. Even after activation, the generated structure mainly contains tortuous micropores with a size of <2 nm, preventing high sulfur content and smooth ion diffusion.
To overcome the aforementioned drawbacks of commercial carbon cloth, Chung et al. investigated the feasibility of employing commercial buckypapers as flexible encapsulation and polysulfide reservoirs with sulfur/carbon slurry being sandwiched in between.143 The buckypaper had a long-range fibrous architecture composed of a CNF skeleton with curved CNTs firmly attached and therefore had excellent ductility and flexibility. After being rolled or folded, the recovered sandwiched electrode (B/S/B) displayed no delamination, indicating good adhesion between sulfur fillings and buckypapers (Fig. 10a–d). Considering the mass of two buckypapers, the actual sulfur content in B/S/B cathodes with an areal loading of 3.2 mgsul cm−2 was estimated to be ∼37 wt%. The robust, ultratough, flexible B/S/B cathodes exhibited an initial capacity of 1010(374) mA h gsul(ele)−1 at 0.2C and retained 51% of initial capacity after 400 cycles. Even after being rolled and folded, the B/S/B cathodes still demonstrated similar cyclability (Fig. 10e). Increasing the areal loading of in-between sulfur to 5.1 mgsul cm−2 led to higher mass fraction of sulfur and areal capacity. He et al. adopted commercial ACFC as the flexible substrate for sulfur slurry infiltration, yielding a foldable sulfur cathode with a high sulfur loading of 3.0–4.5 mgsul cm−2 and a capacity of 979 mA h gsul−1, preserved after 200 cycles at 0.2C.144 Regrettably, the areal density of ACFC was not given; thus, it was difficult to fairly evaluate the performance with regard to the whole electrode weight. According to the data reported by Elazari et al.141 and Zhang et al.,142 the areal density of an ACFC is around 13–14 mg cm−2. Thus, the capacity based on the whole electrode would likely be lower than 300 mA h gele−1 unless the weight of ACFC can be decreased by half.
 | ||
| Fig. 10 Commercial carbon cloth/paper-based flexible sulfur cathodes. Photographs of (a and b) cycled and (c and d) uncycled B/S/B cathodes under and after rolling/folding. (e) Battery performance of the cell employing rolled/folded cycled B/S/B cathode (same as in c and d). Reproduced with permission.143 Copyright 2015, Wiley-VCH. | ||
In summary, unlike broad applications in flexible LIBs, there are few examples of commercial carbon cloth/paper-based flexible cathodes for Li–S batteries. Low cost, mature fabrication technology, excellent electrical conductivity, and high mechanical strength constitute the main advantages of commercial carbon cloth/paper for use in flexible electrochemical energy storage. In particular, wide availability makes commercial carbon cloth/paper notably competitive for practical applications. However, its low specific surface area turns to be the major drawback because sulfur-based materials are especially insulating among existing electrode materials and demand large conductive surface for efficient redox reactions. Therefore, either additional high-surface-area conductive agents are necessary or the sulfur content should be maintained low. Both of these obviously reduce the overall capacity considering the whole electrode.
Several exfoliation strategies have been reported to improve the areal or volumetric specific capacitances of flexible supercapacitors based on carbon cloths,145 but the increase in specific surface area is probably insufficient for facilitating high-capacity sulfur cathodes. To enhance commercial carbon cloth/paper-based flexible cathodes, rational hybridization with other functional materials is more feasible to maximize its advantages of long-range conductivity and flexibility while circumventing the issue of low surface area. Corresponding contents will be summarized and discussed in (Section 2.1.1.5).
2.1.1.4. Carbonized polymer-based flexible cathodes. Polymer is typically a soft material that can be deformably or structurally altered at a room-temperature thermal energy scale, and it is inherently flexible due to its unique mesoscopic physical structure. On the other hand, polymers are normally polymerized from organic monomers and therefore many of them can be easily carbonized into conductive carbon materials at appropriate temperature and atmosphere. By inheriting the flexibility from polymers and electrical conductivity from carbon materials, carbonized polymer presents to be a highly tunable and attractive prototype electrode material for flexible electrochemical energy storage devices.
Carbonized polymer-based flexible cathodes for Li–S batteries can be broadly classified into two classes based on their difference in fabrication methods. One is the flexible cathode comprising carbonized electrospun polymer nanofibers (PNFs). The other are those carbonized from an existing or preformed flexible polymer matrix (FPM).
According to (Section 2.1.1.1), CNTs have been regarded as a superior building block for flexible electrodes owing to their fascinating features because of 1D nanostructures. However, bulk application of CNTs is always impeded by the controllable mass production of CNTs and the complexity of their assembly into macroscopic electrodes. On the contrary, electrospinning, a drawing process based on electrostatic interactions, seems to provide the simplest way to generate ultrathin fibers that are exceptionally long in length, uniform in diameter, and diverse in composition.146 More importantly, electrospinning is a continuous process and therefore quite suitable for high-volume production. Basically, 1D nanofibers are formed via uniaxial stretching or elongation of a viscoelastic jet derived from a polymer solution or melt during a typical electrospinning process. Underlying formation mechanisms, as well as approaches for structural and morphological control, have been comprehensively summarized in Li and Xia's review article.146 The applications of electrospun 1D nanofibers in electrochemical energy storage have also been reviewed previously.147,148
Focusing on Li–S batteries, electrospinning has already widely been employed to fabricate electrode, electrolyte, and separator materials. Earliest works can be traced to 2011, when Tsutsumi et al.149 and Ji et al.150 reported electrospun sulfur fibers and electrospun porous CNF–S composites, respectively. In particular, the latter one demonstrated that electrospinning was quite efficient in generating PNFs, which can be subsequently carbonized into 1D CNFs with desirable electrochemical performance. Like in the fabrication of commercial carbon fibers, the most widely employed polymer precursor is PAN.151 Other precursors such as polystyrene (PS) and poly(methyl methacrylate) (PMMA) can also be introduced to generate different porous structures in carbonized electrospun PNFs. Owing to multiple advantages in controlling an ideal 1D morphology, internal porosity, electrical conductivity, and composition during both electrospinning and carbonizing procedures, carbonized electrospun PNFs show great potential for flexible electrodes in Li–S batteries. More importantly, these PNFs can be directly assembled into flexible mats on various substrates after being extruded, so it is unnecessary to further filtrate them or trigger their self-assembly by evaporating a large amount of solvents. This saves time and energy, making this approach a prospect for industrialization.
Yu's group developed a series of electrospun composite nanofibers for flexible Li–S and analogous alkali metal–chalcogen batteries.152–157 In 2014, they reported the first flexible Li–S battery cathode based on carbonized electrospun PNFs.152 The fabrication procedures contained step-by-step (1) co-dissolution of CNTs with PAN in precursor solution, (2) electrospinning of the precursor, (3) stabilization and carbonization, (4) filtration with potassium hydroxide (KOH), and (5) activation at 800 °C, resulting in a porous flexible CNF–CNT composite (PCNF–CNT) film (Fig. 11a). The introduction of CNTs and KOH activation rendered resultant composite nanofibers with higher electrical conductivity and more abundant micropores than pristine PCNFs and CNFs, respectively. Meanwhile, the ideal electrospun 1D morphology was maintained, endowing as-obtained film electrodes with good mechanical flexibility and a 3D interconnected framework with shortened electron/ion paths. Sulfur could be physically confined in these micropores after thermal impregnation, exerting a unique “quasi-solid-state” electrochemistry in carbonate-based electrolytes.158–161 At a current density of 0.05 A gsul−1 (∼0.03C), the flexible S/PCNF–CNT cathode retained a reversible capacity of 637(255) mA h gsul(ele)−1 after 100 cycles, much higher than that of S/PCNF (334/130 mA h gsul/ele−1). The rate performance of S/PCNF–CNT was also much better than its counterpart without CNT addition. The fabrication is quite scalable and extendable for making high-performance flexible electrodes from carbonized electrospun PNFs.
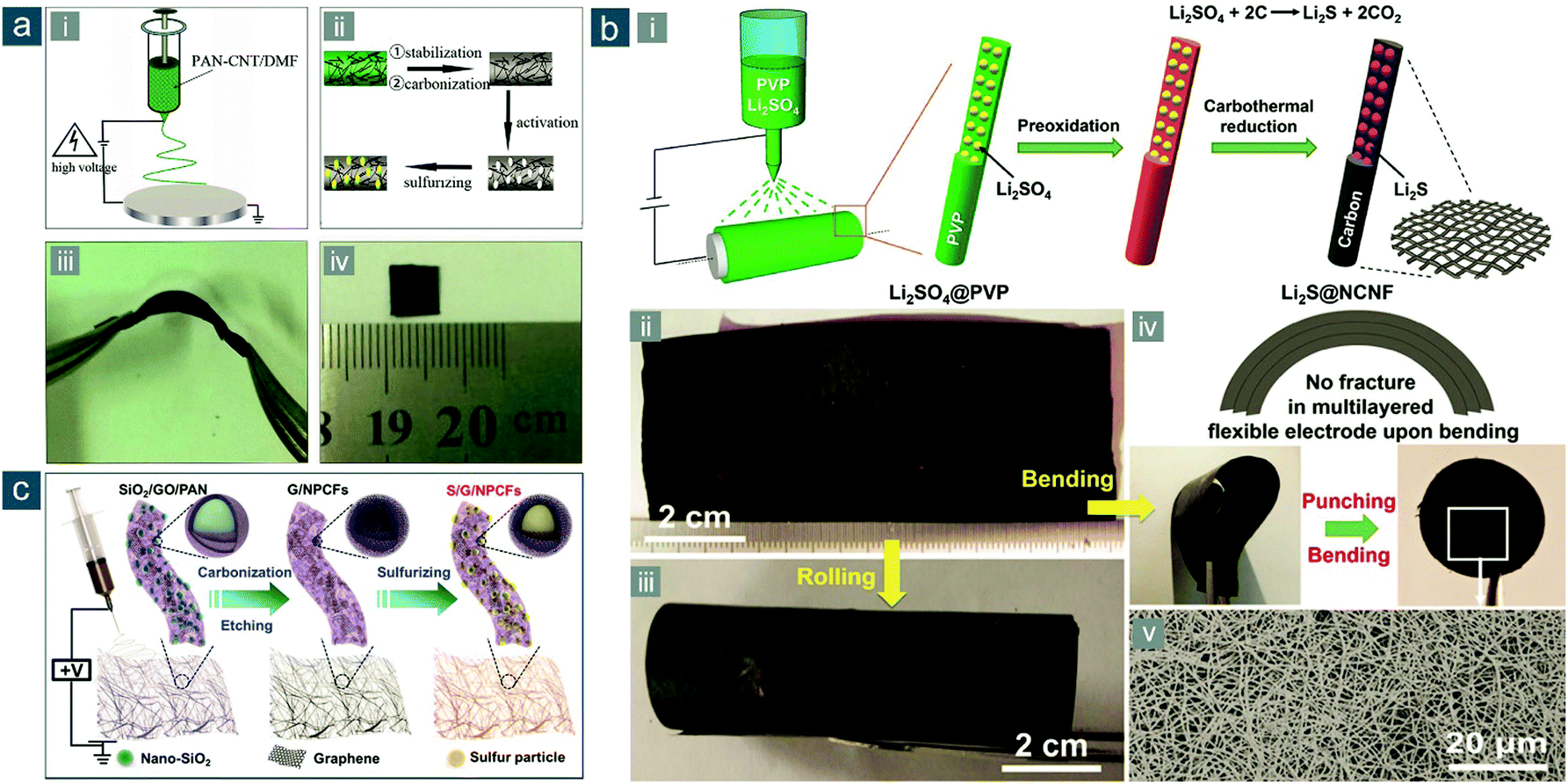 | ||
| Fig. 11 Flexible sulfur cathodes based on carbonized electrospun PNFs. (a) Electrospun PCNF–CNT film: (i and ii) schematics for the synthesis of sulfur/PCNF–CNT electrode and (iii and iv) photographs of the flexible films. Reproduced with permission.152 Copyright 2014, Royal Society of Chemistry. (b) Electrospun Li2S@NCNF paper: (i) schematic for the synthesis of flexible Li2S@NCNF paper electrodes and photographs of Li2S@NCNF in (ii) flat and (iii) bent states, and (iv) a multilayered and punched disc electrode; (v) SEM image of Li2S@NCNF. Reproduced with permission.164 Copyright 2017, Wiley-VCH. (c) Schematic for the fabrication of sulfur/G/NPCF electrode through electrospinning. Reproduced with permission.166 Copyright 2017, Royal Society of Chemistry. | ||
After that, methods of sulfur loading, kinds of polymer precursors, and properties of other functional components have been regulated during fabrication for further enhancing the performance of flexible cathode based on carbonized electrospun PNFs. To replace the tedious melt-diffusion process, Fu et al. designed a self-inhibiting, gradient sulfur cathode via sulfur vapor condensation within a binder-free electrospun CNF web.162 The unique gradient sulfur distribution resulted in different functional regions in the cathode, enabling a high sulfur loading (2.6 mgsul cm−2), a high sulfur content (65 wt%), and a stabilized capacity of 700(455) mA h gsul(ele)−1 after 20 cycles at 0.1 A gsul−1 (∼0.06C). Goodenough and coworkers explored the use of vulcanized rubber instead of elemental sulfur as the active material and loaded it on a flexible electrospun CNF network, demonstrating a high rate capability as a capacity of ∼880 (∼370) mA h gsul(ele)−1 after ∼25 cycles at 5.0C.163
Polymer precursors can be altered to achieve high heteroatom-doping levels and uniform distribution of embedded particles. Qiu and colleagues reported a facile and low-cost synthetic strategy for flexible Li2S-nanoparticle-decorated nitrogen-doped CNF (Li2S@NCNF) paper electrodes through electrospinning of polyvinylpyrrolidone (PVP)/lithium sulfate (Li2SO4) solution and subsequent carbonization coupled with carbothermal reduction (Fig. 11b-i).164 Large-area and robust paper electrodes were obtained, which could be rolled and stacked (Fig. 11b-ii–iv), consisting electrospun nanofibers with ultrafine Li2S nanoparticles (<10 nm) homogeneously embedded (Fig. 11b-v). Owing to the small grain size of Li2S and chemical affinities of NCNF to polysulfides/Li2S, the flexible Li2S@NCNF electrode, with a high Li2S loading of 3.0 mg cm−2, exhibited a high initial capacity of 720(364) mA h gLi2S(ele)−1 at 0.2C and 83% capacity retention after 50 cycles. Moreover, by stacking Li2S@NCNF papers, the Li2S loadings could be easily scaled up to 9.0 mg cm−2 without significant drop in performance.
Besides single-phase polymer precursors, multiphase polymer precursors possibly endow electrospun nanofibers with some fascinating structures and properties. Lou and coworkers explored a dual-phase polymer precursor of PAN and PS for electrospinning fabrication of lotus root-like multichannel carbon (LRC) nanofibers.165 During the carbonization, PS was thermally decomposed into volatile compounds, leaving parallel channels (average diameter of 60 nm) within the shell of carbonized PAN. The large void offered by these parallel channels allowed higher sulfur mass content, while the shell contained abundant micropores, enabling confinement of small sulfur molecules or serving as a physical barrier to polysulfide dissolution. After being loaded with sulfur at 300 °C, the freestanding LRC/S mat was further coated with a thin, compact, and pliable layer of ethylenediamine-functionalized rGO (EFG), forming a ‘pie-like’ paper electrode (denoted as LRC/S@EFG). The LRC/S@EFG electrode exhibited remarkable electrochemical performance, including a high sulfur loading (3.6 mgsul cm−2), a high sulfur content (68 wt%), a high capacity (1314/894 mA h gsul/ele−1) at 0.2C, and high stability (78% capacity retention after 200 cycles). Moreover, the sulfur loading could be easily scaled up to 10.8 mgsul cm−2 by stacking the paper electrodes, with no obvious performance degradation.
The introduction of functional additives and fillers during electrospinning is a facile and tunable strategy for adjusting structure and properties of carbonized nanofibers. Very recently, Song et al. proposed an elaborate synthesis of graphene-embedded graphitic nitrogen-doped mesoporous CNFs (G/NPCFs) and prepared a high-strength, flexible cathode consisting of these NPCFs (Fig. 11c).166 In their synthesis, rGO and nano-silica (SiO2) were added in the precursor solution, providing multiple and synergistic effects on nanofiber strength and graphitization degree. As a result, the S/G/NPCF electrode exhibited excellent flexibility and foldability, as well as outstanding electrochemical performance especially at high rates. At 5.0C, a high initial capacity of 558(296) mA h gsul(ele)−1 was obtained and 76.5% capacity retention after 500 cycles, which is quite impressive and much better than those of control samples without rGO and/or nano-SiO2. Yang et al. employed much larger SiO2 particles as the template during electrospinning synthesis of PAN-based CNFs and obtained distinctive N-doped hollow carbon bead strings for flexible sulfur cathodes with a high sulfur content of 76 wt%, which delivered a reversible capacity of 1249(949) mA h gsul(ele)−1.167 These works indicate that engineering sophisticated additives or fillers is very attractive for their ability to fabricate various electrospun micro-/nanostructures that largely benefit electrochemical performance.
Besides carbonized electrospun PNFs, another flexible electrode prototype is carbonized polymer in form of pre-formed flexible monoliths like paper, fabric, foam, and elastomer. This kind of flexible electrode is denoted as carbonized FPM, with obvious advantages such as simple fabrication, low cost, and diverse architectures. To fully exert these advantages while preparing flexible electrode materials, the selected FPM should be naturally abundant, economically cheap, texturally carbonizable, morphologically fibrous, and mechanically elastic.
One of the most widely explored materials for FPM carbonization is cellulose. Cellulose, defined as a polysaccharide consisting of a linear chain of hundreds to thousands of D-glucose units connected by β(1 → 4) linkages, is the most abundant and renewable polymer on earth, with approximately 75–100 billion tons produced annually worldwide.168 It is an essential component of lignocellulosic plant. Some species of bacteria are also able to secrete it, and this is denoted as bacterial cellulose (BC).169 With almost infinite intramolecular hydrogen bonding, single cellulose chains can be assembled together to form ultralong and very strong fibrils, which are major constituents in many natural plant fibers such as cotton and hemp.
Cotton is regarded as the purest natural form of cellulose. Cotton fibers are fairly uniform in diameter (12–20 μm) and generally possess a length of several centimeters, corresponding to a large aspect ratio of ∼103. Hence, it can be easily spun into various flexible fabrics. Further carbonization of these fabrics results in flexible conductive electrode materials. Miao et al. first adopted a binder-free carbon fiber cloth-S electrode with a high sulfur loading (6.7 mgsul cm−2) and a high capacity (1156/673 mA h gsul/ele−1) at 0.3 mA cm−2 (∼C/40), in which carbon fiber cloth was prepared from artificially woven cotton threads in compact bundles.170 Probably due to the limited specific surface area of carbonized cotton fiber, sulfur fully covered the conductive surface during melt diffusion, leading to decrease in electrical conductivity from 1.58 to 0.37 S cm−1 and therefore an ultralow cyclic rate was applied in this demonstration.
To overcome this likely drawback in surface area, Fang et al. developed a simple, low-cost, and scalable approach based on hollow carbon fiber foams (HCFFs) carbonized from more fluffy natural cotton instead of tightknit cotton cloth (Fig. 12a-i).171 Through slurry coating, sulfur, homogeneously mixed with high-surface-area conductive agents containing carbon blacks and CNTs rather than in pristine state, was infiltrated in the 3D interconnected, long-range conductive, flexible HCFF network (Fig. 12a-ii), allowing exceptionally high sulfur loadings from 6.2 to 21.2 mgsul cm−2. The use of extra conductive agents gave a high mass fraction for sulfur in the whole electrode (74 wt%) with a sulfur loading of 21.2 mgsul cm−2, but the electrical conductivity was not sacrificed drastically, corresponding to 46% of decrease in square resistance (from 12 to 6.5 Ω □−1). As a result, an impressive capacity of 1100(814) mA h gsul(ele)−1 was obtained at 3.55 mA cm−2 (0.1C), corresponding to an areal capacity of 23.3 mA h cm−2; the cycling stability was also promising with ∼70% capacity retention after 150 cycles, which was ascribed to substantial electrolyte uptake by HCFFs to improve polysulfide immobilization. Owing to its superiority in loading and localization of a huge amount of sulfur/lithium (poly)sulfides, such a flexible carbonized cotton electrode was also employed as scaffolds for statically and dynamically stable Li–polysulfide batteries, as Chung et al. demonstrated.172
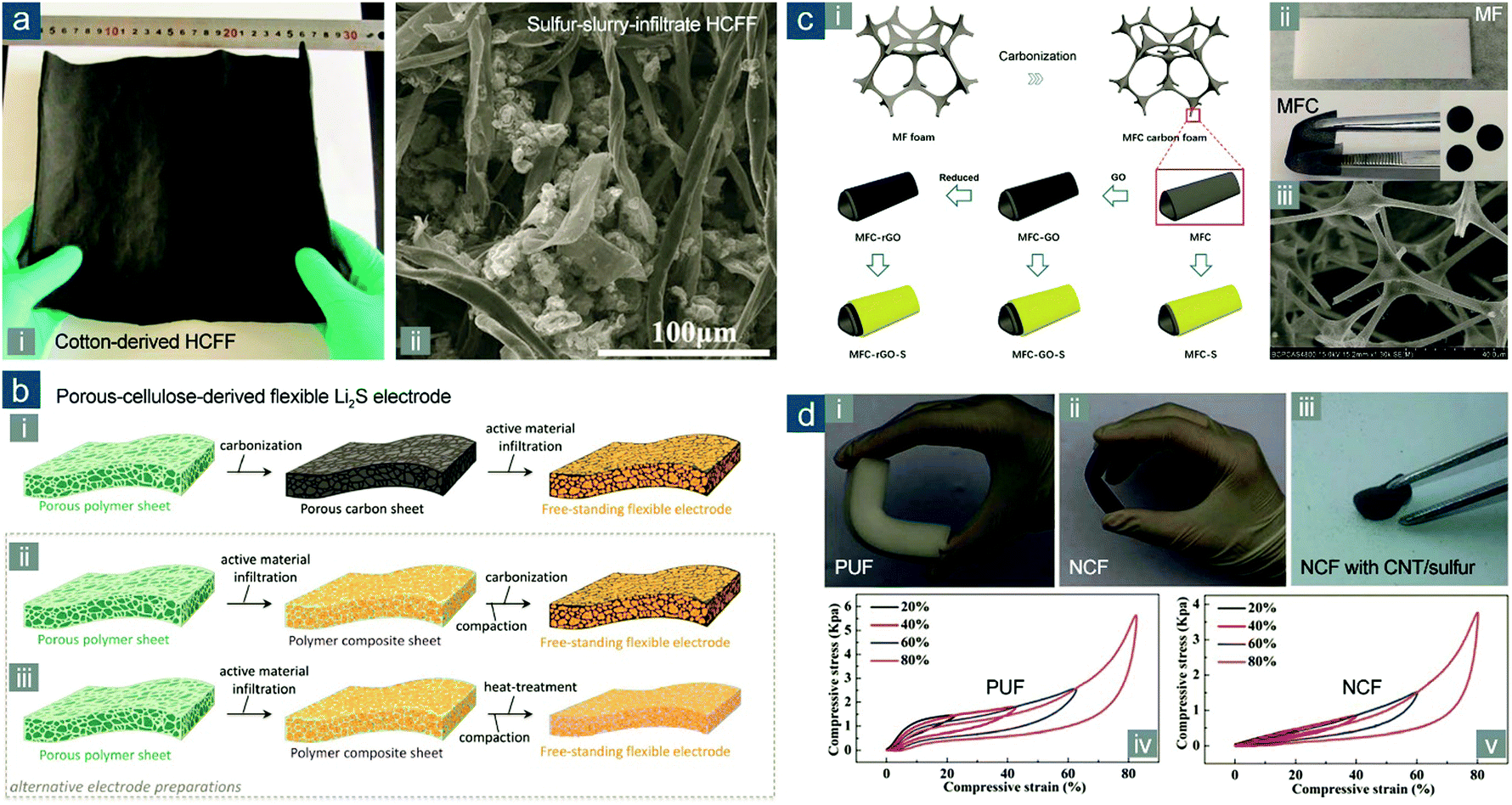 | ||
| Fig. 12 Flexible sulfur cathodes based on carbonized FPMs. (a) Cotton-derived HCFF: (i) photograph of large-area HCFF and (ii) SEM image of HCFF with sulfur slurry infiltration. Reproduced with permission.171 Copyright 2016, Wiley-VCH. (b) Schematic for the preparation of flexible electrodes from porous polymer sheets: (i) conventional and (ii and iii) alternative processes. Reproduced with permission.175 Copyright 2016, Wiley-VCH. (c) MFC-based flexible electrodes: (i) schematic for the fabrication; (ii) images of MF and MFC; (iii) SEM image of MFC. Reproduced with permission.180 Copyright 2016, Royal Society of Chemistry. (d) PUF-based flexible electrodes: photographs of bent (i) PUF, (ii) NCF, and (iii) NCF with CNT/sulfur composites loaded; compressive stress–strain curves of (iv) PUF and (v) NCF. Reproduced with permission.181 Copyright 2016, Royal Society of Chemistry. | ||
Besides cotton, paper is another typical flexible material derived from moist fibers of cellulose pulp, but it is not as pure as cotton, because many additional chemicals are involved during papermaking.173 Commercial wool pulp-based paper is less superior in both strength and durability to cotton paper/textile, but also notably less dependent on fibril length of cellulose for gaining essential mechanical properties. This makes it more economically efficient by using cheaper raw materials. More importantly, paper materials are always thinner and lighter than cotton cloths, holding greater promise for miniaturized, portable, and wearable devices. In this regard, Hu and Cui provided a perspective on energy and environmental nanotechnology in conductive paper and textiles, including flexible electrochemical energy storage.174
Aiming at efficiently preparing carbonized FPMs, Yushin and coworkers proposed an alternative synthetic scheme where active materials were first infiltrated on porous polymer sheets (e.g., paper-like porous cellulose sheets), and the polymer composite sheets were then carbonized to obtain freestanding electrode sheets with desirable electrical conductivity (Fig. 12b).175 Such synthesis was highly promising for preparing flexible electrodes based on active materials that are incompatible with carbon in surface affinities (e.g., Li2S). It was technically viable to achieve small grain size and good uniformity in these polar active materials within a hydrophilic polymer matrix (e.g., cellulose) and then to improve electrical conductivity through carbonization. The as-prepared carbon-coated nano-Li2S/carbonized porous cellulose sheet electrode exhibited good flexibility to endure bending. The electrochemical performance was also outstanding, including high capacities (1640 and 1262 mA h gsul−1 at 0.05 and 0.5C, respectively) and an exceptional cycling stability (97% retention capacity during 100 cycles at 0.5C). Through a similar synthetic tactic, Tu's group fabricated an integrated carbon-coated nano-Li2S on carbon paper, starting with a different precursor of Li2SO4 soaked in napkin paper.176 The initial capacity was as high as 820 mA h g−1 based on Li2S (295 mA h gele−1) at 0.1C.
BC, another class of cellulose, possesses a larger cellulose content, a greater polymerization degree, and higher crystallinity than plant-derived cellulose.169 Thus, BC is more advantageous than FPM for carbonization. Zheng and Sun's group prepared a 3D flexible carbonized BC (CBC) aerogel, derived from a BC hydrogel, for fabricating a flexible sulfur cathode with a highly interconnected nanofibrous network, good electrical conductivity, and desirable mechanical durability.177 After sulfur precipitation from CS2 solution, the sulfur content was as high as 81 wt%; while considering the use of an additional thin CBC interlayer, the content was still ∼72 wt%. With a CBC interlayer, the flexible CBC/S cathode exhibited an enhanced initial capacity (976/∼700 mA h gsul/ele−1) and cycling stability (63.5% capacity retention after 300 cycles) at 0.8 A gsul−1 (∼0.48C).
Fan and coworkers further reported a BC-derived and N,O-codoped carbon nanoribbon (CNR) aerogel as a binder-free conductive matrix by pyrolyzing BC aerogels in ammonia atmosphere.178 The CNR/polysulfide catholyte gel electrode held great promise as it enabled both high sulfur content (90 wt%) and areal loading (6.4 mgsul cm−2) without compromising on capacity and cycling performance (905/815 mA h gsul/ele−1, 96% of initial capacity retained after 100 cycles at 0.2C). More importantly, they systematically compared BC-derived CNRs with plant cellulose-derived carbon microfibers and indicated the superiority of using BC as the precursor for carbonized FPM, which was ascribed to the thinner diameter of BC nanofibrils (<100 nm, in sharp contrast to 10–20 μm of carbonized plant cellulose), higher specific surface area of CNRs, and larger doping contents.
Other than the above biomass-derived FPMs like cotton, paper, and BC, there are many artificial thermosetting FPMs, such as melamine and polyurethane foams, which are readily molded, structurally elastic, thermally non-collapsing, commercially available, and chemically dopant-rich. Therefore, they can be easily converted into flexible, conductive, and functionalized carbon monoliths via direct pyrolysis without extra tedious procedures. This is an effective and efficient way to utilize commercial raw materials. Mi et al. first reported the synthesis of 3D N-doped flexible carbon foam/S cathode through pyrolysis of melamine foams (MFs) and investigated the influence of carbonization temperature.179 As-obtained carbon foams (MFCs) exhibited a highly extended and sparse macroporous skeleton built from interconnected carbon fibers, which afforded a mechanically bendable framework and abundant free volume to load sulfur/carbon slurry. MFCs pyrolyzed at a lower temperature of 800 °C showed the best performance (632/228 mA h gsul/ele−1 at 0.2C) owing to the preservation of doped nitrogen during pyrolysis. Xu and coworkers further decorated the pliable MFCs with GO or rGO for improving electrical conductivity and polysulfide adsorption (Fig. 12c).180 Sulfur precipitated from the disproportionation of sodium polysulfides in acid, instead of sulfur/carbon slurry as in the work of Mi et al.,179 was coated on MFC skeletons to achieve a higher sulfur content of 75 wt%. As a result, the capacity of flexible MFC–rGO–S electrode was as high as 654(491) mA h gsul(ele)−1 after 100 cycles at 0.1C.
Yang and colleagues explored an alternative raw material, polyurethane foam (PUF), which is extensively used in modern industry (accounting for ∼5% of annual consumption of plastics worldwide), for the synthesis of flexible elastic N-doped carbon foams (NCFs).181 After simple pyrolysis, the NCF retained the high porosity and elasticity of pristine PUF (Fig. 12d-i–iii). At different applied strains of 20, 40, 60, and 80%, both PUF and NCF could sustain large-strain compression and bending and recovered most of their volume elastically (Fig. 12d-iv,v). Serving as both 3D current collectors and interlayers, the NCF endowed CNT/S cathodes with high capacity of 1124 mA h gsul−1 at 0.5C, with 80% capacity retention after 100 cycles. But the use of binder and CNT unfortunately resulted in a relatively low overall sulfur fraction in the integrated electrode (23 wt%) compared to that in other binder-free, flexible electrodes. More progress can be expected in the near future.
There is the third class of carbonized FPMs besides carbonized biomass-derived and artificial thermosetting FPMs. This is achieved by templated fabrication of FPMs from organic precursors rather than direct use of existing or commercial FPMs. Using N-containing ionic liquids as the organic precursor and glass microfiber filer paper as the template, Schneider et al. described the synthesis of freestanding, continuous carbon films with a high N-doping content and hierarchical porous structure for binder-free composite carbon/sulfur cathodes with sulfur loadings of 2.5–8.5 mgsul cm−2.182 Stable areal capacities of 2.7 mA h cm−2 were achieved. Aiming at a facile, low-cost, and high-yield fabrication of N-doped porous CNFs, Zhou et al. first prepared a Zn2+ ethylene glycol/pyridine complex mixture and then carbonized it followed by etching Zn with acid.183 As-obtained N-doped CNFs were uniform in fibrous morphology, rich in both N dopants and micro-/mesoporosity, and capable of being filtrated into highly flexible films. With 72 wt% sulfur melted in the N-doped CNF film, a high initial capacity of 1170(842) mA h gsul(ele)−1 was obtained at 0.2C, with 73% capacity retention after 200 cycles. Even when sulfur loading was increased from 1.5 to 3.0 and 4.5 mgsul cm−2, the outstanding electrochemical performance exhibited little degradation.
In summary, carbonized polymers, either electrospun PNFs or FPMs, have some fascinating merits for flexible sulfur-based electrodes: (1) highly mature technologies for high-volume production and large-scale application, which are compatible with traditional textile, paper, polymer, and carbon fiber industries, (2) abundant and cheap raw materials from either capacity-excess polymers and organic chemicals (e.g., PAN, PS, thermosetting plastics, and ethylene glycol) or renewable and recyclable biopolymers (e.g., cellulose), (3) good inheritance of flexibility from polymer precursors, (4) enhanced electrical conductivity through carbonization, and (5) tremendous possibilities for regulating micro-/nanostructure, carbon texture, functional groups, porosity, and assembly during fabrication.
More specifically, electrospinning is very efficient in generating 1D nanostructures, while the use of existing 2D or 3D flexible materials such as fabric and paper largely simplifies the synthesis procedures. To balance flexibility and electrical conductivity, as well as heteroatom residues, carbonization temperature is regarded as a key parameter. It is crucial but not easy to select ideal polymer precursors and synthesis methods. Hence, it is foreseeable that physical chemistry and materials chemistry during carbonization of various polymer precursors will be the cornerstone for this research area, for not only flexible Li–S batteries and its analogues, but also for electrochemical energy storage systems, where carbonized polymer electrode materials play an increasingly important role.
2.1.1.5. Hybridized carbon-based flexible cathodes. Single-component carbonaceous materials always suffer from inherent problems that are difficult for solving, limiting their overall performance as flexible electrode materials. For example, graphene sheets and CNTs tend to pack together, forming restacked graphite and CNT bundles with largely reduced external accessible surface area and pore volume for ion/active material storage. Micro-/mesoporous carbon has high accessible surface area and abundant porous structure, but the electrical conductivity is inferior due to a large number of defects and amorphous sp3-hybridized carbon. Besides, most micro-/mesoporous carbon structures are more particulate than fibrous, so they are commonly unable to form self-supported structures. In this regard, deliberate combination of several nanocarbon building blocks or engineering of hybridized multicomponent carbon is of exceptional interest to achieve a holistic design of high-performance flexible sulfur-based electrodes. Such a strategy can be broadly divided into several categories based on the dimensions of componential carbon materials: 1D/2D, 1D/3D, and 2D/3D hybridization.
One prototype is 1D/2D hybridization, featured in CNT/graphene hybrid nanostructures. In fact, particulate CNT/graphene hybrids, with either CNTs spaced/pillared between graphene sheets184–187 or graphene grown/crafted on outer/inner walls of CNTs,188,189 have long been proved to show extraordinary capability in supercapacitors,184 batteries,185,186 and in catalysis187–189 due to larger exposure of active surface/sites and better interfacial charge transport than pure graphene or CNTs. They have consequently been adopted in Li–S batteries, exhibiting better or even unexpected performance.185,186,190–192 Based on the success of these particulate counterparts, macroscopic CNT/graphene hybrids, mostly in the shape of films, arise for flexible sulfur-based electrodes. Differentiated by the main body of the hybrid, this prototype can be divided into (1) modifying 1D by 2D, denoted as (1 + 2)D and (2) modifying 2D by 1D denoted as (2 + 1)D.
Start with the former, (1 + 2)D, as discussed in (Section 2.1.1.1), 1D nanocarbon such as CNTs possess extraordinary electrical conductivity, chemical stability, and mechanical rigidness, and in most cases, sulfur is deposited on the outer wall without sufficient protection and thus subject to rapid dissolution. The protection, however, can be afforded by conformally coating a graphene skin. Yang et al. adopted a two-step electrochemical deposition method to coat carbon fibers with sulfur spheres and graphene sequentially.193 In the presence of an outer graphene coating, the freestanding carbon fiber/S electrode exhibited enhanced capacity retention. However, as indicated in (Section 2.1.1.3), the specific surface area of carbon fibers is too low to afford sufficient conductive surface for high-sulfur-content cathodes. In order to increase sulfur content, CNTs or CNFs are more preferably adopted than carbon fibers. For example, low-specific-surface-area carbon fibers can be easily replaced by electrospun PCNFs as Chu et al. demonstrated, resulting in a flexible rGO-coated PCNF/S paper electrode that delivered a decent capacity of 624(364) mA h gsul(ele)−1 after 200 cycles at 0.1C.194 Based on a well-developed ultrasonication-assisted method for fabricating SACNT-based flexible sulfur electrodes, Wang and colleagues intentionally introduced graphene as a 2D supplement to 1D SACNTs, obtaining a 3D hierarchical SACNT/graphene conductive framework for flexible sulfur cathodes (Fig. 13a).195 In the composite with an optimal SACNT/graphene mass ratio of 1:1, graphene fully enfolded the composite, serving as a block layer, while underneath the graphene coating, SACNT, graphene, and sulfur were uniformly dispersed with abundant porosity. Such a synergistic combination of the three contributed to higher strength of the flexible composite and better electrochemical performance with a high initial capacity of 1048(524) mA h gsul(ele)−1 at 1.0C, capacity retention of 59% after 1000 cycles, and good rate capability at up to 10C. Similar strategy for high-sulfur-loading Li–polysulfide batteries was proposed by preparing CNF/rGO hybrid electrode, enabling a very high areal sulfur loading of 20.3 mgsul cm−2.196
 | ||
| Fig. 13 1D/2D hybridized flexible sulfur cathodes. (a) Schematic for the synthesis of sulfur (S)–SACNT/graphene (G) composite. Reproduced with permission.195 Copyright 2015, Royal Society of Chemistry. (b) Schematic for the preparation of flexible MGP–S paper and (c) image of bent MGP–S paper. Reproduced with permission.197 Copyright 2013, Royal Society of Chemistry. (d) Schematic for the synthesis of rGO/S and CNT–rGO/S composite films and their electrical conductivities (s), tensile strengths (σt), and Young's moduli (E) are listed. Reproduced with permission.198 Copyright 2015, American Chemical Society. | ||
Moving to the other one, (2 + 1)D, as summarized in (Section 2.1.1.2), surface functional groups, mainly oxygenated ones like hydroxyl, carbonyl, carboxyl, and epoxy groups, have profound influence on determining dispersion and solvation states of graphene/GO in solution and therefore further porosity, electrical conductivity, and polysulfide adsorptivity of the final graphene assemblies. All these factors translate into the electrochemical and mechanical performance of graphene-based flexible electrodes, so they have to be regulated. On the contrary, it is technically complicated to achieve an optimal content and species distribution of oxygen groups because they vary significantly depending on the graphite precursor, preparation methods, and processing parameters. Such a conflict in balancing electrical and mechanical properties requires an alternative approach. In this regard, 1D nanomaterials are introduced as enhancers for modifying 2D nanomaterials (e.g., graphene-based flexible electrodes). Stemming from the rapidly growing research of 2D nanomaterials, this (2 + 1)D strategy has attracted more attention than (1 + 2)D.
Primary purpose of introducing 1D nanomaterials is to use these large-aspect-ratio fibers for bridging small pieces of graphene sheets into a single freestanding film. Wang's group pioneered the study of utilizing a mesoporous graphene paper (MGP), where sulfur was immobilized in mesopores, as a flexible electrode (MGP–S) for Li–S batteries.197 In this case, hollow SiO2 spheres were used as hard templates for creating highly uniform mesoporosity (a pore size of ∼30 nm) and a large mesopore volume (1.25 cm3 g−1) in MGP, though its highly protuberant nature may prohibit its efficient stacking into a robust film, thereby requiring the assistance of CNF reinforcement (Fig. 13b). Along with large storage space for sulfur and lithiated products, as well as good structural integrity (Fig. 13c), the flexible MGP–S paper electrode exhibited a very high initial capacity of 1393(766) mA h gsul(ele)−1 at 0.1C. As one of the earliest efforts toward fabricating flexible Li–S batteries, this work notably demonstrated the potential of (2 + 1)D hybridization strategy for fabricating flexible sulfur-based electrodes.
Since CNTs usually possess better electrical conductivity than chemically derived graphene and can effectively prevent the restacking of graphene sheets through intercalation, they not only reinforce the freestanding film, but also promote charge transfer and reaction kinetics. Wu and Liu's group prepared highly flexible and conductive CNT–rGO/S composite films as cathodes by first synthesizing sulfur particles wrapped by rGO through simultaneous generation of sulfur and coreduction of GO by hydroiodic acid (HI) and Na2S2O3, and subsequently codispersing and infiltrating them into CNTs (Fig. 13d).198 After CNT incorporation, electrical conductivity, tensile strength, and Young's modulus increased from 47 S cm−1, 45.4 MPa, and 6.7 GPa to 102 S cm−1, 72.0 MPa, and 9.7 GPa, respectively. The pliable CNT–rGO/S cathode delivered a peak capacity of 912(483) mA h gsul(ele)−1 and maintained 772(409) mA h gsul(ele)−1 after 100 cycles at 0.2C, both higher than that without CNTs. More importantly, mechanical properties of the cycled electrode were evaluated, suggesting that a high tensile strength of 62.3 MPa, ∼87% that of uncycled electrode, was preferably preserved.
In order to achieve a better balance between oxygen functional groups and electrical conductivity, Shi and Yu's group employed mildly reduced and less defective GO (mrLGO) to replace conventional GO (CGO) for fabricating flexible and binder-free rGO/S/CNT composite films for Li–S batteries.199 Effects of both GO precursors (LGO vs. CGO), reduction methods (mild reduction vs. HI reduction), and the addition of CNTs were systematically investigated. It was noted that the mrLGO film possessed a much higher conductivity (28.8 S cm−1) than the mrCGO film (5.85 S cm−1) and lower carbon/oxygen atomic ratio of 4.29 than that of HI-rLGO (6.94), thus attaining a desirable balance. Such a balance endowed flexible mrLGO/S/CNT cathode with a considerably enhanced cycling stability (1000/530 mA h gsul/ele−1 after 200 cycles at 0.2C against to 598/311 mA h gsul/ele−1 for HI-rLGO/S/CNT), despite slightly lower initial capacity. The introduction of CNTs was further suggested to improve conductivity and reaction kinetics of all rGO/S films. Attributed to such a positive effect on charge transfer, CNTs were also introduced to a freestanding 3D graphene–Li2S (3DCG–Li2S) aerogel for realizing an exceptionally high Li2S content in the whole electrode (81.4 wt%).200 As He et al. demonstrated, 3DCG–Li2S delivered a high initial capacity of 1052(856) mA h gLi2S(ele)−1 at 0.2C with 94.5% capacity retention after 300 cycles, much better than that of the sample without CNT, indicating the synergistic effect between CNTs and graphene in building highly conductive, flexible, and binder-free electrode materials.
Very recently, Liu and Zhou's group designed a bifunctional hierarchical carbon network based on CNTs and graphene, in which both the concepts of (2 + 1)D and (1 + 2)D were involved.201 GO films incorporated with nickel acetate were first obtained by vacuum-filtration-assisted self-assembly, during which an ultrathin compact GO skin formed on the bottom of macroporous GO films. Subsequently, the hierarchical GO films underwent a CVD process, upon which GO was reduced and nickel acetate decomposed to form nickel (Ni) catalysts for CNT growth on graphene. The introduction of CVD-grown CNTs significantly improved specific surface area of the hierarchical carbon film, thus providing a high sulfur loading and good electrical contact while in situ formed ultrathin graphene shells were preserved, serving as a physical barrier to protect polysulfides from shuttling. Such a rational design of hierarchical freestanding electrodes combined the advantages of both (2 + 1)D and (1 + 2)D strategies, thus leading to superior performance such as a high sulfur loading of 3.6 mgsul cm−2, a capacity of 1184(699) mA h gsul(ele)−1 at 0.05C, and outstanding cycling stability with capacity retention of 81.2% after 200 cycles at 1.0C.
1D/2D hybridization is very effective to fully exert the advantages of 1D and 2D nanomaterials and realize possible synergistic enhancements. However, unlike 1D and 2D nanomaterials (e.g., CNTs/CNFs and graphene, respectively) that can solely be used for flexible electrodes regardless whether the performance is satisfactory or not, 3D nanoporous or particulate carbon such as carbon blacks, carbon spheres, and non-fibrous micro-/mesoporous carbon cannot form self-supported macrostructure without the assistance of polymer binders. 1D or 2D nanomaterials are indispensable as robust mechanical supports and long-range conductive skeletons to make it possible for 3D carbon structures to be involved in the design of flexible and binder-free sulfur cathodes. Therefore, the other two categories, i.e., 1D/3D and 2D/3D hybridization, come into being.
Zhang and coworkers developed a series of 1D/3D hybridized nanocarbon-based flexible sulfur electrodes with good flexibility and electrochemical performance.202–205 On the basis of their original work, where hierarchical ultralong CNT-interpenetrated MWCNT/S paper electrodes were prepared through a dispersion–filtration method,95 MWCNT/S composites were replaced by various sulfur-infiltrated 3D nanostructured porous carbon materials with feature sizes in a broad range from several hundreds of nanometers to almost ten micrometers (Table 1), indicating high versatility and applicability of ultralong CNTs for constructing flexible electrodes. On one hand, these materials were unable to be self-supportive, but when incorporated in ultralong CNT scaffolds, very robust films could be obtained without sacrificing mechanical flexibility compared to the ultralong CNT/MWCNT film. On the other hand, these materials basically exhibited higher specific surface area, more abundant micro-/mesoporosity, and surface functionalities than MWCNTs and thus were more ideal hosts to facilitate sulfur cathodes. This strategy was generic and versatile, allowing these flexible electrodes to be useful platforms for diverse investigations of multiple effects, as listed in Table 1.
| Supported 3D building block | Feature size | Tensile strength (MPa) | Electrical conductivity (S cm−1) | Initial capacity (mA h gsul/ele−1) | Research highlight |
|---|---|---|---|---|---|
| Interconnected hollow graphitic nanoshell (GS)–S202 | 500–1000 nm | 2.1 at a strain of 9.3% | N. A. |
• 1346/606 at 0.1C
• 535/241 at 10C |
• Systematically compared small-size GSs and large-size graphene flakes
• Suggested that smaller-size 3D building blocks (i.e., GS) have better contact with 1D conductive backbones (i.e., ultralong CNT), contributing to better electrochemical performance |
| Wrinkled NG/S granular203 | 1–2 μm | 1.1–6.8 depending on compaction ratio | 6.4–25.0 depending on compaction ratio | 1311/695 at 0.05C (46.6% compaction ratio) |
• Systematically investigated the influences of compaction ratio on mechanical strength, conductivity, porosity, electrolyte uptake, and electrochemical performance of flexible electrodes
• Suggested that calendaring is a critical step for controlling the properties and performance of flexible electrodes |
| Hexagonal graphitic carbon nanocage (gCNC)/S204 |
• 1–5 μm in diameter
• ∼80 nm in thickness |
2.75 at a strain of 7.3% | 62.9 |
• 1354/812 at 0.1C
• 750/450 at 5.0C |
• Developed an in situ approach for synthesizing highly interlinked all-carbon nanoarchitecture
• Suggested the dominant role of interfacial junctions between multiple building blocks in a flexible hybridized electrode |
| Porous graphene microsphere (GMS)–S205 | 5–10 μm | N. A. | 27.0 (GMS only) | 1066/554 at ∼0.03C | • Suggested the possibility of using very large particles for flexible electrodes |
Among these works, the effort of Huang et al. suggested the dominant role of interfacial junction between different building blocks in improving charge transfer and ensuring high-power performance (Fig. 14).204 During the synthesis, ultralong CNTs and magnesium oxide (MgO) templates were first dispersed and filtrated into a composite film, in which ultralong CNTs intercrossed with MgO flakes firmly. Then after high-temperature CVD and removal of MgO, highly graphitic carbon nanocages were replicated from MgO and ultralong CNTs interpenetrated into each flake of the carbon nanocages, resulting in a flexible all-carbon interlinked architecture (Fig. 14a–c). This in situ formed hybrid film had an exceptional electrical conductivity of 62.9 S cm−1, much higher than that of mechanically co-filtrated ultralong CNT/nanocage films (18.7 S cm−1). As a result, the in situ formed CNT/nanocage scaffolds enabled the flexible sulfur cathode to deliver superb high-power performance. A high discharge capacity of 750(450) mA h gsul(ele)−1 was achieved at a high current density of 8.35 A gsul−1 (5.0C) (Fig. 14d). The benefit of the strongly coupled interface between each carbon component was also proved by Wu et al., by directly growing 3D CNT forests on commercial cloths to afford rapid electron transport paths.206
 | ||
| Fig. 14 1D/3D hybridized flexible sulfur cathodes. (a) Schematic of the sulfur cathode with CNT/carbon nanocage scaffold. (b) SEM image of sulfur/CNT/carbon nanocage composite. (c) Image of flexible cathode film. (d) Rate performance of sulfur electrode based on pure CNT, pure carbon nanocage, and CNT/carbon nanocage scaffolds. Reproduced with permission.204 Copyright 2014, Royal Society of Chemistry. | ||
Besides nanoscale interfacial charge transfer, macroscopic regularity strongly impacts the performance of 1D/3D hybridized flexible electrodes. Sun et al. reported an aligned and laminated carbon hybrid cathode that was highly flexible and accommodated a large amount of sulfur (71 wt%), in which ordered mesoporous carbon CMK-3/S composite (CMK-3@S) particles were alternately laminated between aligned CNT sheets with a small thickness (∼20 nm) and a high conductivity (102–103 S cm−1).207 Such a hybrid electrode delivered a high initial capacity of 1226(870) mA h gsul(ele)−1 at 0.1C with 75% capacity retention after 100 cycles, and stable capacities of around 400(280) mA h gsul(ele)−1 at 2.0C during 1000 cycles. This performance was much better than that of hybrid CNT/CMK-3@S electrodes without a laminated structure or with randomly agglomerated CNTs, indicating the beneficial effect of the ordered assembly.
The role of graphene in 2D/3D hybridized structure is essentially similar to that of CNTs in the 1D/3D one: providing sufficient mechanical adhesion and electrical conduction to 3D carbon constituents that lack flexibility. Wu et al. synthesized a series of freestanding graphene-based hierarchical porous carbon (GPC) films for flexible sulfur-based cathodes (Fig. 15a).153 Using GO as the 2D template and support, thin layers of microporous carbon were coated on both sides of GO after hydrothermal carbonization and KOH activation, resulting in graphene-based microporous carbon (GMC) sheets. The GMC sheets were further coated with sulfur, functionalized by surface positive charge, assembled with negatively charged GO that underwent subsequent reduction, and vacuum filtrated into GPC films with a thickness of 100 μm and a conductivity of 3.25 S cm−1. The GMC contained rich micropores that could store small sulfur molecules, providing much stronger physical confinement than graphene. As a consequence, the pliable GPC film exhibited excellent cycling performance with capacities stabilized at 1030(422) and 626(357) mA h gsul(ele)−1 at 0.2C when sulfur content was 41 and 57 wt%, respectively. On the contrary, graphene-based film electrodes displayed rapid capacity decay due to severe polysulfide dissolution without micropore confinement. The GPC–S cathode film was further assembled in flexible Li–S batteries using tape as the packaging material, displaying comparable electrochemical performance in both flat and bent states (Fig. 15b and c).
 | ||
| Fig. 15 2D/3D hybridized flexible sulfur cathodes. Freestanding GPC-based film: (a) schematic for the fabrication; (b) the second charge–discharge profiles for GPC–sulfur cathode films in bent and flat states at 0.5C; (c) cycle performance for GPC–sulfur cathode films at 0.5C and (inset) photograph of a bent cell encapsulated in an argon-filled glass bottle. Reproduced with permission.153 Copyright 2015, Royal Society of Chemistry. Graphene (G)–NDHCS–S hybrid paper: (d) schematic for the fabrication of NDHCS–S composite and G–NDHCS–S paper; (e) images of G–NDHCS–S paper in flat, bent, and recovered states; (f and g) top-view and (h) cross-sectional SEM images of G–NDHCS–S paper. Reproduced with permission.208 Copyright 2015, Wiley-VCH. | ||
Manthiram and coworkers also demonstrated the 2D/3D hybridization concept by designing a dual-confined flexible sulfur cathode based on N-doped double-shelled hollow carbon spheres (NDHCS) and graphene.208 Sulfur was first encapsulated by NDHCS (denoted as NDHCS–S), and NDHCS–S composites were further filtrated with graphene to obtain flexible films (Fig. 15d and e), in which graphene not only served as a mechanical and conductive support but also offered extra confinement to polysulfides by conformal wrapping (Fig. 15f–h). 2D graphene and 3D NDHCS synergistically contributed to dual confinement and extraordinary electrochemical performance, which was manifested as a high initial discharge capacity of 1360(843) mA h gsul(ele)−1 at 0.2C, excellent rate capability of 600(372) mA h gsul(ele)−1 at 2.0C, and sustainable cycling stability (62% capacity retention) for 200 cycles, all significantly better than those of pristine graphene–S films. The performance was very promising when considering the high sulfur loading of 3.9 mgsul cm−2. Chen's group also reported the synthesis of “bubble-like” interconnected carbon fabrics (ICF), in which sulfur nanodots (2–5 nm in size) were stitched, further fabricated a freestanding sulfur cathode enabled by rGO encapsulation.209 The synergistic effect between porous ICF and rGO ensured high utilization and stable operation of this freestanding sulfur cathode, even when sulfur content and areal loading were as high as 70 wt% and 2.9 mgsul cm−2, respectively. A high initial capacity of 1149(804) mA h gsul(ele)−1 was achieved at 0.1C with 78% capacity retention after 200 cycles. These works adequately proved that 3D porous carbon with suitable chemical affinities can afford unique physical and chemical immobilization to sulfur and polysulfides,102,158,210–213 which can barely be met by 1D CNTs or 2D graphene. Thus, it will significantly promote flexible Li–S batteries if these 3D carbon materials can be delicately introduced into flexible sulfur cathodes.
In summary, hybridized carbon-based flexible cathodes can (1) furthest exert the desirable merits (e.g., high conductivity, high surface area, tunable micro-/mesoporous structure, rich surface functionalities, and excellent flexibility) of different componential carbon materials (e.g., 1D CNTs/CNFs, 2D graphene, and 3D nano-/nanoporous carbon), (2) rationally integrate these properties together, (3) overcome some intrinsic drawbacks of each carbon building block through complementary enhancements, and (4) even bring synergistic effects if suitable hybridization strategies are adopted. 1D and 2D nanomaterials serve as enhancers for each other in their hybrids, while in 1D/3D and 2D/3D hybridization scenarios, 1D or 2D nanomaterials are introduced mainly to make use of 3D-architectured carbon that has high accessible surface area, large pore space, and strong confinement to sulfur species but is normally regarded as an “impossible” single constituent of a flexible electrode in the absence of sticky additives. Future direction for hybridized carbon-based flexible sulfur cathodes might be unveiled under the exploration of novel carbon nanomaterials and advanced fabrication methods.
2.1.2.1. Polymer-glued flexible composite cathodes. Although carbonaceous materials possess indispensable advantages such as light weight, extraordinary electrical conductivity, and large surface area as well as superior chemical, thermal, and mechanical stability, a majority of them are sp2/sp3-hybridized carbon bonds, leaving limited room for effectively regulating composition and properties. Heteroatom doping and surface functionalization help to modulate them to some extent, but it is difficult to achieve precise control over these two approaches, especially considering the complex physical chemistry and materials chemistry of carbonaceous materials. Therefore, uniting carbon with one or several extra phases of materials with well identified compositions, molecular structures, and physicochemical properties is regarded as a straightforward materials chemistry route for boosting composite performance. Polymer, as briefly introduced in (Section 2.1.1.4) as an inherent soft material, has attracted tremendous attention for producing flexible composites, which are under continuous investigations in diverse areas such as engineering, medical, and energy science. In this section, polymer-glued flexible composite electrodes, in which polymers and other components are homogeneously dispersed, will be discussed primarily and are differentiated from those being discussed in the next section, i.e., polymer-supported ones, in which polymer and other components are spatially separated.
As indicated in (Section 2.1.1.5), there are varieties of carbonaceous materials that cannot be directly used for flexible energy storage devices, such as carbon blacks, carbon spheres, and non-fibrous micro-/mesoporous carbon, owing to their structurally rigid nature and/or small aspect ratio. They can be integrated in flexible electrodes through the assistance of low-dimensional carbon nanomaterials like CNTs and graphene, but the manufacture of this hybridized carbon is always complicated. An alternative solution is to apply a polymeric binder to glue particulate carbon and afford integral flexibility. Though the use of a binder, mostly non-active, brings extra “dead” weight and inevitably lowers the gross gravimetric and/or volumetric energy density of an electrode, the binder makes the fabrication process fairly simple and compatible with a continuous coating technology that is widely employed in conventional battery and supercapacitor industries. Moreover, the increase in weight might not be as considerable as it appears to be if the polymer-glued flexible composite electrode is flexible and truly freestanding, because the use of heavy metal current collectors can still be avoided.
In this regard, Kaskel and colleagues developed a solvent-free procedure to fabricate flexible, high-capacity micro-/mesoporous carbon–S nanocomposite cathodes using poly(tetrafluoroethylene) (PTFE) as the gluing binder.214 Porous carbon–S nanocomposites were first prepared via a melt-diffusion method, and then the composite powder, conductive agents (herein MWCNTs), and PTFE were homogenized together through intensive grounding, during which PTFE was fibrillated under strong shearing forces and agglomerated single particles to form larger aggregates. These aggregates were further rolled out upon heating, rendering freestanding cathode foils with a large area and a thickness of ∼80 μm. The solvent-free processed nanocomposite cathode exhibited capacities stabilized at 740(314) mA h gsul(ele)−1 at 0.1C for over 160 cycles, better than that of a conventional blade-coated cathode with the same composition. No organic solvent or water was involved during the electrode preparation. Such very dry electrodes exhibited enhanced electrochemical performance. It could be further enhanced by adopting higher-surface-area porous carbon, together with higher sulfur content, to achieve higher capacities based on electrode weight. Similarly, Li et al. also employed PTFE to prepare flexible electrodes consisting of waste biomass-derived mesoporous carbon/CNT composites, which were further evaluated in both supercapacitors and Li–S batteries.215
Aqueous binders have also been employed for polymer-glued flexible composite electrodes owing to their good processability and environmental friendliness. Ni et al. reported a facile route for synthesizing ultrathin and flexible composite films based on rGO-wrapped sulfur particles with the assistance of an aqueous binder, sodium alginate (SA), which simultaneously served as a surfactant and an adhesive agent.216 The SA-glued film electrode exhibited a high reversible capacity of 1341(818) mA h gsul(ele)−1 at 0.1C and retained 823(502) mA h gsul(ele)−1 at 0.5C after 100 cycles, both better than those of the physically mixed rGO/S film. In order to secure an excellent electrical conductivity along with exceptional mechanical stiffness, Mitra and coworkers further mixed SA with polyaniline to obtain a hybrid binder that glued rGO/Mn3O4/S nanocomposite particles into pliable electrode films, which exhibited a high capacity of 1015(538) mA h gsul(ele)−1 at 5.0 A gsul−1 (∼3.0C) and capacity retention of 71% after 500 cycles.217
Besides the configuration with a single composite sheet, the flexible cathode can also be prepared by sandwiching sulfur composites between two pieces of binder-glued freestanding carbon foils (Fig. 16a). Such a concept was first initiated by Song et al., who developed a milling–roll pressing–drying synthetic scheme to fabricate large-area, flexible, porous carbon films consisting of Ketjen black and PTFE, and sandwiched ball-milled carbon black/sulfur cathode between two pieces of carbon films (Fig. 16b).218 The porous carbon film could effectively capture dissolved polysulfides and reduce contact resistance, leading to a drastic increase in capacity from 777 to 1495 mA h gsul−1 at 0.2C when the sulfur loading was 1.5 mgsul cm−2. However, the actual capacity based on the whole electrode decreased due to high areal density of porous carbon films (∼3.0 mg cm−2). Such a detrimental effect could be offset by increasing the areal sulfur loading to 4.0 mgsul cm−2, while the high-loading sandwiched electrode could be stably cycled with a capacity of ∼1000(340) mA h gsul(ele)−1 for 150 cycles at 0.5C. Guo and coworkers also realized a similar sandwich-structured flexible sulfur cathode by a facile slurry casting technique to sequentially coat polyvinylidene fluoride (PVDF) binder-containing carbon, sulfur, and carbon slurry on a polymer separator (Fig. 16c).219 The carbon layer was much lighter (0.38–0.52 mg cm−2) than the sulfur layer (1.0–1.4 mg cm−2), thus resulting in a higher sulfur content of ∼49 wt% than that in work of Song et al.218 The integrated sandwich sulfur electrode (denoted as CSC) was also highly flexible (Fig. 16d) and exhibited a capacity of 730(358) mA h gsul(ele)−1 retained after 500 cycles at 0.6C and good rate capability of 620(304) mA h gsul(ele)−1 at 5.0C. Such enhanced performance was ascribed to excellent electrical conductivity, tolerance to volume fluctuation, and mitigation of polysulfide diffusion by carbon layers.
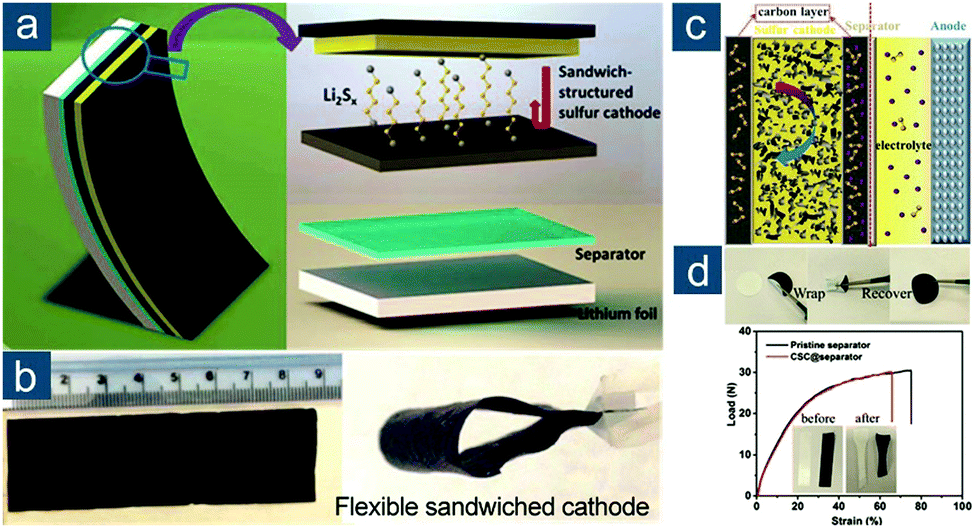 | ||
| Fig. 16 Polymer-glued sandwiched flexible sulfur cathodes. (a) Schematic of Li–S cell with a sandwiched cathode. (b) Images of flexible sandwiched electrodes in flat (left) and bent states (right). Reproduced with permission.218 Copyright 2014, Royal Society of Chemistry. (c) Schematic of Li–S cell with integrated CSC cathode. (d) Images and load-strain curves of the pristine separator and CSC@separator electrodes. Reproduced with permission.219 Copyright 2016, Wiley-VCH. | ||
Besides gluing, another predominant role of polymer is to functionalize and enhance flexible all-carbon scaffolds by introducing additional benefits such as polysulfide adsorption or promotion of reaction kinetics. Kong et al. modified flexible nano-S/SACNT electrode by 4 wt% PVP (well known for its capability of binding polysulfides), attaining a thin encapsulation shell of PVP around both sulfur nanoparticles and SACNTs.220 As a result, the PVP-encapsulated nano-S/SACNT electrode exhibited considerably enhanced cycling stability with a capacity of 856(402) mA h gsul(ele)−1 preserved after 200 cycles at 1.0C, 94% higher than that of an unmodified electrode. Moreover, the PVP-modified film exhibited 838% increase in tensile strength (13.6 MPa) compared to the pristine one (1.45 MPa), which was ascribed to the improved dispersion of SACNTs with the assistance of amphiphilic PVP surfactants. Yu et al. proposed a multi-level polysulfide-interception strategy by integrating NG/sulfur composites with two hydroxyl functionalized nanofibrillated cellulose (NFC)/CNT bottom/top layers into a flexible sandwiched electrode, which was further densified by roll pressing for practical use.221 The introduction of hydrophilic NFC as well as the nitrogen dopants in graphene endowed the electrode with a remarkable ability to localize polysulfides. With sulfur loadings of 3.6 and 8.1 mgsul cm−2, the sandwiched electrodes exhibited high initial capacities of 968(532) and 936(496) mA h gsul(ele)−1 at 0.5C with capacity retention of 58% and 33% after 500 and 1000 cycles, respectively. Zhang and coworkers proposed a quinonoid-imine-enriched nanostructured polymer mediator that bound polysulfides strongly and facilitated electrochemical kinetics dramatically, and further incorporated it into a freestanding and conductive scaffold consisting of ultralong CNTs to form a flexible sulfur cathode with a high areal loading of 3.3 mgsul cm−2, achieving high capacities of 1330(560) and 1120(470) mA h gsul(ele)−1 at 0.2 and 1.0C (1.1 and 5.5 mA cm−2), respectively, as well as desirable capacity retention of 70% for 200 cycles at 1.0C.222
Chen's group reported a rational design toward high-rate performance Li–S batteries by incorporating a multifunctional polymeric additive, poly(anthraquinonyl sulfide) (PAQS), in nano-S/rGO composite thin films (denoted as nano-S:rGO:PAQS) (Fig. 17).223 PAQS was predicted to possess strong binding to lithium (poly)sulfide species (Fig. 17a). In the work, PAQS was mixed with a nano-S:rGO dispersion and vacuum filtered (Fig. 17b), resulting in a freestanding film with a layered and porous structure (Fig. 17c and d). The as-obtained nano-S:rGO:PAQS film exhibited excellent flexibility (Fig. 17e). The addition of PAQS was critical to this design as it not only restricted polysulfide diffusion through strong anchoring to gain a prolonged cycling lifetime, but also facilitated nanoscale ion transport at high rates. Compared to other samples with PVP, without polymer addition, or supported on Al foils, the pliable nano-S:rGO:PAQS thin film electrode possessed notably better electrochemical performance, including the highest initial capacity of 1323(635) mA h gsul(ele)−1, the highest capacity retention of 85% over 100 cycles, and the highest Coulombic efficiency of 97% without LiNO3 addition at 0.25C (Fig. 17f), as well as excellent rate performance of 615(295) mA h gsul(ele)−1 at 8.0C and an ultralong cycling life of 1200 cycles at 0.5C.
 | ||
| Fig. 17 Multifunctional polymer-glued flexible sulfur cathodes. (a) Theoretically calculated most stable configurations of PAQS interacted with Li2S and Li–S−. (b) Schematic of nano-S:rGO:PAQS composite thin film cathode. (c and d) Cross-sectional SEM images and (e) images of flexible nano-S:rGO:PAQS film. (f) Cycling performance of nano-S/rGO (on aluminium foils), nano-S:rGO (film), nano-S:rGO:PVP (film), and nano-S:rGO:PAQS (film) cathodes at 0.25C. Reproduced with permission.223 Copyright 2015, American Chemical Society. | ||
Other than these lithium-ion conducting polymers, electrically conductive polymers also facilitate reaction kinetics of flexible composite electrodes. Milroy and Manthiram adopted a conductive, elastic, and electroactive nanocomposite binder composed of polypyrrole and PU (PPyPU) to reinforce sulfur cathode supported on flexible carbon felt substrates.224 The rational combination of PPyPU and carbon felt enabled the electrode to withstand repeated deformation of bending/rolling and exhibit more favorable reaction kinetics than that using insulating PVDF binder. As-obtained flexible sulfur cathode, with a sulfur loading of 3.2 mgsul cm−2, delivered stable capacities of ∼1000 mA h gsul−1 at C/3 rate for over 100 cycles; however, the capacity based on the weight of the whole electrode could not be appropriately estimated because of the lack in necessary data, i.e., the areal density of carbon felts. Nevertheless, this work demonstrated great promise for practical utility due to the simplicity of one-pot synthesis and availability of raw materials at industrial scale. Wang et al. also employed a poly(3,4-ethylene-dioxythiophene):poly(styrene sulfonate) conductive binder to glue graphene encapsulated hollow sulfur particles for flexible sulfur composite cathodes, yielding an excellent mechanical flexibility and a high initial capacity of 1060(678) mA h gsul(ele)−1 at 0.1C.225
All the above examples mainly focus on enabling or enhancing the use of carbon/sulfur composites in flexible sulfur-based cathodes through the incorporation of multifunctional polymers. Sulfur itself, on the contrary, can also form flexible composite materials with polymers but in absence of other components. Kumta and colleagues reported an electrospinning synthesis of flexible sulfur wires (Flex-SWs) and yarns, which was expected to be an attractive platform for miniaturized device applications such as textile batteries.226 The polymer (PS)–S interface served as a physical barrier to reduce dissolution of polysulfides, thus enabling stable cycling of Flex-SW cathode (after being coated with a lithium-ion conductor) with a capacity of ∼650 mA h gsul−1 at 1.22 mA cm−2 (∼0.18C) and an ultralow cyclic decay rate of ∼0.003% for 50 cycles. The sulfur content was not high (30 wt%) in Flex-SWs, but it could be possibly improved through the incorporation of conductive polymers during electrospinning.
In summary, there have been extensive investigations on polymer-glued flexible composite electrodes, in which polymers are introduced to achieve sufficient level of mechanical flexibility and adhesion. Advantages of these polymer-reinforced electrodes include (1) simple, scalable, and continuous fabrication techniques such as slurry coating and roll pressing, which are scalable and highly compatible with current battery or supercapacitor plants; (2) cheap and off-the-shelf raw materials like commercial binders (e.g., PTFE, PVDF, and SA) and carbon materials (e.g., carbon blacks and carbon felts), making it less dependent on expensive carbon nanomaterials (e.g., CNTs and graphene); and (3) some unique attributes endowed by polymer additives (e.g., polysulfide affinity and electrical/ionic conductivities). However, the disadvantages cannot be ignored either: extra binders always account for 10–20 wt% of electrode weight, resulting in relatively low sulfur mass fraction (<50 wt%) in the whole electrode, which is detrimental to the gravimetric energy density of an actual device. Therefore, it is believed that polymer-glued flexible composite electrodes can be improved by decreasing the amount of polymer binders without sacrificing mechanical tenacity or designing electroactive or electrically conductive polymer additives.
2.1.2.2. Polymer-supported flexible composite cathodes. Different from aforementioned polymer-glued flexible composite electrodes, in which polymer and other components are homogeneously dispersed, polymer-supported flexible composite cathodes are described in this section. These cathodes are featured with spatial separation between a polymer substrate and sulfur composites and the polymer is mainly utilized for mechanical support.
Note that a porous polymer membrane naturally exists in rechargeable lithium batteries. Therefore, it can be directly used as flexible substrates for the deposition of conductive and active materials. Li and Cheng's group proposed a flexible integrated structure of sulfur and graphene on a polypropylene (PP) separator for Li–S batteries (Fig. 18a).227 A thin graphene layer was first deposited on the PP separator (denoted as G@PP separator) through a large-scale slot coating technique, not only allowing better adhesion of sulfur layer on its rough surface but also acting as an upper current collector and a physical barrier for polysulfide utilization and localization. The PP separator afforded the integrated electrode (denoted as S–G@PP) excellent mechanical robustness with a fracture stress and a strain of 30 MPa and 65%, respectively, both much larger than those of a sulfur-coated graphene film (G–S, 6.0 MPa and 3.3%) (Fig. 18b). More importantly, the electrical conductivity of S–G@PP remained around 8.0 S cm−1 for over 50000 cycles of repeated bending (Fig. 18c). Besides the excellent flexibility, the integrated S–G@PP electrode exhibited favorable electrochemical performance, including a high capacity of 1278(∼588) mA h gsul(ele)−1 at 0.3 A gsul−1 (∼0.18C), superior rate performance of 512(∼236) mA h gsul(ele)−1 at 12 A gsul−1 (∼7.2C), and stable cycling with capacity retention of 71% for 500 cycles.
 | ||
| Fig. 18 Polymer-supported flexible sulfur cathodes. Flexible integrated S–G@PP: (a) schematic of electrode configuration using integrated S–G@PP; (b) stress–strain curves of S–G@PP and (inset) G–S films; (c) electrical conductivity (measured upon repeated bending for 50000 cycles) of flexible S–G@PP electrode. Reproduced with permission.227 Copyright 2014, Wiley-VCH. Flexible S–PDMS/GF: (d) schematic for the fabrication of PDMS/GF and S–PDMS/GF electrodes; (e) stress–strain curves of PDMS/GF and S–PDMS/GF; (f) electrical conductivity (measured upon repeated bending for 22000 cycles) of flexible S–PDMS/GF electrode. Reproduced with permission.232 Copyright 2014, Elsevier. | ||
Another prototype is polymer-supported graphene or graphite foam (GF). The controllable CVD synthesis of GFs, replicated from 3D catalytic porous metal foams like Ni foams, was first developed by Cheng and Ren's group in 2011.228 A GF is composed of 3D interconnected framework of high-quality CVD-grown few-layer graphene, possessing many fascinating properties such as a high electrical conductivity of 10–20 S cm−1 (for bulk GFs; 1.36 × 104 S cm−1 estimated for graphene within GFs), an extremely light weight (∼0.10 mg cm−2 with a thickness of ∼100 μm), and an ultrahigh porosity of ∼99.7%. Moreover, it is pliable and bendable so as to be widely employed as electrode materials in flexible energy storage devices.229,230 In Li–S battery research, Xi et al. also demonstrated the great potential of GF/S composites as binder-free electrodes, which, however, lacked sufficient flexibility.231 This might be attributed to the fragile skeleton of GFs after being loaded with a huge amount of sulfur.
To address this issue and realize a truly flexible GF-based sulfur cathodes, Zhou et al. first deposited a thin layer of poly(dimethyl siloxane) (PDMS) on one side of graphene within GFs and then infiltrated a well-mixed sulfur/carbon slurry into the PDMS/GF, obtaining a freestanding, 3D interconnected cathode with high sulfur loadings of up to 10.1 mgsul cm−2 (Fig. 18d).232 The elastic coating of PDMS rendered the composite framework with remarkable flexibility and it could be bent into arbitrary shapes without fracture. After being filled by the sulfur slurry (denoted as S–PDMS/GF), the composite foam exhibited an enhanced stiffness with the modulus increasing from 0.06 to 0.42 MPa, but the maximum elastic strain decreasing from ∼180% to 20%, which was still sufficiently good (Fig. 18e). Dynamic electrical properties were further probed to be almost unchanged (∼1.25 S cm−1) during 22000 cycles of bending (Fig. 18f). With an extremely high sulfur loading of 10.1 mgsul cm−2, the flexible S–PDMS/GF delivered remarkable capacities of ∼1330(∼665) and ∼450(∼225) mA h gsul(ele)−1 at 0.3 and 6.0 A gsul−1 (∼0.18 and 3.6C), respectively, and was able to be reversibly cycled for 1000 cycles. Such a concept of designing a polymer-supported GF-based flexible composite cathode was further explored by He and Chen's group to fabricate highly flexible PDMS-encapsulated 3D Li2S/GF electrodes, which exhibited a capacity of 775(388) mA h gLi2S(ele)−1 after 300 cycles at 0.2C, corresponding to capacity retention of 87.7%.233
In summary, the number of reports for polymer-supported flexible composite electrodes is not as large as those for polymer-glued ones. Compared to these homogeneously mixed polymer composites, the role of polymers in the supported flexible electrodes is more concentrated on mechanical support. As a consequence, the polymer-supported flexible composite cathode is much simpler in its structure and fabrication owing to the use of commercially available flexible polymer substrates and scalable coating techniques. These two favorable characteristics give rise to fruitful opportunities for polymer-supported flexible composite cathodes, which are under less intensive investigation at present. These opportunities include but are not limited to (1) making use of more elastic polymer substrates to realize a stretchable Li–S battery; (2) adopting polymer encapsulation that is either transparent, biocompatible, or stimulus responsive for developing multifunctional, smart, and flexible Li–S batteries; (3) replacing the coating technique by printing, which has been explored for various sulfur electrodes by Manthiram's group,234 to fabricate electrode arrays on arbitrary substrates for potential miniaturized device applications.
2.1.2.3. Inorganic-based flexible composite cathodes. Unlike carbon and polymers, bulk inorganic materials are normally incapable of constructing flexible electrodes alone, because they are inflexible and a lot of them are electrically insulating. Therefore, in most cases, inorganic active materials are composited with carbon and/or polymers to attain reliable mechanical and electrochemical properties for high-performance flexible energy storage devices.10 In Li–S batteries, elemental sulfur is the major active material that reversibly stores electricity, but inorganic materials, as various functional enhancers, still play a pivotal role in enhancing sulfur composite cathodes as a broad range of inorganic components, such as transition metal oxides,235–242 hydroxides,243,244 sulfides,245–248 carbides,249,250 and nitrides,251–253 have proved:
(1) Almost all inorganic materials, connected by ionic or polar covalent bonds, possess favorable polar surfaces with strong binding affinities to polar polysulfide intermediates and therefore are beneficial for polysulfide adsorption and shuttle inhibition.35,254
(2) Some of them, mostly electrically conductive and polar ones (e.g., cobalt disulfides (CoS2),246 titanium monocarbides250), have the ability to enhance electrochemical reaction kinetics of both polysulfide interconversion and Li2S formation, resulting in low polarization, high energy efficiencies, and good rate capabilities.
(3) Several examples (e.g., indium-doped tin oxides,238 molybdenum disulfides,248 and CoS2246) have proved to regulate the spatial deposition of Li2S on stronger binding sites and reduce the size of Li2S nuclei/deposits, making Li2S precipitates less prone to loss contact through detachment.
(4) Quite a few cases have witnessed a transition of discharge reaction pathways from solution to surface (e.g., Magnéli-phase titanium oxide,236 MXene-phase titanium carbides249) or even a surface-bound thiosulfate/polythionate redox (e.g., manganese dioxides (MnO2)239).
(5) A series of transition metal sulfides have been demonstrated to efficiently catalyze Li2S oxidation upon charging, which guarantees rechargeability.247
Because of these desirable benefits, building inorganic-based polar hosts is burgeoning as one of the most popular and important strategies for Li–S batteries. Correspondingly, inorganic-based flexible composite electrodes are attracting increasing interest for flexible Li–S batteries. Most of them consist of inorganic nanostructure-decorated carbon, for which synthetic methods are essentially the same as discussed in (Section 2.1.1). The primary purpose of introducing inorganic components is to improve adsorption of polysulfides and mitigate their rampant dissolution. Zeng et al. prepared Cu-embedded PCNFs through electrospinning, obtaining a freestanding sulfur cathode (denoted as S@PCNFs-Cu) with good flexibility.154 The synergistic effects from the physical confinement in PCNFs and chemical immobilization offered by embedded Cu nanoparticles prevented dissolution of polysulfides during cycling, thereby leading to much improved cycling performance of 680(354) mA h gsul(ele)−1 after 100 cycles at 0.05 A gsul−1 (∼0.03C), against to 396(154) mA h gsul(ele)−1 for S@PCNFs. Zhang et al. subjected tetrabutyl titanate-treated commercial filter papers to carbonization and prepared titanium dioxide (TiO2)-grafted carbon paper (CP@TiO2) electrodes, which were further loaded with sulfur after being drop casted with S/CS2 solution.255 The decoration by TiO2 helped to trap polysulfides, but showed no significant influence on the flexibility of CP films. As a consequence, the flexible CP@TiO2-sulfur electrode exhibited favorable capacity retention of 850(340) mA h gsul(ele)−1 after 200 cycles at 0.5C. Similarly, Wang and colleagues embedded ultrafine TiO2 nanoparticles into NPCFs via their previously reported electrospinning method, yielding a prolonged cycling performance of 618(340) mA h gsul(ele)−1 after 500 cycles at 1.0C.256 Xiong and colleagues hydrothermally grew tungsten disulfide (WS2) nanosheets on a carbon cloth to obtain a freestanding C@WS2/S cathode, which delivered a very high capacity of 1581(174) mA h gsul(ele)−1 at 0.1C and a remarkably long cycling life of 1500 cycles with capacity retention of 89% at 2.0C.257 The superior cycling performance was ascribed to the intrinsic adsorptivity of WS2 to polysulfides, as well as the dense growth and vertical alignment of WS2 nanosheets to fully expose the abundant adsorptive sites. Xu and Manthiram reported a hybrid paper electrode consisting of hollow cobaltosic sulfide (Co3S4) polyhedral, ACNFs, and sulfur powder.258 This paper electrode was prepared through vacuum filtration, showing good flexibility without cracking on bending. A high initial capacity of 953(505) mA h gsul(ele)−1 at 1.0C was achieved with a sulfur loading of 2.5 mgsul cm−2 and the capacity was remained at 610(323) mA h gsul(ele)−1 after 450 cycles. Even with a high loading of 13.5 mgsul cm−2, the Co3S4/ACNF/S electrode delivered a high areal capacity of 13 mA h cm−2 at 0.3C.
Besides these inorganic-based flexible cathodes made from solid sulfur materials, there are a huge class of freestanding, binder-free, and flexible inorganic/carbon hybrids reported for dissolved polysulfide catholytes. In these hybrid electrode scaffolds, inorganics materials are either directly grown on carbon substrates to build integrated electrodes (e.g., CNT/ACNF@MnO2 paper,259 Co3O4 nanowire (NW) arrays on carbon cloth,260 electrospun CNFs decorated with indium-doped tin oxide238 and chloride nanoparticles261) or synthesized elsewhere and subsequently supported on freestanding carbon architectures (e.g., TiO2 NWs embedded in graphene film,262 rhenium disulfide263 or phosphorene264 on CNF paper). All these designs achieve good flexibility, electrode integrity, conductivity, and polysulfide immobilization simultaneously, although polysulfide catholytes in the liquid state are not very suitable for flexible device applications.
In order to realize dual physical/chemical confinements to polysulfide dissolution/diffusion, inorganic materials have also been employed as ideal encapsulation of flexible sulfur cathodes. Yu et al. demonstrated that hydrothermally synthesized flexible rGO–S aerogels were modified by an ultrafine oxide layer through an atomic layer deposition (ALD) method.265 The ALD-coated oxide layer acted as both a chemical trap and a physical barrier to minimize the contact between polysulfides and electrolyte, significantly improving capacity retention from 63% to 89% with a capacity of 845(465) mA h gsul(ele)−1 obtained at 0.2C after 100 cycles. Similarly, Chen et al. employed ALD to preferentially grow a thin encapsulation layer of alumina (Al2O3) on Li2S particles rather than graphene in a freestanding, binder-free Li2S–rGO sponge electrode.266 The integrated Al2O3–Li2S–rGO electrode delivered a highly reversible capacity of 736(427) mA h gLi2S(ele)−1 at 0.2C after 150 cycles, 59% higher than that without Al2O3. Such an enhanced cycling stability could also be ascribed to dual physical/chemical confinements.
Engineering desirable hollow micro-/nanostructures for flexible inorganic-based composite materials is an intriguing and straightforward way to prove the superiority of these polar materials in encapsulating sulfur species and promoting their conversion.267 Cui and coworkers designed a 3D interconnected graphene cage with an ultrathin and sulfiphilic coating of electrodeposited nickel phosphosulfides (Ni–P–S@G cage) for freestanding Li2S cathodes.268 The incorporation of Ni–P–S not only promoted adsorption of polysulfides but also favored impregnation of Li2S with small sizes and uniform distribution, which further contributed to lowering the energy barrier and reducing polarization, while the combination of graphene cages and conductive Ni–P–S ensured a 3D interconnected conductive framework without any other assistance. As a result, the Li2S@Ni–P–S@G cage electrode outperformed all other control samples with only graphene cages or nickel sulfides/phosphides, displaying an outstanding electrochemical performance such as high capacities of 980(594) and 543(329) mA h gLi2S(ele)−1 at 0.1 and 4.0C, respectively, a good cycling stability for over 300 cycles with an extremely low cyclic decay rate of ∼0.076%, and a high-loading capability of Li2S (up to 5.2 mgLi2S cm−2).
Very recently, Lin and Peng's group reported a facile confinement conversion strategy to fabricate highly foldable metal–organic framework (MOF)/CNT thin films for flexible sulfur cathodes (Fig. 19).269 CNTs interpenetrated through MOF crystals and interwove the electrode into a stratified structure to provide both conductivity and structural integrity, while MOFs offered strong sulfur confinement via both inherent topotactic porous structure and possible chemical affinities of central metal ions (Fig. 19a). The type of MOF crystals could be easily tuned by altering the precursors, making the foldable MOF/CNT film an ideal material platform to investigate encapsulation effects of MOFs at high sulfur loadings of up to 11.3 mgsul cm−2 and in special working conditions like bending (Fig. 19b). In all these cases, the foldable MOF/CNT/S electrode yielded stable cycling for at least 50 cycles (Fig. 19c). Most predominantly, a high areal capacity of 3.5 mA h cm−2 sustained for more than 200 cycles, corresponding to a specific capacity of ∼760(532) mA h gsul(ele)−1 (Fig. 19d). The foldable MOF/CNT composite films were further carbonized into various porous carbon/CNT films, which were also demonstrated to be superior for flexible sulfur cathodes.270,271
 | ||
| Fig. 19 Inorganic-based flexible sulfur cathodes. (a) Schematic for the confinement-conversion synthesis of S8-loaded MOF/CNT composite thin films from metal hydroxide nanostrand (MHN)/CNT precursors. (b) Photographs of foldable HKUST-1/CNT thin film and corresponding Li–S pouch cells lighting a light-emitting diode (LED) at different bending angles. Cycling performance of (c) Li–S pouch cell at different bending angles and (d) the cell with a sulfur loading of 4.57 mgsul cm−2. Reproduced with permission.269 Copyright 2017, Nature Publishing Group. | ||
Besides the abovementioned inorganic/carbon hybrid electrodes, porous metal foams also showed potential as flexible substrates for sulfur cathodes without the assistance of carbon. Chen and coworkers prepared monodisperse sulfur nanodots (2 nm in average size) on pliable Ni foams through a room-temperature electrodeposition method with sulfur loadings ranging from 0.21 to 4.79 mgsul cm−2.272 With an optimal loading of 0.45 mgsul cm−2, the flexible cathode delivered a very high initial capacity of 1458 mA h gsul−1. Nevertheless, the huge density of Ni foams (estimated to be ca. 20–30 mg cm−2) restrained the actual capacity to lower than 30 mA h gele−1, which undoubtedly asked for lighter materials. Moreover, the backbone of porous metal foam was rigid despite its macroscopic flexibility, raising the risk of rupture after long-term mechanical deformation.
In summary, inorganic materials are not as good as nanocarbon or polymers in providing desirable mechanical flexibility. However, the incorporation of polar inorganics has been proved to be very efficient in enhancing electrochemical performance of sulfur cathodes. Therefore, judiciously designing flexible composite materials that inherit the advantages of both inorganic components (e.g., polysulfide binding affinity) and nanocarbon (e.g., electrical conductivity, structural integrity, and mechanical flexibility) is regarded as a holistic strategy toward high-performance flexible sulfur cathodes. The hybridization of inorganics and carbon will also render synergistic enhancement in enhancing polysulfide immobilization and conversion as long as ideal structures and composites are rationally engineered. Future progress leads to new chemistry and understanding of the synthesis, electrochemical/chemical redox, and interfacial interaction.
2.2. Flexible lithium-based anodes
Lithium metal is another cornerstone material of Li–S batteries due to its exceptionally high theoretical gravimetric and volumetric specific capacities of 3862 mA h g−1 and 2062 mA h cm−3, respectively, as well as a very negative potential of −3.040 V vs. the standard hydrogen electrode.273,274 Lithium foils, with a thickness of 50–200 μm, are common anodes in both regular and flexible Li–S batteries. Like other metal thin foils, lithium foils indeed exhibit a certain degree of flexibility when subjected to mild mechanical deformation. However, at harsher conditions such as bending and twisting, lithium metal is too soft (with a Mohs hardness of only 0.6) and plastic (with a ductility of ∼50–70%) to be recovered unless very thin lithium foils and lithium wires are employed, the fabrication of which is very challenging though. The stress induced by local extrusion results in permanent distortion such as wrinkling, creasing, or buckling.The undesirable distortion, in turn, is detrimental to stable and safe operation of Li–S batteries and other lithium metal batteries, owing to a few intrinsic intractable issues of lithium metal anodes: (1) infinite volume change, originating from the “hostless” feature of lithium metal during plating/stripping; (2) compositionally non-uniform, structurally unstable, and temporally variable solid electrolyte interphase (SEI), which is referred to as an electrically insulating but ionically conducting layer at lithium/electrolyte boundary; (3) lithium dendritic growth, induced by constant and dynamic changes in SEI and transport behaviors across the lithium/electrolyte boundary; and (4) vigorous and exothermic reactions between lithium and electrolyte components, potentially leading to unwanted gas evolution, electrolyte depletion, and thermal runaway.274–277
The aforementioned issues had halted the commercialization of lithium metal anodes for a long time. But with the recent emergence of lithium metal batteries that have higher energy densities than LIBs, research on lithium metal anodes is being revived277 and has led to a revolution in electrode structures,278,279 interfaces,280–283 and electrolyte recipes.284–286 Many strategies have been explored and most of them suggest that a composite lithium-based anode performs better than pristine lithium metal. In particular, engineering of a 3D nanostructured lithium host and stable SEI or artificial protection is suggested to be very effective and efficient. The combination of these two strategies is believed to pave a viable path toward reliable lithium anodes. For the design of flexible lithium-based anodes, the key consequently lies in the efficient construction of flexible lithium hosts and flexible interfaces, which will be discussed separately in this section.
Unlike the abundance of flexible sulfur-based cathodes, flexible lithium-based anodes are rarely reported because of the difficulties in handling highly reactive lithium metal and lack of suitable host materials. Cui and coworkers designed a layered lithium–rGO composite electrode by infusing molten lithium into an rGO film with nanoscale interlayer gaps (Fig. 20a–d).290 The rGO film possessed several remarkable advantages for sealing lithium metal including (1) superior structural stability to mitigate the volume fluctuation, (2) favorable “superlithiophilicity”, which was endowed by both residue functional groups and the nanoscale capillary effect, to facilitate uniform infusion and electrochemical deposition of lithium metal, and (3) the top rGO cap layer as an artificial interface to stabilize SEI. Moreover, the lithium–rGO film showed excellent flexibility to be coiled and twisted (Fig. 20e). Because of the lightweight of rGO host, this flexible composite lithium anode exhibited a high capacity of up to 3390 mA h gele−1, corresponding to ∼88% of the theoretical value. At a current density and capacity of 3.0 mA cm−2 and 1.0 mA h cm−2, respectively, a low overpotential of ∼80 mV was retained for more than 2.5 × 105 s, along with dendrite-free lithium deposition. Yu and Yao's group developed a flexible scaffold consisting of Cu NWs (Fig. 20f), which allowed as high as 7.5 mA h cm−2 of lithium to be plated without dendritic growth.291 In contrast to planar Cu foils, the interconnected Cu NW network exhibited a larger surface area, thus considerably decreasing ion flux density and enhancing homogeneity of lithium-ion-flux distribution (Fig. 20g). As a result, lithium can be uniformly plated and a high Coulombic efficiency of 98.6% was maintained during 200 cycles at 1.0 mA cm−2. The flexible Cu NW hosts were further enhanced by electrodepositing a lithiophilic Ni layer on Cu to fabricate Cu–Ni core–shell NW networks, which demonstrated a high capacity of 1882 mA h gele−1.292
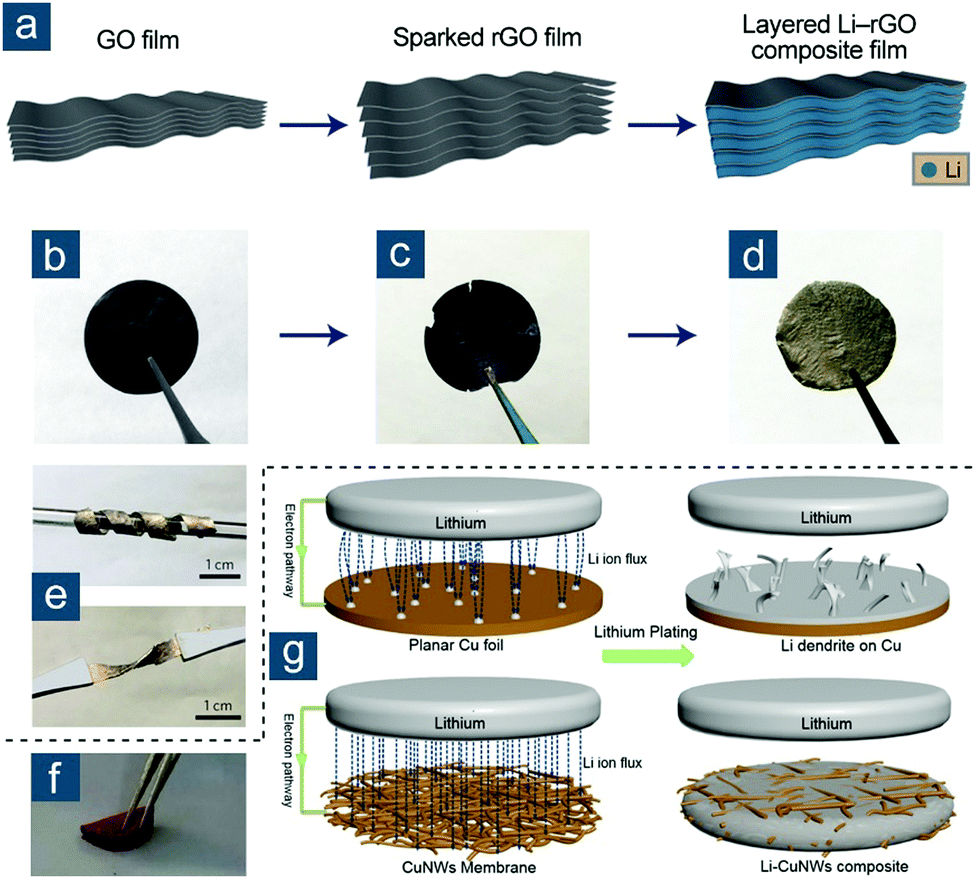 | ||
| Fig. 20 Flexible lithium hosts. Flexible layered lithium (Li)–rGO composite electrode: (a) schematic for the synthesis from GO film (left) to sparked rGO film (middle) to layered Li–rGO film (right) and (b–d) corresponding photographs; (e) photographs of layered Li–rGO strip coiled around a glass rod (upper) and twisted by two tweezers (below) to show superior flexibility. Reproduced with permission.290 Copyright 2016, Nature Publishing Group. Flexible lithium scaffold consisting of copper NWs (CuNWs): (f) photograph of flexible and bendable CuNW membrane; (g) schematic of lithium-ion-flux distribution and lithium metal plating models on (upper) planar Cu foil and (below) CuNW membrane. Reproduced with permission.291 Copyright 2016, American Chemical Society. | ||
Other host materials, although lacking in unambiguous demonstration of flexibility in corresponding studies, have long been seen as flexible substrates in energy storage applications and demonstrated for lithium metal anodes. These materials include pliable porous metal meshes,293 graphene monoliths,294 electrospun PNFs/CNFs,295–298 and various surface-modified CNFs/CNTs.299–301 Nevertheless, strict validation of their flexibility, especially when they are incorporated with lithium, is required. Since there are many flexible Li2S-based cathodes, non-metallic anode could be an alternative to lithium metal anode and fabricating flexible nonmetallic anodes is easier. In fact, either intercalated compounds (e.g., graphite) or alloys (e.g., lithiated silicon/germanium/tin) have inherent “host” structure (i.e., the graphene plane for graphite and the nonmetal atoms for alloys) to accommodate lithium and thus can broadly be classified into “lithium hosts”. For example, silicon, regarded as one of the most important high-capacity anode materials, has a theoretical capacity of 2011 mA h g−1 when fully lithiated (Li4.4Si), just below that of lithium metal. Flexible silicon anodes have been pursued with substantial progress,302,303 so it will be very interesting and straightforward to build flexible Li–S full cells based on the Li2S/Si pair. Nevertheless, it should also be noted that the gravimetric energy density will be concurrently compromised by ∼40% (theoretically) due to the decrease in both specific capacity and working voltage.
One approach for SEI regulation is to tune the electrolyte recipe.59,276,284,286 Nevertheless, liquid electrolyte engineering is not very suitable for flexible batteries because of the potential risk of electrolyte leakage as we mentioned before. Another strategy is to protect the lithium metal by an “artificial” interface.280,281,283 For flexible lithium metal anodes, such an artificial interface should meet several critical features: (1) good flexibility; (2) high ionic conductivity but low electrical conductivity; (3) spatial homogeneities in composition, structure, ion-transport resistance, and mechanical properties; (4) strong adhesion to lithium metal; and (5) superior chemical and thermal stabilities.
Cui and colleagues pioneered the concept of flexible artificial interface by placing an interconnected hollow carbon nanosphere film on the lithium metal.280 Such a film was synthesized by carbon coating a colloidal multilayer opal structure of PS particles, which could be facilely removed upon heating (Fig. 21a). The as-obtained film was composed of a thin amorphous carbon layer, which was chemically stable against lithium metal and possessed low ion-transferring impedance. More importantly, the amorphous carbon had a Young's modulus of ∼200 GPa. In combination with the unique interconnected hollow structure (Fig. 21b), a superior flexibility of this film was obtained (Fig. 21c and d). The static interface between this film and electrolyte segregated the vulnerable lithium from the electrolyte, thus leading to a high Coulombic efficiency of 99% that was stably preserved for over 150 cycles at 1.0 mA cm−2. Since then, various flexible artificial interfaces were proposed, including polymer (e.g., PDMS,305 blended Nafion/polyvinylidene difluoride,306 poly(ethyl α-cyanoacrylate)307), adaptive polymer/supramolecule (e.g., a hydrogen-bonding self-healing polymer (SHP),308 boron-mediated cross-linked PDMS),309 and polymer/inorganic composites (e.g., cuprous nitride (Cu3N)/styrene butadiene (SBR),310 a film of SiO2@PMMA core/shell nanospheres).311 Among them, Zheng et al. demonstrated a soft and flowable SHP coating that is highly viscoelastic, pinhole-free, and self-healing to guide uniform lithium deposition.308 The dynamic adaptiveness was endowed by a unique supramolecular structure with a branched diacid/triacid backbone and myriad hydrogen bonding sites (Fig. 21e). As a consequence, the SHP exhibited liquid-like characteristics and superior stretchability (Fig. 21f), allowing quick and complete relaxation after being strained but with no crack. Even at a high current density of 5.0 mA cm−2, dense deposition of dendrite-free lithium was obtained under protection of SHP. Liu et al. further proved such a concept by designing a dynamic polymer, i.e., silly putty (boron-mediated cross-linked PDMS), that can be reversibly switched between its “solid” and “liquid” properties in response to the rate of lithium growth.309 Uniform surface coverage and dendrite suppression were afforded. These adaptive polymers are very attractive due to their dynamic responses to morphological changes in interfaces; however, they have to be swelled by liquid electrolytes to conduct lithium ions, restraining the practicality of being paired with solid electrolytes. To improve inherent ion conductivity of the artificial interface, Liu et al. designed a rationally hybridized interface of mechanically strong lithium-ion-conducing Li3N nanoparticles embedded within a flexible matrix of SBR.310 Li3N, one of the fastest lithium-ion conductors with an ionic conductivity of ∼10−4–10−3 S cm−1 at room temperature, was in situ generated through spontaneous reaction between presynthesized Cu3N nanoparticles in the SBR matrix and lithium metal (Fig. 21g). The synergistic effect afforded by ion-conducting Li3N and flexible SBR enabled Li|Cu half cells to stably operate with an average Coulombic efficiency of 97.4% for 100 cycles at 1.0 mA cm−2. More importantly, such a flexible protective film was further conformally coated on a 3D lithiophilic host to fabricate a lithium metal anode with specific and areal capacities of ∼2000 mA h gele−1 and 10 mA h cm−2, respectively, which was further paired with a lithium titanate counter electrode (3.0 mA h cm−2) and exhibited performance comparable to a 750 μm-thick lithium foil but much better than a 50 μm-thick lithium foil and electrodeposited lithium (10 mA h cm−2) without protection. The aforementioned efforts are remarkable as they demonstrated highly effective dendrite suppression and highly efficient lithium metal anodes. Nevertheless, the feasibility of adopting them in flexible Li–S batteries has to be further examined in a polysulfide-rich environment and with specifically designed lithium hosts. The presence of corrosive polysulfides will notably influence the mechanical and physicochemical properties of these flexible artificial interfaces.
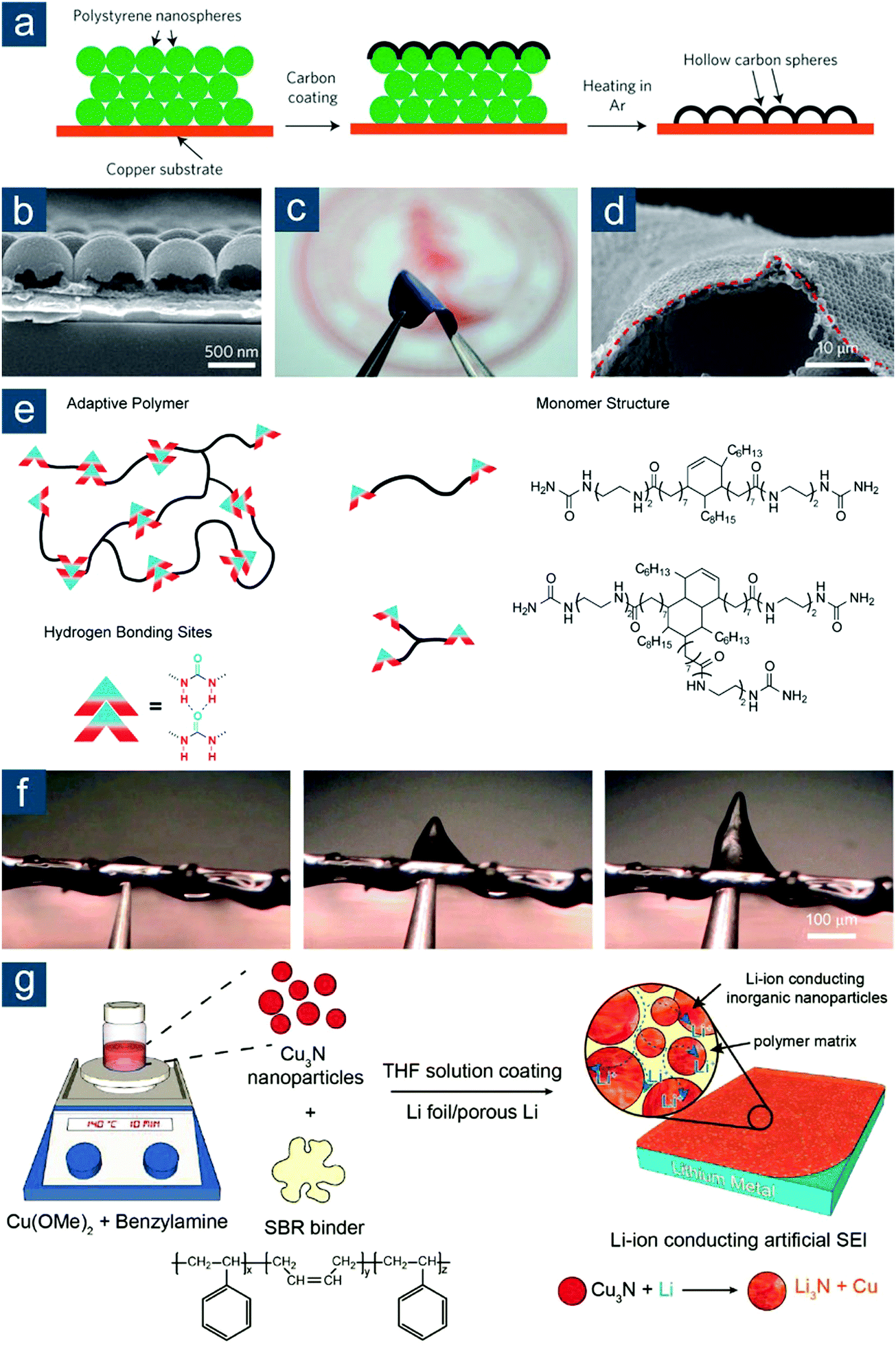 | ||
| Fig. 21 Flexible interfaces. Interconnected hollow carbon spheres: (a) schematic for the preparation of Cu electrode modified by hollow carbon sphere film; (b) cross-sectional SEM image showing hollow spheres; (c and d) digital and SEM images showing flexibility. Reproduced with permission.280 Copyright 2014, Nature Publishing Group. Adaptive SHP coating: (e) molecular structure of SHP with black lines showing fatty diacid/triacid backbones and red-blue boxes showing urea hydrogen-bonding sites; (f) pictures showing mechanical piercing of SHP. Reproduced with permission.308 Copyright 2016, American Chemical Society. Hybrid Cu3N/SBR artificial SEI: (g) schematic for the fabrication. Reproduced with permission.310 Copyright 2016, Wiley-VCH. | ||
In summary, flexible lithium-based anode is in its infancy, dramatically different from its counter electrode. There are also scarce reports of a flexible lithium-based anode employed in a flexible Li–S device, except for Xie et al.'s effort of building a Li–S cell with flexible CNT films adopted on both sides.312 Although flexibility of each electrode and the whole cell was not strictly proved, it paved a possible path toward flexible Li–S full cells. Despite the very few examples discussed in this section, they all revealed the great promise of designing flexible hosts and interfaces for flexible lithium-base anodes. Based on the current progress, several considerations are provided for the future development: (1) the flexibility of not only the host material/interface but also the whole composite electrode after lithium impregnation should be quantitatively evaluated; (2) the utilization (i.e., Coulombic efficiency) and cycling life of lithium metal anode ought to be further enhanced; (3) the interfacial compatibility between the lithium metal anode and the electrolyte, whether in liquid or solid state, must be impeccably dealt with. To realize these, multidisciplinary efforts from electrochemistry, materials chemistry, organic chemistry, physics, nanotechnology, and engineering science, as well as multidomain advancements in electrodes, interfaces, and electrolytes are eagerly required.
2.3. Flexible solid-state electrolytes
To build a reliable and durable Li–S battery, a flexible solid-state or quasi-solid-state electrolyte is indispensable. In conventional Li–S batteries, liquid electrolytes are usually employed, mostly with ether solvents (e.g., 1,2-dimethoxyethane (DME), 1,3-dioxolane (DOL)), which are able to solubilize polysulfides.55 Conventional carbonate-based LIB electrolytes and room-temperature ionic liquids, which are either polysulfide-incompatible (carbonates)313 or sparingly polysulfide-soluble (ionic liquids),314 have been adopted in some special cases. However, the use of these flammable and fluidic liquid electrolytes strictly limits the design of flexible batteries considering the difficulty in robust cell packaging, and probably raises the risk of electrolyte leakage and fire. Thus, solid-state electrolytes (SSEs) are preferable for flexible Li–S batteries due to their inherent safety and reliability.56 Moreover, the challenge of polysulfide shuttle can be addressed through SSEs that either kinetically retard the diffusion of polysulfides or thermodynamically prevent their dissolution.Corresponding to the major constituent, SSEs can be classified into polymer electrolytes, which contain two subcategories of solid polymer electrolytes (SPEs) and gel polymer electrolytes (GPEs), and inorganic electrolytes.55 Aiming at flexible Li–S batteries, polymer electrolytes are preferred because inorganic ones are mainly rigid unless they are composited with flexible substrates or matrices.
SPEs are solvent-free polymer electrolytes in which lithium salts are dissolved and lithium ions migrate via local segmental motions of polymer chains. Moreover, they normally possess good mechanical compliance and interfacial stability against lithium metal. Along with their easy processability into pliable films, they are especially promising for flexible Li–S batteries. The most widely explored polymer matrix of SPEs is poly(ethylene oxide) (PEO), a high-molecular-weight analogue to linear ether solvents with alternate ether groups (–O–) to complex with lithium cations and conduct them. However, PEO is highly crystalline at ambient temperatures; consequently, the segmental movements of PEO chains are restricted, resulting in very low ionic conductivities of 10−7–10−8 S cm−1 for PEO-based SPEs. As a consequence, the cell with a PEO-based SPE has to be operated at a temperature above the melting point of PEO (ca. 63 °C), usually 70–90 °C, which is normally unacceptable for flexible device applications. To overcome this issue, there are two major approaches: introducing an extra liquid phase to plasticize SPEs, namely using GPEs; compositing SPEs with functional fillers to increase the amorphous region.
To maintain mechanical flexibility and shape compliance, one concept is to engineer the inorganic filler. Zhang indicated that increasing the weight fraction of inorganic fillers, i.e., SiO2 aerogels, from 20 to 50 wt% enabled instant adsorption of liquid electrolytes by PEO/SiO2 (50 wt%) membranes without dimensional shrinkage, proving that a large amount of non-swellable inorganic fillers helped to stabilize composite GPEs.317 These composite GPEs possessed an ionic conductivity of ∼10−4 S cm−1 at 30 °C when incorporated with 62 wt% of liquid electrolytes. Li–S cells employing the PEO/SiO2 (50 wt%) composite GPE delivered a high initial capacity of ∼800 mA h gsul−1 at 0.2 mA cm−2. Another promising concept for strengthening GPEs is crosslinking. Choudhury et al. reported a highly stretchable GPE by crosslinking a polyepichlorohydrin (a chlorinated PEO analogue) network using ethylene thiourea and MgO as crosslinkers.318 The effect of ethylene thiourea addition and temperature on mechanical strength and ionic conductivity was thoroughly investigated. The best ionic conductivity of 2.4 × 10−4 S cm−1 was obtained at 25 °C, along with a tensile strength of above 1.2 MPa at an excellent elongation of >600%. The highest capacity was ∼700 mA h gsul−1 at 0.2C.
Alternative materials to PEO were also explored for flexible GPEs. Nair et al. prepared nanocellulose–laden composite GPEs using a thermally induced polymerization process.319 Nanoscale microfibrillated cellulose was introduced as a reinforcing agent to a crosslinked acrylate-based matrix, improving mechanical properties of GPEs and rendering an opaque, freestanding, robust, and non-tacky membrane. This GPE membrane possessed a high ionic conductivity of 1.2 × 10−3 S cm−1 at 20 °C, contributing to favorable electrochemical performance such as a high initial capacity of 1280 mA h gsul−1 at 0.1C and almost unchanged capacity of 730 mA h gsul−1 at 1.0C for over 60 cycles. Kang and He's group described an in situ synthetic strategy for a pentaerythritol tetraacrylate (PETEA)-based GPE with an extremely high ionic conductivity of 1.13 × 10−2 S cm−1 at 25 °C, which was around one to two orders of magnitude higher than those reported for PEO-based GPEs (Fig. 22a and b).320 Moreover, PETEA-based GPEs facilitated the formation of a flexible and conformal passivation layer on the sulfur cathode, inhibiting the shuttle effect and preserving strongly integrated electrolyte/electrode interface (Fig. 22c). As a result, the Li–S cell employing a simple sulfur/carbon black cathode and the PETEA-based GPE exhibited a much enhanced cycling stability and rate performance than a liquid-electrolyte cell, including high capacities of 1210 and 601 mA h gsul−1 at 0.2 and 1.0C, respectively, and a decent capacity retention of 82% (corresponding to a capacity of 530 mA h gsul−1) after 400 cycles at 0.5C. Attributed to its quadruple symmetric geometry, PETEA can be easily crosslinked and polymerized into a robust network, affording favorable mechanical properties for flexible Li–S batteries. A Li–S pouch cell employing the PETEA-based GPE was fabricated and further operated in either flat or bent states, both exhibiting steady capacities of >800 mA h gsul−1 at 0.1C (Fig. 22d and e). In contrast, the cell using liquid electrolytes failed to endure mechanical deformation, as suggested by the drastic capacity decay. To further improve mechanical pliability, Liu et al. fabricated an acrylate-based hierarchical electrolyte through in situ gelation of a PETEA-based GPE into an electrospun PMMA scaffold.321 The freestanding GPE film was quite flexible and had a high ionic conductivity of 1.02 × 10−3 S cm−1, which was attributed to the structural similarity and synergistic compatibility between PETEA and PMMA. The ester-rich structure also displayed excellent polysulfide anchoring ability. Therefore, very promising capacity retention of 80 and 92% was obtained at 0.3 and 3.0C after 500 cycles, corresponding to much higher capacities of 792 and 574 mA h gsul−1 than those obtained on cells with liquid electrolytes, respectively. Wang and coworkers also exerted the advantages of chemical crosslinking in preparing a freestanding trimethylolpropane ethoxylate triacrylate GPE film and further combined it with a CNF interlayer to obtain enhanced electrochemical performance with an initial capacity of 1177 mA h gsul−1 at 0.1C and capacity retention of 65% after 300 cycles at 1.0C.322
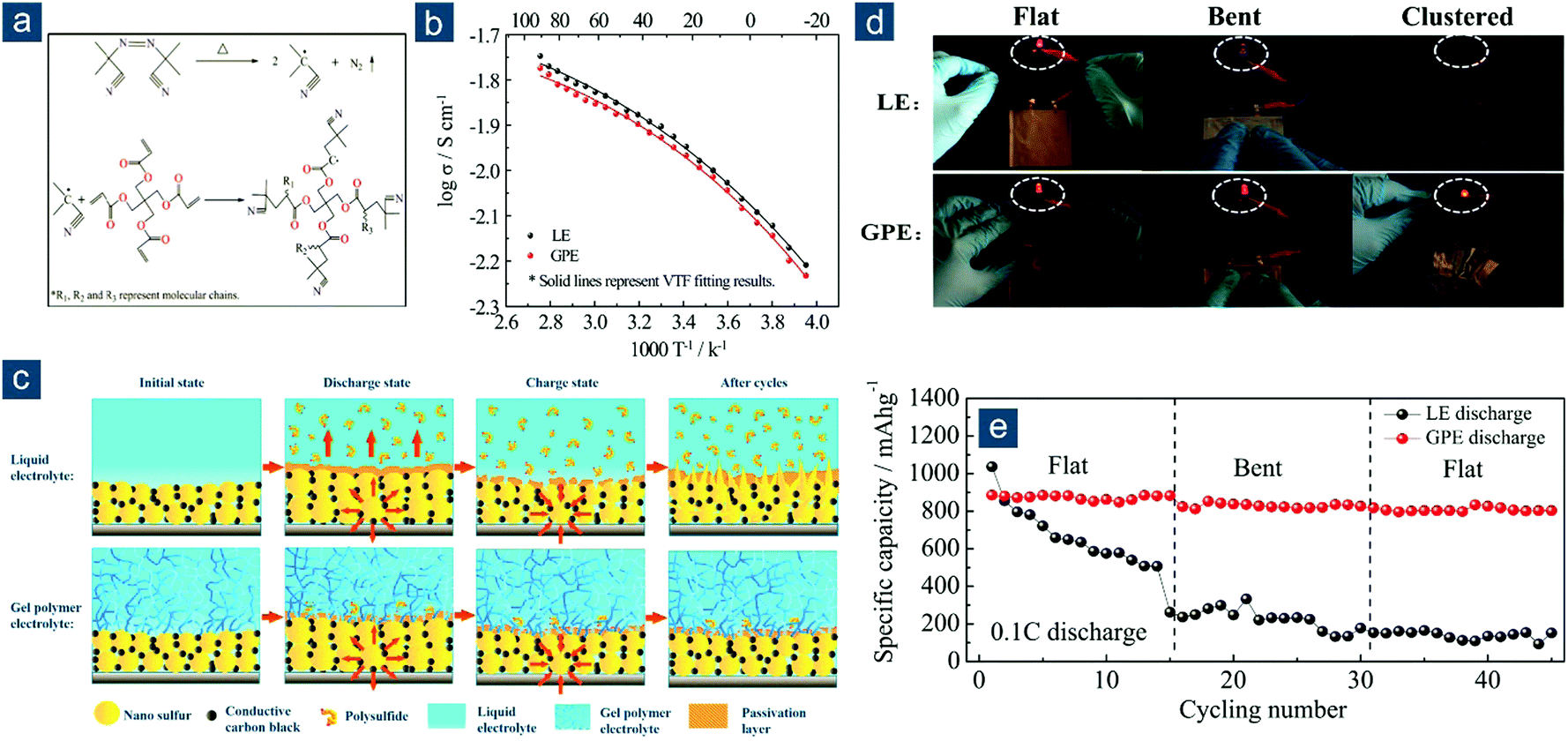 | ||
| Fig. 22 Flexible GPEs for Li–S batteries. (a) Schematically illustrated polymerization mechanism of PETEA monomers. (b) Ionic conductivities for PETEA-based GPE and blank liquid electrolyte (LE, 1 mol L−1 LiTFSI/DOL:DME (1:1 by volume) with 1 wt% LiNO3) at various temperatures. (c) Schematically illustrated immobilization mechanism for polysulfides by capitalizing on PETEA-based GPE. (d) Photographs of red LEDs lit by flexible Li–S pouch cells using LE or PETEA-based GPE under various deformative states. (e) Cycling performances of flexible Li–S pouch cells in flat and bent states at 0.1C. Reproduced with permission.320 Copyright 2016, Elsevier. | ||
It is imperative to improve ionic conductivity of SPEs by delicately designing functional fillers. Lin et al. proposed a concept of utilizing heterocharged halloysite nanotubes (HNTs) as modifiers for preparing flexible and high-ionic-conductivity PEO/LiTFSI SPEs (Fig. 23a).326 Lithium salts could be easily dissociated with lithium cations adsorbed on the negatively charged outer SiO2 surface and anions accommodated in the hollow interior by positively charged Al2O3 walls (Fig. 23b). With 10 wt% HNTs incorporated in PEO/LiTFSI SPE (15:1 mass ratio), an exceptional ionic conductivity of 1.11 × 10−4 S cm−1 was obtained at 25 °C, corresponding to at least two orders of magnitude enhancement (Fig. 23c). Besides, the lithium ion transference number was raised from 0.1 to 0.4. The mechanical strength was predominantly increased from 1.25 to 2.28 MPa at a strain of 400% (Fig. 23d). All these features suggested the great promise of PEO/LiTFSI/HNT composite SPE for flexible Li–S batteries. The Li–S cell employing a polyaniline/carbon/sulfur cathode and the 10 wt% HNT/PEO/LiTFSI SPE steadily delivered capacities of 745 ± 21 mA h gsul−1 at 0.1C for 100 cycles at an ambient temperature of 25 °C.
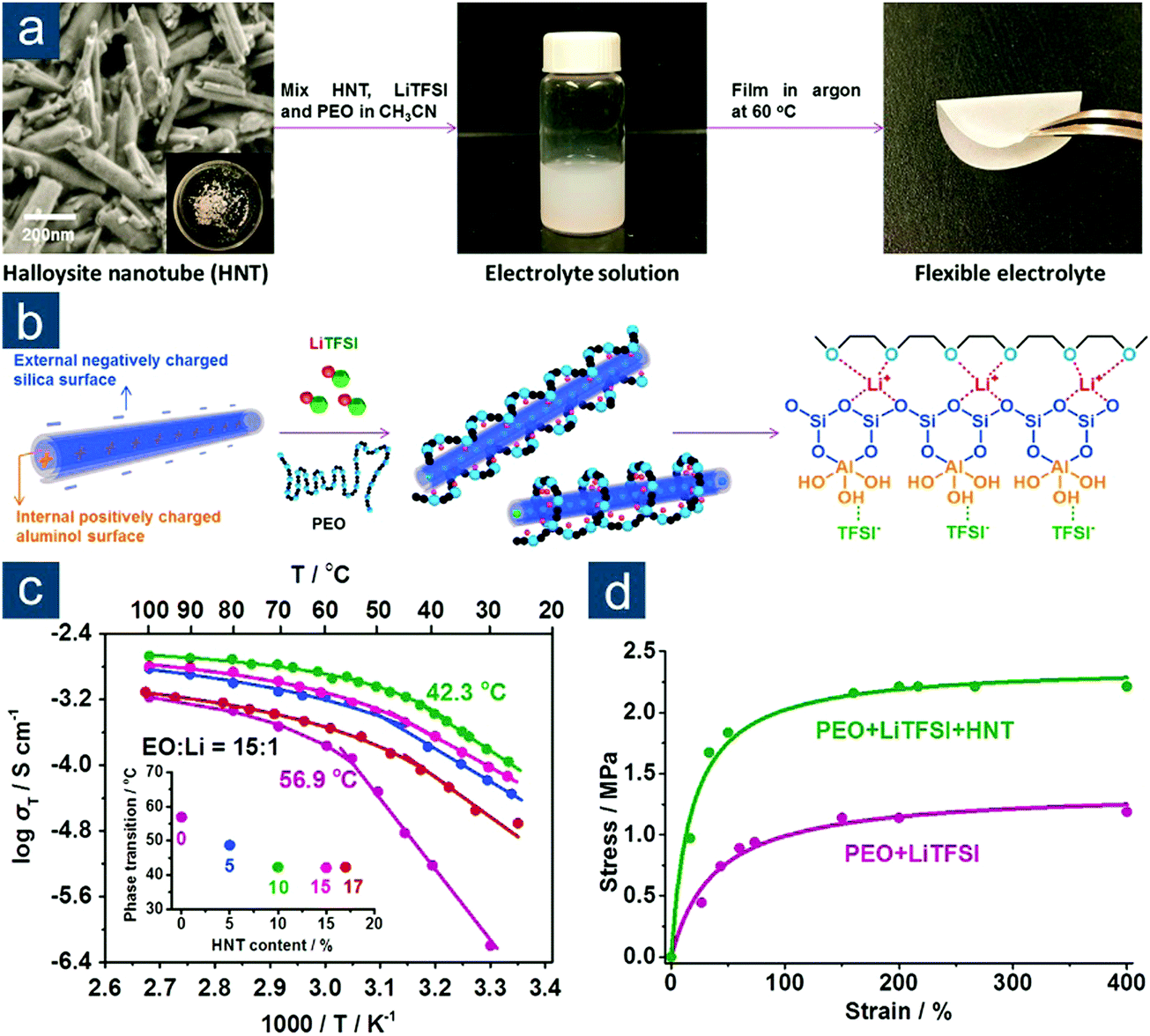 | ||
| Fig. 23 Flexible SPEs for Li–S batteries. (a) Fabrication of HNT-modified flexible PEO/LiTFSI SPE. (b) Schematically illustrated mechanism of HNT addition for enhanced ionic conductivity. (c) Ionic conductivities of PEO/LiTFSI/HNT SPEs with different HNT contents at EO:Li = 15:1 (mass ratio) as a function of temperature and (inset) phase transition temperature as a function of HNT content obtained after fitting. (d) Stress–strain curves of PEO/LiTFSI/HNT and PEO/LiTFSI SPEs. Reproduced with permission.326 Copyright 2016, Elsevier. | ||
Besides the functional additives, other polymer matrices beyond PEO have also been explored for yielding high-ionic-conductivity SPEs. Liu and Wang's group reported a unique SPE based on biomass-derived starch, which was further crosslinked with γ-(2,3-epoxypropoxy)propyltrimethoxysilane.327 Starch is a natural polysaccharide with abundant ether groups similar to PEO and thus capable of transporting ions reversibly. The crosslinking not only enhanced the stretchability of as-obtained SPE films but also helped maintain low crystallinity to realize efficient segmental motions. As a result, the flexible starch/LiTFSI SPE exhibited quite a decent ionic conductivity of 3.39 × 10−4 S cm−1 at 25 °C, almost three orders of magnitude higher than that of PEO/LiTFSI SPE. Moreover, with a wide temperature range of 20–100 °C, the ionic conductivity of starch/LiTFSI changed not as considerably as that of PEO/LiTFSI, demonstrating the superior adaptivity of starch-based SPEs to operating conditions. The key to such a high room-temperature ionic conductivity was attributed to the large amount (40 wt%) of lithium salts that can be completely dissolved in the crosslinked starch matrix without phase segregation. The lithium ion transference number was also as high as 0.8. Along with excellent oxidative stability as well as the good compatibility with lithium metal anodes the starch/LiTFSI SPE rendered Li–S batteries with remarkable cycling stability with steady capacities of 864 ± 16 mA h gsul−1 at 0.1C for 100 cycles at 25 °C.
In summary, safety and reliability are always the core requirements for practical batteries. Especially for flexible batteries, these requirements are even more eagerly desired because they have to be operated under mechanical deformation for long-term services. Hence, high-performance pliable SSEs are highly recommended for flexible batteries because they can easily adopt various shapes, configurations, and deformative modes, exhibiting inherent safety and promising versatility. In regard to the types of SSEs (GPE, SPE, and inorganic SSE), current issues for developing suitable flexible SSEs, particularly those in flexible Li–S batteries, primarily focus on several aspects: (1) insufficient lithium ionic conductivity, in particular for SPEs, which ought to be improved to ∼10−3–10−2 S cm−1 at ambient temperature; (2) unfavorable mechanical flexibility and compliance, especially for rigid or fragile inorganic SSEs and plasticized GPEs; (3) compromised safety due to the presence of liquid components, specifically for GPEs; (4) poor physical contact and interfacial instability at the two electrode/electrolyte interfaces, with the latter one shown by many inorganic SSEs such as sulfides and those containing reducible high-valence metal cations (e.g., Ti4+); and (5) incompatibility with the Li–S chemistry, e.g., highly reactive polysulfides and lithium metal, which has been revealed in several burgeoning studies.334,335 Nevertheless, the blooming progress in flexible SSEs will eventually pave the way to high-energy-density, safe, and flexible Li–S batteries.
3. Flexible materials for analogous flexible alkali metal–chalcogen batteries
Over the past decades, research enthusiasm for Li–S battery is growing steeply. Success in addressing its fundamental and practical problems has led to the exploration of new electrode materials that share an analogous multielectron chemistry. More concretely, alkali metals, i.e., Group IA elements except hydrogen, are expected to embody the role of lithium metal as the anode due to their comparably negative redox potentials to that of lithium, while chalcogens, i.e., Group VIA elements, are alternative cathode materials because of their relatively small electrochemical equivalents and high redox potentials among the same-row elements, which are endowed by the nature of two-electron redox and the second highest electronegativities, respectively. Structures and basic physicochemical properties of major elements in Group IA and VIA, which have so far been utilized for electrochemical energy storage, i.e., Na, K, Se, and Te, are listed in Table 2 for comparison with those of lithium and sulfur. The only exception is oxygen because it is gaseous at room temperature, against the other solid materials, and therefore possesses distinctive chemistry and demands unusual cell configurations when used as cathode materials for rechargeable batteries.| Group | Element | Electron configurationa | Electronegativityb | Crystal structure | Density (g cm−3) | Electrical conductivity (S cm−1) | Melting point (°C) | Crustal contentf (ppm) |
|---|---|---|---|---|---|---|---|---|
| a [He] = 1s2; [Ne] = [He]2s22p6; [Ar] = [Ne]3s23p6; [Kr] = [Ar]3d104s24p6. b Pauling scale. c Body-centered cubic (bcc). d The most thermodynamically stable phase at room temperature. e Measured at 25 °C. f Data obtained from http://WebElements.com. | ||||||||
| IA | Li | [He]2s1 | 0.98 | bccc | 0.534 | 1.1 × 105e |
180.5 | 20 |
| Na | [Ne]3s1 | 0.93 | bcc | 0.968 | 2.1 × 105e |
97.8 | 23000 |
|
| K | [Ar]4s1 | 0.82 | bcc | 0.862 | 1.4 × 105e |
63.5 | 15000 |
|
| IVA | S | [Ne]3s23p4 | 2.58 | Orthorhombic (α-S)d | 2.07 (α-S)d | 5 × 10−30 (at 20 °C) | 115.2 | 420 |
| Monoclinic (β-S) | 1.96 (β-S) | |||||||
| Se | [Ar]3d104s24p4 | 2.55 | Hexagonald | 4.81d | 1 × 10−5 | 221.0 | 0.05 | |
| Te | [Kr]4d105s25p4 | 2.10 | Hexagonald | 6.24d | 2.5 | 449.5 | 0.001 | |
The gravimetric/volumetric energy densities of various batteries are arranged in Fig. 1c. Because of heavy atomic weight, potassium, selenium, and tellurium have much more inferior gravimetric specific capacities, while the high densities of selenium (4.81 g cm−3) and tellurium (6.24 g cm−3), with the most thermodynamically stable hexagonal crystal structures (h-Se and h-Te), contribute to desirable volumetric specific capacities. Considering the output voltage, it can be found that only five out of nine redox pairs, i.e., Li–S, Li–Se, Li–Te, Na–S, and Na–Se, can afford acceptable energy densities in terms of either weight, volume, or both. Therefore, the following content will basically center on the four alkali metal–chalcogen batteries other than Li–S batteries.
In this section, we will start with flexible Li–Se batteries since selenium is the most extensively studied sulfur analogue for secondary batteries. Then, quite a few examples of flexible Li–Te batteries will be introduced, followed by discussions of flexible Na–S and Na–Se batteries employing the sodium metal anode. Similar to the status of flexible Li–S batteries, almost all efforts are made for flexible cathode materials. Finally, this section will end with a brief comparison between flexible alkali metal–chalcogen batteries and their Li–S counterpart.
3.1. Flexible Li–Se batteries
Selenium, right below sulfur in the periodic table, is a rare element usually used in pigment and semiconductor industries. Compared to sulfur, selenium holds several major merits for serving as cathode materials: (1) an approximately 1024 orders magnitude higher electrical conductivity, (2) room-temperature stable, chain-like allotrope h-Se that is more electroactive and more easily stabilized through spatial confinement (in sharp contrast to room-temperature metastable β-S), and (3) compatibility with conventional, cheap carbonate-based LIB electrolytes when physically confined in porous substrates.336 Hence, it is anticipated that selenium will exhibit a better utilization rate, cycling stability, and rate capability than sulfur. Besides, despite its inferior gravimetric specific capacity of 679 mA h g−1 compared to sulfur (1672 mA h g−1), h-Se has a comparable volumetric specific capacity of 3265 mA h cm−3 (sulfur, 3461 mA h cm−3), and it is, therefore, suitable for most applications that are volume sensitive, like portable electronics and electrical vehicles. Theoretically, the Li–Se battery, utilizing h-Se and lithium metal as the cathode and anode, respectively and outputting an average voltage of 2.0 V, affords high gravimetric and volumetric energy densities of 1155 W h kg−1 and 2528 W h L−1, respectively, both better than current those of LIB technologies.In 2012, Abouimrane and Amine's group first demonstrated a class of selenium and sulfur selenides as cathode materials for lithium and sodium rechargeable batteries.337 Since then, the development of selenium cathode has been significantly expedited with considerable progress, which has been summarized in two perspectives contributed by Guo's336 and Amine's groups.338 Until 2014, Ye and Dai's group reported the first flexible selenium cathode based on rGO and CNTs in light of the great success of flexible sulfur-based cathodes.339 Molten selenium was first coated on CNTs at 260 °C, and after cooling, as-obtained CNT@Se composites were further sonicated with GO and co-filtrated to produce a flexible freestanding composite film, in which GO was subsequently reduced by hydrazine vapor. Both rGO and CNTs not only functioned as flexible scaffolds but also facilitated long-range electron and ion transport, endowing the rGO–CNT@Se film cathode with excellent cycling stability (315/95 mA h gsel/ele−1). Note that both the utilization (46%) and mass fraction (30 wt%) of selenium were not high, which might be attributed to the lack of effective selenium confinement. He et al. also reported a freestanding rGO/CNT/Se ternary cathode with CNT@Se sandwiched between rGO to afford superior electrical contact. As a result, high capacities of 633(323) and 436(222) mA h gsel(ele)−1 at 0.2 and 4.0C (1.0C = 679 mA gsel−1, similarly for selenium hereinafter), respectively, were achieved both better than those of the CNT@Se cathode.340
In order to improve performance and selenium content, mesoporous carbon nanoparticles (MCNs) were introduced to replace CNTs for selenium accommodation and further embedded in the flexible rGO framework.341 Other than the use of MCNs, the whole synthetic route was the same as in the original work of Han et al.339 MCNs, with a small size of 50 nm and favorable mesopore dominance, effectively eliminated agglomeration into bulk selenium and confined selenium in the conductive scaffold with a much improved selenium content of 62 wt%. According to prior reports of Se–micro-/mesoporous carbon composite cathodes,342,343 electroactive selenium chains were stabilized in mesopores or smaller micropores, enabling high utilization and good cycling stability. As a consequence, the flexible Se/MCN–rGO paper demonstrated an ultrahigh selenium utilization of 97% at 0.1C, i.e., 655(406) mA h gsel(ele)−1, and an exceptionally long cycling life with 89% capacity retention after 1300 cycles at 1.0C. Considering the high content of selenium, this work is regarded as one of the most remarkable achievements for flexible Li–Se batteries.
Similar to flexible sulfur-based cathodes, carbonized electrospun PNFs also hold great promise for flexible selenium cathodes. By intentionally introducing a well-known porogen, triblock polymer Pluronic F127, in PAN precursor, Yu and Zhu's group synthesized electrospun PCNFs with profoundly increased porosity and specific surface area compared to pristine PCNFs synthesized in absence of F127. These composite PCNFs are denoted as f-PCNFs, and they maintained good flexibility after selenization (Fig. 24a–d).155 Consequently, much less crystalline selenium was present in PCNFs than in f-PCNFs, which led to a remarkably higher capacity and initial Coulombic efficiency (Fig. 24e, 643/322 mA h gsel/ele−1 and 56.9% for Se@PCNFs at 0.05 A gsel−1 (∼0.074C), against 405/203 mA h gsel/ele−1 and 34.9% for Se@f-PCNFs). This could be ascribed to the suppression of side reactions between free polyselenides produced from bulk selenium and carbonate electrolytes. In addition, owing to excellent encapsulation of selenium in the 1D conductive porous skeleton, flexible Se@PCNF cathode also manifested an almost non-fading cycling performance with a capacity of 516(270) mA h gsel(ele)−1 preserved after 900 cycles at 1.0 A gsel−1 (∼1.5C). By employing the same electrospun PCNF–CNT as demonstrated in flexible Li–S batteries, flexible selenium PCNF–CNT was fabricated in a similar way, obtaining a reversible capacity of 638(223) mA h gsel(ele)−1 after 80 cycles at 0.05 A gsel−1 (∼0.074C).156 It is interesting to note that the selenium utilization (94%) was much higher than that of sulfur (38%) when the same PCNF–CNT conductive backbone was adopted, demonstrating the huge benefit of using much more conductive selenium.
 | ||
| Fig. 24 Flexible selenium cathodes. (a and b) Schematics for the synthesis of selenium (Se)@PCNF electrodes. (c and d) Photographs of flexible Se@PCNF electrode. Cycling performance of flexible Se@PCNF and Se@f-PCNF cathodes in (e) Li–Se and (f) Na–Se batteries at 0.05 A gsel−1. Reproduced with permission.155 Copyright 2014, Wiley-VCH. | ||
3.2. Flexible Li–Te batteries
Tellurium, the last nonradioactive element in the chalcogen family, is a well-known semiconductor with the highest electrical conductivity of 2.5 S cm−1 among all nonmetallic materials. Despite its relatively low gravimetric specific capacity of 420 mA h g−1 that results from its heavy atomic weight, tellurium possesses a comparable volumetric specific capacity of 2621 mA h cm−3 to those of sulfur and selenium. Moreover, tellurium, as an electrically conducting active material, demands for much less carbon additives in the electrode. The reduction in lightweight carbon benefits both gravimetric and volumetric specific capacities. With an estimated output voltage of 1.8 V, a Li–Te battery offers theoretical gravimetric and volumetric energy densities of 682 W h kg−1 and 2078 W h L−1, respectively. The first attempt to build a Li–Te battery was made in Wang's lab.344 By using a tellurium/porous carbon composite cathode and a carbonate electrolyte, the Li–Te battery exhibited an average voltage of ∼1.5 V and a reversible capacity of 224 mA h gtel−1 at 0.05 A gtel−1 (∼0.12C, 1.0C = 420 mA gtel−1, similarly for tellurium hereinafter) after 1000 cycles (87% retention). Considering the relatively low voltage and promising volumetric capacity, Guo and coworkers demonstrated the application of tellurium/carbon composites as anode materials for LIBs, suggesting an extremely high tellurium utilization of 98% and a long-term cycling stability.345Tellurium is of particular interest for flexible electrode materials because of its two most favorable features: (1) extraordinary electrical conductivity that is even comparable to carbon and (2) inherent chirality of helical Te chains in the h-Te crystal that favors the formation of 1D Te nanostructures along the c-axis, i.e., [001] direction.346 Therefore, freestanding films consisting of ultralong Te NWs can be directly used for carbon-free, freestanding tellurium electrodes. Ding et al. fabricated a freestanding Te mat through vacuum filtration of Te NWs with a diameter of 7 nm grown in the [001] direction (Fig. 25a-i–iii).347 Such a high anisotropic 1D Te nanostructure possessed multiple advantages such as fully exposed Te zig-zag chains to lithium ions and a high electrical conductivity of ∼6.7 S cm−1 in the direction perpendicular to the c-axis (Fig. 25a-iv–vi). Moreover, a novel electrolyte using dimethyl sulfoxide as the solvent was employed, raising the working voltage from ∼1.7 V in carbonate or ether electrolytes to 2.03 V. The combination of Te NWs with the new electrolyte rendered a flexible tellurium cathode with desirable capacity of ∼144 mA h gtel/ele−1 at 0.1 A gtel−1 (∼0.24C) that was stably and reversibly attained for over 80 cycles, corresponding to a volumetric energy density of 1800 W h L−1 (Fig. 25a-vii). Yu and Liu's group employed the freestanding Te NW mat as a sacrificial template to synthesize a microporous CNT film, which was then inserted between a Te NW cathode and a separator to improve conductivity and intercept dissolved lithium telluride in dimethyl sulfoxide solvent.348 To achieve better encapsulation, the Te NW mat adhered to the CNT film was melted and fully infiltrated in the micropores of CNT, giving rise to a substantial improvement of capacity to 402(201) mA h gtel(ele)−1 at 0.1C and enhanced cycling stability. In order to improve rate performance, He and Chen's group further explored a flexible tellurium cathode derived from a 3D hierarchical aerogel with Te NWs wrapped homogeneously by rGO (Fig. 25b).349 The synthetic method was similar to that described in their previous report on 3DCG–Li2S. With 63 wt% tellurium, the flexible rGO/Te NW electrode was found to have high capacities of 418(263) and 174(110) mA h gtel(ele)−1 at 0.2 and 10C, respectively, as well as pronounced long-cycle performance at a high rate of 1.0C. The successful implementation of Te NWs renders flexible tellurium electrodes quite distinctive from its sulfur and selenium counterparts.
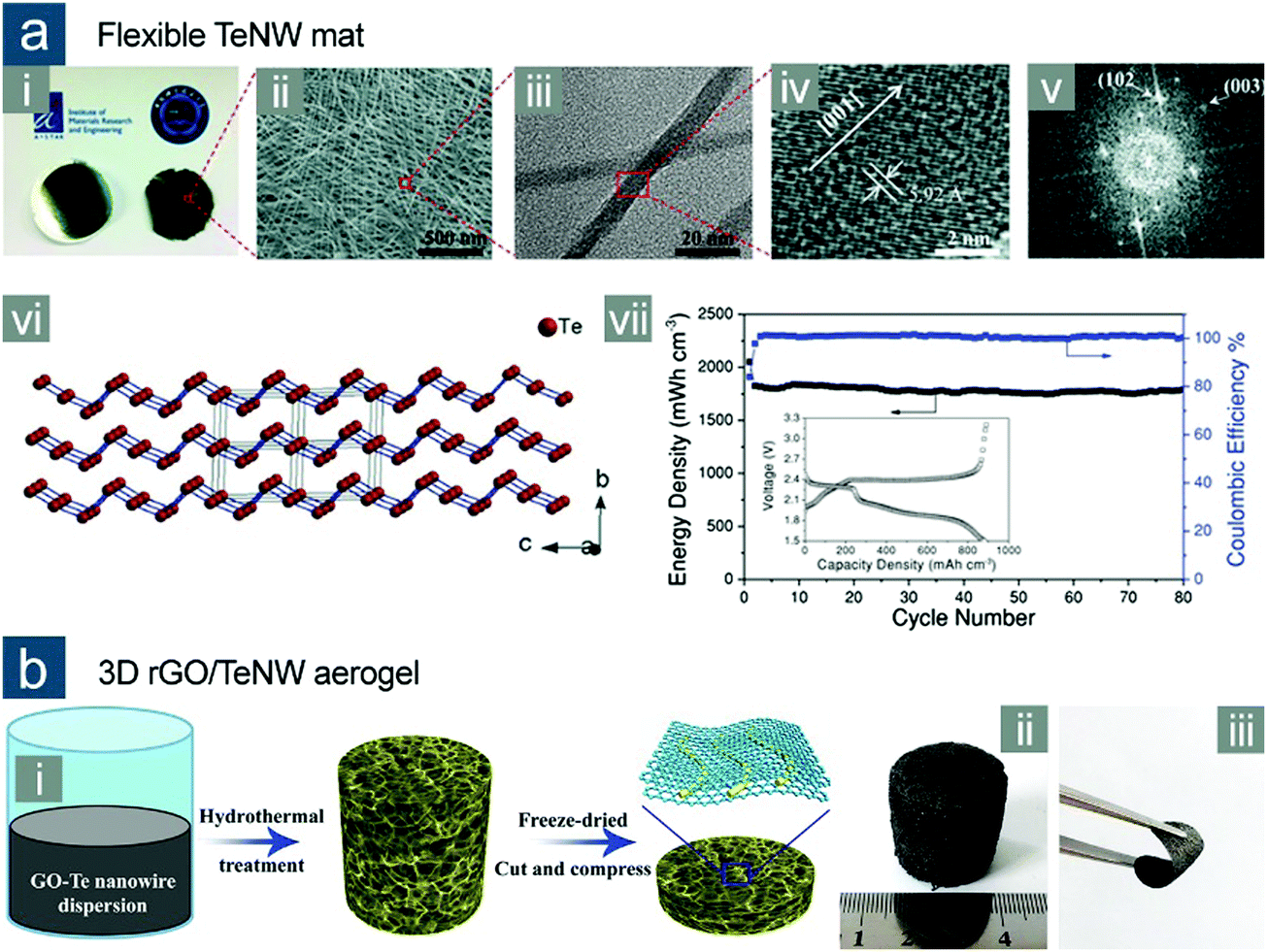 | ||
| Fig. 25 Flexible tellurium cathodes based on tellurium NWs (TeNWs). (a) Flexible, carbon-free TeNW mat: (i) photograph; (ii) SEM and (iii) TEM morphology images; (iv, v) nanoscale crystalline structure of freestanding TeNW mat; (vi) simulated crystal structure of h-Te; (vii) cycling performance of TeNW mat at 0.1 A gtel−1. Reproduced with permission.347 Copyright 2015, Wiley-VCH. (b) 3D rGO/TeNW aerogel: (i) schematic for fabrication of 3D rGO/TeNW aerogel and its derived flexible electrodes; photographs of (ii) 3D rGO/TeNW aerogel and (iii) flexible 3D rGO/TeNW electrode. Reproduced with permission.349 Copyright 2016, American Chemical Society. | ||
3.3. Flexible Na–S/Se batteries
With the expanding demand for electrical vehicles and smart grid, increasing demand for lithium can be foreseen, raising concerns about lithium supply and concurrent geopolitical issues related to non-uniform global distribution of lithium.15 To tackle potential crisis of lithium shortage, alternative chemistries dealing with earth abundant elements such as sodium, potassium, and magnesium, are being explored worldwide. Among them, sodium-based chemistries and technologies are the most researched, because sodium has the highest crustal content of 23000 ppm among all alkali and alkali earth elements and faces only moderate shortcomings of gravimetric/volumetric capacities (1166 mA h g−1 and 1128 mA h cm−3, respectively, for sodium metal), redox potential (0.33 V less negative), and kinetic barriers (unlike multivalent metals such as Mg and Al) compared to lithium. Consequently, sodium-based energy storage technologies are expected to be cheaper and more sustainable substituents to current lithium-based ones.
Sulfur has been regarded as one of sodium's “golden partners” when used for energy storage. High-temperature (HT) Na–S batteries have already been commercialized for large-scale stationary applications.14 In a typical HT Na–S cell, both sodium metal and sulfur are molten at an operating temperature of about 300 °C and separated by sodium ion-conducting β-alumina ceramics. However, the high operating temperature impedes the mobile and discrete applications of HT Na–S batteries. Besides, about only one third of the theoretical capacity of sulfur can be exerted with Na2S3 as the final discharge product, which is determined by phase equilibrium at this temperature.350 The presence of molten sodium and sulfur also leads to safety, reliability, and maintenance issues. Therefore, room-temperature (RT) Na–S and other Na–chalcogen batteries have become the main research targets in recent years.351
Compared to Li–S batteries, RT Na–S batteries face even more formidable challenges: (1) owing to the larger ionic radius of sodium compared to lithium, the electrode kinetics of Na–S batteries is more sluggish; (2) higher instability of sodium compared to lithium in organic solvents, leading to more serious gas evolution and electrolyte depletion issues; (3) sodium is more reactive in contact with polysulfides, resulting in severer shuttle effect; and (4) sodium ion is a milder Lewis acid than lithium ion, possibly inducing drastic differences in SEI composition/structure or phase stabilities of their sulfide or polysulfide species.351 These issues have to be preferentially considered when designing RT Na–S batteries, especially flexible ones.
Due to the above challenges, there have been few studies about flexible RT Na–S batteries. Ahn and colleagues reported the first flexible RT Na–S battery by adopting a sulfurized PAN (SPAN) nanofiber web as the cathode material, in which monodisperse sulfur was covalently bond to the polymeric backbone, fully addressing the shuttle effect.352 PAN nanofibers were prepared through electrospinning and further pyrolyzed with or without the presence of sulfur, producing SPAN or HT-PAN webs, respectively. More interestingly, the SPAN web exhibited good flexibility and bendability, in contrast to the brittle and rigid HT-PAN. With 41 wt% sulfur, the pliable SPAN cathode manifested a reversible capacity of 756(310) mA h gsul(ele)−1 at 0.01C and maintained stable capacities above 649(266) mA h gsul(ele)−1 at 0.1C for 200 cycles. In order to increase areal sulfur loading in flexible sulfur cathodes for RT Na–S batteries, Lu et al. fabricated a flexible carbonized cotton textile–S cathode with a sulfur content of 24 wt% and an areal loading of 2 mgsul cm−2, which delivered an initial capacity of 390(94) mA h gsul(ele)−1 at 0.1C in a tetraglyme electrolyte.353 Such an electrode, prepared from large-scale cotton textile, can be readily used in a flexible RT Na–S pouch cell, with barely any performance degradation on bending.
As indicated in (Section 3.1), selenium cathodes are less tortured by shuttle phenomenon than sulfur cathodes, likely because it is easier to confine the stable chain-like selenium allotropes in porous hosts. Therefore, flexible Na–Se batteries can obtain better electrochemical performance than flexible RT Na–S batteries. Using the same materials, i.e., electrospun F127-templated PCNFs155 and in situ selenized PCNF–CNTs,156 Yu and colleagues demonstrated flexible Na–Se batteries. For example, when paired with a sodium metal anode, the flexible Se@PCNF delivered an initial capacity of 600(300) mA h gsel(ele)−1 at 0.05 A gsel−1 (∼0.074C), corresponding to a high selenium utilization of 88% (Fig. 24f). Such a utilization was comparable to that of cathodes paired with lithium and much higher than that of typical sulfur cathodes paired with sodium, demonstrating the superiority of selenium for electrochemical energy storage (Fig. 24e and f). However, it should also be noted that the cycling stability of as-proved flexible Na–Se battery was worse than its Li–Se counterpart applying the same cathode materials, suggesting the significant role of sodium metal anode in Na–chalcogen batteries.
In summary, analogous flexible alkali metal–chalcogen batteries generally involve the same materials and synthetic methods as flexible Li–S batteries. The only exception is Te NW, which is inherently conductive and self-standing to act as a carbon-free flexible electrode material. The basic chemistry and principles of alkali metal–chalcogen batteries are analogous, but some fascinating features of selenium, tellurium, and sodium render them a somewhat promising or at least interesting candidate electrode material for future flexible energy storage devices: (1) much higher electrical conductivities of selenium and tellurium than of sulfur, along with higher stabilities of their partially lithitated/sodiated products in organic, especially carbonate, electrolytes, usually endow selenium/tellurium electrodes with better utilization, rate capability, and cycling stability; (2) despite relatively low gravimetric specific capacities, selenium and tellurium possess much higher density than sulfur, giving comparable volumetric specific capacities that are pivotal in many intriguing but volume-constrained applications; (3) sodium is much more abundant on earth than lithium and sodium precursors are considerably cheaper.
Despite the above advantages, there are major concerns about the scarcity of selenium and tellurium. These two elements, especially tellurium, are ironically among the rarest with only 0.05 and 0.001 ppm crustal contents, respectively. However, their promise, at least selenium's, might not be as desperate as expected. On one hand, the consumption of selenium is far less than sulfur, so its natural reserve is foreseen to be adequate for next several decades.336 On the other hand, the cost and reserve issue of selenium can be partially addressed by employing sulfur selenides as the active materials, which combine complementary attributes of conductive but expensive selenium and high-capacity, cheap but insulating sulfur.354,355 In fact, Yu and coworkers have already demonstrated several flexible S0.6Se0.4/CNF electrodes for flexible Li/Na–chalcogen batteries, representing a more sustainable way of utilizing selenium for electrochemical energy storage.157,356 More efforts in developing flexible sulfur selenide-based cathodes are required for reliable flexible alkali metal–chalcogen batteries. Similar to flexible Li–S batteries, more efficient flexible electrolytes and anodes other than currently developed flexible cathode materials are also requested.
4. Device integration of flexible Li–S batteries
Aforementioned advancements mainly focus on individual material or cell component. However, these components have to be integrated and packaged into a compact and pliable device; otherwise it will never be possible to realize an actual flexible Li–S battery. Encapsulation, i.e., packaging, materials thus play a key role in this regard. Presently, there are only 18 out of 103 previous papers that report assembled flexible Li–S batteries (Fig. 26a). Based on cell configurations and shapes, these flexible Li–S batteries (with one exception of a flexible Na–S battery353) can be categorized into (1) flexible pouch cell, (2) foldable pouch cell, and (3) cable/fiber-type cell (Table 3).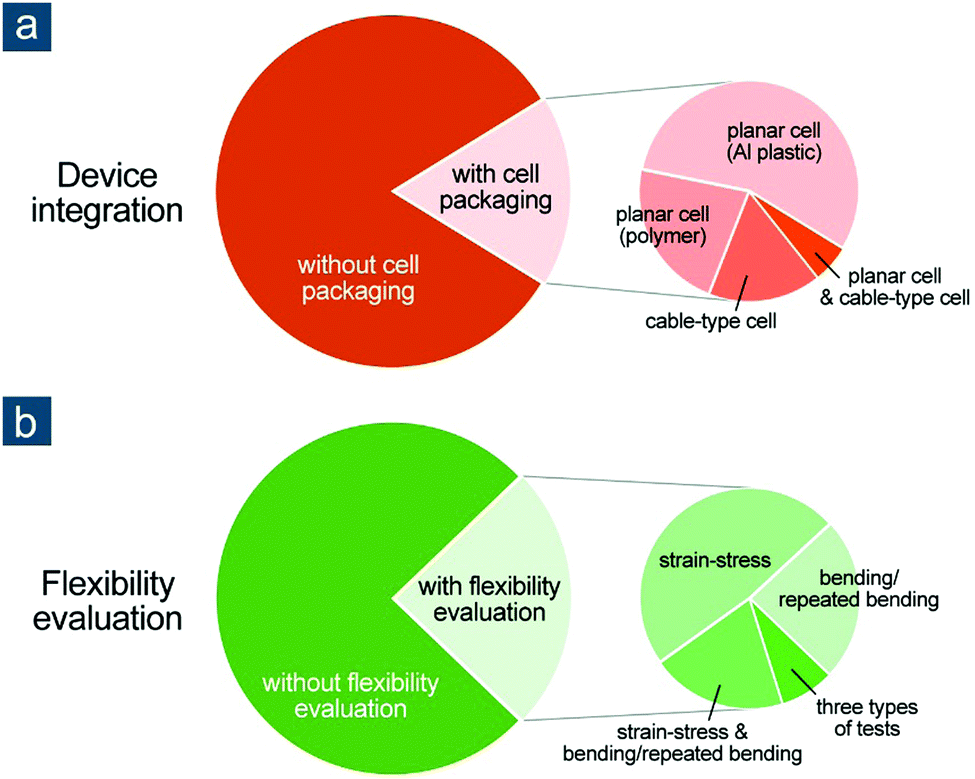 | ||
| Fig. 26 Statistical analyses of flexible Li–S batteries, regarding (a) device integration and (b) flexibility evaluation. Details about the integrated cell prototypes and the evaluation methods of flexibility are listed in Tables 3 and 4, respectively. | ||
| Category | Cathode material | Anode material | Encapsulation material | Cell type | Bending testa | Bending parameterb | Bending demonstration |
|---|---|---|---|---|---|---|---|
| a Galvanostatic discharge–charge curve: GDC. b Control parameters for bending tests include bending angle φ and bending radius ρ. | |||||||
| Flexible pouch cell | Li2S@NCNF164 | Li foil | Al plastic | Pouch | LED | ||
| Electrospun S/G/NPCF166 | Li foil | Al plastic | Pouch | LED panel | |||
| Sandwiched NFC/CNT/NG/S221 | Li foil | Al plastic | Pouch | LED | |||
| C/S composites on G@PP separator227 | Li foil | Al plastic | Pouch | Cycling | N. A. | LED panel | |
| Electrospun S/TiO2/G/NPCF256 | Li foil | Al plastic | Pouch | LED panel | |||
| MOF-derived carbon/CNT/S271 | Li foil | Al plastic | Pouch | LED | |||
| Carbonized cotton textile–S353 | Na foil | Al plastic | Pouch | Cycling | N. A. | LED panel | |
| GPC–S153 | Li foil | Tape | Pouch | Cycling | N. A. | LED | |
| S–PDMS/GF232 | Li foil | PDMS | Pouch | LED | |||
| Li2S–PDMS/GF233 | Li foil | PDMS | Pouch | LED | |||
| Foldable pouch cell | Foldable MOF/CNT/S269 | Li foil | Al plastic | Pouch (two shapes) | Cycling | φ: 45, 90, 135, 180° | LED |
| Foldable DWCNT/S358 | Patterned Li foil | Al plastic | Pouch | Cycling | φ: 180° | LED | |
| Tandem CNF/SWCNT/S359 | Li foil | Al plastic | Pouch | GDC | φ: 90, 180° | ||
| Aligned CNT/CMK-3@S207 | Li foil | PDMS | Pouch | Cycling | φ: 60, 180° | LED panel | |
| Cable/fiber-type cell | GO/CMK-3@S/CNT fiber362 | Li wire | Heat-shrinkable polyolefin tube | Cable |
• LED, 30 min
• Five-cable-embedded textile |
||
| Nanostructured rGO–S paper125 | Li foil | Al plastic | Pouch | Cycling | N. A. | LED panel | |
| Li wire | Heat-shrinkable polyolefin tube | Cable | Cycling | N. A. | LED panel | ||
| rGO/CNT/S fiber363 | Li wire | Heat-shrinkable polyolefin tube | Cable | Cycling | ρ: 6.5 mm | LED, 4 h | |
| rGO/S/stainless steel fiber364 | Li wire | Heat-shrinkable polyolefin tube | Cable | LED | |||
For flexible and foldable pouch cells, the most widely used encapsulation material is Al plastic (Table 3). Al plastic is composed of (1) an Al interlining, which is the impermeable barrier to both internal electrolytes and external humidity, (2) a polymer lining (usually polyolefin) firmly attached to the inner surface of Al, sealing the whole cell, and (3) another polymer layer (usually with a high melting point) for compactness and protection. Its price is not high and its fabrication is simple, rendering it one of the most common packaging shells for LIBs and Li–S pouch cells. However, Al plastic is not fully flexible to withstand rigorous mechanical deformation especially when the thickness of assembled cell is high. After being bent or folded, Al plastic is always observed with creases or wrinkling, which may potentially cause safety issues from shell deformation and perforation.11 Therefore, softer yet robust materials are preferred, such as elastic PDMS or scotch tape. The typical fabrication procedures of a flexible Li–S pouch cell is schematically illustrated in Fig. 27a, in which it can be found that all cell components are laminated together and further sandwiched between two pieces of a pliable PDMS (or Al plastic) film with two metal tags placed between each PDMS film and the cathode or anode.207 Owing to the good elasticity of PDMS, thin-film Li–S battery with PDMS encapsulation can be bent or even twisted without shell cracking or cell failure (Fig. 27b–f). To realize both a high sulfur content and high areal sulfur loading, binder- and current-collector-free electrodes are adopted, mostly consisting of non-weldable carbon nanomaterials. This raises a problem of connecting the electrode to the external circuit. In laboratory experiments, metal tags are used as the electrode lug but only provide weak connection through simple physical contact.152,207,232 In mature flexible battery prototypes, rational design of electrode lugs is expected. One possible solution is to replace the metal tags by metal nanowires to decrease contact resistance through nanoscale interfacial contact.
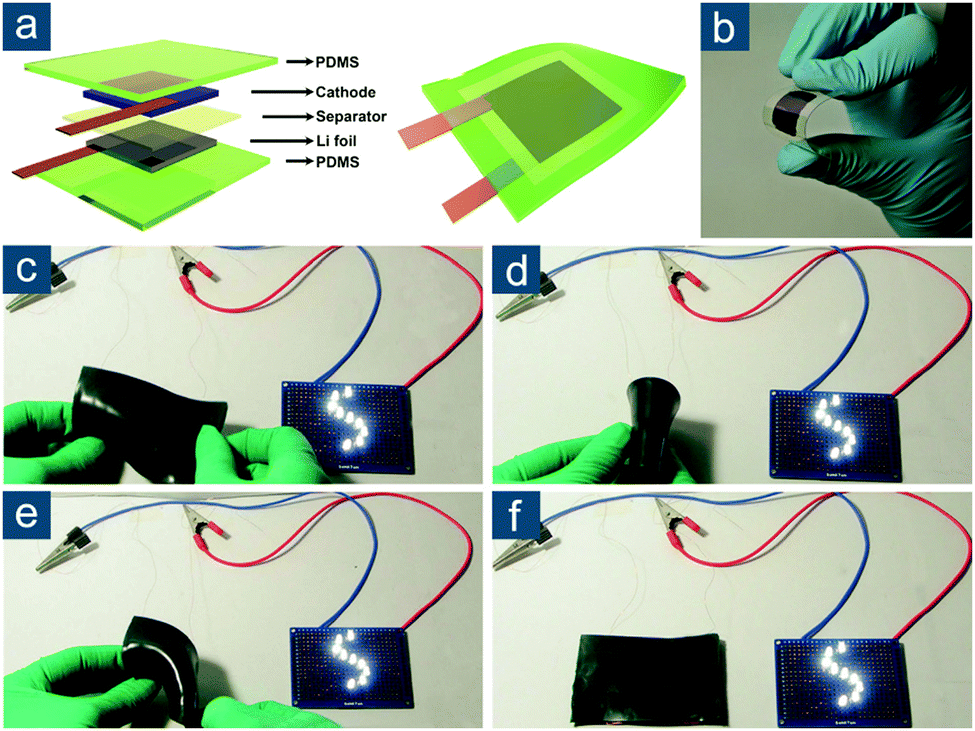 | ||
| Fig. 27 Cell packaging of flexible Li–S batteries. (a) Schematics of flexible Li–S battery: exploded (left) and general (right) views. Photographs of (b) bent Li–S cell and (c–f) flexible film battery (consisting of two serially connected Li–S cells) lighting up nine LEDs when being bent and recovered. Reproduced with permission.207 Copyright 2015, Wiley-VCH. | ||
Other than the common flexible Li–S pouch cells, we will introduce two special flexible Li–S battery prototypes: the foldable planar Li–S battery and cable-type Li–S battery. Before that, we will first summarize several appropriate methods for evaluating flexibility, which is actually the most distinguishable part of flexible battery research from conventional Li–S battery research but is unfortunately paid much less attention.
4.1. Evaluation of flexibility
Compared to regular electrodes and electrochemical cells, it is necessary for flexible electrodes and energy storage devices to evaluate not only electrochemical performance but also flexibility. Quantitative measurements and analyses are crucial for guiding electrode and cell design. Unfortunately, in current studies on flexible Li–S batteries, photographs of bent or curved electrodes are mostly employed to prove “flexibility”. This method is more qualitative than quantitative, and it is barely clear how strong a deformative force the electrode/cell can withstand, how differently deformation influences various properties of the electrode/cell, and whether the electrode/cell can survive from repeated or cyclic mechanical deformations. All these questions can only be answered by applying appropriate quantitative methods for flexibility evaluation.To date, only 25 out of 103 previous papers involve one or several approaches to evaluate flexibility (Fig. 26b). All these approaches and corresponding quantitative results of flexibility are summarized in Table 4. Generally, these approaches can be classified as follows:
| Cathode material | Stress–strain test | Bending test | Repeated bending test | Other tests | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mode | Stress (MPa) | Strain (%) | Sulfur load (wt%/mgsul cm−2) | Dependent variablea | Independent variableb | Dependent variablea | Independent variableb | Repetitions | ||
| a Normalized sheet/fiber resistance: R/R0; open circuit voltage: OCV; electrochemical impedance: Im; electrical conductivity: σ. b Independent variables for bending tests include: bending length ratio L/L0, bending angle φ, and bending radius ρ. | ||||||||||
| Mesoporous SACNT–nanoS97 | Stretch | 3.65 | ∼4.3 | None | ||||||
| 2.50 | ∼4.6 | 51/∼1.9 | ||||||||
| 1.59 | ∼4.3 | 59/∼1.9 | ||||||||
| 0.99 | ∼3.5 | 69/∼1.9 | ||||||||
| Electrospun S/PCNF–CNT152 | Stretch | 0.27 (with CNT) | 2.8 | 40/0.8 | ||||||
| 0.47 | 3.0 | 39/0.8 | ||||||||
| PUF-derived NCF/S181 | Compress | ∼0.0055/∼0.0037 (PUF/NCF) | 80 | None | ||||||
| SACNT/G/S195 | Stretch | 2.14 | ∼5.0 | 50/N. A. | ||||||
| CNT–rGO/S198 | Stretch | 72.0 | ∼0.7 | 53/1.9 | ||||||
| CNT/GS–S202 | Stretch | 2.1 | 9.3 | 45/1.1 | ||||||
| CNT/NG/S203 | Stretch | 6.8 | 2.5 | 53/4.7 | ||||||
| CNT/gCNC/S204 | Stretch | 2.75 | 7.3 | None | ||||||
| C/S/C sandwich on PP separator219 | Stretch | ∼30 N (w/o C/S/C) | ∼75 | None | ||||||
| ∼29 N | ∼68 | 49/1.2 | ||||||||
| SACNT@PVP–nanoS220 | Stretch | 13.6 | 15 | 47/1.0 | ||||||
| Sandwiched NFC/CNT/NG/S221 | Stretch | 19.5 | ∼5.5 | 55/3.6 | ||||||
| 16.6 | ∼6.0 | 53/8.1 | ||||||||
| MOF-derived carbon/CNT/S271 | Stretch | 2.46 | ∼1.3 | 70/3.0 | ||||||
| Nanostructured rGO–S paper125 | Stretch | 2.50 | ∼4.6 | 51/∼0.6 | R/R0 | L/L0: ∼0.05–1.0 | ||||
| Carbonized cotton textile–S353 | 1.59 | ∼4.3 | 59/1.0 | R/R0 | φ: 0–180° | |||||
| Electrospun S/G/NPCF166 | Stretch | 0.99 | ∼3.5 | 69/1.1 | OCV | φ: 45, 90, 180° | ||||
| Aligned CNT/CMK-3@S207 | OCV | φ: 0–180° | ||||||||
| Foldable DWCNT/S358 | OCV | N. A. | ||||||||
| Foldable MOF/CNT/S269 | Im | φ: 45, 90, 135, 180 | ||||||||
| B/S/B sandwich143 | Im | N. A. | Cycling after rolling | |||||||
| AAO-templated S–CNT94 | Stretch | 10.0 | 9.0 | 23/2.5 | σ | ρ: ∼1.5 mm | 12000 |
|||
| C/S composites on G@PP separator227 | Stretch | 30 (with PP) | 65 | ∼46/∼1.8 | σ | ρ: ∼1.5 mm | 50000 |
Interface adhesion | ||
| 6 (w/o PP) | 3.3 | N. A. | ||||||||
| S–PDMS/GF232 | Stretch | 0.13 (with C/S) | ∼50 | 50/10.1 | σ | ρ: ∼1.5 mm | 22000 |
|||
| 0.11 | ∼180 | None | ||||||||
| PPyPU-glued C/S composites on carbon felt224 | N. A. | N. A. | 50 | |||||||
| GO/CMK-3@S/CNT fiber362 | Stretch | ∼31–34 | ∼12.5–15 | 68/N. A. | OCV | φ: 0–180° | R/R0 | ρ: 2.5 cm | 1000 | |
| rGO/CNT/S fiber363 | Stretch | 20 | ∼2.9 | 45/N. A. | R/R0 | ρ: 3.5, 7.0, 10.0 mm | R/R0 | L/L0: 0.5 | 100 | |
(1) Stress–strain tests. Usually the stress–strain curve is obtained through stretching the sample along the direction perpendicular to its cross section and simultaneously measuring the applied stress and real-time strain response. Through this approach, tensile strength, maximum strain, Young's modulus, and dynamic mechanical response of an electrode can be obtained. In only one report, compression instead of stretching was used, mainly to capture volume recovery properties.181 It is also interesting to find that most stress–strain curves were obtained on electrodes loaded with sulfur, which was suggested to be detrimental to mechanical strength. Therefore, the presented data is quite relevant to reveal the flexibility of actual electrodes tested in batteries.
(2) Bending tests. Rather than a picture showing an electrode is “bendable”, measuring different properties and/or evaluating performance of a bent electrode provides proper and precise understanding of its flexibility. Dependent variables include electrical conductivity and sheet/fiber resistance of the electrode as well as the open circuit voltage and electrochemical impedance of the cell and independent variables include bending length ratio, bending angle, and bending radius, all schematically illustrated in Fig. 28a. The bending response can be studied by controlling these test parameters. Note that the measurements listed in Table 4 were obtained on bending.
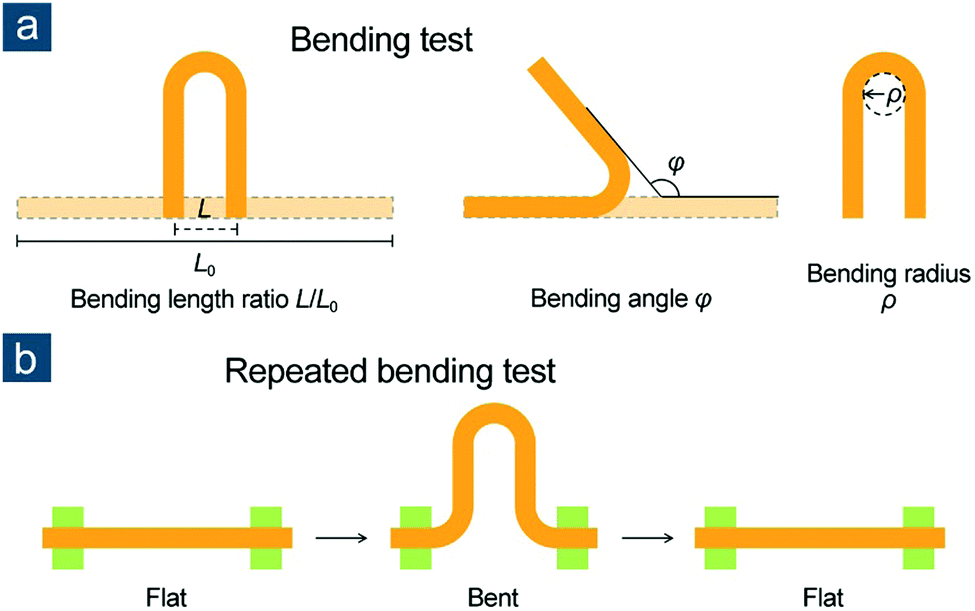 | ||
| Fig. 28 Bending tests for evaluating flexibility. (a) Schematics illustrating bending length ratio L/L0, bending angle φ, and bending radius ρ. (b) Schematic of the repeated bending test. | ||
(3) Repeated bending tests. Different from above one-time bending tests, repeated bending tests mainly aim at the restorative ability of flexible electrodes suffering from tremendous mechanical deformation. Electrical conductivity or sheet/fiber resistance of the electrode is measured after certain numbers of bending/stretching cycles (Fig. 28b). These tests are favorable for proving the efficiency and persistence of flexible electrodes during long-term service.
Besides these three major approaches, other tests may be needed. For example, adhesion strength will have to be assessed if the electrode materials are supported on a flexible substrate. The pull-off227 or single lap shear357 tests can be used for this measurement.
4.2. Foldable planar Li–S batteries
Innovation of portable electronics leads to fully deformable and shape-conformable devices, requiring for commensurate innovation in lightweight and flexible power sources. Not only does the electrochemical performance have to be improved, but also extraordinary device flexibility needs to be guaranteed to adapt to more complex deformative conditions beyond simple bending. Desirable foldability, twistability, and stretchability are therefore strongly demanded, requiring both flexible materials and exotic cell configurations. One of the major obstacles for developing foldable planar Li–S batteries is the use of metal current collectors (e.g., Al foils for cathode and Cu foils for anode), which are flexible and recoverable when mildly bent (with small bending angles or large bending radii), but suffer from plastic deformation when folded (with a bending angle of 180° and small bending radii). This issue also appears in lithium metal foils, which are softer and more plastic. Another challenge is the weak adhesion of active materials to current collectors, inevitably leading to delamination or fracture of the electrode layer upon folding.In order to overcome the above challenges, Koratkar and coworkers first reported a foldable Li–S battery by using superelastic and foldable double-walled CNT (DWCNT) films as current collectors for both the sulfur cathode and lithium anode (Fig. 29).358 The CNT film possessed a high electrical conductivity of ∼800 S cm−1, a light weight of 0.6–0.8 mg cm−1, and a thickness of ∼3 μm, thus benefiting a rapid reaction kinetics and high gravimetric/volumetric energy densities. Molecular dynamic simulation indicated that the intercrossed DWCNT bundles, as the basic structural units of the interpenetrated DWCNT film, could endure huge deformation with a bending angle of up to 180°, while a single DWCNT would break at a bending angle of ∼120° (Fig. 29a). Besides the use of superelastic DWCNT films, the electrode structure was further engineered with a checkboard pattern by using a criss-cross mask (Fig. 29b and c). In this way, both the sulfur cathode and lithium anode formed discontinuous films tightly attached to DWCNT current collectors so that folding took place predominantly in the pure CNT region, drastically reducing the probability of electrode delamination. As a consequence, the Li–S battery with patterned electrodes on superelastic DWCNT current collectors was insensitive to folding (Fig. 29d and e), maintaining 88% of its initial capacity (754/339 mA h gsul/ele−1) after 100 folding cycles at 0.5C. Chang et al. designed a freestanding tandem sulfur cathode with alternately stacked CNF/S and CNF/single-walled CNT layers to boost its electrochemical performance with a high sulfur loading of 16 mgsul cm−2 and demonstrated foldable Li–S batteries based on such a tandem cathode and a lithium foil anode, which exhibited nearly unchanged capacities when folded.359 Other cathodes such as MOF/CNT/S films269 and aligned CNT/CMK-3@S207 were also paired with lithium foils to fabricate foldable Li–S batteries, i.e., those bent at an angle of 180°. Nevertheless, the durability of plastic lithium to repeated folding should be validated during longer cycling.
 | ||
| Fig. 29 Foldable Li–S batteries. (a) Molecular dynamics simulations of CNT folding: (left) three-DWCNT bundle and (right) single DWCNT with the color map as a representation of different atomistic strain. (b) Schematic of prototype foldable Li–S battery with active materials (lithium and sulfur) patterned onto DWCNT current collector films in a checkerboard pattern. Photographs of (c) checkerboard pattern of sulfur/DWCNT cathode and (d and e) folded cell used to light an LED. Reproduced with permission.358 Copyright 2015, American Chemical Society. | ||
4.3. Flexible cable-type Li–S batteries
In the case of next-generation flexible power sources, one of the main challenges is their limited shape versatility, mainly in planar form, which hinder their implementation in flexible electronic devices of various shapes.360 Especially for wearable devices, motion is hardly regular, inducing omnidirectional mechanical deformation, which is extremely complex and unpredictable, far different from simple bending or folding. Thus, highly adaptive and conformal power accessories are eagerly demanded. Recently, cable-type rechargeable batteries have been attracting considerable interest because of their excellent spinnability and knittability just like cloth fibers. Hence, they can be readily integrated in flexible textiles and power wearable electronics. Lee et al. reviewed the recent progress of cable-type LIBs, showing the great promise of these cable-type energy storage devices for future flexible and wearable electronics.361Development of cable-type Li–S batteries is thereby booming due to their overwhelming advantages in terms of energy density and cost over cable-type LIBs. Compared to flexible planar Li–S batteries, cable-type Li–S batteries are drastically different in cell configuration and corresponding materials. Typically, a lithium wire instead of a foil is employed as the anode. The cathode can be a fiber, either paralleled to or twisted on a lithium wire; alternatively, it can also be a film coaxially coated on the lithium wire. To prevent short circuit, a separator is placed between them if liquid electrolytes are employed. Alternatively, the two electrodes can also be spatially segregated by gel polymer electrolytes or SSEs. Most commonly, the outermost shell is a heat-shrinkable polyolefin tube, with two ends sealed with adhesives, to realize compact encapsulation through heating of the cable-type cell.
Like flexible planar Li–S batteries, the most exciting advancement for cable-shaped Li–S batteries lies on the controllable synthesis of high-performance fiber-type cathode materials. Peng and coworkers pioneered the research on cable-type Li–S batteries (Fig. 30).362 In this primary study, the fibrous cathode was built by embedding sulfur-encapsulated particles, GO-coated CMK-3@S, in the aligned CNT fiber through wet spinning (Fig. 30a and b). The application of spinnable CNT arrays/sheets is highly versatile for synthesizing fibrous electrode materials. Owing to the ultralight weight of CNT sheets (areal density of 1.4 × 10−3 mg cm−2), a high mass content of sulfur (68 wt%) was uniformly dispersed in the hybrid fiber, resulting in narrow distribution of linear density (0.17–0.20 mg cm−1). Therefore, the hybrid fiber cathode delivered a high initial capacity of 1051(715) mA h gsul(ele)−1 in a conventional Li–S battery, with a linear capacity of 0.12–0.14 mA h cm−1. The fiber cathode and lithium wire anode were further encapsulated in a plastic tube with each electrode at the opposite end, exhibiting superb flexibility under either bent or twisted states (Fig. 30c). More interestingly, a fabric integrated with five cable-type Li–S batteries was prepared, unambiguously proving the utility of this GO/CMK-3@S/CNT fiber cathode for wearable applications (Fig. 30d).
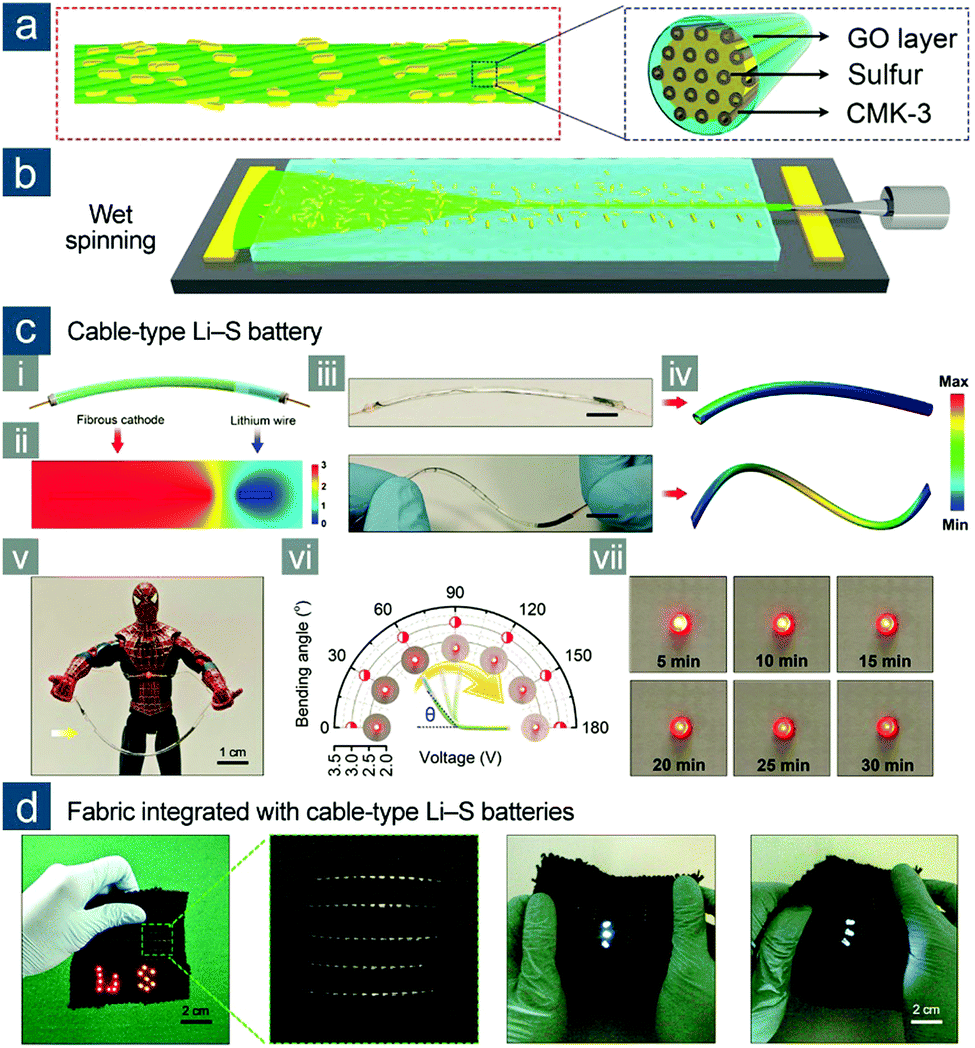 | ||
| Fig. 30 Flexible cable-type Li–S batteries. (a) Schematics of (left) fibrous sulfur cathode based on aligned CNT fiber and (right) GO-coated CMK-3@S particles embedded in CNT fiber. (b) Schematic of wet-spinning setup. (c) Demonstration of cable-type Li–S battery: (i) schematic of cable-type Li–S battery; (ii) potential distribution in the battery. (iii and iv) photographs and stress distribution of the battery in bending and twisting states; (v) bent battery lighting up a red LED; (vi) OCVs of the battery being bent from 0° to 180° with photographs displaying a red LED lit up by the battery bent at different angles; (vii) a red LED lit up by cable battery for 30 min. (d) Fabric integrated with five cable-type Li–S batteries, working under stretching and twisting deformations. Reproduced with permission.362 Copyright 2016, Wiley-VCH. | ||
After that, other cable-shaped Li–S batteries have been demonstrated successively, employing different cathode materials and cell configurations. Cao et al. wrapped a lithium wire with a polyolefin separator and a nanostructured rGO–S film in sequence, obtaining a coaxial cable-shaped Li–S battery.125 A high initial capacity of 1360(762) mA h gsul(ele)−1 was delivered at 0.1C, comparable to that of a coin cell. After 25 cycles without bending, the cable-type Li–S battery was cycled upon bending with only slight difference in performance. Kim and colleagues adopted a facile, one-pot wet spinning method to prepare ultralight composite fibers consisting of aligned rGO/CNT filled with 45 wt% of sulfur (denoted as rGO/CNT/S).363 The alignment of rGO and CNT was ascribed to liquid crystalline nature of high-concentration GO precursors, rendering excellent mechanical and electrical properties. Very long composite fibers (up to 4 m) could be continuously fabricated. The cable Li–S battery, in which an rGO/CNT/S fiber was placed parallel to a lithium wire, could light a LED for 4 h and endure repeated bending for 30 cycles. Huang and Ma's group reported dip-coating synthesis of a fibrous sulfur cathode using industrially available stainless steel fiber as the current collector to imbibe rGO/S composites via capillary action.364 However, the cycling performance of as-prepared cable-type Li–S battery should be further improved.
5. Summary and prospects
We have reviewed the recent progress of flexible Li–S and analogous alkali metal–chalcogen batteries. Owing to the knowledge of materials chemistry, electrochemistry, energy science, nanoscience, and engineering mechanics, great improvements have been achieved in flexible cathodes based on sulfur and other chalcogens like selenium and tellurium with respect to their outstanding electrochemical and mechanical performance. It can be clearly seen that among the 103 chosen works on flexible Li–S batteries and analogues, the average content, specific capacities based on active material and the whole electrode, areal loading, and areal capacity are 52.5%, 1003 and 521 mA h g−1, 3.75 mg cm−2, and 3.54 mA h cm−2, respectively (Fig. 31). In a typical Li–S pouch cell using a conventional sulfur cathode (70 wt% sulfur content) that is double-side coated on an Al foil (∼4.0 mg cm−2), such a high content of sulfur in the whole electrode (including the Al foil) can only be achieved when the areal sulfur loading is as high as 8.0 mgsul cm−2, at which it is very challenging to achieve a very high utilization ratio of the active material. Notably, flexible cathode materials, especially the binder- and current-collector-free ones, are much less dependent on areal loading of the active material to obtain a high gravimetric energy density of the whole electrode. Thus, a single flexible chalcogen cathode will be light and thin enough for miniaturized flexible applications while enabling higher energy density than LIB cathodes. Compared to the achievements made so far with respect to the cathode, research on flexible lithium metal anodes is still in its infancy, but a few fascinating results have proven the great potential of developing highly flexible and high-efficiency lithium metal anodes. Besides the flexible electrode materials, flexible and reliable SSEs are also indispensable for securing better safety and durability. Innovations in unique flexible Li–S battery prototypes are strongly predicted to create more fruitful opportunities for emerging applications such as foldable and wearable devices.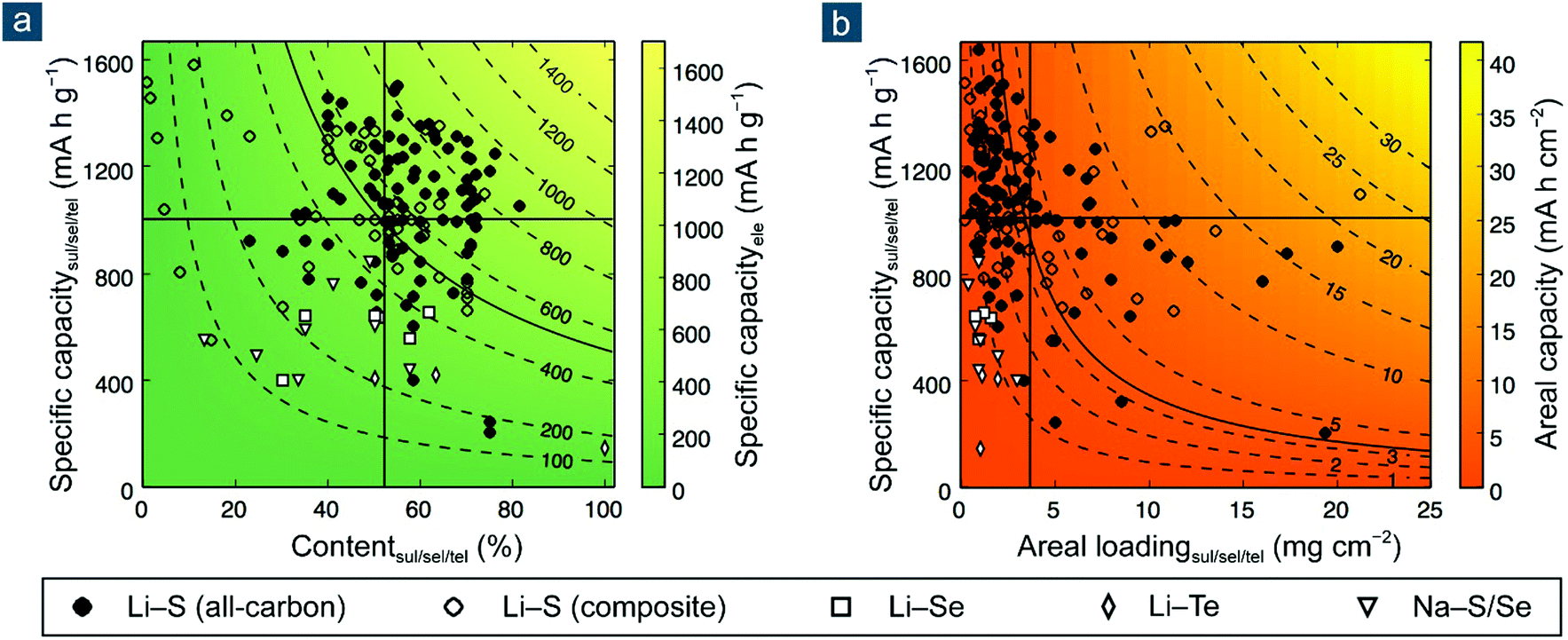 | ||
| Fig. 31 Summary of electrochemical performance of flexible alkali metal–chalcogen batteries. (a) Specific capacity based on mass of active material, i.e., sulfur (sul), selenium (sel), and tellurium (tel), vs. content of active material. Color map shows specific capacity based on mass of electrode (ele). (b) Specific capacity based on mass of active material vs. areal loading of active material. Color map shows areal capacity. In each figure, there are three solid lines, corresponding to the mean values of x-axis (vertical), y-axis (horizontal), and color bar (isoline), respectively, and several dashed isolines with the exact value shown aside. Solid circle, hollow circle, open square, open diamond, and open triangle refer to flexible Li–S batteries with all-carbon cathode scaffolds, Li–S batteries with composite cathode scaffolds, Li–Se, Li–Te, and Na–S/Se batteries, respectively. | ||
Materials families, dedicated to flexible Li–S and analogous alkali metal–chalcogen batteries, have rapidly expanded, ranging from carbonaceous materials to polymers and inorganic materials. It has to be pointed out that when used for flexible energy storage, they exhibit pros and cons. Take the flexible cathode as an example. CNT films exhibit superior conductivity and flexibility, while the complex fabrication procedures and difficulties in continuous production and high-volume dispersion in solutions hinder their large-scale applications. GO and rGO membranes can be easily fabricated from uniform GO/rGO solutions, possessing extraordinary mechanical pliability, but it is usually challenging to balance sheet conductivity, porosity, and flexibility, which requires delicate regulation of reduction and assembly. The industrial production of GO is also engineeringly and environmentally intractable. Electrospun PNFs, commercial raw materials, and biomass are cheap, easily available, and suitable for bulk fabrication, but complicated and energy-consuming post-treatments such as high-temperature carbonization and chemical activation are normally required to realize favorable conductivity, porosity, and specific surface area, partially offsetting their intrinsic merits with respect to cost and abundance. Unlike traditional intercalated electrode materials, sulfur and other chalcogens are basically operated via multielectron transfer and multiple intermediates, thus requiring the cathode scaffolds not only with satisfactory electrical/ionic conductivities but also capabilities of controllably manipulating the transport and reaction phenomena of intermediates. A single-component material will probably not be able to meet all requirements. Therefore, the assembly of different components into a composite flexible cathode can shed new light for overcoming the challenges faced by a single constituent. The same idea is also appropriate for flexible lithium metal anodes and flexible SSEs.
For further improvement in battery performance and practical applications of flexible Li–S and analogous alkali metal–chalcogen batteries, several major concerns and limitations are observed, suggesting that there is substantial room for future advancement:
(1) The content of active material (i.e., sulfur, selenium, and tellurium) in the flexible cathodes should be further enhanced with its utilization ratio (i.e., specific capacity based on the weight of active materials) being well maintained, aiming at higher energy density in regard to the whole battery. Although the flexible cathode is notably better than conventional ones in specific electrode capacities with the same loading of active material, it is noted that the highest specific electrode capacity realized so far is still lower than 1000 mA h gele−1 (Fig. 31a). One of the limitations is that the mechanical flexibility is compromised if sulfur content is increased. Thus, more efficient 3D conductive and mechanical networks are required to accommodate more active materials but maintain preferable flexibility.
(2) The pore structure of flexible cathode must be well engineered. Generally, the flexible electrode has abundant void spaces to allow active materials with sufficiently access to ions, enabling high specific capacity and good rate performance of sulfur. However, the excessive pores have to be filled with electrolytes, reducing both overall gravimetric and volumetric energy densities of the whole cell. A balance between utilization and rate capability of active materials and practical energy densities should be considered.
(3) The performance of flexible lithium metal anodes and other alkali metal anodes needs to be improved, including flexibility, utilization (i.e., Coulombic efficiency), content of alkali metal in the whole composite, and cycling life. To realize this, two critical issues, namely dendritic growth and imperfect SEI, must be unambiguously addressed. The structure and composition of the alkali metal anode itself as well as its interaction and synergy with the electrolyte need deeper understanding and further rational design. The revival of the lithium metal anode, as well as the burgeoning sodium metal anode, will eventually revolutionize the whole research area of energy storage. It has to be noted that challenges associated with the alkali metal itself are much more tremendous and formidable than those of how to make it flexible.
(4) Safe, reliable, high-ionic-conductivity, and flexible SSEs are the ultimate option for flexible Li–S batteries and analogues because of the presence of highly flammable and dangerous alkali metals. Before being deployed in flexible batteries, issues of low ionic conductivity, insufficient mechanical strength, and interfacial contact have to be solved. Designing integrated cathode/SSE/anode architectures is highly desired yet very challenging. The development of flexible solid-state alkali metal–chalcogen full cells is still immature; thus, implications and inspirations from other more developed solid-state batteries such as flexible solid-state LIBs and Zn–air batteries will be very valuable.229,281,365,366 The use of flexible SSEs in flexible batteries, as well as the very likely successful experience during the development, will eventually drive the transformation of conventional Li–S and analogous alkali metal–chalcogen batteries to solid-state or quasi-solid-state configurations since they are inherently safer.
(5) Special applications need to be explored for other flexible alkali metal–chalcogen batteries, because they generally have inferior energy densities than Li–S batteries. Selenium and tellurium are much more expensive than sulfur, but possess higher electrical conductivities, which probably contribute to better rate performance in a volume-sensitive condition. The main advantage of using sodium is the cost of both sodium metal and sodium salts.
(6) Methods for flexibility evaluation should be established, verified, and standardized. The tests at a pouch-cell scale in dynamic bending/folding conditions will provide more accurate phenomena. Operando characterization of a working electrode in a battery is strongly suggested to gain more information about stress/strain distribution, detailed electrochemistry at the interfaces of electrode/electrolyte, ion diffusion pathways, and redistribution of active materials in the flexible battery.
(7) More prototype flexible batteries are expected to be formulated with additional functionalities, e.g., stretchability, transparency, wearability, implantability, biocompatibility, and self-power ability. These functionalities allow the prototype flexible batteries to be adopted in various applications like photodetectors, biosensors, and electronic skins, and integrated with flexible energy-harvesting devices like flexible photovoltaics and flexible piezoelectric/triboelectric nanogenerators.367
(8) Low-cost and large-scale production of flexible materials and devices will be preferred. Advanced printing techniques such as 3D printing will be very fascinating for micro-/nanoscale flexible batteries.368
(9) Thermal and strain management are important. On one hand, the strain in the device arises from not only each component but also their interfaces. Internal stress and resistance need to be well controlled, which requires an integrated design of the whole device. On the other hand, the temperature has a profound influence on the performance of each component (e.g., ionic conductivity and mechanical properties of flexible SPEs; diffusivity and reactivity of polysulfides). Good thermal management helps stabilize the transport and reaction phenomena within the cell and homogenize the spatial differences in concentrations, reaction rates, and so on.
(10) The safety and environmental issues of flexible Li–S and analogous alkali metal–chalcogen batteries should be considered when practical applications are explored. The standardization, interfaces between flexible power sources and power-consuming equipment, battery management system, and recycling of flexible batteries should also be considered to develop an acceptable business model and bulk applications in a sustainable society. The fundamental understandings of chemistry, physics, materials science, engineering, and social sciences are therefore strongly required to provide a reasonable solution and roadmap.
Aiming at higher energy densities, along with acceptable durability and safety, flexible batteries, now mainly based on LIB chemistries, are standing at a crossroad, where the original road to flexible LIBs, though a foreseen thoroughfare, may have an underwhelming end, while the other road to flexible alkali metal–chalcogen batteries, though very likely full of thistles and thorns, leads to a brighter future. Although the concept of Li–S battery has been researched for more than 40 years, great achievements have been made toward its application only recently. It is believed that with the combined efforts in nanomaterials and electrochemistry, more strategies will be realized in building high-energy-density, safe, and reliable flexible Li–S batteries and analogous systems. These insightful ideas, raised during the exploration, will eventually benefit the understanding and development of conventional alkali metal–chalcogen batteries, as well as other electrochemical systems based on multielectron chemistry.
Acknowledgements
This work was supported by the National Key Research and Development Program (No. 2016YFA0202500, 2016YFA0200102, and 2015CB932500) and Natural Scientific Foundation of China (No. 21676160). We thank Xin-Yan Liu, Xin-Bing Cheng, Ge Zhang, Jin Xie, Ying-Zhi Sun, Ze-Wen Zhang, Chen-Zi Zhao, Rui Zhang, Dai-Wei Wang, Zhe Yuan, Pei-Yan Zhai, Rui Xu, and Meng Zhao for the helpful discussion.References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22, E28–E62 CrossRef CAS PubMed.
- X. Q. Zhang, X. B. Cheng and Q. Zhang, J. Energy Chem., 2016, 25, 967–984 CrossRef.
- Q. Cao and J. A. Rogers, Adv. Mater., 2009, 21, 29–53 CrossRef CAS.
- S. Bae, H. Kim, Y. Lee, X. F. Xu, J. S. Park, Y. Zheng, J. Balakrishnan, T. Lei, H. R. Kim, Y. I. Song, Y. J. Kim, K. S. Kim, B. Ozyilmaz, J. H. Ahn, B. H. Hong and S. Iijima, Nat. Nanotechnol., 2010, 5, 574–578 CrossRef CAS PubMed.
- D. H. Kim, J. Viventi, J. J. Amsden, J. L. Xiao, L. Vigeland, Y. S. Kim, J. A. Blanco, B. Panilaitis, E. S. Frechette, D. Contreras, D. L. Kaplan, F. G. Omenetto, Y. G. Huang, K. C. Hwang, M. R. Zakin, B. Litt and J. A. Rogers, Nat. Mater., 2010, 9, 511–517 CrossRef CAS PubMed.
- D. H. Kim, N. S. Lu, R. Ma, Y. S. Kim, R. H. Kim, S. D. Wang, J. Wu, S. M. Won, H. Tao, A. Islam, K. J. Yu, T. I. Kim, R. Chowdhury, M. Ying, L. Z. Xu, M. Li, H. J. Chung, H. Keum, M. McCormick, P. Liu, Y. W. Zhang, F. G. Omenetto, Y. G. Huang, T. Coleman and J. A. Rogers, Science, 2011, 333, 838–843 CrossRef CAS PubMed.
- M. Kaltenbrunner, T. Sekitani, J. Reeder, T. Yokota, K. Kuribara, T. Tokuhara, M. Drack, R. Schwodiauer, I. Graz, S. Bauer-Gogonea, S. Bauer and T. Someya, Nature, 2013, 499, 458–463 CrossRef CAS PubMed.
- B. C. K. Tee, A. Chortos, A. Berndt, A. K. Nguyen, A. Tom, A. McGuire, Z. L. C. Lin, K. Tien, W. G. Bae, H. L. Wang, P. Mei, H. H. Chou, B. X. Cui, K. Deisseroth, T. N. Ng and Z. N. Bao, Science, 2015, 350, 313–316 CrossRef CAS PubMed.
- G. M. Zhou, F. Li and H. M. Cheng, Energy Environ. Sci., 2014, 7, 1307–1338 CAS.
- H. Gwon, J. Hong, H. Kim, D. H. Seo, S. Jeon and K. Kang, Energy Environ. Sci., 2014, 7, 538–551 CAS.
- K. K. Fu, J. Cheng, T. Li and L. B. Hu, ACS Energy Lett., 2016, 1, 1065–1079 CrossRef CAS.
- W. Liu, M. S. Song, B. Kong and Y. Cui, Adv. Mater., 2017, 29, 1603436 CrossRef PubMed.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- N. Nitta, F. X. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- D. Aurbach, B. D. McCloskey, L. F. Nazar and P. G. Bruce, Nat. Energy, 2016, 1, 16128 CrossRef CAS.
- A. Manthiram, Y. Z. Fu and Y. S. Su, Acc. Chem. Res., 2013, 46, 1125–1134 CrossRef CAS PubMed.
- L. Ma, K. E. Hendrickson, S. Y. Wei and L. A. Archer, Nano Today, 2015, 10, 315–338 CrossRef CAS.
- Z. W. Seh, Y. M. Sun, Q. F. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605–5634 RSC.
- L. Wen, F. Li and H. M. Cheng, Adv. Mater., 2016, 28, 4306–4337 CrossRef CAS PubMed.
- J. T. Di, X. H. Zhang, Z. Z. Yong, Y. Y. Zhang, D. Li, R. Li and Q. W. Li, Adv. Mater., 2016, 28, 10529–10538 CrossRef CAS PubMed.
- L. L. Peng, Y. Zhu, H. S. Li and G. H. Yu, Small, 2016, 12, 6183–6199 CrossRef CAS PubMed.
- Y. X. Yin, S. Xin, Y. G. Guo and L. J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS PubMed.
- A. Manthiram, S. H. Chung and C. X. Zu, Adv. Mater., 2015, 27, 1980–2006 CrossRef CAS PubMed.
- G. Zhang, Z. W. Zhang, H. J. Peng, J. Q. Huang and Q. Zhang, Small Methods, 2017, 1, 1700134 CrossRef.
- D. Bresser, S. Passerini and B. Scrosati, Chem. Commun., 2013, 49, 10545–10562 RSC.
- M. K. Song, E. J. Cairns and Y. G. Zhang, Nanoscale, 2013, 5, 2186–2204 RSC.
- X. Fang and H. S. Peng, Small, 2015, 11, 1488–1511 CrossRef CAS PubMed.
- R. J. Chen, T. Zhao and F. Wu, Chem. Commun., 2015, 51, 18–33 RSC.
- W. M. Kang, N. P. Deng, J. G. Ju, Q. X. Li, D. Y. Wu, X. M. Ma, L. Li, M. Naebe and B. W. Cheng, Nanoscale, 2016, 8, 16541–16588 RSC.
- Q. Pang, X. Liang, C. Y. Kwok and L. F. Nazar, Nat. Energy, 2016, 1, 16132 CrossRef CAS.
- M. Liu, X. Y. Qin, Y. B. He, B. H. Li and F. Y. Kang, J. Mater. Chem. A, 2017, 5, 5222–5234 CAS.
- X. L. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- Y. Yang, G. Y. Zheng and Y. Cui, Chem. Soc. Rev., 2013, 42, 3018–3032 RSC.
- D. W. Wang, Q. C. Zeng, G. M. Zhou, L. C. Yin, F. Li, H. M. Cheng, I. R. Gentle and G. Q. M. Lu, J. Mater. Chem. A, 2013, 1, 9382–9394 CAS.
- L. Borchardt, M. Oschatz and S. Kaskel, Chem. – Eur. J., 2016, 22, 7324–7351 CrossRef CAS PubMed.
- M. P. Yu, R. Li, M. M. Wu and G. Q. Shi, Energy Storage Mater., 2015, 1, 51–73 CrossRef.
- J. Liang, Z. H. Sun, F. Li and H. M. Cheng, Energy Storage Mater., 2016, 2, 76–106 CrossRef.
- S. Rehman, K. Khan, Y. F. Zhao and Y. L. Hou, J. Mater. Chem. A, 2017, 5, 3014–3038 CAS.
- H. L. Wang, Y. Yang, Y. Y. Liang, J. T. Robinson, Y. G. Li, A. Jackson, Y. Cui and H. J. Dai, Nano Lett., 2011, 11, 2644–2647 CrossRef CAS PubMed.
- L. W. Ji, M. M. Rao, H. M. Zheng, L. Zhang, Y. C. Li, W. H. Duan, J. H. Guo, E. J. Cairns and Y. G. Zhang, J. Am. Chem. Soc., 2011, 133, 18522–18525 CrossRef CAS PubMed.
- G. Y. Zheng, Y. Yang, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2011, 11, 4462–4467 CrossRef CAS PubMed.
- L. F. Xiao, Y. L. Cao, J. Xiao, B. Schwenzer, M. H. Engelhard, L. V. Saraf, Z. M. Nie, G. J. Exarhos and J. Liu, Adv. Mater., 2012, 24, 1176–1181 CrossRef CAS PubMed.
- M. Q. Zhao, Q. Zhang, J. Q. Huang, G. L. Tian, J. Q. Nie, H. J. Peng and F. Wei, Nat. Commun., 2014, 5, 3410 Search PubMed.
- Z. Y. Wang, Y. F. Dong, H. J. Li, Z. B. Zhao, H. B. Wu, C. Hao, S. H. Liu, J. S. Qiu and X. W. Lou, Nat. Commun., 2014, 5, 5002 CrossRef CAS PubMed.
- T. Z. Hou, W. T. Xu, X. Chen, H. J. Peng, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2017, 56, 8178–8182 CrossRef CAS PubMed.
- L. Kong, H. J. Peng, J. Q. Huang and Q. Zhang, Nano Res., 2017 DOI:10.1007/s12274-017-1652-x.
- S. H. Chung and A. Manthiram, J. Mater. Chem. A, 2013, 1, 9590–9596 CAS.
- H. J. Peng, W. T. Xu, L. Zhu, D. W. Wang, J. Q. Huang, X. B. Cheng, Z. Yuan, F. Wei and Q. Zhang, Adv. Funct. Mater., 2016, 26, 6351–6358 CrossRef CAS.
- S. S. Zhang, J. Power Sources, 2013, 231, 153–162 CrossRef CAS.
- S. Zhang, K. Ueno, K. Dokko and M. Watanabe, Adv. Energy Mater., 2015, 5, 1500117 CrossRef.
- Z. Lin and C. D. Liang, J. Mater. Chem. A, 2015, 3, 936–958 CAS.
- D. Aurbach, E. Pollak, R. Elazari, G. Salitra, C. S. Kelley and J. Affinito, J. Electrochem. Soc., 2009, 156, A694–A702 CrossRef CAS.
- Z. Lin, Z. C. Liu, W. J. Fu, N. J. Dudney and C. D. Liang, Adv. Funct. Mater., 2013, 23, 1064–1069 CrossRef CAS.
- L. M. Suo, Y. S. Hu, H. Li, M. Armand and L. Q. Chen, Nat. Commun., 2013, 4, 1481 CrossRef PubMed.
- C. W. Lee, Q. Pang, S. Ha, L. Cheng, S. D. Han, K. R. Zavadil, K. G. Gallagher, L. F. Nazar and M. Balasubramanian, ACS Cent. Sci., 2017, 3, 605–613 CrossRef CAS PubMed.
- R. G. Cao, W. Xu, D. P. Lv, J. Xiao and J. G. Zhang, Adv. Energy Mater., 2015, 5, 1402273 CrossRef.
- C. Huang, J. Xiao, Y. Y. Shao, J. M. Zheng, W. D. Bennett, D. P. Lu, L. V. Saraf, M. Engelhard, L. W. Ji, J. Zhang, X. L. Li, G. L. Graff and J. Liu, Nat. Commun., 2014, 5, 3015 Search PubMed.
- C. Z. Zhao, X. B. Cheng, R. Zhang, H. J. Peng, J. Q. Huang, R. Ran, Z. H. Huang, F. Wei and Q. Zhang, Energy Storage Mater., 2016, 3, 77–84 CrossRef.
- X. B. Cheng, C. Yan, J. Q. Huang, P. Li, L. Zhu, L. D. Zhao, Y. Y. Zhang, W. C. Zhu, S. T. Yang and Q. Zhang, Energy Storage Mater., 2017, 6, 18–25 CrossRef.
- J. Q. Huang, Q. Zhang and F. Wei, Energy Storage Mater., 2015, 1, 127–145 CrossRef.
- Y. S. Su and A. Manthiram, Chem. Commun., 2012, 48, 8817–8819 RSC.
- J. Q. Huang, Q. Zhang, H. J. Peng, X. Y. Liu, W. Z. Qian and F. Wei, Energy Environ. Sci., 2014, 7, 347–353 CAS.
- S. H. Chung and A. Manthiram, Adv. Funct. Mater., 2014, 24, 5299–5306 CrossRef CAS.
- H. B. Yao, K. Yan, W. Y. Li, G. Y. Zheng, D. S. Kong, Z. W. Seh, V. K. Narasimhan, Z. Liang and Y. Cui, Energy Environ. Sci., 2014, 7, 3381–3390 CAS.
- J. Q. Huang, T. Z. Zhuang, Q. Zhang, H. J. Peng, C. M. Chen and F. Wei, ACS Nano, 2015, 9, 3002–3011 CrossRef CAS PubMed.
- J. Balach, T. Jaumann, M. Klose, S. Oswald, J. Eckert and L. Giebeler, Adv. Funct. Mater., 2015, 25, 5285–5291 CrossRef CAS.
- H. J. Peng, D. W. Wang, J. Q. Huang, X. B. Cheng, Z. Yuan, F. Wei and Q. Zhang, Adv. Sci., 2016, 3, 1500268 CrossRef PubMed.
- T. Z. Zhuang, J. Q. Huang, H. J. Peng, L. Y. He, X. B. Cheng, C. M. Chen and Q. Zhang, Small, 2016, 12, 381–389 CrossRef CAS PubMed.
- P. Y. Zhai, H. J. Peng, X. B. Cheng, L. Zhu, J. Q. Huang, W. C. Zhu and Q. Zhang, Energy Storage Mater., 2017, 7, 56–63 CrossRef.
- H. J. Peng, J. Q. Huang, X. B. Cheng and Q. Zhang, Adv. Energy Mater., 2017, 7, 1700260 CrossRef.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- M. F. L. De Volder, S. H. Tawfick, R. H. Baughman and A. J. Hart, Science, 2013, 339, 535–539 CrossRef CAS PubMed.
- Q. Zhang, J. Q. Huang, M. Q. Zhao, W. Z. Qian and F. Wei, ChemSusChem, 2011, 4, 864–889 CrossRef CAS PubMed.
- Q. Zhang, J. Q. Huang, W. Z. Qian, Y. Y. Zhang and F. Wei, Small, 2013, 9, 1237–1265 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- W. J. Ma, L. Song, R. Yang, T. H. Zhang, Y. C. Zhao, L. F. Sun, Y. Ren, D. F. Liu, L. F. Liu, J. Shen, Z. X. Zhang, Y. J. Xiang, W. Y. Zhou and S. S. Xie, Nano Lett., 2007, 7, 2307–2311 CrossRef CAS PubMed.
- S. S. Fan, M. G. Chapline, N. R. Franklin, T. W. Tombler, A. M. Cassell and H. J. Dai, Science, 1999, 283, 512–514 CrossRef CAS PubMed.
- X. C. Gui, J. Q. Wei, K. L. Wang, A. Y. Cao, H. W. Zhu, Y. Jia, Q. K. Shu and D. H. Wu, Adv. Mater., 2010, 22, 617–621 CrossRef CAS PubMed.
- M. Zhang, K. R. Atkinson and R. H. Baughman, Science, 2004, 306, 1358–1361 CrossRef CAS PubMed.
- S. H. Ng, J. Wang, Z. P. Guo, G. X. Wang and H. K. Liu, Electrochim. Acta, 2005, 51, 23–28 CrossRef CAS.
- S. W. Lee, N. Yabuuchi, B. M. Gallant, S. Chen, B. S. Kim, P. T. Hammond and Y. Shao-Horn, Nat. Nanotechnol., 2010, 5, 531–537 CrossRef CAS PubMed.
- M. Kaempgen, C. K. Chan, J. Ma, Y. Cui and G. Gruner, Nano Lett., 2009, 9, 1872–1876 CrossRef CAS PubMed.
- L. B. Hu, J. W. Choi, Y. Yang, S. Jeong, F. La Mantia, L. F. Cui and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21490–21494 CrossRef CAS PubMed.
- K. L. Jiang, Q. Q. Li and S. S. Fan, Nature, 2002, 419, 801 CrossRef CAS PubMed.
- S. Dörfler, M. Hagen, H. Althues, J. Tübke, S. Kaskel and M. J. Hoffmann, Chem. Commun., 2012, 48, 4097–4099 RSC.
- C. Barchasz, F. Mesguich, J. Dijon, J. C. Leprêtre, S. Patoux and F. Alloin, J. Power Sources, 2012, 211, 19–26 CrossRef CAS.
- Y. S. Su, Y. Z. Fu and A. Manthiram, Phys. Chem. Chem. Phys., 2012, 14, 14495–14499 RSC.
- K. K. Jin, X. F. Zhou, L. Z. Zhang, X. Xin, G. H. Wan and Z. P. Liu, J. Phys. Chem. C, 2013, 117, 21112–21119 CAS.
- G. M. Zhou, D. W. Wang, F. Li, P. X. Hou, L. C. Yin, C. Liu, G. Q. Lu, I. R. Gentle and H. M. Cheng, Energy Environ. Sci., 2012, 5, 8901–8906 CAS.
- Z. Yuan, H. J. Peng, J. Q. Huang, X. Y. Liu, D. W. Wang, X. B. Cheng and Q. Zhang, Adv. Funct. Mater., 2014, 24, 6105–6112 CrossRef CAS.
- L. Sun, M. Y. Li, Y. Jiang, W. B. Kong, K. L. Jiang, J. P. Wang and S. S. Fan, Nano Lett., 2014, 14, 4044–4049 CrossRef CAS PubMed.
- L. Sun, D. T. Wang, Y. F. Luo, W. B. Kong, Y. Wu, L. N. Zhang, K. L. Jiang, Q. Q. Li, Y. H. Zhang, J. P. Wang and S. S. Fan, ACS Nano, 2016, 10, 1300–1308 CrossRef CAS PubMed.
- K. L. Jiang, J. P. Wang, Q. Q. Li, L. A. Liu, C. H. Liu and S. S. Fan, Adv. Mater., 2011, 23, 1154–1161 CrossRef CAS PubMed.
- L. Qie and A. Manthiram, Adv. Mater., 2015, 27, 1694–1700 CrossRef CAS PubMed.
- Y. Zhao, F. X. Yin, Y. G. Zhang, C. W. Zhang, A. Mentbayeva, N. Umirov, H. X. Xie and Z. Bakenov, Nanoscale Res. Lett., 2015, 10, 450 CrossRef PubMed.
- G. M. Zhou, E. Paek, G. S. Hwang and A. Manthiram, Nat. Commun., 2015, 6, 7760 CrossRef CAS PubMed.
- Q. Pang, J. T. Tang, H. Huang, X. Liang, C. Hart, K. C. Tam and L. F. Nazar, Adv. Mater., 2015, 27, 6021–6028 CrossRef CAS PubMed.
- T. Z. Hou, X. Chen, H. J. Peng, J. Q. Huang, B. Q. Li, Q. Zhang and B. Li, Small, 2016, 12, 3283–3291 CrossRef CAS PubMed.
- H. J. Peng, T. Z. Hou, Q. Zhang, J. Q. Huang, X. B. Cheng, M. Q. Guo, Z. Yuan, L. Y. He and F. Wei, Adv. Mater. Interfaces, 2014, 1, 1400227 CrossRef.
- M. Wu, Y. Cui and Y. Z. Fu, ACS Appl. Mater. Interfaces, 2015, 7, 21479–21486 CAS.
- A. Bhargav, S. V. Patil and Y. Z. Fu, Sustainable Energy Fuels, 2017, 1, 1007–1012 Search PubMed.
- A. Mentbayeva, A. Belgibayeva, N. Umirov, Y. G. Zhang, I. Taniguchi, I. Kurmanbayeva and Z. Bakenov, Electrochim. Acta, 2016, 217, 242–248 CrossRef CAS.
- G. J. Hu, Z. H. Sun, C. Shi, R. P. Fang, J. Chen, P. X. Hou, C. Liu, H. M. Cheng and F. Li, Adv. Mater., 2017, 29, 1603835 CrossRef PubMed.
- Y. Z. Fu, Y. S. Su and A. Manthiram, Angew. Chem., Int. Ed., 2013, 52, 6930–6935 CrossRef CAS PubMed.
- H. J. Peng, J. Q. Huang, X. Y. Liu, X. B. Cheng, W. T. Xu, C. Z. Zhao, F. Wei and Q. Zhang, J. Am. Chem. Soc., 2017, 139, 8458–8466 CrossRef CAS PubMed.
- M. Li, W. Wahyudi, P. Kumar, F. Wu, X. Yang, H. Li, L. J. Li and J. Ming, ACS Appl. Mater. Interfaces, 2017, 9, 8047–8054 CAS.
- S. Kim, H. Song and Y. Jeong, Carbon, 2017, 113, 371–378 CrossRef CAS.
- R. Ummethala, M. Fritzsche, T. Jaumann, J. Balach, S. Oswald, R. Nowak, N. Sobczak, I. Kaban, M. H. Rümmeli and L. Giebeler, Energy Storage Mater., 2017 DOI:10.1016/j.ensm.2017.04.004.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- F. Bonaccorso, L. Colombo, G. H. Yu, M. Stoller, V. Tozzini, A. C. Ferrari, R. S. Ruoff and V. Pellegrini, Science, 2015, 347, 1246501 CrossRef PubMed.
- W. Lv, Z. J. Li, Y. Q. Deng, Q. H. Yang and F. Y. Kang, Energy Storage Mater., 2016, 2, 107–138 CrossRef.
- W. C. Ren and H. M. Cheng, Nat. Nanotechnol., 2014, 9, 726–730 CrossRef CAS PubMed.
- A. C. Ferrari, F. Bonaccorso, V. Fal'ko, K. S. Novoselov, S. Roche, P. Boggild, S. Borini, F. H. L. Koppens, V. Palermo, N. Pugno, J. A. Garrido, R. Sordan, A. Bianco, L. Ballerini, M. Prato, E. Lidorikis, J. Kivioja, C. Marinelli, T. Ryhanen, A. Morpurgo, J. N. Coleman, V. Nicolosi, L. Colombo, A. Fert, M. Garcia-Hernandez, A. Bachtold, G. F. Schneider, F. Guinea, C. Dekker, M. Barbone, Z. P. Sun, C. Galiotis, A. N. Grigorenko, G. Konstantatos, A. Kis, M. Katsnelson, L. Vandersypen, A. Loiseau, V. Morandi, D. Neumaier, E. Treossi, V. Pellegrini, M. Polini, A. Tredicucci, G. M. Williams, B. H. Hong, J. H. Ahn, J. M. Kim, H. Zirath, B. J. van Wees, H. van der Zant, L. Occhipinti, A. Di Matteo, I. A. Kinloch, T. Seyller, E. Quesnel, X. L. Feng, K. Teo, N. Rupesinghe, P. Hakonen, S. R. T. Neil, Q. Tannock, T. Lofwander and J. Kinaret, Nanoscale, 2015, 7, 4598–4810 RSC.
- H. Gwon, H. S. Kim, K. U. Lee, D. H. Seo, Y. C. Park, Y. S. Lee, B. T. Ahn and K. Kang, Energy Environ. Sci., 2011, 4, 1277–1283 CAS.
- L. L. Zhang, X. Zhao, M. D. Stoller, Y. W. Zhu, H. X. Ji, S. Murali, Y. P. Wu, S. Perales, B. Clevenger and R. S. Ruoff, Nano Lett., 2012, 12, 1806–1812 CrossRef CAS PubMed.
- Y. X. Xu, Z. Y. Lin, X. Q. Huang, Y. Liu, Y. Huang and X. F. Duan, ACS Nano, 2013, 7, 4042–4049 CrossRef CAS PubMed.
- Y. N. Meng, Y. Zhao, C. G. Hu, H. H. Cheng, Y. Hu, Z. P. Zhang, G. Q. Shi and L. T. Qu, Adv. Mater., 2013, 25, 2326–2331 CrossRef CAS PubMed.
- J. Jin, Z. Y. Wen, G. Q. Ma, Y. Lu, Y. M. Cui, M. F. Wu, X. Liang and X. W. Wu, RSC Adv., 2013, 3, 2558–2560 RSC.
- J. Cao, C. Chen, Q. Zhao, N. Zhang, Q. Q. Lu, X. Y. Wang, Z. Q. Niu and J. Chen, Adv. Mater., 2016, 28, 9629–9636 CrossRef CAS PubMed.
- S. W. Luo, M. J. Yao, S. Lei, P. Z. Yan, X. Wei, X. T. Wang, L. L. Liu and Z. Q. Niu, Nanoscale, 2017, 9, 4646–4651 RSC.
- X. L. Li, X. R. Wang, L. Zhang, S. W. Lee and H. J. Dai, Science, 2008, 319, 1229–1232 CrossRef CAS PubMed.
- D. V. Kosynkin, A. L. Higginbotham, A. Sinitskii, J. R. Lomeda, A. Dimiev, B. K. Price and J. M. Tour, Nature, 2009, 458, 872–876 CrossRef CAS PubMed.
- Y. Liu, X. Z. Wang, Y. F. Dong, Y. C. Tang, L. X. Wang, D. Z. Jia, Z. B. Zhao and J. S. Qiu, Chem. Commun., 2016, 52, 12825–12828 RSC.
- G. M. Zhou, L. C. Yin, D. W. Wang, L. Li, S. F. Pei, I. R. Gentle, F. Li and H. M. Cheng, ACS Nano, 2013, 7, 5367–5375 CrossRef CAS PubMed.
- C. Lin, C. J. Niu, X. Xu, K. Li, Z. Y. Cai, Y. L. Zhang, X. P. Wang, L. B. Qu, Y. X. Xu and L. Q. Mai, Phys. Chem. Chem. Phys., 2016, 18, 22146–22153 RSC.
- C. Wang, X. S. Wang, Y. J. Wang, J. T. Chen, H. H. Zhou and Y. H. Huang, Nano Energy, 2015, 11, 678–686 CrossRef CAS.
- C. Wang, X. S. Wang, Y. Yang, A. Kushima, J. T. Chen, Y. H. Huang and J. Li, Nano Lett., 2015, 15, 1796–1802 CrossRef CAS PubMed.
- K. Han, J. M. Shen, S. Q. Hao, H. Q. Ye, C. Wolverton, M. C. Kung and H. H. Kung, ChemSusChem, 2014, 7, 2545–2553 CrossRef CAS PubMed.
- G. M. Zhou, E. Paek, G. S. Hwang and A. Manthiram, Adv. Energy Mater., 2016, 6, 1501355 CrossRef.
- G. M. Zhou, S. F. Pei, L. Li, D. W. Wang, S. G. Wang, K. Huang, L. C. Yin, F. Li and H. M. Cheng, Adv. Mater., 2014, 26, 625–631 CrossRef CAS PubMed.
- S. F. Pei and H. M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- J. B. Donnet, T. K. Wang, S. Rebouillat and J. C. M. Peng, Carbon fibers, Marcel Dekker, New York, 1998 Search PubMed.
- B. Liu, J. Zhang, X. F. Wang, G. Chen, D. Chen, C. W. Zhou and G. Z. Shen, Nano Lett., 2012, 12, 3005–3011 CrossRef CAS PubMed.
- L. F. Shen, Q. Che, H. S. Li and X. G. Zhang, Adv. Funct. Mater., 2014, 24, 2630–2637 CrossRef CAS.
- R. Elazari, G. Salitra, A. Garsuch, A. Panchenko and D. Aurbach, Adv. Mater., 2011, 23, 5641–5644 CrossRef CAS PubMed.
- S. S. Zhang and D. T. Tran, J. Power Sources, 2012, 211, 169–172 CrossRef CAS.
- S. H. Chung, C. H. Chang and A. Manthiram, Small, 2016, 12, 939–950 CrossRef CAS PubMed.
- N. He, L. Zhong, M. Xiao, S. J. Wang, D. M. Han and Y. Z. Meng, Sci. Rep., 2016, 6, 33871 CrossRef CAS PubMed.
- D. S. Yu, S. L. Zhai, W. C. Jiang, K. L. Goh, L. Wei, X. D. Chen, R. R. Jiang and Y. Chen, Adv. Mater., 2015, 27, 4895–4901 CrossRef CAS PubMed.
- D. Li and Y. N. Xia, Adv. Mater., 2004, 16, 1151–1170 CrossRef CAS.
- S. Cavaliere, S. Subianto, I. Savych, D. J. Jones and J. Roziere, Energy Environ. Sci., 2011, 4, 4761–4785 CAS.
- S. J. Peng, G. R. Jin, L. L. Li, K. Li, M. Srinivasan, S. Ramakrishna and J. Chen, Chem. Soc. Rev., 2016, 45, 1225–1241 RSC.
- H. Tsutsumi and K. Sunada, in Metal/Air, Metal/Sulfur, and Metal/Water Batteries, ed. Y. ShaoHorn, N. Dudney and T. Fuller, 2011, vol. 35, pp. 49–54 Search PubMed.
- L. W. Ji, M. M. Rao, S. Aloni, L. Wang, E. J. Cairns and Y. G. Zhang, Energy Environ. Sci., 2011, 4, 5053–5059 CAS.
- M. Inagaki, Y. Yang and F. Y. Kang, Adv. Mater., 2012, 24, 2547–2566 CrossRef CAS PubMed.
- L. C. Zeng, F. S. Pan, W. H. Li, Y. Jiang, X. W. Zhong and Y. Yu, Nanoscale, 2014, 6, 9579–9587 RSC.
- C. Wu, L. J. Fu, J. Maier and Y. Yu, J. Mater. Chem. A, 2015, 3, 9438–9445 CAS.
- L. C. Zeng, Y. Jiang, J. Xu, M. Wang, W. H. Li and Y. Yu, Nanoscale, 2015, 7, 10940–10949 RSC.
- L. C. Zeng, W. C. Zeng, Y. Jiang, X. Wei, W. H. Li, C. L. Yang, Y. W. Zhu and Y. Yu, Adv. Energy Mater., 2015, 5, 1401377 CrossRef.
- L. C. Zeng, X. Wei, J. Q. Wang, Y. Jiang, W. H. Li and Y. Yu, J. Power Sources, 2015, 281, 461–469 CrossRef CAS.
- Y. Yao, L. C. Zeng, S. H. Hu, Y. Jiang, B. B. Yuan and Y. Yu, Small, 2017, 13, 1603513 CrossRef PubMed.
- B. Zhang, X. Qin, G. R. Li and X. P. Gao, Energy Environ. Sci., 2010, 3, 1531–1537 CAS.
- J. C. Guo, Y. H. Xu and C. S. Wang, Nano Lett., 2011, 11, 4288–4294 CrossRef CAS PubMed.
- S. Xin, L. Gu, N. H. Zhao, Y. X. Yin, L. J. Zhou, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2012, 134, 18510–18513 CrossRef CAS PubMed.
- E. Markevich, G. Salitra, Y. Talyosef, F. Chesneau and D. Aurbach, J. Electrochem. Soc., 2017, 164, A6244–A6253 CrossRef CAS.
- K. Fu, Y. P. Li, M. Dirican, C. Chen, Y. Lu, J. D. Zhu, Y. Li, L. Y. Cao, P. D. Bradford and X. W. Zhang, Chem. Commun., 2014, 50, 10277–10280 RSC.
- B. C. Yu, J. W. Jung, K. Park and J. B. Goodenough, Energy Environ. Sci., 2017, 10, 86–90 CAS.
- M. L. Yu, Z. Y. Wang, Y. W. Wang, Y. F. Dong and J. S. Qiu, Adv. Energy Mater., 2017, 7, 1700018 CrossRef.
- Z. Li, J. T. Zhang, Y. M. Chen, J. Li and X. W. Lou, Nat. Commun., 2015, 6, 8850 CrossRef CAS PubMed.
- X. Song, S. Q. Wang, Y. Bao, G. X. Liu, W. P. Sun, L. X. Ding, H. K. Liu and H. H. Wang, J. Mater. Chem. A, 2017, 5, 6832–6839 CAS.
- D. Yang, W. Ni, J. L. Cheng, Z. P. Wang, T. Wang, Q. Guan, Y. Zhang, H. Wu, X. D. Li and B. Wang, Appl. Surf. Sci., 2017, 413, 209–218 CrossRef CAS.
- H. L. Zhu, W. Luo, P. N. Ciesielski, Z. Q. Fang, J. Y. Zhu, G. Henriksson, M. E. Himmel and L. B. Hu, Chem. Rev., 2016, 116, 9305–9374 CrossRef CAS PubMed.
- Y. Huang, C. L. Zhu, J. Z. Yang, Y. Nie, C. T. Chen and D. P. Sun, Cellulose, 2014, 21, 1–30 CrossRef CAS.
- L. X. Miao, W. K. Wang, K. G. Yuan, Y. S. Yang and A. B. Wang, Chem. Commun., 2014, 50, 13231–13234 RSC.
- R. P. Fang, S. Y. Zhao, P. X. Hou, M. Cheng, S. G. Wang, H. M. Cheng, C. Liu and F. Li, Adv. Mater., 2016, 28, 3374–3382 CrossRef CAS PubMed.
- S. H. Chung, C. H. Chang and A. Manthiram, ACS Nano, 2016, 10, 10462–10470 CrossRef CAS PubMed.
- S. J. Eichhorn, C. A. Baillie, N. Zafeiropoulos, L. Y. Mwaikambo, M. P. Ansell, A. Dufresne, K. M. Entwistle, P. J. Herrera-Franco, G. C. Escamilla, L. Groom, M. Hughes, C. Hill, T. G. Rials and P. M. Wild, J. Mater. Sci., 2001, 36, 2107–2131 CrossRef CAS.
- L. B. Hu and Y. Cui, Energy Environ. Sci., 2012, 5, 6423–6435 Search PubMed.
- F. X. Wu, E. B. Zhao, D. Gordon, Y. R. Xiao, C. C. Hu and G. Yushin, Adv. Mater., 2016, 28, 6365–6371 CrossRef CAS PubMed.
- D. H. Wang, D. Xie, T. Yang, Y. Zhong, X. L. Wang, X. H. Xia, C. D. Gu and J. P. Tu, J. Power Sources, 2016, 331, 475–480 CrossRef CAS.
- Y. Huang, M. B. Zheng, Z. X. Lin, B. Zhao, S. T. Zhang, J. Z. Yang, C. L. Zhu, H. Zhang, D. P. Sun and Y. Shi, J. Mater. Chem. A, 2015, 3, 10910–10918 CAS.
- S. Q. Li, T. Mou, G. F. Ren, J. Warzywoda, Z. D. Wei, B. Wang and Z. Y. Fan, J. Mater. Chem. A, 2017, 5, 1650–1657 CAS.
- L. W. Mi, W. D. Xiao, S. Z. Cui, H. W. Hou and W. H. Chen, Dalton Trans., 2016, 45, 3305–3309 RSC.
- Y. B. An, Q. Z. Zhu, L. F. Hu, S. K. Yu, Q. Zhao and B. Xu, J. Mater. Chem. A, 2016, 4, 15605–15611 CAS.
- Z. X. Cao, J. Zhang, Y. M. Ding, Y. L. Li, M. J. Shi, H. Y. Yue, Y. Qiao, Y. H. Yin and S. T. Yang, J. Mater. Chem. A, 2016, 4, 8636–8644 CAS.
- A. Schneider, C. Suchomski, H. Sommer, J. Janek and T. Brezesinski, J. Mater. Chem. A, 2015, 3, 20482–20486 CAS.
- W. D. Zhou, B. K. Guo, H. C. Gao and J. B. Goodenough, Adv. Energy Mater., 2016, 6, 1502059 CrossRef.
- Z. J. Fan, J. Yan, L. J. Zhi, Q. Zhang, T. Wei, J. Feng, M. L. Zhang, W. Z. Qian and F. Wei, Adv. Mater., 2010, 22, 3723–3728 CrossRef CAS PubMed.
- M. Q. Zhao, X. F. Liu, Q. Zhang, G. L. Tian, J. Q. Huang, W. C. Zhu and F. Wei, ACS Nano, 2012, 6, 10759–10769 CrossRef CAS PubMed.
- C. Tang, Q. Zhang, M. Q. Zhao, J. Q. Huang, X. B. Cheng, G. L. Tian, H. J. Peng and F. Wei, Adv. Mater., 2014, 26, 6100–6105 CrossRef CAS PubMed.
- G. L. Tian, M. Q. Zhao, D. S. Yu, X. Y. Kong, J. Q. Huang, Q. Zhang and F. Wei, Small, 2014, 10, 2251–2259 CrossRef CAS PubMed.
- R. T. Lv, T. X. Cui, M. S. Jun, Q. Zhang, A. Y. Cao, D. S. Su, Z. J. Zhang, S. H. Yoon, J. Miyawaki, I. Mochida and F. Y. Kang, Adv. Funct. Mater., 2011, 21, 999–1006 CrossRef CAS.
- Y. G. Li, W. Zhou, H. L. Wang, L. M. Xie, Y. Y. Liang, F. Wei, J. C. Idrobo, S. J. Pennycook and H. J. Dai, Nat. Nanotechnol., 2012, 7, 394–400 CrossRef CAS PubMed.
- R. J. Chen, T. Zhao, J. Lu, F. Wu, L. Li, J. Z. Chen, G. Q. Tan, Y. S. Ye and K. Amine, Nano Lett., 2013, 13, 4642–4649 CrossRef CAS PubMed.
- H. J. Peng, J. Q. Huang, M. Q. Zhao, Q. Zhang, X. B. Cheng, X. Y. Liu, W. Z. Qian and F. Wei, Adv. Funct. Mater., 2014, 24, 2772–2781 CrossRef CAS.
- S. Y. Yuan, Z. Y. Guo, L. N. Wang, S. Hu, Y. G. Wang and Y. Y. Xia, Adv. Sci., 2015, 2, 1500071 CrossRef PubMed.
- Z. Z. Yang, H. Y. Wang, X. B. Zhong, W. Qi, B. Y. Wang and Q. C. Jiang, RSC Adv., 2014, 4, 50964–50968 RSC.
- R. X. Chu, J. Lin, C. Q. Wu, J. Zheng, Y. L. Chen, J. Zhang, R. H. Han, Y. Zhang and H. Guo, Nanoscale, 2017, 9, 9129–9138 RSC.
- L. Sun, W. B. Kong, Y. Jiang, H. C. Wu, K. L. Jiang, J. P. Wang and S. S. Fan, J. Mater. Chem. A, 2015, 3, 5305–5312 CAS.
- S. C. Han, X. Pu, X. L. Li, M. M. Liu, M. Li, N. Feng, S. Dou and W. G. Hu, Electrochim. Acta, 2017, 241, 406–413 CrossRef CAS.
- X. D. Huang, B. Sun, K. F. Li, S. Q. Chen and G. X. Wang, J. Mater. Chem. A, 2013, 1, 13484–13489 CAS.
- Y. Chen, S. T. Lu, X. H. Wu and J. Liu, J. Phys. Chem. C, 2015, 119, 10288–10294 CAS.
- R. Li, M. Zhang, Y. R. Li, J. Chen, B. W. Yao, M. P. Yu and G. Q. Shi, Phys. Chem. Chem. Phys., 2016, 18, 11104–11110 RSC.
- J. R. He, Y. F. Chen, W. G. Lv, K. C. Wen, C. Xu, W. L. Zhang, W. Qin and W. D. He, ACS Energy Lett., 2016, 1, 820–826 CrossRef CAS.
- W. Deng, X. F. Zhou, Q. L. Fang and Z. P. Liu, J. Mater. Chem. A, 2017, 5, 13674–13682 CAS.
- L. Zhu, H. J. Peng, J. Y. Liang, J. Q. Huang, C. M. Chen, X. F. Guo, W. C. Zhu, P. Li and Q. Zhang, Nano Energy, 2015, 11, 746–755 CrossRef CAS.
- P. Y. Zhai, J. Q. Huang, L. Zhu, J. L. Shi, W. Zhu and Q. Zhang, Carbon, 2017, 111, 493–501 CrossRef CAS.
- J. Q. Huang, H. J. Peng, X. Y. Liu, J. Q. Nie, X. B. Cheng, Q. Zhang and F. Wei, J. Mater. Chem. A, 2014, 2, 10869–10875 CAS.
- J. L. Shi, H. J. Peng, L. Zhu, W. C. Zhu and Q. Zhang, Carbon, 2015, 92, 96–105 CrossRef CAS.
- X. W. Wu, H. Xie, Q. Deng, H. X. Wang, H. Sheng, Y. X. Yin, W. X. Zhou, R. L. Li and Y. G. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 1553–1561 CAS.
- Q. Sun, X. Fang, W. Weng, J. Deng, P. N. Chen, J. Ren, G. Z. Guan, M. Wang and H. S. Peng, Angew. Chem., Int. Ed., 2015, 54, 10539–10544 CrossRef CAS PubMed.
- G. M. Zhou, Y. B. Zhao and A. Manthiram, Adv. Energy Mater., 2015, 5, 1402263 CrossRef.
- F. Wu, Y. S. Ye, J. Q. Huang, T. Zhao, J. Qian, Y. Y. Zhao, L. Li, L. Wei, R. Luo, Y. X. Huang, Y. Xing and R. J. Chen, ACS Nano, 2017, 11, 4694–4702 CrossRef CAS PubMed.
- N. Jayaprakash, J. Shen, S. S. Moganty, A. Corona and L. A. Archer, Angew. Chem., Int. Ed., 2011, 50, 5904–5908 CrossRef CAS PubMed.
- J. X. Song, T. Xu, M. L. Gordin, P. Y. Zhu, D. P. Lv, Y. B. Jiang, Y. S. Chen, Y. H. Duan and D. H. Wang, Adv. Funct. Mater., 2014, 24, 1243–1250 CrossRef CAS.
- H. J. Peng, J. Y. Liang, L. Zhu, J. Q. Huang, X. B. Cheng, X. F. Guo, W. P. Ding, W. C. Zhu and Q. Zhang, ACS Nano, 2014, 8, 11280–11289 CrossRef CAS PubMed.
- C. Tang, B. Q. Li, Q. Zhang, L. Zhu, H. F. Wang, J. L. Shi and F. Wei, Adv. Funct. Mater., 2016, 26, 577–585 CrossRef CAS.
- S. Thieme, J. Bruckner, I. Bauer, M. Oschatz, L. Borchardt, H. Althues and S. Kaskel, J. Mater. Chem. A, 2013, 1, 9225–9234 CAS.
- H. X. Li, Y. Gong, C. P. Fu, H. H. Zhou, W. J. Yang, M. L. Guo, M. B. Li and Y. F. Kuang, J. Mater. Chem. A, 2017, 5, 3875–3887 CAS.
- W. Ni, J. L. Cheng, X. D. Li, Q. Guan, G. X. Qu, Z. Y. Wang and B. Wang, RSC Adv., 2016, 6, 9320–9327 RSC.
- A. Ghosh, R. Manjunatha, R. Kumar and S. Mitra, ACS Appl. Mater. Interfaces, 2016, 8, 33775–33785 CAS.
- J. X. Song, Z. X. Yu, T. Xu, S. R. Chen, H. Sohn, M. Regula and D. H. Wang, J. Mater. Chem. A, 2014, 2, 8623–8627 CAS.
- H. Q. Wang, W. C. Zhang, H. K. Liu and Z. P. Guo, Angew. Chem., Int. Ed., 2016, 55, 3992–3996 CrossRef CAS PubMed.
- W. B. Kong, L. Sun, Y. Wu, K. L. Jiang, Q. Q. Li, J. P. Wang and S. S. Fan, Carbon, 2016, 96, 1053–1059 CrossRef CAS.
- M. P. Yu, J. S. Ma, M. Xie, H. Q. Song, F. Y. Tian, S. S. Xu, Y. Zhou, B. Li, D. Wu, H. Qiu and R. M. Wang, Adv. Energy Mater., 2017, 7, 1602347 CrossRef.
- C. Y. Chen, H. J. Peng, T. Z. Hou, P. Y. Zhai, B. Q. Li, C. Tang, J. Q. Huang and Q. Zhang, Adv. Mater., 2017, 29, 1606802 CrossRef PubMed.
- H. W. Chen, C. H. Wang, Y. F. Dai, S. Q. Qiu, J. L. Yang, W. Lu and L. W. Chen, Nano Lett., 2015, 15, 5443–5448 CrossRef CAS PubMed.
- C. Milroy and A. Manthiram, Adv. Mater., 2016, 28, 9744–9751 CrossRef CAS PubMed.
- Z. Y. Wang, J. L. Cheng, W. Ni, L. Z. Gao, D. Yang, J. M. Razal and B. Wang, J. Power Sources, 2017, 342, 772–778 CrossRef CAS.
- P. J. Hanumantha, B. Gattu, P. M. Shanthi, S. S. Damle, Z. Basson, R. Bandi, M. K. Datta, S. Park and P. N. Kumta, Electrochim. Acta, 2016, 212, 286–293 CrossRef CAS.
- G. M. Zhou, L. Li, D. W. Wang, X. Y. Shan, S. F. Pei, F. Li and H. M. Cheng, Adv. Mater., 2015, 27, 641–647 CrossRef CAS PubMed.
- Z. P. Chen, W. C. Ren, L. B. Gao, B. L. Liu, S. F. Pei and H. M. Cheng, Nat. Mater., 2011, 10, 424–428 CrossRef CAS PubMed.
- N. Li, Z. P. Chen, W. C. Ren, F. Li and H. M. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17360–17365 CrossRef CAS PubMed.
- J. L. Liu, L. L. Zhang, H. B. Wu, J. Y. Lin, Z. X. Shen and X. W. Lou, Energy Environ. Sci., 2014, 7, 3709–3719 CAS.
- K. Xi, P. R. Kidambi, R. J. Chen, C. L. Gao, X. Y. Peng, C. Ducati, S. Hofmann and R. V. Kumar, Nanoscale, 2014, 6, 5746–5753 RSC.
- G. M. Zhou, L. Li, C. Q. Ma, S. G. Wang, Y. Shi, N. Koratkar, W. C. Ren, F. Li and H. M. Cheng, Nano Energy, 2015, 11, 356–365 CrossRef CAS.
- J. R. He, Y. F. Chen, W. Q. Lv, K. C. Wen, P. J. Li, F. Qi, Z. G. Wang, W. L. Zhang, Y. R. Li, W. Qin and W. D. He, J. Power Sources, 2016, 327, 474–480 CrossRef CAS.
- C. Milroy and A. Manthiram, Chem. Commun., 2016, 52, 4282–4285 RSC.
- Z. W. Seh, W. Y. Li, J. J. Cha, G. Y. Zheng, Y. Yang, M. T. McDowell, P. C. Hsu and Y. Cui, Nat. Commun., 2013, 4, 1331 CrossRef PubMed.
- Q. Pang, D. Kundu, M. Cuisinier and L. F. Nazar, Nat. Commun., 2014, 5, 4759 CrossRef CAS PubMed.
- X. Y. Tao, J. G. Wang, Z. G. Ying, Q. X. Cai, G. Y. Zheng, Y. P. Gan, H. Huang, Y. Xia, C. Liang, W. K. Zhang and Y. Cui, Nano Lett., 2014, 14, 5288–5294 CrossRef CAS PubMed.
- H. B. Yao, G. Y. Zheng, P. C. Hsu, D. S. Kong, J. J. Cha, W. Y. Li, Z. W. Seh, M. T. McDowell, K. Yan, Z. Liang, V. K. Narasimhan and Y. Cui, Nat. Commun., 2014, 5, 3943 CAS.
- X. Liang, C. Hart, Q. Pang, A. Garsuch, T. Weiss and L. F. Nazar, Nat. Commun., 2015, 6, 5682 CrossRef PubMed.
- Q. Fan, W. Liu, Z. Weng, Y. M. Sun and H. L. Wang, J. Am. Chem. Soc., 2015, 137, 12946–12953 CrossRef CAS PubMed.
- X. Y. Tao, J. G. Wang, C. Liu, H. T. Wang, H. B. Yao, G. Y. Zheng, Z. W. Seh, Q. X. Cai, W. Y. Li, G. M. Zhou, C. X. Zu and Y. Cui, Nat. Commun., 2016, 7, 11203 CrossRef CAS PubMed.
- Z. Li, J. T. Zhang, B. Y. Guan, D. Wang, L. M. Liu and X. W. Lou, Nat. Commun., 2016, 7, 13065 CrossRef CAS PubMed.
- X. Q. Niu, X. L. Wang, D. H. Wang, Y. Li, Y. J. Zhang, Y. D. Zhang, T. Yang, T. Yu and J. P. Tu, J. Mater. Chem. A, 2015, 3, 17106–17112 CAS.
- H. J. Peng, Z. W. Zhang, J. Q. Huang, G. Zhang, J. Xie, W. T. Xu, J. L. Shi, X. Chen, X. B. Cheng and Q. Zhang, Adv. Mater., 2016, 28, 9551–9558 CrossRef CAS PubMed.
- Z. W. Seh, J. H. Yu, W. Y. Li, P. C. Hsu, H. T. Wang, Y. M. Sun, H. B. Yao, Q. F. Zhang and Y. Cui, Nat. Commun., 2014, 5, 5017 CrossRef CAS PubMed.
- Z. Yuan, H. J. Peng, T. Z. Hou, J. Q. Huang, C. M. Chen, D. W. Wang, X. B. Cheng, F. Wei and Q. Zhang, Nano Lett., 2016, 16, 519–527 CrossRef CAS PubMed.
- G. M. Zhou, H. Z. Tian, Y. Jin, X. Y. Tao, B. F. Liu, R. F. Zhang, Z. W. Seh, D. Zhuo, Y. Y. Liu, J. Sun, J. Zhao, C. X. Zu, D. S. Wu, Q. F. Zhang and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 840–845 CrossRef CAS PubMed.
- H. B. Lin, L. Q. Yang, X. Jiang, G. C. Li, T. R. Zhang, Q. F. Yao, G. Y. Zheng and J. Y. Lee, Energy Environ. Sci., 2017, 10, 1476–1486 CAS.
- X. Liang, A. Garsuch and L. F. Nazar, Angew. Chem., Int. Ed., 2015, 54, 3907–3911 CrossRef CAS PubMed.
- H. J. Peng, G. Zhang, X. Chen, Z. W. Zhang, W. T. Xu, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2016, 55, 12990–12995 CrossRef CAS PubMed.
- Z. M. Cui, C. X. Zu, W. D. Zhou, A. Manthiram and J. B. Goodenough, Adv. Mater., 2016, 28, 6926–6931 CrossRef CAS PubMed.
- T. G. Jeong, D. S. Choi, H. Song, J. Choi, S. A. Park, S. H. Oh, H. Kim, Y. Jung and Y. T. Kim, ACS Energy Lett., 2017, 2, 327–333 CrossRef CAS.
- Z. H. Sun, J. Q. Zhang, L. C. Yin, G. J. Hu, R. P. Fang, H. M. Cheng and F. Li, Nat. Commun., 2017, 8, 14627 CrossRef PubMed.
- Q. Pang, X. Liang, C. Y. Kwok and L. F. Nazar, J. Electrochem. Soc., 2015, 162, A2567–A2576 CrossRef CAS.
- Z. Zhang, Q. Li, K. Zhang, W. Chen, Y. Q. Lai and J. Li, J. Power Sources, 2015, 290, 159–167 CrossRef CAS.
- X. Song, T. Gao, S. Q. Wang, Y. Bao, G. P. Chen, L. X. Ding and H. H. Wang, J. Power Sources, 2017, 356, 172–180 CrossRef CAS.
- T. Y. Lei, W. Chen, J. W. Huang, C. Y. Yan, H. X. Sun, C. Wang, W. L. Zhang, Y. R. Li and J. Xiong, Adv. Energy Mater., 2017, 7, 1601843 CrossRef.
- H. H. Xu and A. Manthiram, Nano Energy, 2017, 33, 124–129 CrossRef CAS.
- H. H. Xu, L. Qie and A. Manthiram, Nano Energy, 2016, 26, 224–232 CrossRef CAS.
- Z. Chang, H. Dou, B. Ding, J. Wang, Y. Wang, X. D. Hao and D. R. MacFarlane, J. Mater. Chem. A, 2017, 5, 250–257 CAS.
- L. Fan, H. L. L. Zhuang, K. H. Zhang, V. R. Cooper, Q. Li and Y. Y. Lu, Adv. Sci., 2016, 3, 1600175 CrossRef PubMed.
- G. M. Zhou, Y. B. Zhao, C. X. Zu and A. Manthiram, Nano Energy, 2015, 12, 240–249 CrossRef CAS.
- J. Gao, L. Li, J. W. Tan, H. Sun, B. C. Li, J. C. Idrobo, C. V. Singh, T. M. Lu and N. Koratkar, Nano Lett., 2016, 16, 3780–3787 CrossRef CAS PubMed.
- L. Li, L. Chen, S. Mukherjee, J. Gao, H. Sun, Z. B. Liu, X. L. Ma, T. Gupta, C. V. Singh, W. C. Ren, H. M. Cheng and N. Koratkar, Adv. Mater., 2017, 29, 1602734 CrossRef PubMed.
- M. P. Yu, A. J. Wang, F. Y. Tian, H. Q. Song, Y. S. Wang, C. Li, J. D. Hong and G. Q. Shi, Nanoscale, 2015, 7, 5292–5298 RSC.
- Y. Chen, S. T. Lu, J. Zhou, X. H. Wu, W. Qin, O. Ogoke and G. Wu, J. Mater. Chem. A, 2017, 5, 102–112 CAS.
- Z. Li, H. B. Wu and X. W. Lou, Energy Environ. Sci., 2016, 9, 3061–3070 CAS.
- G. M. Zhou, J. Sun, Y. Jin, W. Chen, C. X. Zu, R. F. Zhang, Y. C. Qiu, J. Zhao, D. Zhuo, Y. Y. Liu, X. Y. Tao, W. Liu, K. Yan, H. R. Lee and Y. Cui, Adv. Mater., 2017, 29, 1603366 CrossRef PubMed.
- Y. Y. Mao, G. R. Li, Y. Guo, Z. P. Li, C. D. Liang, X. S. Peng and Z. Lin, Nat. Commun., 2017, 8, 14628 CrossRef PubMed.
- Y. Z. Liu, G. R. Li, J. Fu, Z. W. Chen and X. S. Peng, Angew. Chem., Int. Ed., 2017, 56, 6176–6180 CrossRef CAS PubMed.
- Y. Z. Liu, G. R. Li, Z. W. Chen and X. S. Peng, J. Mater. Chem. A, 2017, 5, 9775–9784 CAS.
- Q. Zhao, X. F. Hu, K. Zhang, N. Zhang, Y. X. Hu and J. Chen, Nano Lett., 2015, 15, 721–726 CrossRef CAS PubMed.
- H. Kim, G. Jeong, Y. U. Kim, J. H. Kim, C. M. Park and H. J. Sohn, Chem. Soc. Rev., 2013, 42, 9011–9034 RSC.
- W. Xu, J. L. Wang, F. Ding, X. L. Chen, E. Nasybutin, Y. H. Zhang and J. G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 CAS.
- X. B. Cheng, R. Zhang, C. Z. Zhao, F. Wei, J. G. Zhang and Q. Zhang, Adv. Sci., 2016, 3, 1500213 CrossRef PubMed.
- M. D. Tikekar, S. Choudhury, Z. Y. Tu and L. A. Archer, Nat. Energy, 2016, 1, 16114 CrossRef CAS.
- D. C. Lin, Y. Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- C. P. Yang, Y. X. Yin, S. F. Zhang, N. W. Li and Y. G. Guo, Nat. Commun., 2015, 6, 8058 CrossRef CAS PubMed.
- R. Zhang, X. B. Cheng, C. Z. Zhao, H. J. Peng, J. L. Shi, J. Q. Huang, J. F. Wang, F. Wei and Q. Zhang, Adv. Mater., 2016, 28, 2155–2162 CrossRef CAS PubMed.
- G. Y. Zheng, S. W. Lee, Z. Liang, H. W. Lee, K. Yan, H. B. Yao, H. T. Wang, W. Y. Li, S. Chu and Y. Cui, Nat. Nanotechnol., 2014, 9, 618–623 CrossRef CAS PubMed.
- X. X. Zeng, Y. X. Yin, N. W. Li, W. C. Du, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2016, 138, 15825–15828 CrossRef CAS PubMed.
- X. B. Cheng, C. Yan, X. Chen, C. Guan, J. Q. Huang, H. J. Peng, R. Zhang, S. T. Yang and Q. Zhang, Chem, 2017, 2, 258–270 CAS.
- N. W. Li, Y. X. Yin, C. P. Yang and Y. G. Guo, Adv. Mater., 2016, 28, 1853–1858 CrossRef CAS PubMed.
- F. Ding, W. Xu, G. L. Graff, J. Zhang, M. L. Sushko, X. L. Chen, Y. Y. Shao, M. H. Engelhard, Z. M. Nie, J. Xiao, X. J. Liu, P. V. Sushko, J. Liu and J. G. Zhang, J. Am. Chem. Soc., 2013, 135, 4450–4456 CrossRef CAS PubMed.
- Y. Y. Lu, Z. Y. Tu and L. A. Archer, Nat. Mater., 2014, 13, 961–969 CrossRef CAS PubMed.
- J. F. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J. G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef CAS PubMed.
- R. Zhang, N. W. Li, X. B. Cheng, Y. X. Yin, Q. Zhang and Y. G. Guo, Adv. Sci., 2017, 4, 1600445 CrossRef PubMed.
- X. B. Cheng, T. Z. Hou, R. Zhang, H. J. Peng, C. Z. Zhao, J. Q. Huang and Q. Zhang, Adv. Mater., 2016, 28, 2888–2895 CrossRef CAS PubMed.
- R. Zhang, X. R. Chen, X. Chen, X. B. Cheng, X. Q. Zhang, C. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2017, 56, 7764–7768 CrossRef CAS PubMed.
- D. C. Lin, Y. Y. Liu, Z. Liang, H. W. Lee, J. Sun, H. T. Wang, K. Yan, J. Xie and Y. Cui, Nat. Nanotechnol., 2016, 11, 626–632 CrossRef CAS PubMed.
- L. L. Lu, J. Ge, J. N. Yang, S. M. Chen, H. B. Yao, F. Zhou and S. H. Yu, Nano Lett., 2016, 16, 4431–4437 CrossRef CAS PubMed.
- L. L. Lu, Y. Zhang, P. Zhao, H. B. Yao, F. Zhou and S. H. Yu, Energy Storage Mater., 2017, 9, 31–38 CrossRef.
- X. Y. Yang, J. J. Xu, D. Bao, Z. W. Chang, D. P. Liu, Y. Zhang and X. B. Zhang, Adv. Mater., 2017, 29, 1700378 CrossRef PubMed.
- X. B. Cheng, H. J. Peng, J. Q. Huang, R. Zhang, C. Z. Zhao and Q. Zhang, ACS Nano, 2015, 9, 6373–6382 CrossRef CAS PubMed.
- Z. Liang, G. Y. Zheng, C. Liu, N. Liu, W. Y. Li, K. Yan, H. B. Yao, P. C. Hsu, S. Chu and Y. Cui, Nano Lett., 2015, 15, 2910–2916 CrossRef CAS PubMed.
- Y. Zhang, B. Y. Liu, E. Hitz, W. Luo, Y. G. Yao, Y. J. Li, J. Q. Dai, C. J. Chen, Y. B. Wang, C. P. Yang, H. B. Li and L. B. Hu, Nano Res., 2017, 10, 1356–1365 CrossRef CAS.
- J. L. Lang, J. A. Song, L. H. Qi, Y. Z. Luo, X. Y. Luo and H. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 10360–10365 CAS.
- S. Matsuda, Y. Kubo, K. Uosaki and S. Nakanishi, ACS Energy Lett., 2017, 2, 924–929 CrossRef CAS.
- Z. Liang, D. C. Lin, J. Zhao, Z. D. Lu, Y. Y. Liu, C. Liu, Y. Y. Lu, H. T. Wang, K. Yan, X. Y. Tao and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2862–2867 CrossRef CAS PubMed.
- Y. Y. Liu, D. C. Lin, Z. Liang, J. Zhao, K. Yan and Y. Cui, Nat. Commun., 2016, 7, 10992 CrossRef CAS PubMed.
- T. T. Zuo, X. W. Wu, C. P. Yang, Y. X. Yin, H. Ye, N. W. Li and Y. G. Guo, Adv. Mater., 2017, 29, 1700389 CrossRef PubMed.
- L. F. Cui, L. B. Hu, J. W. Choi and Y. Cui, ACS Nano, 2010, 4, 3671–3678 CrossRef CAS PubMed.
- B. Wang, X. L. Li, X. F. Zhang, B. Luo, M. H. Jin, M. H. Liang, S. A. Dayeh, S. T. Picraux and L. J. Zhi, ACS Nano, 2013, 7, 1437–1445 CrossRef CAS PubMed.
- J. Y. Zheng, H. Zheng, R. Wang, L. B. Ben, W. Lu, L. W. Chen, L. Q. Chen and H. Li, Phys. Chem. Chem. Phys., 2014, 16, 13229–13238 RSC.
- B. Zhu, Y. Jin, X. Z. Hu, Q. H. Zheng, S. Zhang, Q. J. Wang and J. Zhu, Adv. Mater., 2017, 29, 1603755 CrossRef PubMed.
- J. Luo, R. C. Lee, J. T. Jin, Y. T. Weng, C. C. Fang and N. L. Wu, Chem. Commun., 2017, 53, 963–966 RSC.
- Z. L. Hu, S. Zhang, S. M. Dong, W. J. Li, H. Li, G. L. Cui and L. Q. Chen, Chem. Mater., 2017, 29, 4682–4689 CrossRef CAS.
- G. Y. Zheng, C. Wang, A. Pei, J. Lopez, F. F. Shi, Z. Chen, A. D. Sendek, H. W. Lee, Z. D. Lu, H. Schneider, M. M. Safont-Sempere, S. Chu, Z. N. Bao and Y. Cui, ACS Energy Lett., 2016, 1, 1247–1255 CrossRef CAS.
- K. Liu, A. Pei, H. R. Lee, B. Kong, N. Liu, D. C. Lin, Y. Y. Liu, C. Liu, P. C. Hsu, Z. A. Bao and Y. Cui, J. Am. Chem. Soc., 2017, 139, 4815–4820 CrossRef CAS PubMed.
- Y. Y. Liu, D. C. Lin, P. Y. Yuen, K. Liu, J. Xie, R. H. Dauskardt and Y. Cui, Adv. Mater., 2017, 29, 1605531 CrossRef PubMed.
- W. Liu, W. Y. Li, D. Zhuo, G. Y. Zheng, Z. D. Lu, K. Liu and Y. Cui, ACS Cent. Sci., 2017, 3, 135–140 CrossRef CAS PubMed.
- K. Y. Xie, K. Yuan, K. Zhang, C. Shen, W. B. Lv, X. R. Liu, J. G. Wang and B. Q. Wei, ACS Appl. Mater. Interfaces, 2017, 9, 4605–4613 CAS.
- J. Gao, M. A. Lowe, Y. Kiya and H. D. Abruna, J. Phys. Chem. C, 2011, 115, 25132–25137 CAS.
- L. X. Yuan, J. K. Feng, X. P. Ai, Y. L. Cao, S. L. Chen and H. X. Yang, Electrochem. Commun., 2006, 8, 610–614 CrossRef CAS.
- J. L. Wang, J. Yang, J. Y. Xie, N. X. Xu and Y. Li, Electrochem. Commun., 2002, 4, 499–502 CrossRef CAS.
- J. Hassoun and B. Scrosati, Angew. Chem., Int. Ed., 2010, 49, 2371–2374 CrossRef CAS PubMed.
- S. S. Zhang, J. Electrochem. Soc., 2013, 160, A1421–A1424 CrossRef CAS.
- S. Choudhury, T. Saha, K. Naskar, M. Stamm, G. Heinrich and A. Das, Polymer, 2017, 112, 447–456 CrossRef CAS.
- J. R. Nair, F. Bella, N. Angulakshmi, A. M. Stephan and C. Gerbaldi, Energy Storage Mater., 2016, 3, 69–76 CrossRef.
- M. Liu, D. Zhou, Y. B. He, Y. Z. Fu, X. Y. Qin, C. Miao, H. D. Du, B. H. Li, Q. H. Yang, Z. Q. Lin, T. S. Zhao and F. Y. Kang, Nano Energy, 2016, 22, 278–289 CrossRef CAS.
- M. Liu, H. R. Jiang, Y. X. Ren, D. Zhou, F. Y. Kang and T. S. Zhao, Electrochim. Acta, 2016, 213, 871–878 CrossRef CAS.
- S. Choi, J. J. Song, C. Y. Wang, S. Park and G. X. Wang, Chem. – Asian J., 2017, 12, 1470–1474 CrossRef CAS PubMed.
- Y. Y. Lu, K. Korf, Y. Kambe, Z. Y. Tu and L. A. Archer, Angew. Chem., Int. Ed., 2014, 53, 488–492 CrossRef CAS PubMed.
- K. Zhu, Y. X. Liu and J. Liu, RSC Adv., 2014, 4, 42278–42284 RSC.
- C. Zhang, Y. Lin and J. Liu, J. Mater. Chem. A, 2015, 3, 10760–10766 CAS.
- Y. Lin, X. M. Wang, J. Liu and J. D. Miller, Nano Energy, 2017, 31, 478–485 CrossRef CAS.
- Y. Lin, J. Li, K. Liu, Y. X. Liu, J. Liu and X. M. Wang, Green Chem., 2016, 18, 3796–3803 RSC.
- R. J. Chen, W. J. Qu, X. Guo, L. Li and F. Wu, Mater. Horiz., 2016, 3, 487–516 RSC.
- W. Liu, N. Liu, J. Sun, P. C. Hsu, Y. Z. Li, H. W. Lee and Y. Cui, Nano Lett., 2015, 15, 2740–2745 CrossRef CAS PubMed.
- W. Liu, S. W. Lee, D. C. Lin, F. F. Shi, S. Wang, A. D. Sendek and Y. Cui, Nat. Energy, 2017, 2, 17035 CrossRef CAS.
- K. Fu, Y. H. Gong, J. Q. Dai, A. Gong, X. G. Han, Y. G. Yao, C. W. Wang, Y. B. Wang, Y. N. Chen, C. Y. Yan, Y. J. Li, E. D. Wachsman and L. B. Hu, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7094–7099 CrossRef CAS PubMed.
- H. W. Zhai, P. Y. Xu, M. Q. Ning, Q. Cheng, J. Mandal and Y. Yang, Nano Lett., 2017, 17, 3182–3187 CrossRef CAS PubMed.
- Y. J. Nam, S. J. Cho, D. Y. Oh, J. M. Lim, S. Y. Kim, J. H. Song, Y. G. Lee, S. Y. Lee and Y. S. Jung, Nano Lett., 2015, 15, 3317–3323 CrossRef CAS PubMed.
- X. W. Yu, Z. H. Bi, F. Zhao and A. Manthiram, Adv. Energy Mater., 2016, 6, 1601392 CrossRef.
- S. F. Wang, Y. Ding, G. M. Zhou, G. H. Yu and A. Manthiram, ACS Energy Lett., 2016, 1, 1080–1085 CrossRef CAS.
- C. P. Yang, Y. X. Yin and Y. G. Guo, J. Phys. Chem. Lett., 2015, 6, 256–266 CrossRef CAS PubMed.
- A. Abouimrane, D. Dambournet, K. W. Chapman, P. J. Chupas, W. Weng and K. Amine, J. Am. Chem. Soc., 2012, 134, 4505–4508 CrossRef CAS PubMed.
- G. L. Xu, J. Z. Liu, R. Arnine, Z. H. Chen and K. Arnine, ACS Energy Lett., 2017, 2, 605–614 CrossRef CAS.
- K. Han, Z. Liu, H. Q. Ye and F. Dai, J. Power Sources, 2014, 263, 85–89 CrossRef CAS.
- J. R. He, Y. F. Chen, W. Q. Lv, K. C. Wen, P. J. Li, Z. G. Wang, W. L. Zhang, W. Qin and W. D. He, ACS Energy Lett., 2016, 1, 16–20 CrossRef CAS.
- K. Han, Z. Liu, J. M. Shen, Y. Y. Lin, F. Dai and H. Q. Ye, Adv. Funct. Mater., 2015, 25, 455–463 CrossRef CAS.
- C. P. Yang, S. Xin, Y. X. Yin, H. Ye, J. Zhang and Y. G. Guo, Angew. Chem., Int. Ed., 2013, 52, 8363–8367 CrossRef CAS PubMed.
- C. Luo, Y. H. Xu, Y. J. Zhu, Y. H. Liu, S. Y. Zheng, Y. Liu, A. Langrock and C. S. Wang, ACS Nano, 2013, 7, 8003–8010 CrossRef CAS PubMed.
- Y. Liu, J. W. Wang, Y. H. Xu, Y. J. Zhu, D. Bigio and C. S. Wang, J. Mater. Chem. A, 2014, 2, 12201–12207 CAS.
- J. Zhang, Y. X. Yin, Y. You, Y. Yan and Y. G. Guo, Energy Technol., 2014, 2, 757–762 CrossRef CAS.
- Z. He, Y. Yang, J. W. Liu and S. H. Yu, Chem. Soc. Rev., 2017, 46, 2732–2753 RSC.
- N. Ding, S. F. Chen, D. S. Geng, S. W. Chien, T. An, T. S. A. Hor, Z. L. Liu, S. H. Yu and Y. Zong, Adv. Energy Mater., 2015, 5, 1401999 CrossRef.
- J. Xu, S. Xin, J. W. Liu, J. L. Wang, Y. Lei and S. H. Yu, Adv. Funct. Mater., 2016, 26, 3580–3588 CrossRef CAS.
- J. R. He, Y. F. Chen, W. Q. Lv, K. C. Wen, Z. G. Wang, W. L. Zhang, Y. R. Li, W. Qin and W. D. He, ACS Nano, 2016, 10, 8837–8842 CrossRef CAS PubMed.
- Z. Y. Wen, Y. Y. Hu, X. W. Wu, J. D. Han and Z. H. Gu, Adv. Funct. Mater., 2013, 23, 1005–1018 CrossRef CAS.
- A. Manthiram and X. W. Yu, Small, 2015, 11, 2108–2114 CrossRef CAS PubMed.
- I. Kim, C. H. Kim, S. H. Choi, J. P. Ahn, J. H. Ahn, K. W. Kim, E. J. Cairns and H. J. Ahn, J. Power Sources, 2016, 307, 31–37 CrossRef CAS.
- Q. Q. Lu, X. Y. Wang, J. Cao, C. Chen, K. N. Chen, Z. F. Zhao, Z. Q. Niu and J. Chen, Energy Storage Mater., 2017, 8, 77–84 CrossRef.
- X. N. Li, J. W. Liang, K. L. Zhang, Z. G. Hou, W. Q. Zhang, Y. C. Zhu and Y. T. Qian, Energy Environ. Sci., 2015, 8, 3181–3186 CAS.
- Y. J. Wei, Y. Q. Tao, Z. K. Kong, L. Liu, J. T. Wang, W. M. Qiao, L. C. Ling and D. H. Long, Energy Storage Mater., 2016, 5, 171–179 CrossRef.
- L. C. Zeng, Y. Yao, J. N. Shi, Y. Jiang, W. H. Li, L. Gu and Y. Yu, Energy Storage Mater., 2016, 5, 50–57 CrossRef.
- K. Wang, S. Luo, Y. Wu, X. F. He, F. Zhao, J. P. Wang, K. L. Jiang and S. S. Fan, Adv. Funct. Mater., 2013, 23, 846–853 CrossRef CAS.
- L. Li, Z. P. Wu, H. Sun, D. M. Chen, J. Gao, S. Suresh, P. Chow, C. V. Singh and N. Koratkar, ACS Nano, 2015, 9, 11342–11350 CrossRef CAS PubMed.
- C. H. Chang, S. H. Chung and A. Manthiram, Mater. Horiz., 2017, 4, 249–258 RSC.
- H. Sun, Y. Zhang, J. Zhang, X. M. Sun and H. S. Peng, Nat. Rev. Mater., 2017, 2, 17023 CrossRef CAS.
- S. Y. Lee, K. H. Choi, W. S. Choi, Y. H. Kwon, H. R. Jung, H. C. Shin and J. Y. Kim, Energy Environ. Sci., 2013, 6, 2414–2423 CAS.
- X. Fang, W. Weng, J. Ren and H. S. Peng, Adv. Mater., 2016, 28, 491–496 CrossRef CAS PubMed.
- W. G. Chong, J. Q. Huang, Z. L. Xu, X. Y. Qin, X. Y. Wang and J. K. Kim, Adv. Funct. Mater., 2017, 27, 1604815 CrossRef.
- R. Q. Liu, Y. J. Liu, J. Chen, Q. Kang, L. L. Wang, W. X. Zhou, Z. D. Huang, X. J. Lin, Y. Li, P. Li, X. M. Feng, G. Wu, Y. W. Ma and W. Huang, Nano Energy, 2017, 33, 325–333 CrossRef CAS.
- J. Fu, J. Zhang, X. P. Song, H. Zarrin, X. F. Tian, J. L. Qiao, L. Rasen, K. C. Li and Z. W. Chen, Energy Environ. Sci., 2016, 9, 663–670 CAS.
- J. Fu, F. M. Hassan, J. D. Li, D. U. Lee, A. R. Ghannoum, G. Lui, M. A. Hoque and Z. W. Chen, Adv. Mater., 2016, 28, 6421–6428 CrossRef CAS PubMed.
- F. R. Fan, W. Tang and Z. L. Wang, Adv. Mater., 2016, 28, 4283–4305 CrossRef CAS PubMed.
- K. Fu, Y. G. Yao, J. Q. Dai and L. B. Hu, Adv. Mater., 2017, 29, 1603486 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2017 |