
From dynamic self-assembly to networked chemical systems

Abstract
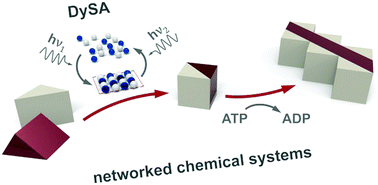
Although dynamic self-assembly, DySA, is a relatively new area of research, the past decade has brought numerous demonstrations of how various types of components – on scales from (macro)molecular to macroscopic – can be arranged into ordered structures thriving in non-equilibrium, steady states. At the same time, none of these dynamic assemblies has so far proven practically relevant, prompting questions about the field's prospects and ultimate objectives. The main thesis of this Review is that formation of dynamic assemblies cannot be an end in itself – instead, we should think more ambitiously of using such assemblies as control elements (reconfigurable catalysts, nanomachines, etc.) of larger, networked systems directing sequences of chemical reactions or assembly tasks. Such networked systems would be inspired by biology but intended to operate in environments and conditions incompatible with living matter (e.g., in organic solvents, elevated temperatures, etc.). To realize this vision, we need to start considering not only the interactions mediating dynamic self-assembly of individual components, but also how components of different types could coexist and communicate within larger, multicomponent ensembles. Along these lines, the review starts with the discussion of the conceptual foundations of self-assembly in equilibrium and non-equilibrium regimes. It discusses key examples of interactions and phenomena that can provide the basis for various DySA modalities (e.g., those driven by light, magnetic fields, flows, etc.). It then focuses on the recent examples where organization of components in steady states is coupled to other processes taking place in the system (catalysis, formation of dynamic supramolecular materials, control of chirality, etc.). With these examples of functional DySA, we then look forward and consider conditions that must be fulfilled to allow components of multiple types to coexist, function, and communicate with one another within the networked DySA systems of the future. As the closing examples show, such systems are already appearing heralding new opportunities – and, to be sure, new challenges – for DySA research.
- This article is part of the themed collection: Chemical systems out of equilibrium
 Please wait while we load your content...
Please wait while we load your content...

