
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface science under reaction conditions: CO oxidation on Pt and Pd model catalysts
Matthijs A.
van Spronsen
 a,
Joost W. M.
Frenken
b and
Irene M. N.
Groot
*c
a,
Joost W. M.
Frenken
b and
Irene M. N.
Groot
*c
aDepartment of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138, USA
bAdvanced Research Center for Nanolithography, P.O. Box 93019, 1090 BA Amsterdam, The Netherlands
cLeiden Institute of Chemistry, Leiden University, P.O. Box 9502, 2300 RA Leiden, The Netherlands. E-mail: i.m.n.groot@lic.leidenuniv.nl
First published on 7th June 2017
Abstract
Platinum and palladium are frequently used as catalytic materials, for example for the oxidation of CO. This is one of the most widely studied reactions in the field of surface science. Although seemingly uncomplicated, it remains an active and interesting topic, which is partially explained by the push to conduct experiments on model systems under relevant reaction conditions. Recent developments in the surface-science methodology have allowed obtaining chemical and structural information on the active phase of model catalysts. Tools of the trade include near-ambient-pressure X-ray photoelectron spectroscopy, high-pressure scanning tunneling microscopy, high-pressure surface X-ray diffraction, and high-pressure vibrational spectroscopy. Interpretation is often aided by density functional theory in combination with thermodynamic and kinetic modeling. In this review, results for the catalytic oxidation of CO obtained by these techniques are compared. On several of the Pt and Pd surfaces, new structures develop in excess O2. For Pt, this requires a much larger excess of O2 than for Pd. Most of these structures also develop in pure O2 and are identified as (surface) oxides. A large body of evidence supports the conjecture that these oxides are more reactive than the corresponding O-covered metallic surfaces under similar conditions, although still debated in the literature. An outlook on this developing field, including directions that move away from CO oxidation towards more complex chemistry, concludes this review.
 Matthijs A. van Spronsen | Matthijs A. van Spronsen received his PhD degree in physics under the directorship of Prof. Joost W. M. Frenken and Dr. Irene M.N. Groot at Leiden University in the Netherlands in 2016, after receiving both his B.S and M.S. degrees in chemistry at the same university. He is currently a postdoctoral researcher in the group of Prof. Friend at the chemistry and chemical biology department at Harvard University. His research interests include behavior of model catalysts under realistic reaction conditions and the development of instruments to probe surfaces in these environments. |
 Joost W. M. Frenken | Joost Frenken is the Director of the Advanced Research Center for Nanolithography (ARCNL) in Amsterdam and a professor of Physics at both universities in Amsterdam (UvA and VU) and at Leiden University. His scientific expertise is in the structure, diffusion, chemical reactions, phase transitions and friction phenomena at surfaces and interfaces, investigated using advanced instruments, developed under his supervision. His achievements have been recognized with several research awards and a membership of the KNAW, The Netherlands Royal Academy of Sciences. Frenken has (co)-initiated two companies, Leiden Probe Microscopy BV and Applied Nanolayers BV. |
 Irene M. N. Groot | Dr Irene Groot obtained her PhD in 2009 from the Leiden Institute of Chemistry. She investigated the adsorption of H2 on platinum and ruthenium using supersonic molecular beam techniques and quantum dynamics calculations. During her postdoctoral stay at the Fritz Haber Institute, Irene investigated CO oxidation on thin metal oxide films at mbar pressures using scanning tunneling microscopy. Irene is now heading her own group at the Leiden Institute of Chemistry, where she uses advanced microscopy techniques at atmospheric pressure and elevated temperatures to investigate heterogeneously catalyzed chemical processes. Examples are hydrodesulfurization, Fischer–Tropsch synthesis, methanol steam reforming, and CO oxidation. |
1 Introduction
Fully aware that this review is not the first (see for example ref. 1–5) and certainly not the last to focus on CO oxidation over Pt and Pd, we will shortly motivate the need for “yet another review” on this topic. The annual number of published papers on this topic has been increasing steadily since the 1970s, as can be seen from Fig. 1. Between 2000 and 2005, the rate of newly appearing research papers increased by approximately a factor of 2.5. These papers originated from two separate scientific fields; from heterogeneous catalysis and from surface science. The former traditionally focuses on how to synthesize and characterize the most active catalysts, while trying to extract details regarding the reaction mechanism, structure of the active sites, etc. The latter, on the other hand, concentrates on model catalysts, resembling one aspect of, but greatly simplifying, the technical catalysts. Its aim is to derive the most detailed level of understanding on the fundamentals of the interaction of molecules with surfaces, including desorption, adsorption, diffusion, reaction, and the possible modification of the composition and structure of the catalyst surface itself.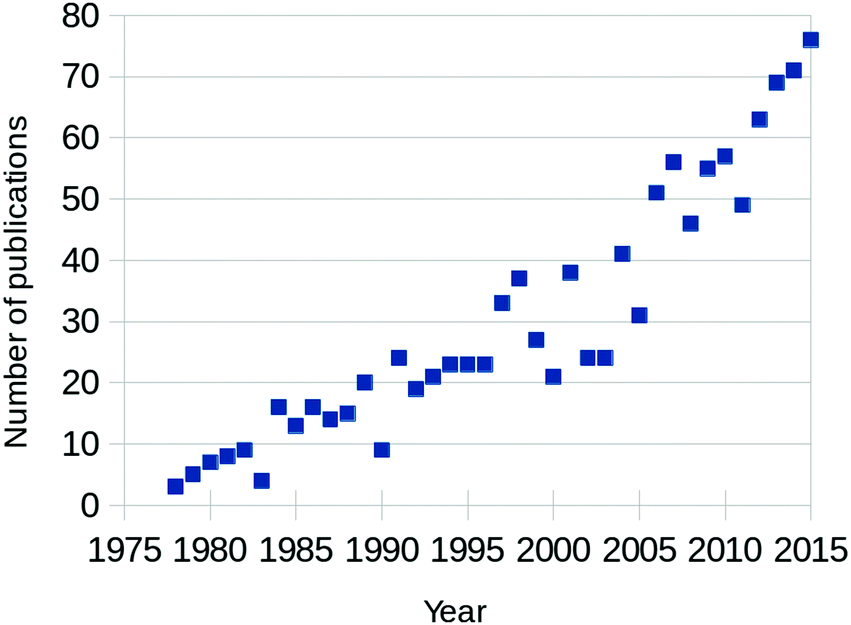 | ||
| Fig. 1 Yearly number of published papers focused on CO oxidation over Pt-group metals, based on the Web of Science database. | ||
Almost concurrently with the increase in publications, technological advancements started pushing the boundaries of analytical methods in surface science to enable operation under chemical conditions. These methods include the integration of a flow reactor with a scanning probe microscopy (SPM) system,6–8 the application of differential pumping stages and electrostatic lenses in near-ambient-pressure (NAP) X-ray photoelectron spectroscopy (XPS),9,10 the use of high-pressure vibrational spectroscopy,11–13 and X-ray diffraction setups.14,15 Simultaneously, theoretical modeling has matured by taking the energies derived from density functional theory (DFT) as input for thermodynamic calculations.16 In this way, theoretical predictions can be made regarding the most stable phase as a function of the reaction conditions. Moreover, DFT can now be linked with microkinetic models, e.g., kinetic Monte Carlo (kMC), and macroscopic gas flow computations.17,18 The combination of all these developments leads to experiments and simulations closer to realistic catalytic conditions, while still yielding very detailed information.
The reaction between CO and O2 is seemingly straightforward. This, together with the wealth of knowledge obtained from studies performed under ultra-high vacuum (UHV) conditions, makes CO oxidation a natural choice to study using state-of-the-art high-pressure or (near) ambient-pressure techniques to obtain insight into the active surface under reaction conditions, which was unattainable under UHV conditions. The ultimate goal here is to understand the active phase of the catalyst under chemical conditions as close to realistic, catalytic applications as possible.
The focus of this review is on the application of newly developed surface-science tools to CO oxidation on model catalysts, mainly single-crystal and polycrystalline surfaces. Instead of starting by summarizing the numerous of studies under UHV conditions, we mainly restrict the discussion to studies at a pressure above 1 Pa and temperatures at or above room temperature (RT). Prior UHV (see, e.g., ref. 3 and 5 for reviews focused on the low-pressure experiments) is included where it helps in the interpretation of the elevated-pressure studies.
CO can be oxidized over Pt and Pd in several ways. In the most simple form, both CO and O2 adsorb onto the metallic surface. The latter adsorbs dissociatively, after which O(ads) can react with CO(ads) to form CO2. This reaction is an example of the Langmuir–Hinshelwood (LH) mechanism and a maximum rate is expected if θCO and θO are equal. The co-adsorption of both reactants could lead to mixed phases or to segregated islands with the reaction occurring at the interface in between. Alternatively, via the Eley–Rideal (ER) mechanism, CO(g) hits the surface and directly reacts with O(ads).
For an optimal reactivity via the LH mechanism, both reactants need to be equally present on the surface. However, the dissociative adsorption of O2 is slightly more involved compared to the adsorption of CO. It is believed to use both physisorbed and precursor states.19 After dissociation, the O atoms are separated by two lattice constants on the Pt(111) surface, thus requiring a large surface area to dissociatively adsorb,20 whereas CO needs only a single free site to adsorb. Therefore, in CO/O2 mixtures, CO competes with O2 for free adsorption sites. The strong adsorption of CO can lead to dense CO overlayers greatly decreasing the O2 adsorption rate. This results in very low CO oxidation activity and is referred to as CO poisoning. This was most clearly demonstrated by Gland and coworkers.21 In their work, no displacement of CO by O(ads), i.e., complete poisoning of the surface, was observed on a Pt(111) surface pre-saturated with CO. Even for pO2 as high as 3 Pa, displacement occurred only after heating to 305 K to thermally desorb some CO.
In specific cases, the active phase of the catalyst is formed under reaction conditions due to exposure to one of the reactants. For example, a noble metal catalyst can form an oxide surface under O2-rich conditions, which could be consumed by CO to form CO2 and an oxygen vacancy, Ov. In a second step, the active surface is restored by filling Ov by O2(g). This mechanism is referred to as the Mars–van Krevelen (MvK) mechanism.22 Instead of an Ov in an oxide lattice, it was proposed that volatile Pt(g) can be created by the reaction of PtO2 with CO.23 The oxide can be restored by the reaction of Pt(g) with O2 forming PtO2(g), which adsorbs on the film. This mechanism can explain metal loss in a catalyst, although the expected rate is very low.
In fact, a similar distinction can be made to the reaction on the metallic surface. In a MvK–LH mechanism, CO initially adsorbs on the surface of the oxide, while in a MvK–ER mechanism CO(g) reacts directly with the lattice oxygen.
The CO oxidation rate on Pt and Pd can be exceedingly high. Above a certain threshold reactivity, one (or both) of the reactants is completely depleted in the near-surface vicinity. The reaction is no longer limited by any of the elementary processes on the surface, but instead by gas-phase diffusion to the catalyst. This is known as the mass-transfer limited (MTL) regime. The conditions under which this occurs are strongly dependent on the sample and reactor geometry.
2 Methodology
2.1 Surface characterization at high pressure
Since heterogeneous catalysis is an interface phenomenon, it is crucial to obtain chemical and structural data on the catalytic surface exposed to chemical conditions. The structure that the surface adopts can depend strongly on the presence of the gas phase and can require a certain degree of mobility to form. The challenging aspect of surface science under reaction conditions is to obtain surface sensitivity, without being influenced too much by the high-pressure gas phase.Surface X-ray diffraction (SXRD)6,24 is inherently sensitive to the outer layers of a material. It exploits the change in the diffraction pattern by the discontinuity of the bulk crystal lattice, which is caused by the surface. This connects the Bragg reflections, the diffraction spots originating from the bulk, via a crystal truncation rod (CTR), which exclusively contains information from the surface. Furthermore, surface sensitivity is enhanced by using grazing incidence angles for the incoming photon flux. The latest development in SXRD is to operate with an X-ray beam at higher energy.110 This squeezes more of the Ewald sphere onto the 2D detector, thereby greatly enhancing the acquisition speed and allowing the study of several structures over time simultaneously.
Several setups have been developed that combine a UHV chamber and a high-pressure cell with Be walls as X-ray transparent windows. They consist of batch-mode25,26 and flow-mode14 reactors.
Since the diffracted intensity scales with the electron density in the material, SXRD is quite insensitive for low-Z atoms, such as O or C. Additionally, as it is a diffraction technique, it requires a certain degree of order in the structures under investigation. Another import caveat of using (high-intensity) X-ray beams is the possibility of overoxidation by artificially increasing the O2 dissociation rate. This has been ascribed to gas-phase O3 formation, followed by O3-assisted oxidation.67 Alternatively, emitted photoelectrons can possibly dissociate (short-lived) O2(ads), as was observed for physisorbed O2 on Pt(111) at 30 K, which partially oxidized the surface to PtO after X-ray irradiation.19
In the case of other photon-based techniques, the wavelength determines the gas-phase sensitivity. For vibrational spectroscopy, gas-phase absorption can completely obscure the signal originating from the adsorbates. To selectively measure this signal either sum-frequency generation (SFG) spectroscopy11 or polarization-modulated (PM) infrared reflectance spectroscopy (IRRAS)† is required.12,13 In SFG, two high-intensity lasers, one visible and the other tunable infrared (IR), are focused on an overlapping spot on the sample. This leads to the generation of an output beam with the sum frequency of the two incoming lasers via a second-order nonlinear process. However, this process is allowed only when inversion symmetry is broken, as on a surface.
Different light polarizations interact differently with a surface, which is the basis of PM IRRAS. Photons with their polarization parallel to the plane of reflection, i.e., p-polarization, interact constructively with the surface. On the other hand, s-polarized photons interact destructively with the surface and therefore contribute no surface signal. In addition, the intensities of both types of polarized photons are dependent on gas-phase absorption, which means that the reflection of s-polarized light exclusively yields gas-phase information, while the difference in reflection between p- and s-polarized light contains surface-specific information. Several systems have been realized that attach a high-pressure cell for PM IRRAS28 or for SFG spectroscopy29,30 to a UHV chamber.
Polarized light is also used in ellipsometry and related techniques, such as ellipso-microscopy for surface imaging (EMSI)31–33 and reflection anisotropy microscopy (RAM).34 With these techniques the dielectric function, especially local variations, of the surface can be studied. These techniques are very surface sensitive and principally without pressure restriction.
Electron-based techniques, such as XPS, experience the greatest difficulty under high-pressure conditions, because of the strong interaction between electrons and the gas phase. Two strategies are adopted to minimize this interaction. Both rely on decreasing the path length of the electrons through the high-pressure gas, either by limiting the high-pressure conditions to a small closed reactor cell with semi-transparent windows, or by placing the sample close to a detector which uses several stages of differential pumping. Both strategies have been used to extend the pressure range of XPS to NAP conditions of up to several tens of hPa.9,10,35–38
The path between the sample and detector (or source) is inherently very small in SPM techniques, rendering them relatively insensitive to pressure. Several scanning tunneling microscopy (STM) setups that allow scanning at high pressure (HP) were developed.7,39–44 In most of the designs, the entire chamber is filled to a certain pressure, in others just the integrated reactor cell inside the microscope. The most difficult part is to design a scan head that has sufficient thermal stability to maintain atomic resolution under high pressures and at elevated temperatures. Recently, a high-pressure STM system (see Fig. 2) and a closely related high-pressure atomic force microscopy (AFM) system, the ReactorSTM and the ReactorAFM, were constructed, where the microscope is integrated with a small flow reactor.6–8 To better relate the local information obtained in scanning probe experiments to the information obtained with averaging tools, such as SXRD, a small SPM head has been realized that can be fitted onto an SXRD chamber (see Fig. 3).15
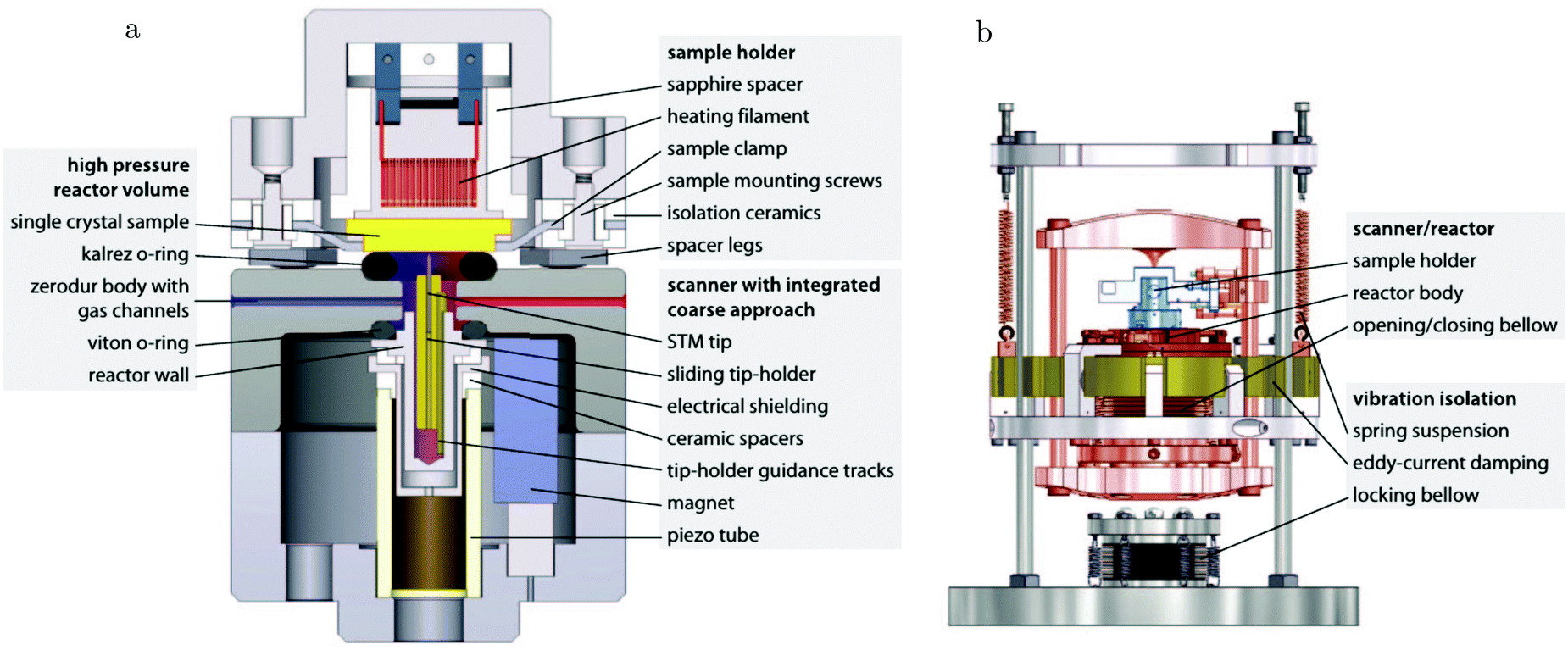 | ||
| Fig. 2 Schematic drawing of the ReactorSTM, an example of a high-pressure flow cell integrated with an STM setup. In this system, the sample can be exposed to a pressure and temperature of up to 600 kPa and 600 K, respectively, while the delicate parts of the microscope remain under UHV and at much lower temperatures. Reproduced from ref. 7 with permission from the American Physical Society, copyright 2014. | ||
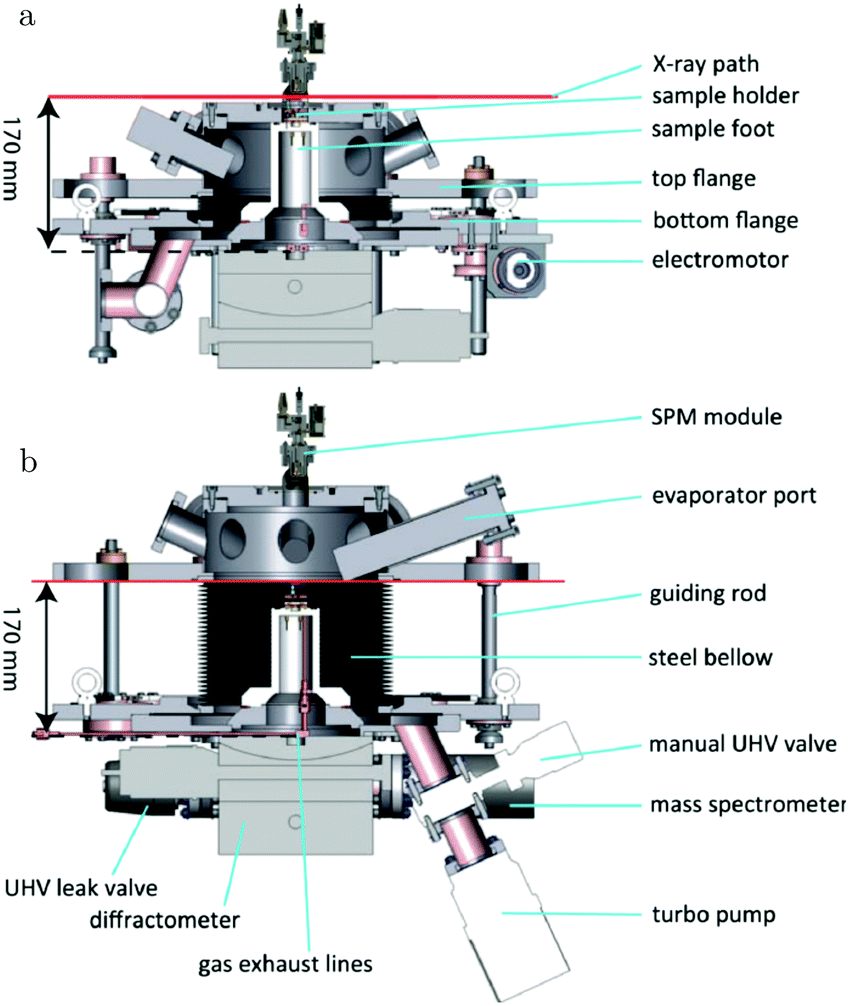 | ||
| Fig. 3 Schematics of the addition of an SPM unit to the SXRD flow reactor described in ref. 14. Reproduced from ref. 15 with permission from the American Physical Society, copyright 2016. | ||
To address the pressure gap from a theoretical point of view, results from DFT, that describe the system on an electronic (microscopic) level, can be linked to thermodynamics. With this method it is possible to address large system sizes and to compare the stability of different surface structures in contact with the surrounding gas phase. The key result obtained in this way is a surface phase diagram, providing insight into possible surface structures over a wide range of temperature and pressure conditions. The crucial quantities that determine the stability of possible surface structures are the total energies of the extended surfaces and of the involved gas phase molecules. For a detailed overview of the combination of DFT with ab initio thermodynamics, see ref. 45.
However, not only thermodynamics, but kinetics also play a pivotal role in obtaining the complete picture of the structure of a catalytic surface under reaction conditions. First-principles kMC simulations are able to deliver this. The first part of this method consists of a description of all elementary reaction steps that constitute the total chemical reaction taking place. The second part consists of the determination of the occurrence, and therefore the relevance, of the individual elementary processes. In kinetic Monte Carlo simulations a master equation is used to coarse-grain the time evolution to the relevant rare-event dynamics, explicitly considering the correlations, fluctuations, and spatial distributions of the chemical species at the catalytic surface. For a detailed overview of kinetic Monte Carlo simulations, see ref. 45 and 46.
2.2 Gas-phase measurements
All these surface-sensitive techniques are generally combined with measurements of the average gas-phase conditions. This can be achieved in several ways. The most frequently employed technique is quadrupole mass spectrometry (QMS). The QMS equipment is housed inside a different chamber to ensure operation under UHV conditions. First, the gas molecules are ionized and reach the detector via a quadrupole mass filter, which provides mass selectivity. The instrument measures different masses as a function of time, of a fraction of the effluent of the high-pressure environment that is leaked into the QMS chamber.Instead of a quadrupole mass filter, gas chromatography (GC) can be used to separate the gas-phase components. After separation, a detector quantitatively measures the different analytes, usually based on the change in thermal conductivity with respect to the carrier gas. Separation by GC is slower than by QMS, which reduces the time resolution.
Since the CO oxidation rate correlates directly with the change in total pressure via
| 2CO + O2 → 2CO2 |
In addition to the average gas composition, more information can be abstracted by measuring the local gas composition. This can be done with planar laser-induced fluorescence (PLIF), which probes the fluorescence light emitted by excited molecules. The energy of excitation is very specific for the molecule of interest. Excitation is done using a laser beam of appropriate wavelength. If not a beam but a light sheet is used, in combination with a 2D detector, spatially resolved partial pressure information can be collected. The application of this technique to operando catalysis research is relatively new and has recently been reviewed by Lundgren and coworkers.47
2.3 Ex situ and titration experiments
A different experimental strategy is to separate the exposure to reaction conditions from the surface measurements. In these ex situ experiments, the model catalyst can be prepared under UHV conditions, exposed to the reaction conditions, while measuring the reactivity allows exploring the reaction kinetics, concluded by characterization of the surface under UHV. This approach does not guarantee to capture the active surface under reaction conditions, but could provide information regarding irreversible changes occurring during reaction conditions, such as C deposition or segregation of bulk impurities.48In titration experiments the model catalyst is exposed to high-pressure conditions to create, e.g., a type of oxide, while the reaction is probed by CO titration. The latter part of the experiments is performed in UHV allowing the use of a wide set of characterization tools.
The separate parts of the ex situ or titration experiments are usually conducted in different chambers. Ideally, the transfer between the chambers does not lead to exposure of the sample to air, because this would lead to additional uncertainty in relating the different stages of the experiment, which could be due to a possible reaction of the sample with oxygen, water vapor, or hydrocarbons.
2.4 Additional experimental considerations
As CO oxidation can be very active, the gas conditions close to the sample can strongly deviate from the average conditions in the reactor. However, the gas phase in the vicinity of the sample determines the surface structure and its reactivity. Under high reactivity conditions, the experimental results are to some extent determined by the properties of the reactor. One of the most important aspects is whether the reactor is operated in flow or batch mode. In the former mode, the reactor volume is actively refreshed within a certain time interval determined by the flow velocity of the gases and the reactor volume. In the latter mode, mixing of the gas phase with a recirculation pump can be used to reduce the difference between the gas phase close to and far away from the sample. Under steady-state conditions, the partial pressures of the reactants are time independent in flow mode, whereas they are time sensitive in batch mode.The choice to use feedback controllers to stabilize experimental parameters, such as the total pressure or temperature, can complicate a direct comparison of the reactivity in different experiments. For flow reactors, a back-pressure controller probes the pressure of the reactor and adjusts a variable flow restrictor to achieve the desired pressure, which effectively increases the inlet pressure of the reactor. This leads to a higher pressure of reactants and an increased reaction rate. On the other hand, a controlled, constant temperature will decrease the reaction rate of CO oxidation compared to operation at constant heating power, because it will decrease the heating power when the sample is (partially) heated due to the exothermic reaction. In some cases, the use of feedback loops can lead to reaction oscillations.49
In this review, all units have been converted to the SI standards (Pa, K, m, kJ mol−1) where needed. However, for clarity, all energies are provided both in kJ mol−1 and in eV per atom or molecule. Total pressures were obtained by adding up the partial pressures of the reactants, excluding any diluents such as He or Ar. In addition, the CO/O2 ratio is given and is defined as 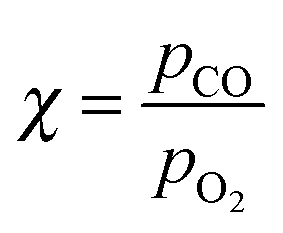 , with χ = 2 for a stoichiometric mixture. Furthermore, vicinal or stepped surfaces are indicated by their Miller indices as well as by the combination of the low-Miller-index planes associated with their terrace and step orientations.50 In the latter notation, the atoms forming the base of the step are not considered as contributing to the terrace. Finally, coverages are reported in monolayers (ML) normalized to the number of atoms in the unreconstructed, metallic surface and doses are reported in Langmuir (L), defined as 1.3 × 10−6 hPa s.
, with χ = 2 for a stoichiometric mixture. Furthermore, vicinal or stepped surfaces are indicated by their Miller indices as well as by the combination of the low-Miller-index planes associated with their terrace and step orientations.50 In the latter notation, the atoms forming the base of the step are not considered as contributing to the terrace. Finally, coverages are reported in monolayers (ML) normalized to the number of atoms in the unreconstructed, metallic surface and doses are reported in Langmuir (L), defined as 1.3 × 10−6 hPa s.
3 Platinum
3.1 Pt(111), CO adsorption
On the Pt(111) surface, CO(ads) structures at higher pressures were recognized using STM by several research groups as superstructures, resulting in moiré patterns (see for example Fig. 4).51–55 The study of Longwitz et al. is the most extensive.53 They report that the exact properties of the moiré pattern depended on pCO, within the studied range of 10−6–103 hPa. The moiré lattice constant increases logarithmically with pCO up to roughly 1 hPa, above which it saturated at 1.2 nm. The rotation of the moiré pattern with respect to the metal lattice remained 30° up to a pCO of 1 Pa, above which it decreased to 23°. The CO coverage showed a similar trend to the moiré lattice constant, saturating at 0.67 ML for pCO > 1 hPa. The proposed structure at this pressure is commensurate and has a unit cell of (√19 × √19)R23.4°-13 CO (Fig. 4d), as derived from atomically resolved images.52,53 The same structure was observed under certain electrochemical conditions.56 Somorjai and coworkers51 also observed a moiré pattern with a similar periodicity in a mixture of 200 hPa CO and 67 hPa O2 at RT, but with the moiré pattern aligned to the substrate lattice. As atomic resolution was lacking, the angles of small triangular islands were used as reference, which should follow the main directions of the Pt(111) surface.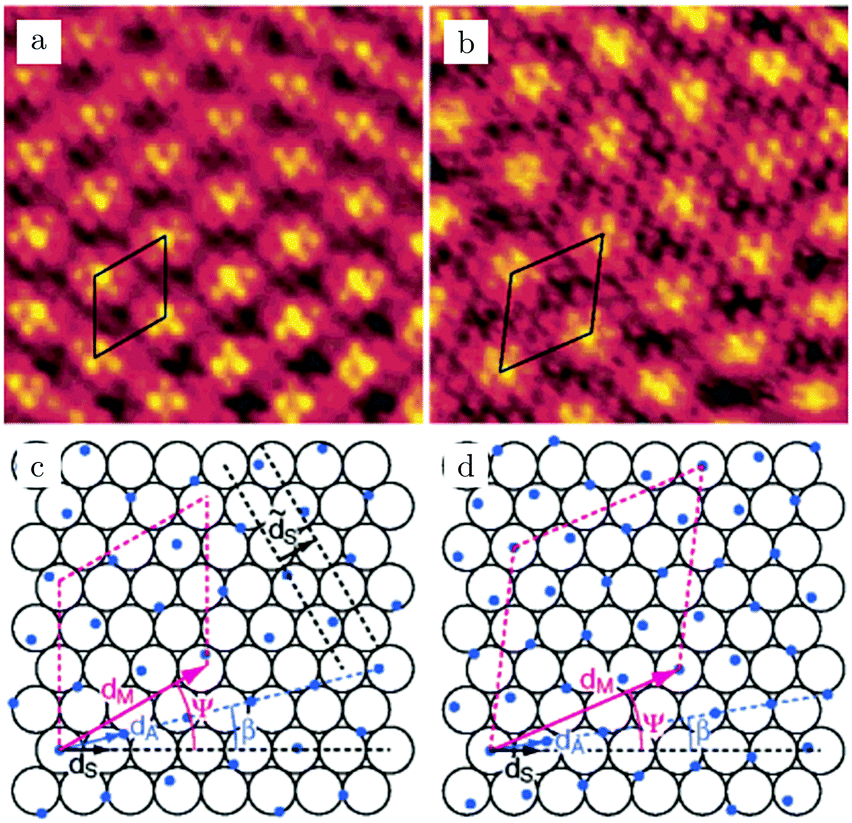 | ||
| Fig. 4 STM images, RT, 5.5 × 5.5 nm2 [(a) 10−2 hPa CO and (b) 958 hPa CO] and ball models (c and d) of two examples of moiré patterns induced by CO overlayers on Pt(111). (d) is a model of the saturation structure of CO(ads)/Pt(111), the (√19 × √19)R23.4°-13 CO structure. Reproduced from ref. 53 with permission from the American Chemical Society, copyright 2004. | ||
The vibrational signatures of CO(ads) as measured by SFG spectroscopy showed pronounced, but complex changes.57 These mainly occurred between 1.3 and 931 hPa. At lower pressures, the signature of atop-bonded CO(ads) dominated the spectra, shifting from 2100 to 2105 cm−1 at a pCO of 1 hPa. At higher pressures, it diminished, resulting in several new features, one of which had a maximum at 2045 cm−1, besides a strong increase of the total background. These changes were explained as the possible formation of incommensurate structures or as CO bonded to Pt steps or kinks. Although CO seems to increase the mobility of Pt atoms at the step and induce the formation of kink sites,55,58,59 large-scale roughening of the Pt(111) surface in CO was not observed at a pressure of around 100 kPa.54 Furthermore, a sputtered Pt(111) surface was remarkably similar to a well-annealed Pt(111) surface; however, it developed these vibrational features at much higher pressures, above 200 hPa instead of 15.3 hPa.60 This favors the assignment to incommensurate overlayers, which would probably be less stable on a rougher surface.
This complex behavior was contradicted by a more recent study by Freund and coworkers.61 After correcting the data for the gas-phase IR absorption, the peak ascribed to CO adsorbed on atop sites was found not to be suppressed. In fact, it increased in intensity corresponding to a coverage of ∼0.7 ML, in perfect agreement with the STM data.52,53 Also, no other signatures were observed, except for a peak at 2085 cm−1 assigned to CO co-adsorbed with H2O, but observed only below RT. The experiments confirmed that bonding to Pt defect sites had no significant influence; it only increased the peak width without introducing peak shifts. Finally, both studies indicated that SFG spectroscopy seems very insensitive to bridge-bonded CO.
3.2 Pt(111), CO oxidation
CO oxidation on Pt(111) has been the focus of several NAP XPS studies. Schnadt et al. used a small flow cell to study this reaction at a constant pressure of 0.15 hPa.38 Their experiment started in a pure O2 atmosphere at 430 K, in which a single, asymmetric feature at a binding energy of 530.0 eV was observed. This species disappeared after co-feeding CO (χ = 0.11), while the temperature was increased to 535 K. Two new XPS features emerged at 531.0 and 532.6 eV, which were assigned to CO(ads) on bridge and atop sites. At the highest temperature, the surface switched back to O-covered, similar to the state in pure O2, although with a lower O coverage. Under these conditions, the QMS data showed typical signs of the MTL regime. The switch from CO to O covered can be easily explained by an increasing rate of CO desorption and oxidation at higher temperatures up to the point where surface sites become available for extensive O2 dissociation. An Arrhenius plot derived from the QMS data shows a superlinear behavior, indicating complex reaction kinetics. In part, this can be explained by the constant-pressure condition of this experiment. While the reaction decreased the total pressure, a controller tried to stabilize the reactor pressure at 0.15 hPa. This leads to a positive feedback between the reactivity and the partial pressure of the reactants. From the limited number of data points and the obscuring effect of the MTL regime, it is unclear whether the reactivity of the O-covered surface was indeed higher. It was at least reactive enough to maintain MTL conditions. Also, from the uncertainty of the differences in core-level shifts and the lack of information regarding the Pt core levels, the O-covered surface cannot reliably be identified as either a (surface) oxide or covered by O(ads).The results by Schnadt et al. were supported by a later NAP XPS study performed at 0.73 hPa with a mixture of χ = 0.1.62 However, the XP spectra recorded in pure CO showed only one peak attributed to adsorbed CO (532.6 eV). In addition, it was much broader, compare the full width at half maximum (FWHM) of 3.4 eV62vs. 1.4 eV,38 and can be explained by a convolution of CO adsorbed at several different binding sites.62 The transition from CO-poisoned to O-rich (single feature at 530.2 eV) was observed between 400 and 450 K, in accordance with the slightly more oxidizing condition.
In addition to synchrotron-based studies, a NAP XPS study employing a lab-based X-ray source was recently published.63 These sources provide X-rays with higher energy than typically used for synchrotron XPS studies and thus have less surface sensitivity. Also, the detection limit is higher (estimated at 0.15 ML63) due to a lower photon flux. Interestingly, no adsorbates were detected on the active catalyst surface under the probed experimental conditions (χ = 0.25–2, p = 0.1 & 1 hPa, and T = 300 to 900–1018 K).
Two regimes were also observed under reaction conditions on a (111)-rich Pt thin film,64 studied using IRRAS and SXRD. For the first, low-activity regime, the presence of CO(ads) was confirmed by a vibrational feature at 2074 cm−1 (batch mode, initial mixture of χ = 1.3 at 18.1 hPa and 680 K). The authors argued that the metallic, low-active phase should be attributed to a partially CO-poisoned surface with islands of O(ads). Their main argument was based on the much lower reaction barrier (45–61 kJ mol−1 or 0.47–0.63 eV) for this surface than found for the fully CO-poisoned surface. Completely poisoned, the rate limiting step is CO desorption and structure-insensitive barriers in the order of 110 kJ mol−1 or 1.14 eV were obtained for a wide range of systems.65 For the surface with mixed adsorbate islands, reactivity can occur at the boundaries between the islands or, possibly, on O-rich islands with co-adsorbed CO molecules. During this regime, the absence of an oxide was inferred from SXRD. No direct proof of the presence of O(ads) was given as it is not easily detectable either by IRRAS or by SXRD.
The low-activity regime abruptly switched to a higher activity regime. In this regime, IRRAS showed that CO was not present on the surface, which left the authors to conclude that the surface was fully covered with O(ads). Although a smooth transition between CO- and O-rich with decreasing χ might be expected, the sharp transition can be the result of the CO oxidation reaction. At low O coverages, reactivity is low and CO can only leave the surface by desorption. As the O coverage increases, the reactivity increases and CO can leave the surface as CO2. In addition, the exothermicity of the reaction can locally raise the temperature of the surface stimulating CO desorption and a further increase in O-coverage, reactivity, and heat production.
In the same study, a 2-nm-thick PtO2 layer was prepared by exposing the Pt(111)-rich film to 18.9 hPa O2 at 600 K and it was reported to be unstable in a mixture of χ = 0.16 at 21.9 hPa and 430 K. It disappeared “instantaneously”, on the order of a single SXRD measurement (hundreds of seconds). In this batch-mode experiment, CO immediately consumed the oxide, leaving the Pt surface in the metallic state. Once most CO had reacted, the catalyst switched to higher reactivity under the more oxidizing conditions and simultaneously PtO2 developed quickly. This experiment shows that PtO2 is reactive for CO oxidation. However, a direct comparison remains difficult due to the relatively low acquisition rate of SXRD and due to the fact that SXRD cannot exclude the coexistence of O(ads) and PtO2.
An operando study combining STM with QMS, performed around 125 kPa,54 confirmed the existence of a higher reactivity phase (see the arrow in Fig. 5). Before the transition (approximately at χ = 0.08 and 423 K), no adsorbates, such as CO superstructures, were observed, most likely indicating that the surface was not CO-poisoned and covered with an adsorbate configuration too dynamical to observe using STM (Fig. 5a). The transition occurred at χ = 0.02 (pCO = 22 hPa) at a temperature of 418 K. Although no atomic resolution was obtained, several interesting details were observed. The surface changed quickly (Fig. 5b), within the time needed for a single STM scan line (∼1 s), to a rough structure of pits and protrusions (Fig. 5c) with height differences irreconcilable with metallic Pt. Furthermore, the STM images show that the full imaged area changed its morphology. The newly developed structure had IV-characteristics hinting at semiconductor behavior. Based on this and on the changed height differences, the author proposed that the structure was oxidic in nature. For the reverse change to the metallic state (Fig. 5d), a slightly higher pCO was needed. This can be explained by the more active surface counter-effecting the increasing pCO, keeping the local gas conditions more oxidizing. This can be inferred from the sharp spike in reactivity (Fig. 5, bottom panel). The reactivity of the oxide was high and increasing due to the increasing CO pressure up to a critical point at which the oxide was no longer stable. After the reduction, the reactivity showed a dip correlating with a switch to the less reactive O-rich surface. The reactivity of the O-rich surface increased with CO pressure to an optimal reactant ratio on the surface, according to LH kinetics (Fig. 5, bottom panel), after which it became increasingly CO poisoned.
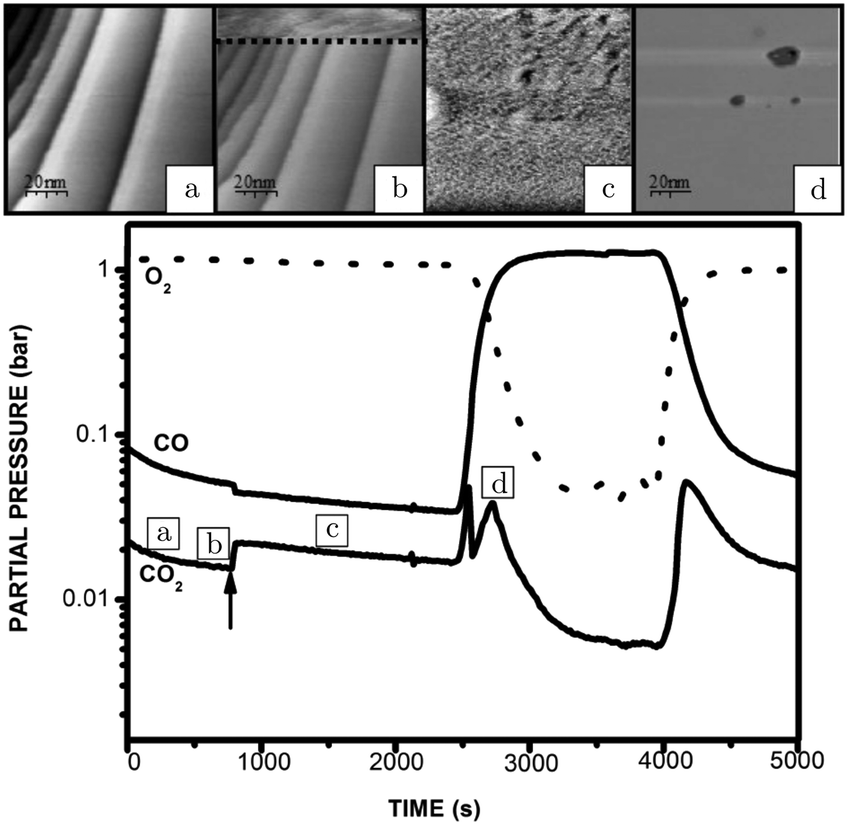 | ||
| Fig. 5 STM images recorded (from bottom to top) at various stages of the experiment, in which the CO and O2 partial pressures were varied at 423 K at a pressure of 125 hPa: (a) reduced metallic surface; (b) during the switch to higher activity; (c) oxidized surface; (d) reduced surface after increasing CO flow. The lower panel presents the QMS data. Reproduced from ref. 54 with permission from the author, copyright 2006. | ||
Under certain conditions and only in some measurements, the direction of the hysteresis was the opposite and, simultaneously, reaction oscillations were observed. The presence of these oscillations depended on the history of the sample and could perhaps be related to a certain required degree of roughness of the surface. Alternatively, it could be driven by some impurity. For Si, Yeates et al.66 established a relation between reaction oscillations under high-pressure conditions on the Pt(111) surface and Si segregation from the bulk. Si is a natural contaminant of Pt and the presence of SiO2 deactivates the surface.63
The SXRD measurements by Ackermann67 connect the higher reactivity to the appearance of a structure with a (2 × 2) periodicity (Fig. 6a). This was measured at 480–500 hPa O2 after adding a pulse of 130–150 hPa CO to a batch reactor with a Pt(111) surface at 495–570 K, which reduced the surface to the metallic state. While CO was being consumed, the surface switched to the (2 × 2) structure and, later, evolved into α-PtO2 (upper panel, Fig. 6b), without affecting the CO oxidation reactivity (lower panel, Fig. 6b). This could mean that the activities of both the (2 × 2) structure and α-PtO2 surpassed the MTL requirement. Hypothetically, α-PtO2 could be a spectator species, because less active sites would be needed to maintain the MTL regime as more CO is consumed. However, this would break down at a certain point when the full surface is oxidized, the point at which the reaction would stop.
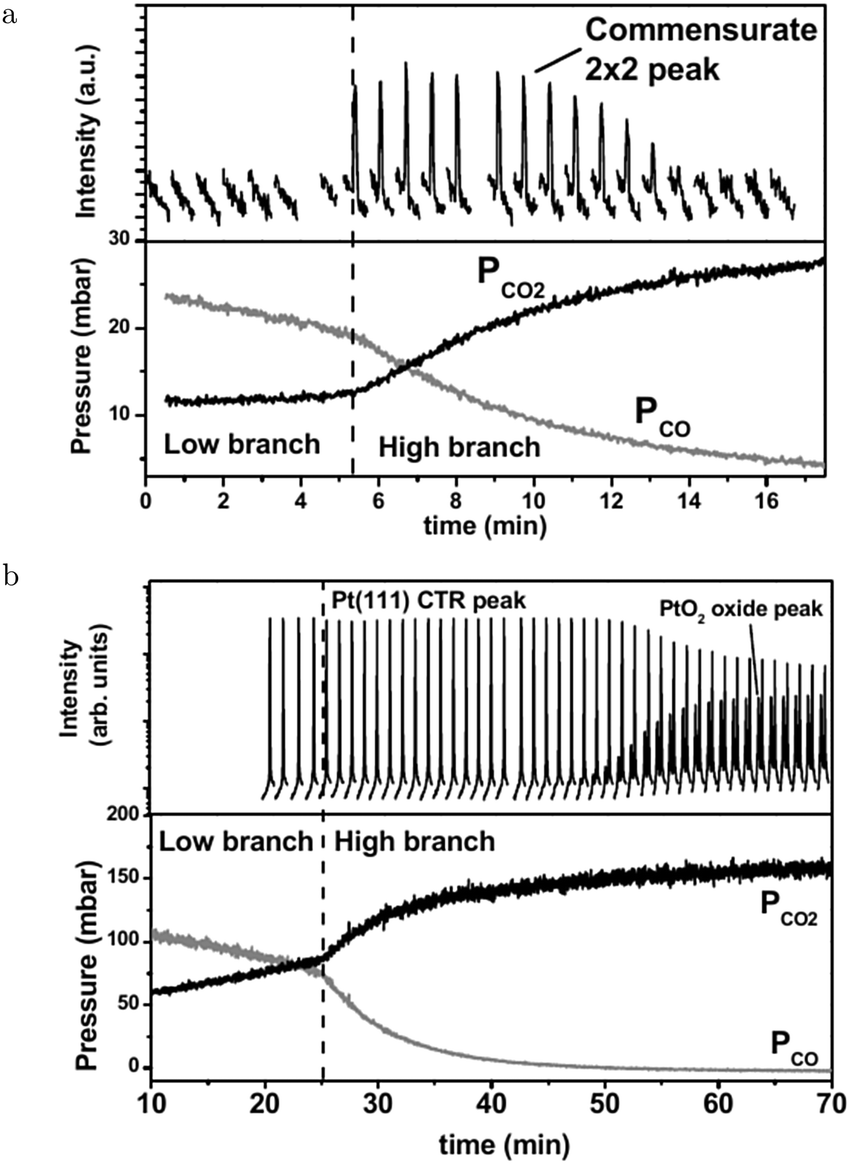 | ||
| Fig. 6 Two sets of measurements in 480–500 hPa O2 after a 130–150 hPa CO pulse at 570 (a) and 495 K (b). Bottom panels present the QMS data. As CO was depleted the reactivity switched to the higher activity regime. Simultaneously, the surface roughened (decrease in Pt(111) CTR) and a commensurate (2 × 2) structure developed. Later in time, PtO2 appeared. Reproduced from ref. 67 with permission from the author, copyright 2007. | ||
The oxide surface with the lowest free energy, the α-PtO2(0001) surface, was found to be inert to CO oxidation based on DFT calculations.68 CO adsorption on this surface is highly activated (174–183 kJ mol−1 or 1.8–1.9 eV), after which the reaction barrier is 48 kJ mol−1 or 0.5 eV. Both the adsorption and the reaction barrier are lowered if an oxygen vacancy is present. However, the oxygen vacancy formation energy is quite high (161 kJ mol−1 or 1.67 eV). Finally, diffusion of CO is also highly activated (135 kJ mol−1 or 1.4 eV). However, a 3D PtO2 cluster cannot consist solely of (0001) surfaces.
The most stable surface orthogonal to the (0001) surface, the (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) surface, is active for CO oxidation. Adsorption of CO on this surface is facile with a barrier of 9.6 kJ mol−1 or 0.1 eV, while the reaction with an oxidic O has a barrier of 29 kJ mol−1 or 0.3 eV.
0) surface, is active for CO oxidation. Adsorption of CO on this surface is facile with a barrier of 9.6 kJ mol−1 or 0.1 eV, while the reaction with an oxidic O has a barrier of 29 kJ mol−1 or 0.3 eV.
In addition, the three-phase boundary between gas/metal/metal-oxide was found to be very reactive.69 CO adsorbed on the metal phase can react with a barrier as low as 8 kJ mol−1 or 0.08 eV. It reacts with an oxidic O atom on the edge of the oxide. The barrier is much lower than that between CO(ads) and O(ads) in the stable co-adsorption phase, p(2 × 2)-(O + CO). The co-adsorption phase is stable at intermediate chemical potentials for CO and O2.
For the PtO2(110) surface, CO can adsorb on a coordinatively unsaturated Pt atom, after which it reacts almost barrierless with an O bridge atom.70 The created vacancy serves as an adsorption site for O2, which can directly react with CO to form CO2 and fill the vacancy. Direct dissociation of O2 was found to be unfeasible.
The exothermicity of CO oxidation was best demonstrated in the work by Somorjai and coworkers.57 In their work, the Pt(111) surface “ignited” at 553–794 K, increasing almost linearly with pCO. After ignition, the sample temperature rapidly increased by 300 K and was self-sustained, i.e., the sample was solely heated by the energy released by the reaction. Moreover, above the ignition point (χ = 0.4, 186 hPa, and 738 K), the apparent reaction barrier decreased from 176 to 59 kJ mol−1 or from 1.8 to 0.61 eV. Also, the order in CO and O2 changed from <0 and >0, respectively, to about 0.5 for both reactants. Interestingly, characteristics of the MTL regime were not observed, which could be related by the massive heat of formation of the ignited sample, stimulating diffusion via convection. During ignition, the vibration spectra changed drastically. Before ignition, only atop-bonded CO was observed, disappearing after ignition. Furthermore, broad, new features were observed after ignition, one of which was assigned to CO bonded to oxidized Pt.
In a titration-type experiment, Kaya and coworkers studied the reactivity of a pre-oxidized (13 hPa O2, 600 s, and cycled between 300–800 K) Pt(111) surface with CO at 1 × 10−5 hPa.71 To measure reactivity, heating beyond 350 K was required. The oxide reacted without forming other oxide structures and the reduced metallic surface became covered with CO(ads).
After re-oxidation (1 × 10−4 hPa O2, 900 s, and 300 K) of a partially reduced PtO2 layer, a mixture of O(ads)/PtO2 was obtained. A different mixture, 4O/PtO2, was formed by annealing the clean Pt(111) surface to 620 K in 0.7 hPa O2. The 4O structure represents a surface oxide with every Pt atom surrounded by 4 O atoms.72 These mixtures were exposed to CO and the order of reactivity was found to be O(ads) > 4O > PtO2.
However, the results of this titration experiment at low CO flux cannot directly be extrapolated to high temperature/pressure conditions, due to orders of magnitude difference in the number of CO molecules impinging on the surface. It was argued that the reaction mechanism changes from LH to MvK on the oxide,27,73–75 which is expected to have a different dependence on the CO flux. It does, however, provide another suggestion that Pt oxides are active for CO oxidation.
3.3 Pt(110), CO adsorption and oxidation
The earliest in situ CO oxidation studies on Pt(110) were performed around two decades ago, employing the optical techniques RAM and EMSI.76,77 Based on the contrast differences in the obtained images, these studies showed that the surface was covered with two types of structures. The measured intensities were assigned based on a comparison to images obtained after exposing the Pt(110) surface to pure gas atmospheres of CO and O2. The two types of structures were identified as a CO-poisoned, inactive phase and an O-rich, active phase. These phases separated into a complex and time-dependent pattern with spiral wavefronts propagating over the surface, as illustrated in Fig. 7. In addition to these complex patterns, the exothermicity of the reaction increased the sample temperature by tens of kelvin. The reported phenomena extended to atmospheric pressure. These studies raise several interesting questions related to the sensitivity of the contrast mechanism in these optical techniques. How much does the signal intensity change upon co-adsorption of CO onto these O-covered islands? Would an oxide be distinguishable from an O-covered island? How does the atomic-scale structure, involving reconstruction and roughness, influence the contrast?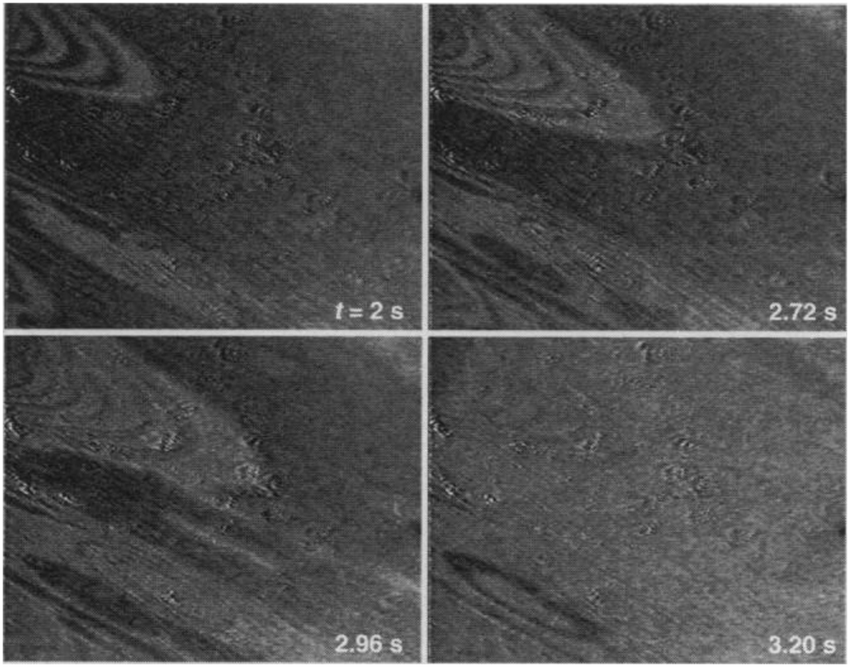 | ||
| Fig. 7 EMSI images showing the transition from a CO-populated (darker) to an O-covered (brighter) Pt(110) surface, caused by a decrease in CO pressure from 7 to 6 Pa at 550 K. Transition occurred via spiral waves. Reproduced from ref. 76 with permission from Science, copyright 1995. | ||
Several operando studies have focused on the structure of the active phase under O2-rich conditions and reported the presence of oxides. An incommensurate, strained PtO2 film, identified via its hexagonal X-ray diffraction pattern, was prepared by Ackermann et al.27 in 500 hPa O2 at 625 K. After formation, the oxide was exposed to CO pulses at the same temperature. Every pulse reduced the intensity of the oxide's Bragg peak by ∼25%, after which it slowly recovered most of its intensity. At the same time, CO2 was produced and CO was consumed. The reactivity was high with a turnover frequency (TOF) estimated at 3 × 103 molecules per (site s) and within the MTL regime. The intensity decrease was explained by an increased roughness in the oxide's structure due to the MvK mechanism. However, this roughness could be expected to increase over time as more CO2 is produced. The observed trend showed the opposite behavior. The intensity decreased instantaneously, followed by a slow recovery. An alternative explanation could be that the CO pulse partially reduced the incommensurate oxide to either an O(ads)-covered surface or a different oxide (vide infra).
In addition to the incommensurate, hexagonal oxide, the authors reported a commensurate oxide with a (1 × 2) periodicity (Fig. 8a). The CTRs of this oxide show that its structure is significantly different from the (1 × 2) missing-row reconstruction, which is the termination of the adsorbate-free surface.78–80 The presence of the diffraction peaks of this oxide was directly correlated with the switch to a high-reactivity regime (Fig. 8b). A possible, DFT-proposed structure of the commensurate oxide is a lifted-row oxide incorporating carbonate ions (Fig. 8c). A structure with a similar periodicity was observed using HP STM after exposing a metallic, unreconstructed, CO-covered Pt(110) surface to an O2-rich mixture (see Fig. 9).74,75
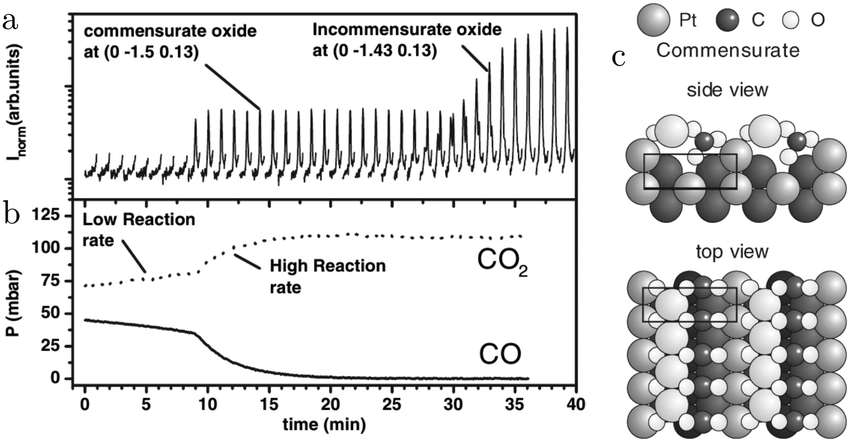 | ||
| Fig. 8 SXRD measurements (a) on Pt(110) in an O2-rich mixture at 625 K plotted with the QMS data (b). After 9 minutes, the reactivity increased and concurrently the commensurate oxide was observed. The DFT-based model of this oxide is plotted in (c). After a time lag, the incommensurate PtO2 was also detected. Reproduced from ref. 27 with permission from the American Physical Society, copyright 2005. | ||
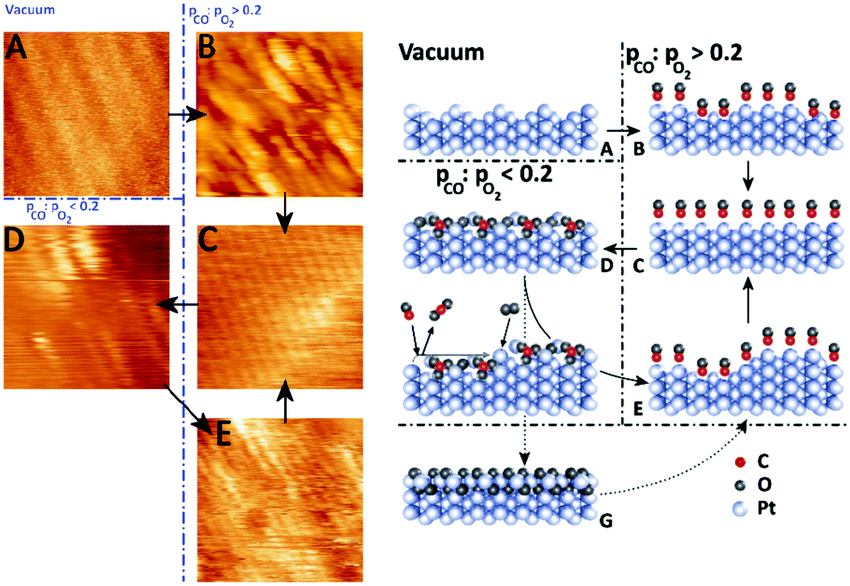 | ||
| Fig. 9 STM images (left panels) and ball models (right panels) showing the transition from a clean (1 × 2) missing-row reconstructed surface (A) to a rough, unreconstructed surface (B), followed by smoothening over time (C). In O2-rich mixtures, the surface formed the (1 × 2) lifted-row surface oxide (D), which roughened over time due to the MvK mechanism. Reduction produced a rough (1 × 1) surface (E). Formation of the incommensurate oxide (G) was not observed in the STM measurements. Reproduced from ref. 74 with permission from Elsevier, copyright 2015. | ||
Only after consuming nearly all CO, the diffraction spots of the incommensurate oxide developed (Fig. 8a). The formation of this incommensurate oxide could explain the development in roughness over time that was observed in an earlier HP STM measurement.73
The experimental conditions in ref. 73 were reproduced by Goodman and coworkers.81 They found a shift to higher reactivity at higher temperature, i.e., 480 instead of 425 K. This could originate from the temperature underestimation in the setup used by Hendriksen et al.82 or due to the fact that gas conditions directly above the sample surface deviate from the average conditions in the reactor. This concentration gradient can differ substantially between a flow and a batch reactor. Furthermore, Goodman and coworkers reported a gradual decrease in CO coverage with increasing temperature. This was measured using PM IRRAS for different CO/O2 ratios at a pCO of 10.6 hPa. This suggests that the surface was gradually covered with O-rich areas with possibly co-adsorbed CO. After the switch to high reactivity, the surface was devoid of CO(ads). The temperature of this switch was dependent on the CO/O2 ratio, with more CO-rich mixtures exhibiting this switch at higher temperatures. After switching, the reactivity showed a transient increase in reactivity, continued by a decrease to a steady state determined by the MTL regime. The different explanations81,83 for this transient “hyper” activity are discussed in the section focused on the Pd(100) surface. Similar transient reactivity maxima (before decreasing to the level dictated by the MTL regime) were also observed for the Pt(111) surface at 0.1 and 1 hPa for χ = 0.25 and χ = 1.63
Oxidation of the Pt(110) surface with atomic O formed a mixture of a chemisorbed overlayer and a surface oxide, visualized by STM.84 Both were formed only after annealing the sample or after dosing at 500 K. The chemisorption structure had a unit cell of (12 × 2)-22O, with PtO2-like stripes along the closed-packed rows. These stripes were expanded with respect to the Pt lattice. This expansion of ∼14% resulted in the ejection of 2 Pt atoms every stripe of 10 atoms long. The ejected Pt atoms became incorporated into adjacent stripes, leading to patches of a 2D structure, labeled as a surface oxide. The surface oxide is metastable according to DFT calculations.
The (12 × 2)-22O structure is the most stable surface termination for a wide range of conditions, based on the Pt(110) surface phase diagram, derived from DFT calculations.68 Furthermore, it can be active for CO oxidation with barriers as low as 9.6–29 kJ mol−1 or 0.1–0.3 eV. These values were derived by using a similar, but much smaller structure with a (6 × 2) unit cell.
Oxidation of the Pt(110) surface in 0.7 hPa O2 between 300 and 473 K resulted in two O species on the surface,85 one of which was identified as chemisorbed O and the other was argued to be a more oxidized species. The former was identified as the (12 × 2)-22O structure and the latter as α-PtO2 by comparing the measured core-level shifts with those calculated by DFT. Both species showed activity towards CO oxidation in titration experiments at 270 K in 10−6 hPa CO, the former being more active by a factor of 1.6–1.7.
The CO-poisoned surface consists of a mixture of CO adsorbed onto bridge and atop sites at RT as inferred from NAP XPS measurements with χ = 1 at 0.5 hPa.86 Bridge-bonded CO desorbed around 373 K, leaving only CO bonded to atop sites.81 Interestingly, this state was active above 393 K and exhibited an additional feature in the C 1s region at a binding energy of 287.9 eV. Because no similar feature was observed in the O 1s spectra, no O accumulated on the surface, although the C 1s binding energy suggests that the deposited C was in a highly oxidized form.
3.4 Pt(100), CO adsorption and oxidation
The atoms in the ideal Pt(100) surface have a lower coordination number compared to those in a Pt(111) surface, 8 versus 9, respectively. This makes the surface free energy of the (100) surface higher than that of the Pt(111) surface. To lower its surface free energy, the cubic Pt(100) surface exhibits a (111)-like, hexagonal reconstruction when it is free from adsorbates.87 Two different varieties of this hexagonal reconstruction exist, the metastable and unrotated Pt(100)-hex, also referred to as Pt(100)-(5 × 20), and the stable Pt(100)-hex-R0.7°. The hex reconstructions are lifted by exposing them to low partial pressures of CO and O2.88,89 The complex and dynamic surface termination leads to complicated behavior under CO reaction conditions at low pressures, such as reaction oscillations and wave patterns.90,91Nonlinear behavior extended to intermediate pressure ranges, studied by Lauterbach and coworkers with EMSI at p = 8 Pa.92 Three differently behaving regimes were reported depending on the temperature and χ (Fig. 10a). Phase I was characterized by large, stable oscillations in reactivity with reaction fronts migrating over the surface upon transition from low to high activity. These large-scale oscillations were not reported at lower pressures and indicate that the majority of the active sites synchronously switched in reactivity. This synchronization can be established by the gas phase, i.e., a highly active site depletes more CO, which decreases the poisoning effect of CO for neighboring sites. Phase II did not show oscillations, but exhibited complicated dynamic patterns on the surface (see Fig. 10b and c). Immeasurably small oscillations on the boundaries between the different patches could not be excluded. More recently, it was argued based on an additional intensity level (darkest patches in Fig. 10c) in the EMSI images that subsurface O was formed.93 Phase III was identified as having unstable reaction oscillations that ceased after minutes in combination with adsorbate islands moving in spatiotemporal patterns. No nonlinear features were observed for parameters outside the regimes in Fig. 10. Although interesting, the information contained in the images is rather limited, because they only provide a contrast difference. From this contrast difference, it is not very trivial to definitively derive structural and/or chemical information.
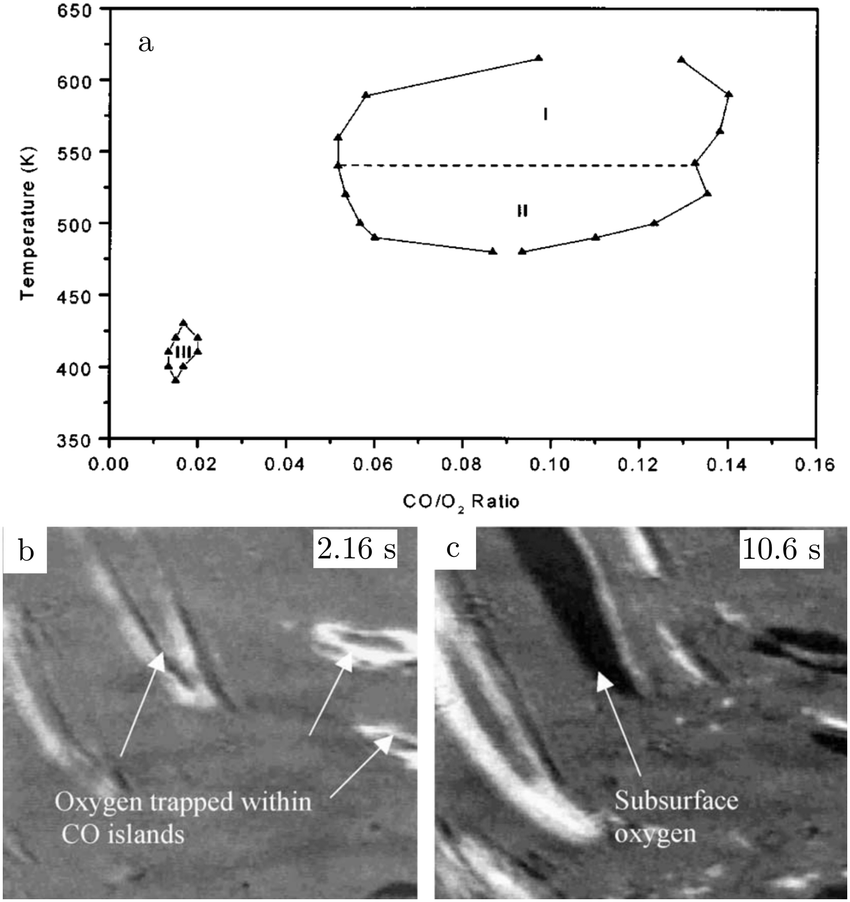 | ||
| Fig. 10 Diagram (a) of conditions probed in the intermediate pressure regime of several Pa, for which nonlinear dynamics was observed. EMSI images (b and c) showing the complex pattern of adsorbate islands on the surface and the formation of chemisorbed O (1 × 1.25 mm2, χ = 0.13, 8 Pa, and 523 K). Reproduced from ref. 92 with permission from Wiley, copyright 2001 (a) and from ref. 93 with permission from Elsevier, copyright 2005 (b and c). | ||
In an earlier publication,94 Lauterbach and coworkers studied the CO oxidation kinetics at 9 Pa and 473 K with χ = 0.1 (phase II, Fig. 10a). After switching the CO flow on and off, an oxygen species accumulated on the surface, as measured by ex situ Auger electron spectroscopy (AES) measurements. This unknown oxygen species, which could be some kind of subsurface O, both decreased the total reactivity of the surface and the susceptibility for CO poisoning. The surface was only deactivated after cycling the reactants. Neither steady-state exposure nor pure O2 exposure led to this deactivation. This may be related to the phase transition between a reconstructed and unreconstructed surface. This produces roughness due to the density mismatch of the two terminations. After exposure, no other contaminants were observed.
The Pt(100) surface remains rather understudied under realistic chemical conditions. In an operando STM study,54 a Pt(100) sample was exposed to CO, O2, and to mixtures thereof at a pressure of 100–125 kPa at 365–423 K. Exposure of the surface induced the lifting of the reconstruction and led to square adatom islands, due to the higher density of the reconstructed surface. Exposure to 116–125 kPa O2 also resulted in square islands, but with a much higher concentration and a higher degree of disorder. These disordered islands showed little evolution over time. Moreover, height histograms showed several step heights incompatible with a metallic Pt(100) surface, a first hint that the surface was oxidized.
Starting in a pure CO atmosphere, the reaction kinetics showed a peak in the CO2 production when the O2 concentration was increased. This was explained by a surface that switched from CO-poisoned to one covered with an optimal CO/O ratio. In the increasingly O2-rich mixture, the reactivity decreased due to lack of CO reaching the O-covered surface. However, very small jumps to higher reactivity were apparent. Based on the similarity to the Pt(111) and Pt(110) surfaces, vide supra, it was argued that the surface had become oxidized. In this case, the oxidized surface would be only slightly more active than the surface covered with chemisorbed O. Furthermore, these jumps were not consistently observed and could have depended on the sample's history, such as the developed roughness by the reaction or remnant roughness induced by the polishing process. These observations are supported by the kinetic measurements in the pressure range of 0.1–800 hPa by Berlowitz et al.48 They concluded that this surface does not form strongly bound oxides in almost pure O2 mixtures (χ of 0.01 to pure O2 at 101 kPa, 400–800 K and 1 hPa at 1150 K). Exposure to these conditions led to samples that were at least as active as clean Pt(100) surfaces.
In addition to CO oxidation, CO can react via the Boudouard reaction
| 2CO(ads) → CO2(g) + C(ads) |
In fact, Somorjai and coworkers95 showed that this surface has a higher activity for the Boudouard reaction than the stepped Pt(755), 5(111) × (100), and the Pt(111) surface [Pt(100) > Pt(755) > Pt(111)]. Adsorption and desorption at 53 hPa of CO is reversible up to 450 K, above which (T > 500 K) the peak attributed to the CO stretch vibration attenuated in the SFG spectra and was red-shifted by 30 cm−1. Also, AES measurements showed significant C deposition. Based on the vibrational-frequency shift, it was concluded that the surface roughened, possibly via the formation of platinum carbonyls.95 The driving force could be lateral repulsion of adsorbed CO molecules. The roughened Pt surface could be the active phase for the Boudouard reaction. The deposited C could act as a stabilizer for the roughness and block sites for CO adsorption.
The higher Boudouard reactivity for the Pt(100) than for the Pt(111) surface was confirmed by DFT calculations in which the effect of the high CO pressure was modeled by compressing two CO layers by fixed slabs of CO and Pt, respectively.96
3.5 Vicinal and polycrystalline Pt surfaces, CO adsorption and oxidation
The (111) surface (of face-centered cubic (fcc) crystals) has two distinct types of steps that differ in the microfacet that the step atoms form. The two kinds of steps form either a (111)-type or a (100)-type facet. The (111) step was recently studied with a combination of SXRD and QMS using a Pt(997), 8(111) × (111), surface.97 The steps increased from single to double step heights, when the sample was heated in a CO/O2 mixture with varying ratios at 10 hPa. This was previously also observed after exposing the steps to O2 under vacuum conditions.98,99
At low temperature, the entire surface was assumed to be CO-covered, leaving not enough neighboring adsorption sites to facilitate O2 dissociation. At higher temperature, the CO coverage was lower due to increased desorption, enabling O2 dissociation. The adsorbed O is likely to first cover the steps. The O-covered steps favor double-step heights. Simultaneously with the step doubling, the CO2 production increased stepwise. Both the doubled step height and the increased reactivity can be the result of the O adsorption on steps, or, as was suggested,97 the double steps themselves could be more reactive than mono-atomic steps. A higher O2 content lowered the temperature for the transition from single to double steps.
The (111) steps in the Pt(332), 5(111) × (111), surface were found to oxidize at an O2 pressure of 1 × 10−6 hPa, based on XPS measurements supported by DFT calculations.72 The proposed model consisted of 1D PtO2 rows that formed on the steps (Fig. 11). This state was labeled 4O, because 4 O atoms surrounded a single Pt atom. These oxide rows were reactive to CO(ads) and DFT showed a barrier of 58 kJ mol−1 or 0.60 eV, 11 kJ mol−1 or 0.11 eV lower than the O(ads) on the terrace, although the O in the former structure is more strongly bonded. However, it was not determined whether indeed monoatomic steps were present under the experimental conditions.
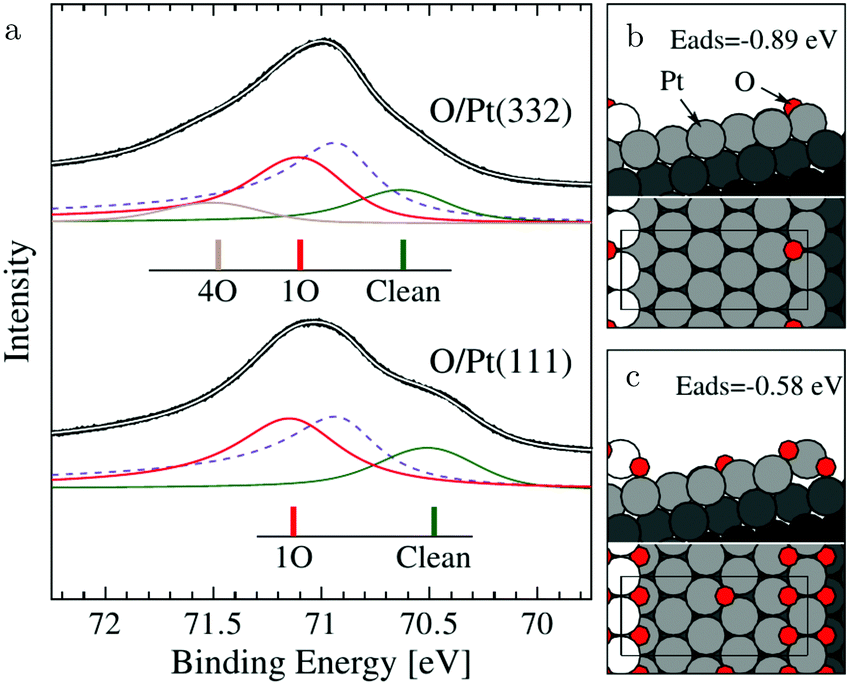 | ||
| Fig. 11 XP spectra (a) of the Pt 4f7/2 region recorded after exposing the Pt(332) and the Pt(111) surfaces to 500 L at 310 K. Models of low (b) and higher (c) O coverage on Pt(332). Reproduced from ref. 72 with permission from the American Physical Society, copyright 2005. | ||
The (100) steps on the other hand behave remarkably differently. No step doubling was observed with SXRD.100 In fact, on the Pt(977), 7(111) × (100), surface, these steps were unstable in either CO or O2-rich mixtures at 200 hPa. This resulted in a surface that formed large facets. This contradicts the naive view that stronger binding to steps by adsorbates can always stabilize vicinal surfaces and shows that the precise atomic configuration of the step is extremely important. The steps were stable only close to stoichiometric ratios of CO and O2 and was maximum reactivity observed. The apparent activation energies for (100)-stepped surfaces, the (977) and the (113), (111) × (100), surfaces respectively, were determined to be 113100 and 100101 kJ mol−1 or 1.17 and 1.04 eV.
The two types of steps behave differently in pure CO atmospheres. Under high vacuum, CO induces doubling of the (100) steps on Pt(755), 5(111) × (100).58 At pressures of around 1 hPa at RT, the Pt(755) surface forms an extended array of triangular clusters,58 which become ordered at slightly elevated temperatures (350–360 K).59 The CO-induced wandering of (100) steps and their doubling in height were confirmed by molecular dynamics (MD) simulations.102 The MD simulations revealed another interesting property of the (100) steps of Pt(211), 2(111) × (100). At the high temperatures of the simulations (1000 K), steps can shift along the step direction and sink into the surface.
No CO-induced doubling was observed for the (111) steps on Pt(332).58 Although for this surface, similar clusters were observed around 1 hPa of CO, they did not uniformly cover the surface and they were parallelogram-shaped.103
Massive restructuring was observed on 50 μm polycrystalline Pt wires in a flow of χ = 0.09 at ∼500 K for a few hours.107 SEM images recorded after the reaction showed triangular facets of 100–500 nm, identified as both (111) and (100). This faceting occurred only after exposing the wire to both reactants simultaneously. The approximate composition in the outer 1 μm was determined to be PtO. These experiments show that even at relatively low temperatures Pt atoms are very mobile. This increased mobility can be caused by the binding to the adsorbates.
Kinetic studies on these wires showed that up to ∼550 K the surface is reaction controlled with barriers of 140–210 kJ mol−1 or 1.45–2.18 eV, indicating a CO-poisoned state. Above this temperature, the reaction was diffusion limited. The reverse transition to the reaction-controlled state occurred 50 K lower.
3.6 CO oxidation over bulk Pt oxides
A specifically interesting type of titration experiments giving insight into the possible reactivity of oxidized Pt-group metals are performed after forming thick, bulk-like oxide layers. Several methods are frequently employed to create these oxides, e.g., DC magnetron sputtering in a reactive O2 atmosphere.108 A Pt oxide film, containing both PtO2 and Pt(OH)4, with a thickness of ∼40 nm was deposited on various substrates by magnetron sputtering. After deposition, it was exposed to either pure CO or to a mixture of χ = 0.43 at a total reactant pressure of 253 hPa. Under both conditions, reactivity was observed above 440 K. In pure CO, only a small spike in CO2 production was observed, while in the mixture, reactivity was sustained, although it decreased for T > 530 K. The fact that sustained reactivity was observed only in the CO/O2 mixture showed that the oxide was an active catalyst, not just a reactant, and a MvK reaction mechanism was proposed. The reactivity decrease at higher temperature was explained as partial decomposition and reduction of the Pt oxide. The apparent activation energies increased from 75 ± 5 to 93 ± 3 kJ mol−1 or from 0.78 ± 0.05 to 0.96 ± 0.03 eV, which was comparable to the value found for a polycrystalline Pt film (103 ± 2 kJ mol−1 or 1.07 ± 0.02 eV), suggesting that the surface was CO-poisoned. Interestingly, some CO2 formation was observed on the reduced surface in pure CO, indicating that the very rough, polycrystalline Pt film was active for the Boudouard reaction.3.7 Conspectus on Pt
The Pt(111) and Pt(110) surfaces can switch to higher reactivity under O2-rich conditions. Under these conditions, signs of MTL regime were observed. Simultaneously, both surfaces form a new structure. For the Pt(111) surface, it is a (2 × 2) structure and for the Pt(110) surface, it is a (1 × 2) structure. A possibility for the latter is a lifted-row surface oxide, in which carbonate ions are an important element. For the former, the Pt(111)-(2 × 2), no atomic structure is known. On both surfaces, PtO2 formed after more CO was depleted. The formation of this oxide had no influence on the reactivity, i.e., the reactivity remained in the MTL regime. This could indicate that PtO2 is active, either via defects in the oxide or via the boundary between the oxide and CO(ads) islands on reduced, metallic Pt.For the Pt(100) surface, complex and dynamic patterns were observed under reaction conditions. However, questions remain, such as, is the active phase under O2-rich conditions chemisorbed O on a metallic surface or a (surface) oxide? Although STM provided an indication that it is a surface oxide, the atomic structure remains completely unknown. Interestingly, the Pt(100) surface is the most reactive surface for the Boudouard reaction.
For vicinal surfaces, the structure and stability is determined by the type of steps. The (100) steps are the most unstable and they are likely to facet and restructure.
4 Palladium
Compared to its 6th period relative, palladium oxidizes more readily and its oxides are more stable than those of Pt. Multiple oxidic structures have been observed, ranging from surface oxides to bulk-like films. Additionally, the Pd oxides tend to be stable at and below RT and can be studied under UHV, leading to more titration-type studies on the activity of Pd oxides. Therefore Pd, even more than Pt, has become the prime showcase for novel in situ/operando surface-science techniques. Most of these studies have focused on the Pd(100) surface, which is discussed first, followed by the other surfaces.4.1 Pd(100) and the √5 surface oxide, CO adsorption and oxidation
Studies using the Pd(100) surface as model catalyst employed a wide range of techniques, such as SXRD,109–113 STM,114 XPS,116,117,129 PLIF,18 and IR spectroscopy.81,119 With X-ray diffraction, 3 different oxides were identified under reaction conditions, depending on the temperature and the CO/O2 ratio.109 The first was characterized as a surface oxide (Fig. 12) with a (√5 × √5)R27° unit cell, which can be regarded as an altered, strained PdO(101) layer,111,120,121 and has an O coverage of 0.8 ML120 (although a PdO(001)/Pd(100) structure has been suggested as well122). The surface oxide contains boundaries between translation domains, roughly every 4–5 nm (see arrows in Fig. 12), which can be explained by the tensile stress in this structure caused by the mismatch between the (√5 × √5) and the PdO(101) unit cells.121 The 2D nature of the surface oxide is concluded based on its diffraction signature (Fig. 13, panel labeled a).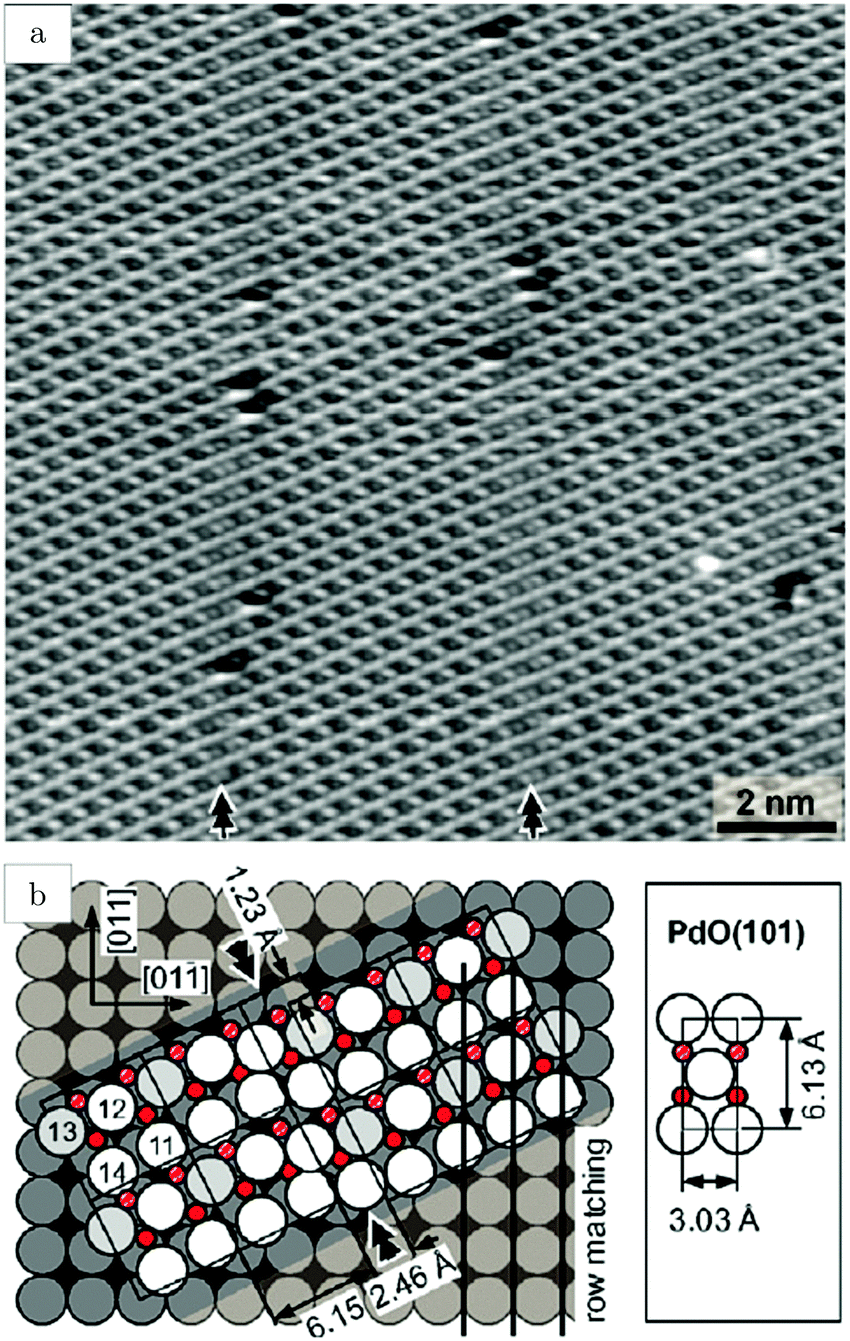 | ||
| Fig. 12 STM image (a) of the (√5 × √5)R27° surface oxide on Pd(100) in UHV. Arrows indicate domain boundaries. Ball model for this oxide (b). Reproduced from ref. 121 with permission from Elsevier, copyright 2007. | ||
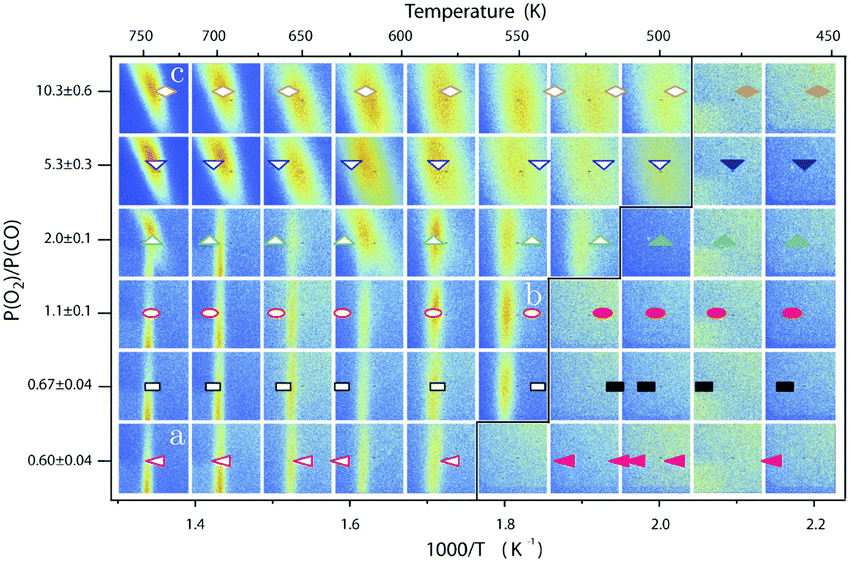 | ||
| Fig. 13 Stability diagram of the Pd(100) surface under reaction conditions for different χ and T, measured using SXRD. Three different oxides (open symbols) can be distinguished: 2D surface oxide (a), epitaxial PdO (b), and a polycrystalline PdO (c), in addition to the reduced, metallic surface (lower right, closed symbols). Reproduced from ref. 109 with permission from the Royal Society of Chemistry, copyright 2011. | ||
The second observed oxide was an epitaxial, but disordered PdO layer with a thickness of 2–3 nm having PdO(101) planes parallel to the Pd(100) surface (Fig. 13, panel labeled b). In addition to PdO(101)∥Pd(100), several researchers proposed PdO(001)∥Pd(100), both under O2-rich reaction conditions114 and in pure O2.67,123,124
This epitaxial film (thickness of 4–6.5 nm)123,124 developed above 1 hPa O2 and 675 K. It consisted of small islands with large fluctuations in height from island to island. Furthermore, a strong kinetic limitation was observed to form the bulk oxide, making the surface oxide stable up to ∼575 K at 100 kPa O2. At higher temperatures, the surface oxide disappeared and the bulk oxide was formed.123 However, in a NAP XPS study, the formation of bulk PdO was already observed above 473 K at 0.5 hPa O2.125
The third observed oxide was a bulk-like PdO that lost epitaxiality with the underlying surface. It appeared in the SXRD measurements as a polycrystalline powder ring (Fig. 13, panel labeled c).
By probing a wide range of conditions, a stability diagram was established (Fig. 13). It shows that roughly below 500–550 K, the Pd(100) surface was reduced. The surface oxide was stable at higher T, most pronounced at ≥670 K, and relatively high χ (0.9–1.7), almost up to stoichiometry. While at low χ, roughly below 0.19, and approximately above 500–550 K, the polycrystalline PdO was present, with higher order at more elevated temperatures. The epitaxial PdO was found at the center of the diagram. Simultaneously with the appearance of any of the oxides (open symbols, Fig. 13), the reactivity switched to the MTL regime.
Gustafson et al.110 used high-energy SXRD to follow the growth of both the surface oxide and the epitaxial bulk oxide while changing the gas composition from χ = 4 and p = 10 hPa to χ = 2 and p = 12 hPa at a constant temperature of 575 K. Concurrently with the formation of the surface oxide (see line II, Fig. 14), the reactivity increased, while the bulk oxide gradually appeared. The epitaxial oxide had a thickness of 5 ± 2 nm and an island width of approximately 47 ± 1 nm.112 It coexisted with the surface oxide thus showing a Stranski–Krastanov, i.e., layer-plus-island, growth mode.
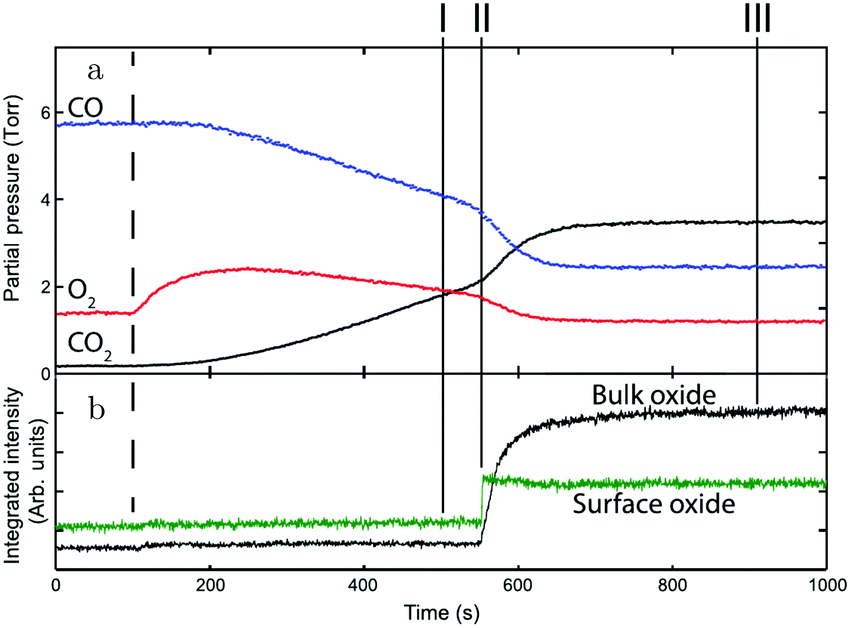 | ||
| Fig. 14 (a) Gas-phase information, while decreasing the CO pressure at 525 K. (b) Intensity of diffraction peaks of the surface oxide (green) and the bulk oxide (black) on Pd(100), recorded with high-energy SXRD. Reproduced from ref. 110 with permission from Science, copyright 2014. | ||
Additional structural information on the transition from a reduced to an oxidized surface can be obtained from a reported work combining HP STM and QMS.114 The surface changed from smooth, metallic to rough with monoatomic islands after the switch, which occurred at roughly χ = 0.05, p = 125 kPa, and T = 408 K. This fast switch happened on the order of a single STM scan line (dashed line, Fig. 15a), typically around or below 1 s. Simultaneously, the reactivity increased by a factor of 1.5 to the MTL regime, although the estimated TOF was rather low [∼10 molecules per (site s)]. The roughness increased over time, resulting in a rough surface consisting of 4–16 nm grains with a polycrystalline appearance (Fig. 15c). The formation of clusters on the onset of bulk oxidation was observed before under UHV conditions using STM.122 In CO-rich mixtures, the oxide reduced to a metallic surface with adatoms and vacancy islands several atoms high/deep. The oxide reduced at a slightly lower CO pressure than at which it was formed, i.e., reversed from that observed on Pt. Also, under certain conditions, spontaneous reaction oscillations were observed.
 | ||
| Fig. 15 STM images (∼125 kPa, 408–433 K, O2-rich mixtures) recorded (a) during the transition from a reduced to an oxidized surface; (b) slightly after; and (c) ∼1 h after the transition. Reproduced from ref. 114 with permission from Elsevier, copyright 2004. | ||
These oscillations were further investigated using SXRD and a roughness-driven model was proposed (Fig. 16a).113 The basic ingredients are the following: (a) the reaction on the oxide induces the continuous formation of roughness due to local reduction/oxidation via a MvK reaction mechanism; (b) the roughness destabilizes the oxide more than it would destabilize a CO-covered, metallic surface, because, for the metallic surface, the CO adsorption energy onto defects is higher, partially stabilizing a rough, metallic surface; (c) the rough oxide switches to a rough, metallic surface above a critical roughness; (d) due to the higher Pd mobility in the metallic state, the surface smoothens over time; (e) the flat metallic surface oxidizes and the cycle starts over. The most important aspect of the measurements was the FWHMs of the metallic and oxidic diffraction peaks (Fig. 16b), combined with the kinetic information (Fig. 16c). The FWHM scales with the roughness. The roughness development showed opposite behavior for the metal and oxide phases, indicating an increasing roughness in the oxide phase and a smoothening in the metallic phase. Recently, the oscillations were re-examined (χ = 0.046, p = 680 hPa, and T = 443 K) using SXRD with a 2D detector and with a higher time resolution of ∼1 s.126 The 2D projection of the Bragg peak was fitted to reveal more, complicated details. During the full oxidation–reduction cycle, the in-plane lattice constant of the surface oxide expanded, while the Bragg peak of the surface oxide slowly changed into a powder ring. In other words, the strained surface oxide transformed into relaxed polycrystalline PdO.
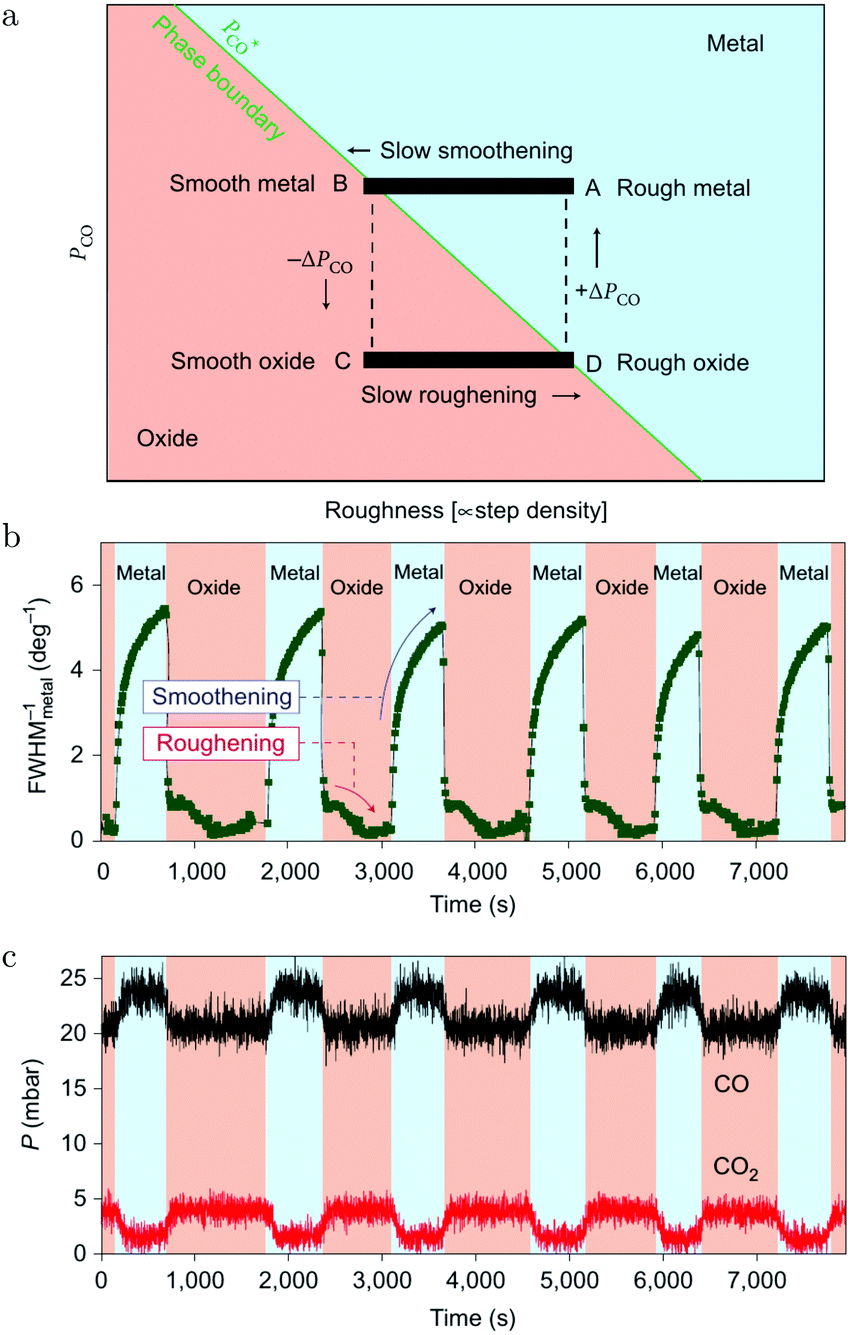 | ||
| Fig. 16 (a) Model of the spontaneous reaction oscillations on Pd(100) cycling through the surface phase diagram, plotted as a function of pCO and roughness. Evolution in time (b) of the FWHM of a metallic diffraction peak (blue-colored regions) and that of the oxide (salmon-colored regions). QMS signals of CO and CO2 (c) as a function of time. Reproduced from ref. 113 with permission from the Nature Publishing Group, copyright 2010. | ||
In addition to the full oxidation–reduction cycles, changes on two different time scales were observed. The first were random intensity drops, occurring predominantly at the end of the oxidation phase (just before full reduction) and mostly two drops per cycle. After the drop, the intensity recovered quickly, indicating a re-growth of the oxide. The newly formed oxide had slightly broader diffraction spots indicating a rougher oxide film with smaller domains. Also, the Bragg peak rotated more towards a powder ring. Finally, rapid oscillations with a modest amplitude were detected, which had a time scale of ∼15 s. During these, the intensity of the oxide Bragg peak momentarily decreased, after which it recovered more slowly. These two faster switches were explained by a partial reduction of the oxide film. The partial reduction preferentially removed the epitaxial oxide grains and was followed by local smoothening. During this partial reduction, not enough surface area was reduced to globally change the gas composition. This left the atmosphere highly oxidizing, leading to re-oxidation in a more polycrystalline form.
CO-Titration experiments on oxidized Pd(100) surfaces are somewhat conflicting. Zheng and Altman reported that the oxidic structures were less reactive to CO and decomposed into more reactive (2 × 2) chemisorption structures.127 Also, the decomposition rate decreased with increasing temperature, which was attributed to a restricted CO lifetime on the oxides. On the other hand, Fernandes et al. showed that the reduction rate increased at higher temperature.128 The √5 surface oxide reacted with CO, forming coexisting regions of surface oxide and metallic, CO-covered islands. Due to the low sticking probability of CO on the surface oxide, CO oxidation was limited to the boundary between these phases. This led to Avrami–Erofeev kinetics, characterized by a slower induction period in which the CO islands nucleated, probably at defect sites in the oxide. This mechanism could be important for a working catalyst when both CO-covered, metallic regions coexist with the surface oxide as was suggested.18,69 Parallelly, the CO flux under reaction conditions can be high enough to facilitate CO adsorption on and reaction with O atoms in the surface oxide.
CO adsorption on Pd oxides was demonstrated by exposing a pre-oxidized Pd(100) surface to 0.5 hPa CO at 393 K.125 CO adsorption induced a change in the core-level shift of the Pd2-fold atoms in the PdO(101) surface of the bulk oxide. They shifted by 0.55 eV to higher binding energy, due to their interaction with CO.
Kondoh and coworkers studied CO adsorption and oxidation with NAP XPS, see Fig. 17a and b, and reported that CO adsorption at 1.3 Pa and RT behaves similarly to lower temperature adsorption under UHV.129 The proposed model for the CO(ads) layer has a unit cell of p(3√2 × √2)R45° with a coverage of 0.67 ML and consists of bridge-bonded CO (Fig. 17d). This CO-saturated surface was the starting point to study CO oxidation (χ = 0.1 and p = 0.29 hPa). After heating to 448 K, the CO coverage was reduced and a p(2√2 × √2)R45° structure with a coverage of 0.5 ML was proposed (Fig. 17c). For both CO structures, reactivity was low, thus both effectively blocked dissociative adsorption of O2.
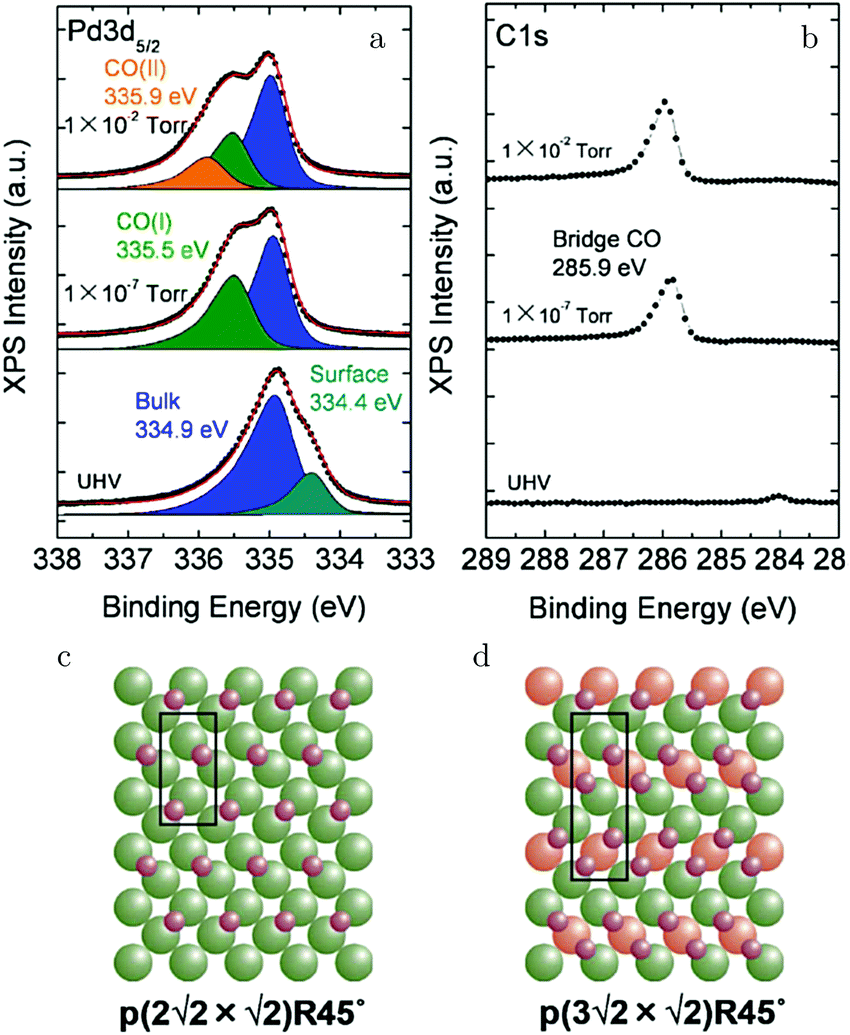 | ||
| Fig. 17 XPS measurements on Pd(100) [Pd 3d5/2 (a) and C 1s (b)] for increasing pCO at RT. Models of the CO(ads) layers for 0.5 ML (c) and 0.67 ML (d). Reproduced from ref. 129 with permission from the American Chemical Society, copyright 2012. | ||
Above 463 K, the reactivity steeply increased and simultaneously the XPS fingerprints of the √5 oxide were observed. Based on the agreement with the expected 1/1 ratio of the two O species (see the lower panel of Fig. 18a), it was excluded that large patches of O(ads) were present. Under these conditions, the reactivity was not in the MTL regime. In fact, it decreased with increasing temperature. This can be explained by a negative apparent activation energy. In this case, at higher temperature, the preference of CO desorption over CO oxidation increases. This is supported by DFT calculations predicting that the barrier for the reaction between CO(ads) and an oxidic O is lower than the energy needed to desorb.130–132 Alternatively, at higher temperature, the CO oxidation rate could become too fast compared to the replenishment rate of O vacancies, leading to a partial decomposition of the surface oxide. This would result in a lower concentration of available O and a lower CO2 production.129 This was proposed based on a decrease in peak intensity corresponding to the upper O atoms in the √5 oxide, which were shown to be solely responsible for the reactivity (yellow, Fig. 18a), when heated to 643 K. Simultaneously, a peak associated with a slightly more reduced state developed in the Pd 3d5/2 spectrum. Although the reaction with CO does tend to destabilize the surface oxide, it seems to contradict with the predicted temperature behavior.131 Using a kMC model, higher stability of the surface oxide with increasing temperature was predicted. Another explanation for the O decrease at higher temperatures could be thermal decomposition. However, this was excluded, because once the CO flow was stopped at constant T, the surface oxidized further, forming PdO.129
 | ||
| Fig. 18 XPS measurements (a) on Pd(100) under CO oxidation conditions for the O 1s region at ∼483 K (lower panel) and at 643 K (upper panel). The broad peak originated from Pd 3p3/2 photoelectrons. (b) Model of the √5 surface oxide with the two different types of O atoms (reactive, 528.8 eV, and inactive, 529.6 eV). Reproduced from ref. 129 with permission from the American Chemical Society, copyright 2012. | ||
In a second NAP XPS study at slightly higher pressure (0.7 instead of 0.30 hPa), the transition to higher reactivity occurred around the same temperature.117 However, the limited temperature range makes it difficult to conclude anything regarding the relation between reactivity and temperature. After decreasing the temperature, the surface returned to CO-covered and poisoned with some small hysteresis, i.e., the reduction occurred 35 K lower in temperature than the oxidation.
Lundgren and coworkers performed similar NAP XPS studies in CO-richer mixtures of χ equal to 0.25 and 1 at 0.67 hPa.116 In the former mixture, the surface also switched from CO-covered to the surface oxide, albeit at a higher activation temperature of 543 K. In the latter mixture, the surface behaved significantly differently. Above the activation temperature of 608 K, chemisorbed O(ads) was the major surface species, and CO2 was easily measured in the gas phase using XPS. In addition to the ratio of reactants, the activation temperature was also dependent on the absolute pressure. In a mixture of χ = 1, it shifted from 523 to 613 K upon increasing the pressure from 1.3 × 10−2 to 1.3 hPa.133
The NAP XPS results were confirmed by an early IRRAS study.134 The reported experiments were conducted in a temperature range of 500–575 K, in a mixture of χ = 2 at 2 hPa. As expected, a high CO coverage was measured, between 0.45 and 0.55 ML, and the apparent activation energy (123 ± 1 kJ mol−1 or 1.28 ± 0.01 eV) of the reaction was nearly identical to the CO adsorption energy. Furthermore, the reaction was found to be negative first order in CO and positive first order in O2, leading to an independence from the total pressure.
The strongest argument against the surface oxide as the active phase on the Pd(100) surface is based on IRRAS measurements by Goodman et al.81 In their work (T = 400–800 K and p = 3–117 hPa), three different phases were identified. The first was explained as a low temperature, CO-poisoned surface with low reactivity. Upon heating the sample, this phase was followed by a “hyperactive” transient phase, after which the TOF decreased to a lower level, while being in the MTL regime, which lasted to the highest probed temperatures. In the third regime, the surface was believed to be deactivated by an oxide. In fact, the IRRAS measurements showed CO adsorbed on oxidized sites at 2142 and 2087 cm−1 for mixtures of χ equal to 0.2 and 0.1. The “hyperactive” regime was explained as a metallic surface covered with O(ads), although no results were presented to support this claim. Similar trends were obtained for the Pd(111) and Pd(110) surfaces.
A peculiarity in the reactivity data led to fierce discussion,83 namely this “hyperactive” phase was solely observed when the sample changed from CO-poisoned to MT-limited, not in the reversed direction. Therefore, it was argued by Van Rijn et al.83 that the “hyperactive” phase was not related to a different surface structure. Instead, it was attributed to a jump in reactivity, much faster than the mixing properties of the reactor. In this case, the catalyst switches instantaneously to the higher reactivity phase, but with the surrounding gas atmosphere still nearly identical to that during the CO-poisoned regime. This leads to a transient peak in the TOF until CO depletion and diffusion to the surface are in equilibrium again.
Furthermore, with SXRD it was confirmed that polycrystalline PdO formed under the conditions for which IRRAS showed CO(ads) on oxidized Pd (χ = 0.1–0.2). For CO-rich mixtures, IRRAS did not reveal these features, while SXRD discovered the presence of epitaxial PdO. The absence of CO(ads) on the latter structure can be explained by either a higher reactivity or weaker binding of CO, hence explaining the absence of any CO in the IRRA spectra.83
Chen et al.119 continued these studies with an IRRAS setup able to reach low wavenumbers, down to 450 cm−1, to probe the metal oxide bonds in PdO. After oxidation of the Pd(100) surface in 13 hPa O2 at 700 K, two new IR features were resolved, at 669 and 615 cm−1. At 450 K, this oxide was neither stable in pure CO at 2.6 hPa nor in a mixture of χ = 1.5 at 60 hPa. The decomposition rate showed similar behavior as reported in ref. 128, with a slow initial rate followed by fast reduction.
Starting from a reduced surface, the oxide vibrational signatures were not observed to develop under reaction conditions (χ = 0.5, 60 hPa, and 525 K), not even after the surface switched to the state of higher activity. This would suggest that PdO was not present on the active Pd(100) surface. However, the reported IR frequencies should most likely be assigned to bulk PdO. Since the IR signatures of the surface oxide remain unknown, in terms of both frequency and IR activity, it is unclear if these would deviate from those of the bulk oxide and whether they would have been observable in the operando IRRAS experiments.
A recent combined kMC/PLIF study further fuels the discussion regarding the active sites on Pd(100) under reaction conditions.18 In this work, the activity of the Pd(100) surface was studied in a mixture of χ = 0.25 and at p = 90 hPa, from 500 to 650 K. The reactivity was modeled with kMC either using the √5 surface oxide or the reduced, metallic surface. The latter was found to reproduce the reactivity of the model catalyst much better, although theory predicts that the √5 surface oxide is the stable structure under these conditions.130 The kMC model showed, furthermore, that while the CO oxidation barrier is rate limiting for the reaction on the metallic surface, the CO adsorption strength is limiting the reactivity of the surface oxide. However, CO binding could be much stronger if O vacancies are present in the surface oxide,135 which would increase the reactivity of the oxide. Alternatively, reactions at the boundary between (metastable) CO(ads) islands and the surface oxide69 could account for the reactivity observed in the PLIF experiment.
 | ||
| Fig. 19 Wulff construction under O2-poor (left) and O2-rich (right) conditions, based on the surface free energies derived from DFT. Insets show the only two surfaces, (100) and (101), constituting the Wulff construction. The size of the constructions is such that the (101) surface area is constant in both cases. Reproduced from ref. 136 with permission from the American Physical Society, copyright 2004. | ||
Nørskov and coworkers confirmed the stability of the PdO(100) and PdO(101) surfaces and studied their interaction with CO.132 The PdO(100) surface does not bind CO, and CO oxidation proceeds via a MvK–ER mechanism. The PdO(101) surface, on the other hand, binds CO strongly (141–142 kJ mol−1 or 1.46–1.47 eV)132,135via the Pd3-fold atoms. Adsorption on atop and bridge sites is degenerate, using the PBE functional,132,135 but atop sites are strongly preferred with the HSE functional. This functional generally describes band gaps better than PBE, which perhaps means it is more accurate for adsorption on band-gap materials.135 In addition, PdO(101) binds O2 even more strongly than CO.132 However, O2 dissociation on PdO(101) is endothermic, making a LH reaction between O(ads) and CO(ads) quite unfavorable. The instability of O(ads)/PdO(101) makes direct CO oxidation by O2 also difficult. Instead, CO and O2 if reacting form carbonate. The barrier for the Mvk-LH reaction between CO(ads) and oxidic O was determined to be 62–64 kJ mol−1 or 0.64–0.66 eV.132,135 This barrier decreases to 54 kJ mol−1 or 0.56 eV for a saturated CO coverage.135 A MvK–ER reaction was also considered but, although a similar barrier was found (68 kJ mol−1 or 0.7 eV),132 was not deemed viable, due to a low pre-exponential factor. This, in turn, is related to the unfavorable loss of entropy during the reaction. For the PdO(100) surface, the MvK–ER mechanism has a lower barrier, 23 kJ mol−1 or 0.24 eV, and, therefore, a higher pre-exponential factor.
Although PdO(100) is the lowest termination for bulk PdO, Seriani et al. showed that PdO(101)/Pd(100) is the surface with the lowest energy under O2-rich conditions.138 It benefits from a good fit with the Pd(100) surface and relatively low surface free energy. Other possibilities are less stable due to a large mismatch with the Pd(100) surface, PdO(100)/Pd(100), or because of the high surface free energy, PdO(001)/Pd(100). Interestingly, CO binds more strongly to 2 layers of PdO(101)/Pd(100) than to a single layer. This effect is reversed by thicker layers. This was mainly attributed to the instability of the PdO(101) bilayer. This instability results in Stranski–Krastanov growth, because at a certain O chemical potential, the most stable structure switches from a single layer PdO(101)/Pd(100) to an infinite number of layers.
An extensive DFT/thermodynamic model predicts the surface phase diagram, see Fig. 20.130 The surface was in equilibrium with a gas phase of fixed composition, i.e., the reaction of CO and O2 was not allowed to occur. In the region of relevant reaction conditions, indicated by the black bar, the 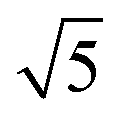 oxide is predicted as the most stable structure, although a phase transition to a CO-covered, reduced surface is in the near vicinity. Noteworthily, no mixed CO(ads)/O(ads) phases are present, due to a stronger O(ads)–CO(ads) repulsion compared to that between the separate adsorbates.
oxide is predicted as the most stable structure, although a phase transition to a CO-covered, reduced surface is in the near vicinity. Noteworthily, no mixed CO(ads)/O(ads) phases are present, due to a stronger O(ads)–CO(ads) repulsion compared to that between the separate adsorbates.
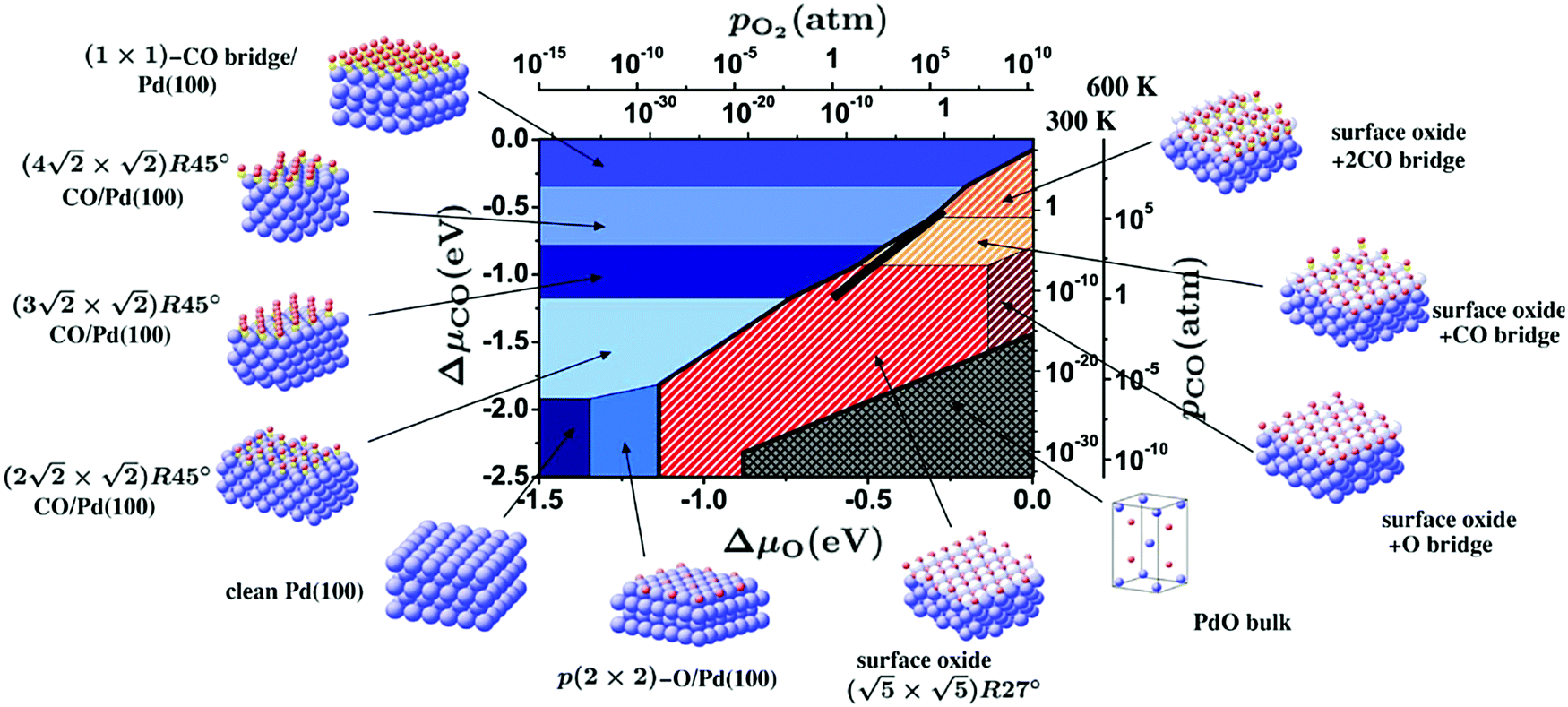 | ||
| Fig. 20 Surface phase diagram of the Pd(100) surface in equilibrium with the gas phase (with the restriction that CO cannot react with O2). The black bar indicates relevant reaction conditions (101 kPa, 300–600 K). Ball models visualize the different structures (metallic Pd, blue; oxidized Pd, gray/light blue; O, red; C, yellow). Reproduced from ref. 130 with permission from the American Physical Society, copyright 2007. | ||
Based on these calculations a kMC model was constructed and calculations were performed for 101 kPa O2 at 300, 400, and 600 K.131 These simulations showed that the surface oxide can also be kinetically stable under reaction conditions. At higher temperatures, the surface oxide becomes even more stable. The CO pressure needed to reduce the structure increases from 10 kPa at 300 K by a factor of 102 at 600 K. At the highest temperature, the reaction is limited by a low CO lifetime on the oxide, due to a high desorption rate.
Alternatively, the O content on the surface can be restored by diffusion of Ov to the subsurface sites.135 This effect is counteracted by CO(ads) which has an increased adsorption energy of 208–219 kJ mol−1 or 2.16–2.27 eV in the presence of an Ov (cf. 141–142 kJ mol−1 or 1.46–1.47 eV for CO adsorbed without the presence of an Ov). Furthermore, CO diffusion is facile, in addition to possible Ov diffusion, with the latter experiencing a higher barrier (23–36 vs. 102 kJ mol−1 or 0.24–0.38 vs. 1.06 eV). Restoring oxygen on the PdO(100) surface is more problematic and it was proposed to occur on metallic Pd after partial reduction.132
4.2 Pd(111) and the √6 surface oxide, CO adsorption and oxidation
Adsorption of CO on Pd(111) did not lead to dissociation or the formation of carbonyls up to 1 hPa and 400 K.139 The coverage of CO(ads) increased with pressure to 0.69 ± 0.02 ML at 1 hPa and 300 K, without showing signs of saturation.35 Adsorption of CO involves hollow, bridge, and atop sites and forms a mixture of the three between 200 and 1000 hPa at 300 K.140 In a different set of experiments, the CO coverage was increased by decreasing the surface temperature from 650 to 210 K at 600 hPa and from 700 to 175 K at 133 hPa. Adsorbed CO shifted gradually from bridge sites to a mixture of 3-fold hollow and atop sites, both with well-defined vibrational signatures.141 This was attributed to a phase transformation of the adsorbates from c(4 × 2)-2CO to (2 × 2)-3CO. However, the expected ratio between atop and bridge CO(ads) of 2/1 in the model of the (2 × 2)-3CO structure was not experimentally reproduced.35,139–141The Pd(111) surface oxidizes similarly to the Pd(100) surface, except that a different surface oxide is formed. The structure and composition of this surface oxide were elucidated by a powerful combination of STM (see Fig. 21), SXRD, XPS, and DFT.142 It has a composition of Pd5O4, having a (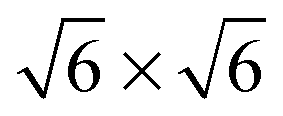 ) unit cell. It consists of two types of Pd atoms, coordinated to 2 and 4 O atoms, respectively, and two types of O atoms, coordinated to 3 and 4 Pd atoms, respectively.144 In addition, a bulk PdO can form, terminated by a PdO(101) surface.143
) unit cell. It consists of two types of Pd atoms, coordinated to 2 and 4 O atoms, respectively, and two types of O atoms, coordinated to 3 and 4 Pd atoms, respectively.144 In addition, a bulk PdO can form, terminated by a PdO(101) surface.143
 | ||
| Fig. 21 (a) STM image (10 nm wide) of Pd5O4 on Pd(111), UHV conditions. (b) Ball model of the surface oxide, based on SXRD and DFT. Reproduced from ref. 142 with permission from the American Physical Society, copyright 2002. | ||
A NAP XPS oxidation study showed that the binding energies of O 1s and Pd 3d did not shift that much from that of O(ads)/Pd(111), but the relative intensities of the different O and Pd species changed significantly.145 Three stages of oxidation were identified, apart from O(ads)/Pd(111). The first was the Pd5O4 surface oxide, stable up to ∼4 × 10−3 hPa at a maximum temperature of 700 K. This structure was characterized by two O species, O3-fold at 529.0 eV and O4-fold at 529.7 eV with a 1/1 ratio, and two Pd species, Pd4-fold at 336.2142–336.3145 eV and Pd2-fold at 335.5 eV142,145 with a 1/4 ratio. The second stage was the formation of a metastable “subsurface” oxide up to 0.4 hPa.145 This increased the Pd4-fold![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd2-fold ratio to 1 and increased the O/Pd ratio.
:Pd2-fold ratio to 1 and increased the O/Pd ratio.
Subsurface O was also reported after exposure to 240 L O2 at temperatures of 600–873 K in UHV.146 This assignment was based on its inertness to co-adsorbed CO. Furthermore, its presence negatively influenced the CO oxidation rate under UHV conditions by decreasing the sticking probability of both reactants. However, the much higher flux of reactants impinging under realistic conditions may mitigate this effect.
Finally, around 1 hPa, the third stage, a PdO bulk oxide appeared.145 In the XP spectra, Pd4-fold shifted from 336.3 to 336.5 eV, similar to the binding energy of PdO/Pd(100).129 Simultaneously, Pd2-fold completely disappeared. In the O 1s spectrum, O3-fold and O4-fold shifted by −0.1 eV and +0.1 eV, respectively, with O4-fold as the dominant species. This was interpreted as an oxide with an O-terminated surface. The activation energies for oxide formation were calculated to be ∼60 kJ mol−1 or 0.62 eV and ∼140 kJ mol−1 or 1.45 eV for the metastable subsurface oxide and the bulk PdO, respectively.
The formation of any subsurface oxide was contradicted by SXRD measurements probing a maximum p of 100 hPa O2.147 They confirmed the formation of the Pd5O4 surface oxide, but observed the growth of epitaxial PdO in different orientations under conditions for which the subsurface oxide was observed in ref. 145. This PdO was oriented such that both the PdO(100) and PdO(101) surfaces were exposed.
Using NAP XPS, Toyoshima et al. investigated the oxidation of Pd(111) and the oxidation of CO.115 Interpretation of the O 1s XP spectra is challenging, because of the overlap with the Pd 3p3/2 peak and because of the wealth of different reported O species. Six were reported after exposing the surface to 0.3 hPa O2 from RT to 773 K. They were assigned as chemisorbed O(ads)/Pd(111), the two distinct O atoms in Pd5O4, chemisorbed O(ads)/Pd5O4, subsurface O, as a precursor for bulk oxidation, and, finally, bulk oxide. Several of these species supposedly have identical binding energies.
However, two important conclusions can be drawn. (1) At 473 K, the Pd(111) surface was CO-poisoned, even under very O2-rich conditions. Also, a non-reactive O species appeared, attributed to subsurface O. (2) At 573 and 673 K, Pd5O4 was observed and most likely covering the complete surface. Concurrently, high activity was observed. This reactivity was correlated with the presence of O3-fold, although this species has an identical binding energy to O(ads)/Pd(111). The O4-fold in Pd5O4 appeared to be unreactive. Moreover, the surface deactivated over time, which could be linked to the formation of yet another O species. This deactivating species appeared only when both CO and O2 were present, but based on the C 1s spectra, a carbonate species was excluded. It was tentatively explained by an oxygen-deficient Pd oxide with O atoms coordinated to even more Pd atoms. Based on the fact that most O species were inert to CO, it was concluded that only the O3-fold of Pd5O4 was active, under the (relatively) low-pressure conditions of this study (0.1–0.8 hPa).
Both the surface oxide and the PdO film can be created under UHV by exposing a Pd(111) surface to an O beam.143 This stability makes it feasible to study their interaction with CO using traditional surface-science techniques, for example, titration experiments. Isothermal measurements with CO at 7 × 10−9 hPa showed that the initial reactivity of PdO is about 7 times higher than the 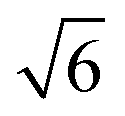 oxide at 450 K, being similar in reactivity to O(ads). Upon continued CO exposure, the oxides depleted, which led to increased reactivity for both structures. The reactivity increase for the
oxide at 450 K, being similar in reactivity to O(ads). Upon continued CO exposure, the oxides depleted, which led to increased reactivity for both structures. The reactivity increase for the 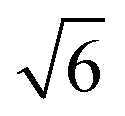 oxide was exponential and more strongly time dependent. This can be explained by a reaction that is being restricted to the interface between CO(ads) islands on the metallic surface and patches of surface oxide. The PdO layer had a linear, modest increase in reactivity, thus indicating that the oxide/CO(ads) boundaries were active in conjunction with an intrinsically active oxide. In addition, IRRA spectra were obtained while pausing the isothermal titration experiments by cooling the sample to 95 K and evacuating the chamber after fixed time intervals. These showed the appearance of a feature at 2090 cm−1, which was assigned to CO adsorbed on top of Pd3-fold next to an Ov. The increased adsorption strength of this CO(ads) is expected to result in a higher CO coverage with an increasing number of vacancies and can explain the observed increase in reactivity of the oxides.
oxide was exponential and more strongly time dependent. This can be explained by a reaction that is being restricted to the interface between CO(ads) islands on the metallic surface and patches of surface oxide. The PdO layer had a linear, modest increase in reactivity, thus indicating that the oxide/CO(ads) boundaries were active in conjunction with an intrinsically active oxide. In addition, IRRA spectra were obtained while pausing the isothermal titration experiments by cooling the sample to 95 K and evacuating the chamber after fixed time intervals. These showed the appearance of a feature at 2090 cm−1, which was assigned to CO adsorbed on top of Pd3-fold next to an Ov. The increased adsorption strength of this CO(ads) is expected to result in a higher CO coverage with an increasing number of vacancies and can explain the observed increase in reactivity of the oxides.
The high reactivity of PdO(101)/Pd(111) was confirmed with temperature-programmed reaction spectroscopy (TPRS); after CO(ads) saturation at 95 K, 71% of CO reacted, forming CO2 with remaining CO desorbing.135 Two main reaction pathways were observed, at 330 and 520 K. Furthermore, IRRA spectra recorded after increasingly higher annealing temperature indicated that the low-temperature pathway involved a reaction between CO(ads) and the pristine PdO(101) surface, resulting in Ov. The remaining CO shifted in adsorption site to atop sites on Pd3-fold in the Ov vicinity. The shift in CO adsorption site led to a broad peak at 2085 cm−1 and this CO reacted between 500 and 550 K.
Interestingly, above 400 K, the oxide was restored by O diffusion from the bulk of the oxide, filling some of the Ov at the surface. After full depletion of the oxide, CO was bonded to several sites not present on the (111) surface, such as small clusters of undercoordinated Pd and (100) or (110) sites. This indicates that the reduction of the oxides produced a rougher, ill-defined surface.
The phase diagram for the Pd(111) surface termination,144 see Fig. 22, shows a strong similarity to that for the Pd(100) surface, see Fig. 20.130 Both show that mixed co-adsorption phases of O(ads) and CO(ads) are not stable, because the CO–O repulsion is larger than that of O–O and CO–CO. Furthermore, the region in the phase diagrams of O(ads) is very narrow, mostly limited in pO2. In particular for the Pd(111) surface, the boundary between CO(ads) and O(ads) is very short, thus making possible coexisting islands of O(ads) and CO(ads) unlikely for a wide range of conditions. For both surfaces, the technological conditions (p = 101 kPa and T = 300–600 K) cover a window (black bar, Fig. 20 and white box, Fig. 22) including a phase transition between CO-covered, reduced Pd, and a surface oxide. For Pd(111), this is the Pd5O4 surface oxide and compared to the 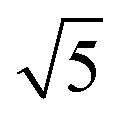 oxide on Pd(100), this surface oxide seems to be more susceptible to reduction under reaction conditions. Since the window of reaction conditions is far away from the region where O(ads) is stable, CO(ads) probably poisons the metallic phase. This would mean that the active structures for CO oxidation are Pd5O4 and CO(ads) on metallic Pd(111), possibly coexisting.
oxide on Pd(100), this surface oxide seems to be more susceptible to reduction under reaction conditions. Since the window of reaction conditions is far away from the region where O(ads) is stable, CO(ads) probably poisons the metallic phase. This would mean that the active structures for CO oxidation are Pd5O4 and CO(ads) on metallic Pd(111), possibly coexisting.
 | ||
| Fig. 22 Phase diagram of the Pd(111) surface in equilibrium with CO and O2 derived from DFT calculations with thermodynamic corrections. Typical reaction conditions are indicated by the white box. Reproduced from ref. 144 with permission from the American Chemical Society, copyright 2014. | ||
On the Pd5O4 surface, CO binds almost 100 kJ mol−1 or 1 eV more weakly than on the Pd(111) surface.144 CO binds most strongly to the bridge between two Pd2-fold atoms. Weak binding with O2 is also predicted, making it unlikely that O2 binds dissociatively on the oxide and that CO oxidation occurs via a LH mechanism of two adsorbed species. The reaction of CO(ads) with an oxidic O atom has a barrier of 87 kJ mol−1 or 0.9 eV, which is higher than that for desorption, leading to very small coverages and low reactivity at higher temperatures. However, the direct reaction between CO(g) and the oxide via an ER mechanism has a very low barrier (5 kJ mol−1 or 0.05 eV) and a microkinetic model taking into account CO coverage and differences in pre-exponential factors indicates that CO(g) is 43 times more reactive than CO(ads), although this was very sensitive to the chosen GGA functional.144
4.3 Pd(110), CO adsorption and oxidation
The Pd(110) surface has received far less interest than the Pd(100) and Pd(111) surfaces. Berlowitz at al. performed a detailed and rather extensive kinetic analysis for the Pd(110) surface in the pressure range of 0.1–800 hPa.48 The reaction orders in O2 and CO were 1.0 ± 0.1 and −1.0 ± 0.1, respectively, down to χ = 0.08 at 525 K. At lower χ, under more oxidizing conditions, the order became positive for CO, while becoming negative for O2.Deactivation was studied under O2-rich conditions (χ = 0.09) at different temperatures. The results were difficult to interpret. At T > 475 K, no deactivation was observed, but below, the activity decreased by a factor of 3–5 on the order of minutes. The same amount of deactivation was reached by pre-oxidizing the surface (1 hPa, 1100 K or 800 hPa, 800 K) and similar temperature-programmed desorption profiles were obtained after the reaction. Based on the similarity between the in situ deactivated samples and those deactivated by pre-oxidation, it was conjectured that an oxide formed that blocked active sites for the reaction. It could be that the deactivation was not observed above >475 K, because above this temperature the oxide became more active than the reduced, CO-covered surface.
In accordance with the other Pd and Pt surfaces, the reactivity was limited at high χ.118 At 525 K, in a batch reactor, χ had to change from the initial value of 0.2 to 0.09, before the transition to the reactive, MTL regime occurred and a transient TOF of 4000 molecules per (site s) was observed, after which it stabilized at 1400 molecules per (site s). The conditions during this transition were very close to the critical points in the order changes.48Ex situ XPS measurements probing the coverage of the active site unveiled that roughly 1 ML O was present.118
The depletion of CO in the vicinity of the surface, leading to the MTL regime, was supported by a combined PLIF/QMS study. It probed the gas phase under reaction conditions (flow reactor, χ = 1 and p = 53 hPa) both close and farther away from the surface.148 An ∼80% and 20–30% decrease in CO pressure was measured, respectively, with the latter in good agreement with the QMS data. The transition temperature was shifted to higher temperature, 638 K, due to the higher pCO. Interestingly, the CO2 PLIF measurement showed also a transient maximum before decreasing to the MTL plateau, comparable to that observed by Goodman and coworkers.118 However, the QMS data revealed no such transient.
A NAP XPS study probed the Pd(110) surface in either pure CO or pure O2 and under reaction conditions.149 The adsorption of CO at RT was studied between 10−7 and 0.1 hPa, but, surprisingly no pressure dependence was found.
Oxygen chemisorption structures were found to be stable up to 0.3 hPa at RT. The observed coverages agreed with the structures (from low to high O2 exposure) having unit cells of c(2 × 4), (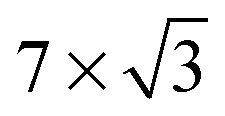 ), and (
), and (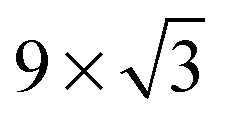 ) (Fig. 23). After heating to 573 K, a new Pd species was found at 336.3 eV attributed to PdO, which seemed to coexist with chemisorbed O.
) (Fig. 23). After heating to 573 K, a new Pd species was found at 336.3 eV attributed to PdO, which seemed to coexist with chemisorbed O.
 | ||
| Fig. 23 Structural models of the two O(ads) chemisorption structures on the Pd(110) surface. Reproduced from ref. 150 with permission from Elsevier, copyright 2008. | ||
In agreement with ref. 118, the CO oxidation rate (χ = 0.1 and 0.29 hPa) was small at low temperature. Between 438 and 473 K, it steeply rose, after which it gradually decreased upon increased heating to 593 K. The transition temperature was rather low compared to that in ref. 118, which could be caused by the lower partial pressure of CO. The decrease in reactivity with temperature above the transition can be explained by a negative apparent activation energy resulting from a lower barrier for the reaction compared to the desorption of (probably) CO. Alternatively, a decreasing sticking coefficient with temperature149 leads to a lower rate of adsorption compared to that of desorption. This will decrease the adsorbate coverage and thus the reactivity.
Based on the XP spectra, Toyoshima et al. propose that under reaction conditions O(ads) is the active species on the surface.149 They indicate that a full layer of the chemisorption structure on Pd(110) had a coverage of almost 0.9 ML, which is comparable to the coverages of the 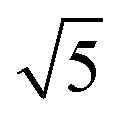 and
and 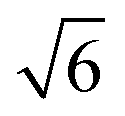 surface oxides on the Pd(100) and Pd(111) surfaces, respectively. The Pd(110) surface is able to adsorb as much O, without forming an oxide. The largest difference is that these chemisorption structures only have O coordinated to two Pd atoms and vice versa.150 Essentially, they consist of close-packed Pd rows, along which the O adsorbs in a zigzag manner, as illustrated in Fig. 23. Due to stress, these rows are slightly rotated with respect to the [10] direction. Every 7th or 9th Pd atom shifts somewhat to keep the close-packed rows in registry with the underlying substrate, resulting in (
surface oxides on the Pd(100) and Pd(111) surfaces, respectively. The Pd(110) surface is able to adsorb as much O, without forming an oxide. The largest difference is that these chemisorption structures only have O coordinated to two Pd atoms and vice versa.150 Essentially, they consist of close-packed Pd rows, along which the O adsorbs in a zigzag manner, as illustrated in Fig. 23. Due to stress, these rows are slightly rotated with respect to the [10] direction. Every 7th or 9th Pd atom shifts somewhat to keep the close-packed rows in registry with the underlying substrate, resulting in (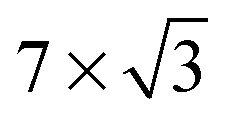 ) and (
) and (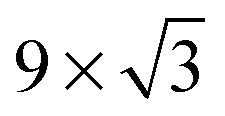 ) unit cells, respectively. DFT calculations predict these chemisorption structures among the lowest in energy at higher O2 chemical potential.151
) unit cells, respectively. DFT calculations predict these chemisorption structures among the lowest in energy at higher O2 chemical potential.151
4.4 Vicinal and polycrystalline Pd surfaces, CO oxidation
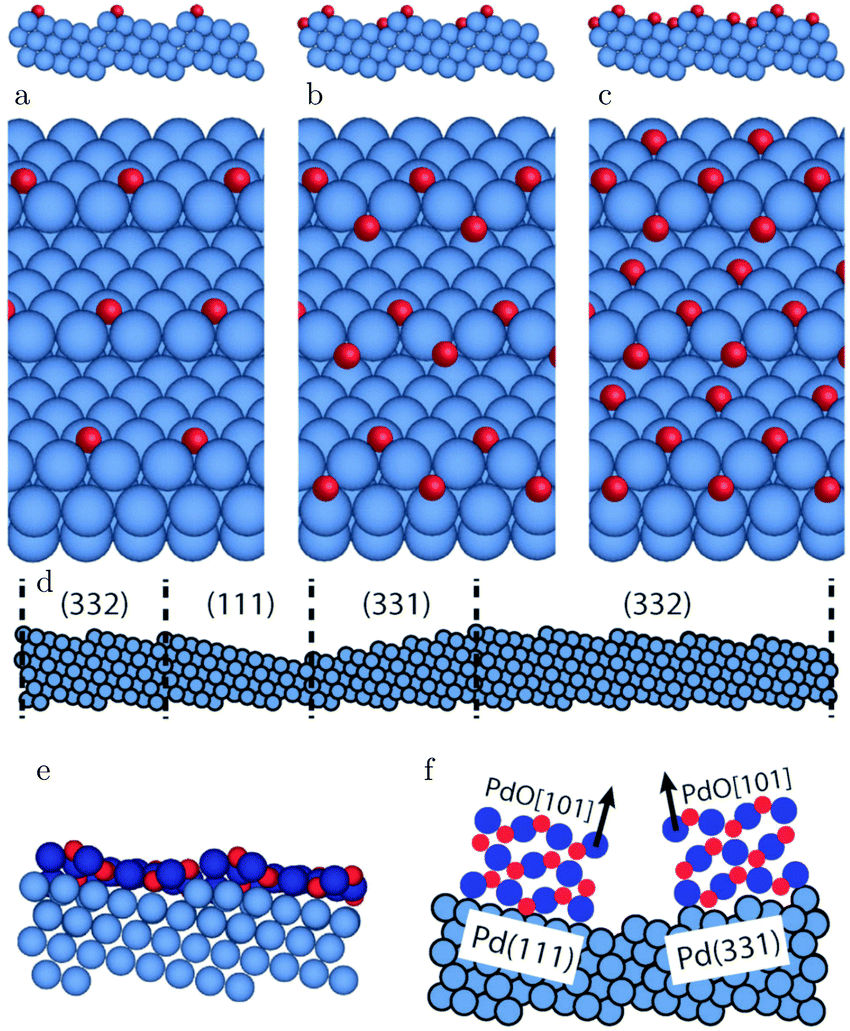 | ||
| Fig. 24 Structural models of the Pd(553) surface for increasingly higher O coverage, derived from operando SXRD measurements: (a)–(c) O(ads) chemisorption onto steps and terraces; (d) faceting under O-rich reaction conditions; (e) PdO(101)-like surface oxide covering the (332) facets; (f) epitaxial PdO on the (111) and (331) facets. Reproduced from ref. 153 with permission from the Royal Society of Chemistry, copyright 2016. | ||
Several other facets were observed, some of which were needed to keep the total inclination parallel to the (553) surface. These were (110), basically bunched (111) steps, and (221), 3(111) × (111). Some (775), 6(111) × (111), facets were also observed. On the (221) and (775) terraces, a surface oxide similar, but distorted, to the one on the (332) facet was observed. Furthermore, few larger (111) facets covered with the Pd5O4 surface oxide were found. Since a mixture of PdO(101)/Pd(332) and Pd5O4/Pd(111) was observed, it is possible that the stability of the two oxides is very similar. Alternatively, the formation of large (111) facets could be kinetically limited at 623 K.
The formation of bulk PdO was recognized between 1 and 100 hPa and simultaneously the (332) and (553) facets disappeared. The PdO diffraction spots showed a 16° angle with the Pd(553) surface, very close to the 15.9° angle between the PdO(201) surface and the c axis of the PdO unit cell. Although the PdO(201) plane was predicted to be parallel to the Pd(553) surface, the actual surface termination of PdO could be different, i.e., it could facet into, e.g., PdO(100) and PdO(101) surfaces. Since the terraces of the Pd(553) surface are 0.1 nm, too small to house a PdO(201) unit cell (1.1 nm), the formation of a rough, disordered oxide was proposed.
The behavior of Pd(553) under CO oxidation conditions was studied with high-energy SXRD at 600 K.153 For χ = 4 at 10 hPa, no changes with respect to the surface under UHV was detected, indicating that this surface is stable in a CO-rich atmosphere. Furthermore, the CO2 production was small. After changing the atmosphere to (small) excess O2, χ = 1.9, the surface faceted. This faceting was roughly similar to that observed in pure O2,152 although the (110) and (221) facets were not detected. Instead (331), 2(111) × (111), facets were found to compensate for the (111) facets and the 5-atom wide terraces of the (332) facets (Fig. 24d). Simultaneously, the CO2 production was MTL. Based on the agreement with the work of Westerström et al.,152 it was proposed that a surface oxide was present (Fig. 24e). After increasing the O2 flow to reach χ = 1, two orientations of epitaxial bulk PdO were uncovered, namely with their (101) planes parallel to the (331) and (111) facets (Fig. 24f). The CO2 production remained limited by the diffusion of CO to the surface and constant, because the pCO did not change. Faceting under O2-rich conditions is consistent with HP STM observations.54
The Pd(1 1 17), 8(100) × (111), surface was neither stable in pure CO nor in an O2-rich mixture as imaged using STM at 50 and 125 kPa and 413–420 K.54 Under the former conditions, the terraces and steps doubled in width/height. Under the latter conditions, both were increased by a factor of 4–5 compared to the clean Pd(1 1 17) surface under UHV. In an increasingly CO-poorer mixture, the reactivity increased stepwise by a factor of 1.6 at a nominal value of χ ≈ 0.03. After this switch, the surface became rough over time and covered with clusters of 4 ± 2 nm. Under certain conditions, spontaneous reaction oscillations were observed on Pd(1 1 17), similarly to those on the Pd(100) surface.
The Pd(112), 2(111) × (100), surface faceted already under UHV at 673 K.154 The faceted surface consisted of a mixture of (113), (111) × (100), and (335), 3(111) × (100), as probed using SXRD. Upon exposing to 6 × 10−7 hPa O2, the unordered mixture of facets transformed into one which had the two facets alternating. A corresponding doubling of the periodicity perpendicular to the steps was observed. At higher pressures, the mixture first segregated into large (113) and (335) facets, marked by tilting of the CTRs. Second, at 1 × 10−4 hPa, the (335) facets transformed into (111) facets and were covered with a structure that had some resemblance to a Pd8O8 surface oxide. This surface oxide is structurally very closely related to PdO(101) and was observed to form under these conditions on an extended Pd(111) surface, albeit metastable as predicted by DFT.155 Finally, formation of epitaxial, bulk PdO with a thickness of 5.6 nm was observed for pressures above 0.1 hPa. The measured surface orientations were (100) [or (101), which has a similar diffraction pattern] and (001). Polycrystalline PdO that lost epitaxiality with the surface was not seen under these conditions. Given the similarities in the oxidation of the Pd(112) and Pd(553) surfaces, similar behavior under CO oxidation can be anticipated.
Recently, Lundgren and coworkers directly and elegantly compared the effect of the two different steps found on vicinal (111) surfaces (of fcc crystals) on the CO oxidation rate (χ = 1 and p = 12 hPa).156 To this end, a curved single crystal was studied with PLIF, yielding the local gas-phase composition, see Fig. 25. The curvature was ±15° around the (111) surface. In this way, it was found that the transition from an inactive, CO-poisoned surface to an active one was 6 K (578 K) lower for B-type, or (111), steps than for A-type, or (100), steps. It was also lower than that for the extended (111) surface. As this transition is determined by the CO desorption rate, it was proposed that the CO adsorption energy was lower for the stepped surfaces than for the (111) surface. This counterintuitive assumption was supported by DFT calculations that focused on the surface stress. It was found that steps provide a facile mechanism to reduce the stress naturally present in the (111) surface. This makes narrow (111) terraces more inert to CO adsorption than extended (111) surfaces. Moreover, it was found that the CO adsorption energy exhibited a stronger coverage dependence for the B-type steps, destabilizing CO at higher coverage, thus explaining the lowest activation temperature. The appealing aspect of this explanation is that, in this case, steps primarily act to alter the activity of the terraces, instead of being active sites themselves. However, given the ease with which vicinal Pd surfaces undergo faceting, the complete explanation could be more complex.
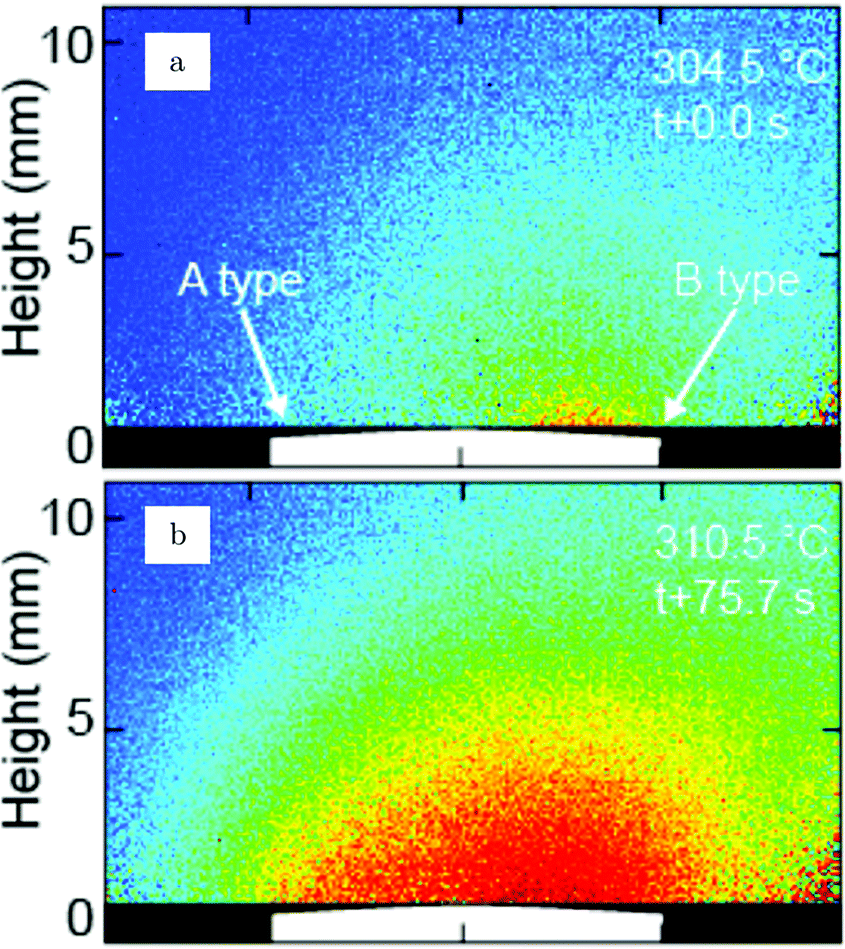 | ||
| Fig. 25 Spatially resolved CO2 PLIF measurements for a curved single crystal of Pd. Central region of the sample is the (111) surface. To the left are the A-type or (100) steps, as in the (112) surface. To the right are the B-type or (111) steps, as in the (553) surface. The first sign of reactivity was observed for the B-type steps (a). At slightly higher temperatures, the A-type steps became active (b). Reproduced from ref. 156 with permission from the American Chemical Society, copyright 2017. | ||
The possibility of an active surface covered with O(ads) was also concluded based on IRRAS measurements on a Pd foil. For the foil, similar results were obtained compared to single crystals. The foil (0.1 mm thick) was studied under reaction conditions (χ = 0.5, p = 8–17 hPa, and T = 500 K) and, initially, CO(ads) was observed and CO2 formation was low. After substantial CO depletion, the reactivity increased and CO(ads) became undetectable, while the vibrational signature of Pd oxide was also absent.119
4.5 Conspectus on Pd
Compared to Pt, the growth and stability of Pd oxides have been characterized more thoroughly. For the Pd(100) surface, the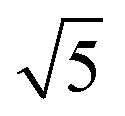 surface oxide, epitaxial PdO, and polycrystalline PdO have been observed under reaction conditions. Similar oxides form on the Pd(111) surface, namely, a
surface oxide, epitaxial PdO, and polycrystalline PdO have been observed under reaction conditions. Similar oxides form on the Pd(111) surface, namely, a 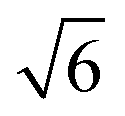 surface oxide and an epitaxial PdO. NAP XPS showed the growth under reaction conditions. An SXRD study on the CO oxidation on Pd(111) complementing the NAP XPS would be desirable. No surface oxide forms on Pd(110), instead an O(ads) structure with comparable O coverage develops. Vicinal Pd surfaces are not very stable and facet under reaction conditions.
surface oxide and an epitaxial PdO. NAP XPS showed the growth under reaction conditions. An SXRD study on the CO oxidation on Pd(111) complementing the NAP XPS would be desirable. No surface oxide forms on Pd(110), instead an O(ads) structure with comparable O coverage develops. Vicinal Pd surfaces are not very stable and facet under reaction conditions.
Simultaneously with the formation of Pd oxides, the reactivity increases significantly. The oxidation of the Pd surfaces can be a result of the active catalyst depleting the gas phase of CO, thus making the local environment highly oxidizing. The surface oxidation can also be the origin of the higher reactivity by providing more reactive O atoms. Titration experiments showed that Pd (surface) oxides are active for CO oxidation, but limited in the CO adsorption strength. Under reaction conditions, defects in the oxide, either intrinsic or created by the MvK reaction mechanism, can bind CO much more strongly and may explain the high reactivity of the oxides. To further answer this question, higher time resolution in operando techniques will be needed in combination with local, rapid gas phase-measurements. Elaborating on this, it remains unclear how the reactivities of the different oxides compare. This is partially caused by the obscuring effect of the MTL regime.
5 Summary and outlook
Summary
Studies on CO oxidation under reaction conditions on Pt and Pd model surfaces were reviewed. To a lesser extent CO adsorption on and oxidation of these metals were also included. Commonly used techniques included SXRD, XPS, IRRAS, STM, and/or DFT.Adsorption of CO is characterized most extensively on Pt(111) and leads to both incommensurate and commensurate overlayers showing moiré patterns due to a mismatch with the Pt lattice. The coverage saturates at 0.7 ML. As similar structures can be prepared by cooling the sample and exposing it to low pressures of CO under UHV, a pressure gap is not present. Under CO oxidation conditions, the surface can form a (2 × 2) reconstruction as well as a PtO2 layer, both present during the high-activity phase of the model catalyst, i.e., in a large excess of O2. The latter was also observed on the Pt(110) surface, developing after the surface had formed a (1 × 2) lifted-row surface oxide. Based on these experiments, it was suggested that the high activity is related to the formation of (surface) oxides. The highest activity of these oxides is observed just below the critical pCO at which they decompose. Under these conditions, they are similarly active as the reduced metallic surface covered with an optimal CO/O ratio, following LH kinetics. For Pt(100), similar kinetics were observed together with roughening of the surface. For Pt(100), the proposed oxide was not nearly as reactive as the most active LH state. However, due to the nobility of Pt, the structural and chemical properties of the (surface) oxides remain largely uncharacterized.
The surface oxides of Pd are more stable than those of Pt. They form at lower pressures and can be studied under UHV. It is well established that a 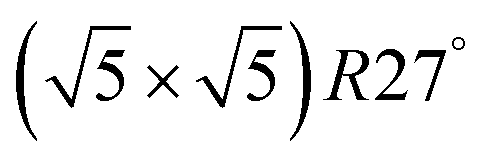 surface oxide forms on Pd(100), which is a modified version of a single PdO(101) layer. On Pd(111), the
surface oxide forms on Pd(100), which is a modified version of a single PdO(101) layer. On Pd(111), the 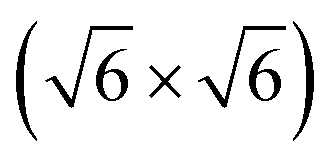 structure has no analogy with any of the surfaces of bulk PdO. At higher O2 pressures, Pd oxidizes to bulk PdO, both epitaxial and polycrystalline. The precise orientation and registry of these PdO layers remain unclear. All these oxides were reported in relation to higher reactivity phases. For the conditions under which polycrystalline PdO/Pd(100) was observed with SXRD, CO adsorption on oxidized Pd was observed with IRRAS.
structure has no analogy with any of the surfaces of bulk PdO. At higher O2 pressures, Pd oxidizes to bulk PdO, both epitaxial and polycrystalline. The precise orientation and registry of these PdO layers remain unclear. All these oxides were reported in relation to higher reactivity phases. For the conditions under which polycrystalline PdO/Pd(100) was observed with SXRD, CO adsorption on oxidized Pd was observed with IRRAS.
In several measurements, most notably in those by Goodman and coworkers, an even higher reactivity phase was reported. This “hyperactive” phase can be explained as a transient response of the gas phase to a rapid switch in activity, after which the TOF decreased to the value corresponding to the MTL regime. An alternative interpretation is that during this phase the most active phase, O(ads)/Pd(100), is present, which deactivates somewhat to the lesser reactive oxide.
In most of these studies, a discontinuous increase in reactivity is witnessed. After this switch, the reaction rate is very high, usually exceeding the gas-phase diffusion and a MTL regime is obtained. This makes it impossible to probe the intrinsic rate of the active surface. The high reactivity leads to very short lifetimes of CO(ads) and, hence, very low CO(ads) coverages. This coverage can easily be lower than the detection limits of techniques that can probe CO, such as IR spectroscopy or XPS. Furthermore, XPS is the only technique available at present that can measure O(ads) under reaction conditions, albeit at much lower pressures. However, for Pt, possible surface oxides are insufficiently described and it is not very clear if the core-level shifts of these oxides differ much from those of O(ads). This makes the distinction between O(ads) and surface oxide difficult. More important is to realize that all these structures are active enough to maintain the conditions for the MTL regime. In this case, it could be more instructive to focus on the stability of a particular structure under reaction conditions.
Outlook
Since the various techniques available for high-pressure surface science tend to be complementary, no single one of them can be used to tell the complete story. Currently, the tools of the trade are still being improved. Examples are the use of high-energy X-rays and larger detectors in SXRD, the development of the ReactorAFM to be able to study the structure and reactivity of bulk oxides too insulating to study with STM,8 or the integration of X-ray scattering with SPM.15 However, there is definitively a need for further instrument development, some of which will be very challenging, such as the full closure of the pressure gap in XPS. Some development should be more straightforward and is directed at combining several techniques to obtain different types of information on the same sample under identical conditions. A very useful combination would be the integration of vibration spectroscopy with SPM and/or with SXRD.Now that many studies have been published using the low-Miller-index planes and some using vicinal surfaces, the trend is to move to more complex model catalysts. Examples are (1) bimetallics, either in the form of a single-crystalline bulk alloy or obtained by depositing small amounts onto a pure single crystal; (2) inverse catalysts, in which the supporting oxide is deposited on the metal; and (3) well-defined, mono-dispersed nanoparticles. These studies are crucial to unveil whether the phenomena observed on single crystals extend to materials resembling more technical catalysts.
Finally, as these techniques gradually mature, the next logical step would be to extend the chemistry. Even in automotive catalysis, which is the traditional motivation to study CO oxidation, several other reactions occur. For example, NO reduction by CO or H274,75,157,158 and NO oxidation when operated in excess O2159–163 are very interesting. In particular for the latter reaction, in situ studies are needed. Other catalytic reactions that are extremely promising to study in the context of fuel-related catalysis are, e.g., hydrodesulfurization for the treatment of fuels using MoS2-based catalysts,164 or Fischer–Tropsch synthesis for the production of clean fuels from CO–H2 mixtures using Fe or Co catalysts.165–167
Acknowledgements
This work is supported by NanoNextNL, a micro and nanotechnology consortium of the Government of the Netherlands and 130 partners.References
- J. Lin, X. Wang and T. Zhang, Chin. J. Catal., 2016, 37, 1805–1813 CrossRef CAS.
- M. Chen, Y. Zheng and H. Wan, Top. Catal., 2013, 56, 1299–1313 CrossRef CAS.
- R. Imbihl, Prog. Surf. Sci., 1993, 44, 185–343 CrossRef CAS.
- J. A. Rodriguez and D. W. Goodman, Surf. Sci. Rep., 1991, 14, 1–107 CrossRef CAS.
- T. Engel and G. Ertl, Adv. Catal., 1979, 28, 1–78 CAS.
- J. W. M. Frenken and I. M. N. Groot, Operando Research in Heterogeneous Catalysis, Springer, 2017, pp. 1–30 Search PubMed.
- C. T. Herbschleb, et al. , Rev. Sci. Instrum., 2014, 85, 083703 CrossRef CAS PubMed.
- S. B. Roobol, M. E. Cañas-Ventura, M. Bergman, M. A. van Spronsen, W. G. Onderwaater, P. C. van der Tuijn, R. Koehler, A. Ofitserov, G. J. C. van Baarle and J. W. M. Frenken, Rev. Sci. Instrum., 2015, 86, 033706 CrossRef CAS PubMed.
- D. F. Ogletree, H. Bluhm, G. Lebedev, C. S. Fadley, Z. Hussain and M. Salmeron, Rev. Sci. Instrum., 2002, 73, 3872–3877 CrossRef CAS.
- H. Bluhm, et al. , J. Electron Spectrosc. Relat. Phenom., 2006, 150, 86–104 CrossRef CAS.
- G. Rupprechter, T. Dellwig, H. Unterhalt and H.-J. Freund, Top. Catal., 2001, 15, 19–26 CrossRef CAS.
- G. Rupprechter and C. Weilach, Nano Today, 2007, 2, 20–29 CrossRef.
- G. Rupprechter and C. Weilach, J. Phys.: Condens. Matter, 2008, 20, 184019 CrossRef.
- R. van Rijn, M. D. Ackermann, O. Balmes, T. Dufrane, A. Geluk, H. Gonzalez, H. Isern, E. de Kuyper, L. Petit, V. A. Sole, D. Wermeille, R. Felici and J. W. M. Frenken, Rev. Sci. Instrum., 2010, 81, 014101 CrossRef CAS PubMed.
- W. G. Onderwaater, P. C. van der Tuijn, R. V. Mom, M. A. van Spronsen, S. B. Roobol, A. Saedi, J. Drnec, H. Isern, F. Carla, T. Dufrane, R. Koehler, B. Crama, I. M. N. Groot, R. Felici and J. W. M. Frenken, Rev. Sci. Instrum., 2016, 87, 113705 CrossRef PubMed.
- K. Reuter, Catal. Lett., 2016, 146, 541–563 CrossRef CAS.
- M. Maestri and A. Cuoci, Chem. Eng. Sci., 2013, 96, 106–117 CrossRef CAS.
- S. Matera, S. Blomberg, M. J. Hoffmann, J. Zetterberg, J. Gustafson, E. Lundgren and K. Reuter, ACS Catal., 2015, 5, 4514–4518 CrossRef CAS.
- Y. S. Kim, A. Bostwick, E. Rotenberg, P. N. Ross, S. C. Hong and B. S. Mun, J. Chem. Phys., 2010, 133, 034501 CrossRef PubMed.
- T. Zambelli, J. V. Barth, J. Wintterlin and G. Ertl, Nature, 1997, 390, 495–497 CrossRef CAS.
- D. J. Burnett, A. T. Capitano, A. M. Gabelnick, A. L. Marsh, D. A. Fischer and J. L. Gland, Surf. Sci., 2004, 564, 29–37 CrossRef CAS.
- P. Mars and D. W. van Krevelen, Chem. Eng. Sci., 1954, 3, 41–59 CrossRef CAS.
- B. V. L'vov and A. K. Galwey, J. Therm. Anal. Calorim., 2013, 111, 145–154 CrossRef.
- I. K. Robinson and D. J. Tweet, Rep. Prog. Phys., 1992, 55, 599 CrossRef CAS.
- P. Bernard, K. Peters, J. Alvarez and S. Ferrer, Rev. Sci. Instrum., 1999, 70, 1478–1480 CrossRef CAS.
- M.-C. Saint-Lager, A. Bailly, P. Dolle, R. Baudoing-Savois, P. Taunier, S. Garaudée, S. Cuccaro, S. Douillet, O. Geaymond, G. Perroux, O. Tissot, J.-S. Micha, O. Ulrich and F. Rieutord, Rev. Sci. Instrum., 2007, 78, 083902 CrossRef PubMed.
- M. D. Ackermann, T. M. Pedersen, B. L. M. Hendriksen, O. Robach, S. C. Bobaru, I. Popa, C. Quiros, H. Kim, B. Hammer, S. Ferrer and J. W. M. Frenken, Phys. Rev. Lett., 2005, 95, 255505 CrossRef CAS PubMed.
- Z. Zhao, T. Diemant, T. Häring, H. Rauscher and R. J. Behm, Rev. Sci. Instrum., 2005, 76, 123903 CrossRef.
- M. Yang, D. C. Tang and G. A. Somorjai, Rev. Sci. Instrum., 2003, 74, 4554–4557 CrossRef CAS.
- S. Liu, A.-a. Liu, R. Zhang and Z. Ren, Rev. Sci. Instrum., 2016, 87, 044101 CrossRef PubMed.
- G. Haas, T. D. Pletcher, G. Bonilla, T. A. Jachimowski, H. H. Rotermund and J. Lauterbach, J. Vac. Sci. Technol., A, 1998, 16, 1117–1121 CAS.
- J. Dicke, P. Erichsen, J. Wolff and H. H. Rotermund, Surf. Sci., 2000, 462, 90–102 CrossRef CAS.
- J. Dicke, H. H. Rotermund and J. Lauterbach, J. Opt. Soc. Am. A, 2000, 17, 135–141 CrossRef CAS PubMed.
- C. Punckt, F. S. Merkt and H. H. Rotermund, New J. Phys., 2007, 9, 213 CrossRef.
- J. Pantförder, S. Pöllmann, J. F. Zhu, D. Borgmann, R. Denecke and H.-P. Steinrück, Rev. Sci. Instrum., 2005, 76, 014102 CrossRef.
- D. F. Ogletree, H. Bluhm, E. D. Hebenstreit and M. Salmeron, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 601, 151–160 CrossRef.
- F. Tao, Chem. Commun., 2012, 48, 3812–3814 RSC.
- J. Schnadt, J. Knudsen, J. N. Andersen, H. Siegbahn, A. Pietzsch, F. Hennies, N. Johansson, N. Mårtensson, G. Öhrwall, S. Bahr, S. Mähl and O. Schaff, J. Synchrotron Radiat., 2012, 19, 701–704 CrossRef CAS PubMed.
- B. J. McIntyre, M. B. Salmeron and G. A. Somorjai, Catal. Lett., 1992, 14, 263–269 CrossRef.
- B. L. Weeks, C. Durkan, H. Kuramochi, M. E. Welland and T. Rayment, Rev. Sci. Instrum., 2000, 71, 3777–3781 CrossRef CAS.
- E. Lægsgaard, L. Österlund, P. Thostrup, P. B. Rasmussen, I. Stensgaard and F. Besenbacher, Rev. Sci. Instrum., 2001, 72, 3537–3542 CrossRef.
- M. Rößler, P. Geng and J. Wintterlin, Rev. Sci. Instrum., 2005, 76, 023705 CrossRef.
- F. Tao, D. Tang, M. Salmeron and G. A. Somorjai, Rev. Sci. Instrum., 2008, 79, 084101 CrossRef PubMed.
- F. Tao, L. Nguyen and S. Zhang, Rev. Sci. Instrum., 2013, 84, 034101 CrossRef PubMed.
- K. Reuter, Ab Initio Thermodynamics and First-Principles Microkinetics for Surface Catalysis, Operando Research in Heterogeneous Catalysis, Springer, 2017, pp. 151–188 Search PubMed.
- K. Reuter, First-Principles Kinetic Monte Carlo Simulations for Heterogeneous Catalysis: Concepts, Status and Frontiers, Modeling Heterogeneous Catalytic Reactions: From the Molecular Process to the Technical System, Wiley-VCH, 2009 Search PubMed.
- S. Blomberg, J. Zhou, J. Gustafson, J. Zetterberg and E. Lundgren, J. Phys.: Condens. Matter, 2016, 28, 453002 CrossRef PubMed.
- P. J. Berlowitz, C. H. F. Peden and D. W. Goodman, J. Phys. Chem., 1988, 92, 5213–5221 CrossRef CAS.
- G. W. Graham, D. König, B. D. Poindexter, J. T. Remillard and W. H. Weber, Top. Catal., 1999, 8, 35–43 CrossRef CAS.
- B. Lang, R. W. Joyner and G. A. Somorjai, Surf. Sci., 1972, 30, 440–453 CrossRef CAS.
- J. A. Jensen, K. B. Rider, M. Salmeron and G. A. Somorjai, Phys. Rev. Lett., 1998, 80, 1228 CrossRef CAS.
- E. K. Vestergaard, P. Thostrup, T. An, E. Lægsgaard, I. Stensgaard, B. Hammer and F. Besenbacher, Phys. Rev. Lett., 2002, 88, 259601 CrossRef PubMed.
- S. R. Longwitz, J. Schnadt, E. K. Vestergaard, R. T. Vang, E. Lægsgaard, I. Stensgaard, H. Brune and F. Besenbacher, J. Phys. Chem. B, 2004, 108, 14497–14502 CrossRef CAS.
- Ş. C. Bobaru, PhD thesis, Leiden University, 2006.
- L. Nguyen, F. Cheng, S. Zhang and F. Tao, J. Phys. Chem. C, 2013, 117, 971–977 CAS.
- I. Villegas and M. J. Weaver, J. Chem. Phys., 1994, 101, 1648–1660 CrossRef CAS.
- X. Su, P. S. Cremer, Y. R. Shen and G. A. Somorjai, J. Am. Chem. Soc., 1997, 119, 3994–4000 CrossRef CAS.
- F. Tao, S. Dag, L.-W. Wang, Z. Liu, D. R. Butcher, H. Bluhm, M. Salmeron and G. A. Somorjai, Science, 2010, 327, 850–853 CrossRef CAS PubMed.
- J. Kim, M. C. Noh, W. H. Doh and J. Y. Park, J. Am. Chem. Soc., 2016, 138, 1110–1113 CrossRef CAS PubMed.
- X. Su, J. Jensen, M. X. Yang, M. B. Salmeron, Y. R. Shen and G. A. Somorjai, Faraday Discuss., 1996, 105, 263–274 RSC.
- G. Rupprechter, T. Dellwig, H. Unterhalt and H.-J. Freund, J. Phys. Chem. B, 2001, 105, 3797–3802 CrossRef CAS.
- A. S. Duke, R. P. Galhenage, S. A. Tenney, P. Sutter and D. A. Chen, J. Phys. Chem. C, 2015, 119, 381–391 CAS.
- S. Krick Calderón, M. Grabau, L. Óvári, B. Kress, H.-P. Steinrück and C. Papp, J. Chem. Phys., 2016, 144, 044706 CrossRef PubMed.
- A. Farkas, K. Zalewska-Wierzbicka, C. Bachmann, J. Goritzka, D. Langsdorf, O. Balmes, J. Janek and H. Over, J. Phys. Chem. C, 2013, 117, 9932–9942 CAS.
- S. M. McClure, M. Lundwall, Z. Zhou, F. Yang and D. W. Goodman, Catal. Lett., 2009, 133, 298–306 CrossRef CAS.
- R. C. Yeates, J. E. Turner, A. J. Gellman and G. A. Somorjai, Surf. Sci., 1985, 149, 175–190 CrossRef CAS.
- M. D. Ackermann, PhD thesis, Leiden University, 2007.
- T. M. Pedersen, W. X. Li and B. Hammer, Phys. Chem. Chem. Phys., 2006, 8, 1566–1574 RSC.
- W. X. Li and B. Hammer, Chem. Phys. Lett., 2005, 409, 1–7 CrossRef CAS.
- X.-Q. Gong, R. Raval and P. Hu, Phys. Rev. Lett., 2004, 93, 106104 CrossRef PubMed.
- D. Miller, H. Sanchez Casalongue, H. Bluhm, H. Ogasawara, A. Nilsson and S. Kaya, J. Am. Chem. Soc., 2014, 136, 6340–6347 CrossRef CAS PubMed.
- J. G. Wang, W. X. Li, M. Borg, J. Gustafson, A. Mikkelsen, T. M. Pedersen, E. Lundgren, J. Weissenrieder, J. Klikovits, M. Schmid, B. Hammer and J. N. Andersen, Phys. Rev. Lett., 2005, 95, 256102 CrossRef CAS PubMed.
- B. L. M. Hendriksen and J. W. M. Frenken, Phys. Rev. Lett., 2002, 89, 046101 CrossRef CAS PubMed.
- M. A. Van Spronsen, G. J. C. Van Baarle, C. T. Herbschleb, J. W. M. Frenken and I. M. N. Groot, Catal. Today, 2015, 244, 85–95 CrossRef CAS.
- M. A. van Spronsen, G. J. C. van Baarle, C. T. Herbschleb, J. W. M. Frenken and I. M. N. Groot, Catal. Today, 2015, 256, 384 CrossRef CAS.
- H. H. Rotermund, G. Haas, R. U. Franz, R. M. Tromp and G. Ertl, Science, 1995, 270, 608–610 CAS.
- H. H. Rotermund, Surf. Sci., 1997, 386, 10–23 CrossRef CAS.
- J. C. Campuzano, A. M. Lahee and G. Jennings, Surf. Sci., 1985, 152/153, 68–76 CrossRef.
- E. C. Sowa, M. A. Van Hove and D. L. Adams, Surf. Sci., 1988, 199, 174–182 CrossRef CAS.
- E. Vlieg, I. K. Robinson and K. Kern, Surf. Sci., 1990, 233, 248–254 CrossRef CAS.
- F. Gao, Y. Wang, Y. Cai and D. W. Goodman, J. Phys. Chem. C, 2009, 113, 174–181 CAS.
- M. A. van Spronsen, PhD thesis, Leiden University, 2016.
- R. van Rijn, O. Balmes, R. Felici, J. Gustafson, D. Wermeille, R. Westerström, E. Lundgren and J. W. M. Frenken, J. Phys. Chem. C, 2010, 114, 6875–6876 CAS.
- W. X. Li, L. Österlund, E. K. Vestergaard, R. T. Vang, J. Matthiesen, T. M. Pedersen, E. Lægsgaard, B. Hammer and F. Besenbacher, Phys. Rev. Lett., 2004, 93, 146104 CrossRef CAS PubMed.
- D. R. Butcher, M. E. Grass, Z. Zeng, F. Aksoy, H. Bluhm, W.-X. Li, B. S. Mun, G. A. Somorjai and Z. Liu, J. Am. Chem. Soc., 2011, 133, 20319–20325 CrossRef CAS PubMed.
- J.-Y. Chung, F. Aksoy, M. E. Grass, H. Kondoh, P. Ross Jr., Z. Liu and B. S. Mun, Surf. Sci., 2009, 603, L35–L38 CrossRef CAS.
- P. Heilmann, K. Heinz and K. Müller, Surf. Sci., 1979, 83, 487–497 CrossRef CAS.
- A. Borg, A.-M. Hilmen and E. Bergene, Surf. Sci., 1994, 306, 10–20 CrossRef CAS.
- R. Martin, P. Gardner and A. M. Bradshaw, Surf. Sci., 1995, 342, 69–84 CrossRef CAS.
- R. Imbihl, M. P. Cox, G. Ertl, H. Müller and W. Brenig, J. Chem. Phys., 1985, 83, 1578–1587 CrossRef CAS.
- R. Imbihl, M. P. Cox and G. Ertl, J. Chem. Phys., 1986, 84, 3519–3534 CrossRef CAS.
- T. P. Lele, T. D. Pletcher and J. Lauterbach, AIChE J., 2001, 47, 1418–1424 CrossRef CAS.
- N. McMillan, T. Lele, C. Snively and J. Lauterbach, Catal. Today, 2005, 105, 244–253 CrossRef CAS.
- J. Dicke, H. H. Rotermund and J. Lauterbach, Surf. Sci., 2000, 454/456, 352–357 CrossRef.
- K. McCrea, J. S. Parker, P. Chen and G. A. Somorjai, Surf. Sci., 2001, 494, 238–250 CrossRef CAS.
- C. Minot, Catal. Today, 2004, 89, 357–362 CrossRef CAS.
- O. Balmes, G. Prevot, X. Torrelles, E. Lundgren and S. Ferrer, ACS Catal., 2016, 6, 1285–1291 CrossRef CAS.
- G. Comsa, G. Mechtersheimer and B. Poelsema, Surf. Sci., 1982, 119, 159–171 CrossRef CAS.
- G. Comsa, G. Mechtersheimer and B. Poelsema, Surf. Sci., 1982, 119, 172–183 CrossRef CAS.
- O. Balmes, G. Prevot, X. Torrelles, E. Lundgren and S. Ferrer, J. Catal., 2014, 309, 33–37 CrossRef CAS.
- L. Piccolo, S. Nassreddine and F. Morfin, Catal. Today, 2012, 189, 42–48 CrossRef CAS.
- J. R. Michalka, A. P. Latham and J. D. Gezelter, J. Phys. Chem. C, 2016, 120, 18180–18190 CAS.
- Z. Zhu, C. Barroo, L. Lichtenstein, B. Eren, C. H. Wu, B. Mao, T. Visart de Bocarmé, Z. Liu, N. Kruse, M. Salmeron and G. A. Somorjai, J. Phys. Chem. Lett., 2014, 5, 2626–2631 CrossRef CAS PubMed.
- H. Härle, A. Lehnert, U. Metka, H.-R. Volpp, L. Willms and J. Wolfrum, Chem. Phys. Lett., 1998, 293, 26–32 CrossRef.
- H. Härle, A. Lehnert, U. Metka, H.-R. Volpp, L. Willms and J. Wolfrum, Appl. Phys. B: Lasers Opt., 1999, 68, 567–572 CrossRef.
- R. Kissel-Osterrieder, F. Behrendt, J. Warnatz, U. Metka, H.-R. Volpp and J. Wolfrum, Proc. Combust. Inst., 2000, 28, 1341–1348 CrossRef CAS.
- A. K. Galwey, P. Gray, J. F. Griffiths and S. M. Hasko, Nature, 1985, 313, 668–671 CrossRef CAS.
- V. Johánek, M. Václavů, I. Matolínová, I. Khalakhan, S. Haviar and V. Matolín, Appl. Surf. Sci., 2015, 345, 319–328 CrossRef.
- R. van Rijn, O. Balmes, A. Resta, D. Wermeille, R. Westerström, J. Gustafson, R. Felici, E. Lundgren and J. W. M. Frenken, Phys. Chem. Chem. Phys., 2011, 13, 13167–13171 RSC.
- J. Gustafson, M. Shipilin, C. Zhang, A. Stierle, U. Hejral, U. Ruett, O. Gutowski, P.-A. Carlsson, M. Skoglundh and E. Lundgren, Science, 2014, 343, 758–761 CrossRef CAS PubMed.
- M. Shipilin, U. Hejral, E. Lundgren, L. R. Merte, C. Zhang, A. Stierle, U. Ruett, O. Gutowski, M. Skoglundh, P.-A. Carlsson and J. Gustafson, Surf. Sci., 2014, 630, 229–235 CrossRef CAS.
- M. Shipilin, J. Gustafson, C. Zhang, L. R. Merte, A. Stierle, U. Hejral, U. Ruett, O. Gutowski, M. Skoglundh, P.-A. Carlsson and E. Lundgren, J. Phys. Chem. C, 2015, 119, 15469–15476 CAS.
- B. L. M. Hendriksen, M. D. Ackermann, R. van Rijn, D. Stoltz, I. Popa, O. Balmes, A. Resta, D. Wermeille, R. Felici, S. Ferrer and J. W. M. Frenken, Nat. Chem., 2010, 2, 730–734 CrossRef CAS PubMed.
- B. L. M. Hendriksen, S. C. Bobaru and J. W. M. Frenken, Surf. Sci., 2004, 552, 229–242 CrossRef CAS.
- R. Toyoshima, M. Yoshida, Y. Monya, Y. Kousa, K. Suzuki, H. Abe, B. S. Mun, K. Mase, K. Amemiya and H. Kondoh, J. Phys. Chem. C, 2012, 116, 18691–18697 CAS.
- S. Blomberg, M. J. Hoffmann, J. Gustafson, N. M. Martin, V. R. Fernandes, A. Borg, Z. Liu, R. Chang, S. Matera, K. Reuter and E. Lundgren, Phys. Rev. Lett., 2013, 110, 117601 CrossRef CAS PubMed.
- V. R. Fernandes, M. van den Bossche, J. Knudsen, M. H. Farstad, J. Gustafson, H. J. Venvik, H. Grönbeck and A. Borg, ACS Catal., 2016, 6, 4154–4161 CrossRef CAS.
- M. S. Chen, Y. Cai, Z. Yan, K. K. Gath, S. Axnanda and D. W. Goodman, Surf. Sci., 2007, 601, 5326–5331 CrossRef CAS.
- M. Chen, X. V. Wang, L. Zhang, Z. Tang and H. Wan, Langmuir, 2010, 26, 18113–18118 CrossRef CAS PubMed.
- M. Todorova, E. Lundgren, V. Blum, A. Mikkelsen, S. Gray, J. Gustafson, M. Borg, J. Rogal, K. Reuter, J. N. Andersen and M. Scheffler, Surf. Sci., 2003, 541, 101–112 CrossRef CAS.
- P. Kostelník, N. Seriani, G. Kresse, A. Mikkelsen, E. Lundgren, V. Blum, T. Šikola, P. Varga and M. Schmid, Surf. Sci., 2007, 601, 1574–1581 CrossRef.
- G. Zheng and E. I. Altman, Surf. Sci., 2002, 504, 253–270 CrossRef CAS.
- E. Lundgren, J. Gustafson, A. Mikkelsen, J. N. Andersen, A. Stierle, H. Dosch, M. Todorova, J. Rogal, K. Reuter and M. Scheffler, Phys. Rev. Lett., 2004, 92, 046101 CrossRef CAS PubMed.
- A. Stierle, N. Kasper, H. Dosch, E. Lundgren, J. Gustafson, A. Mikkelsen and J. N. Andersen, J. Chem. Phys., 2005, 122, 044706 CrossRef CAS PubMed.
- R. Westerström, et al. , Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 115440 CrossRef.
- W. G. Onderwaater, O. Balmes, S. B. Roobol, M. A. van Spronsen, J. Drnec, F. Carla, R. Felici and J. W. M. Frenken, Catal., Struct. React., 2017, 3, 89–94 CrossRef.
- G. Zheng and E. I. Altman, J. Phys. Chem. B, 2002, 106, 1048–1057 CrossRef CAS.
- V. R. Fernandes, J. Gustafson, I.-H. Svenum, M. H. Farstad, L. E. Walle, S. Blomberg, E. Lundgren and A. Borg, Surf. Sci., 2014, 621, 31–39 CrossRef CAS.
- R. Toyoshima, M. Yoshida, Y. Monya, K. Suzuki, B. S. Mun, K. Amemiya, K. Mase and H. Kondoh, J. Phys. Chem. Lett., 2012, 3, 3182–3187 CrossRef CAS PubMed.
- J. Rogal, K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 205433 CrossRef.
- J. Rogal, K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 155410 CrossRef.
- J. T. Hirvi, T.-J. J. Kinnunen, M. Suvanto, T. A. Pakkanen and J. K. Nørskov, J. Chem. Phys., 2010, 133, 084704 CrossRef PubMed.
- S. Blomberg, J. Zetterberg, J. Gustafson, J. Zhou, C. Brackmann and E. Lundgren, Top. Catal., 2016, 59, 478–486 CrossRef CAS.
- J. Szanyi and D. W. Goodman, J. Phys. Chem., 1994, 98, 2972–2977 CrossRef CAS.
- F. Zhang, L. Pan, T. Li, J. T. Diulus, A. Asthagiri and J. F. Weaver, J. Phys. Chem. C, 2014, 118, 28647–28661 CAS.
- J. Rogal, K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 075421 CrossRef.
- J. R. McBride, K. C. Hass and W. H. Weber, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 5016 CrossRef CAS.
- N. Seriani, J. Harl, F. Mittendorfer and G. Kresse, J. Chem. Phys., 2009, 131, 054701 CrossRef PubMed.
- V. V. Kaichev, I. P. Prosvirin, V. I. Bukhtiyarov, H. Unterhalt, G. Rupprechter and H.-J. Freund, J. Phys. Chem. B, 2003, 107, 3522–3527 CrossRef CAS.
- G. Rupprechter, H. Unterhalt, M. Morkel, P. Galletto, T. Dellwig and H.-J. Freund, Vacuum, 2003, 71, 83–87 CrossRef CAS.
- E. Ozensoy, D. C. Meier and D. W. Goodman, J. Phys. Chem. B, 2002, 106, 9367–9371 CrossRef CAS.
- E. Lundgren, G. Kresse, C. Klein, M. Borg, J. N. Andersen, M. De Santis, Y. Gauthier, C. Konvicka, M. Schmid and P. Varga, Phys. Rev. Lett., 2002, 88, 246103 CrossRef CAS PubMed.
- F. Zhang, T. Li, L. Pan, A. Asthagiri and J. F. Weaver, Catal. Sci. Technol., 2014, 4, 3826–3834 CAS.
- Z. Duan and G. Henkelman, ACS Catal., 2014, 4, 3435–3443 CrossRef CAS.
- G. Ketteler, D. F. Ogletree, H. Bluhm, H. Liu, E. L. D. Hebenstreit and M. Salmeron, J. Am. Chem. Soc., 2005, 127, 18269–18273 CrossRef CAS PubMed.
- R. J. Wrobel, S. Becker and H. Weiss, J. Phys. Chem. C, 2015, 119, 5386–5394 CAS.
- N. Kasper, P. Nolte and A. Stierle, J. Phys. Chem. C, 2012, 116, 21459–21464 CAS.
- S. Blomberg, C. Brackmann, J. Gustafson, M. Aldén, E. Lundgren and J. Zetterberg, ACS Catal., 2015, 5, 2028–2034 CrossRef CAS PubMed.
- R. Toyoshima, M. Yoshida, Y. Monya, K. Suzuki, K. Amemiya, K. Mase, B. S. Mun and H. Kondoh, J. Phys. Chem. C, 2013, 117, 20617–20624 CAS.
- R. Westerström, C. J. Weststrate, A. Resta, A. Mikkelsen, J. Schnadt, J. N. Andersen, E. Lundgren, M. Schmid, N. Seriani, J. Harl, F. Mittendorfer and G. Kresse, Surf. Sci., 2008, 602, 2440–2447 CrossRef.
- M. Kralj, T. Pertram, N. Seriani, F. Mittendorfer, A. Krupski, C. Becker and K. Wandelt, Surf. Sci., 2008, 602, 3706–3713 CrossRef CAS.
- R. Westerström, J. Gustafson, A. Resta, A. Mikkelsen, J. N. Andersen, E. Lundgren, N. Seriani, F. Mittendorfer, M. Schmid, J. Klikovits, P. Varga, M. D. Ackermann, J. W. M. Frenken, N. Kasper and A. Stierle, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 155410 CrossRef.
- M. Shipilin, J. Gustafson, C. Zhang, L. R. Merte and E. Lundgren, Phys. Chem. Chem. Phys., 2016, 18, 20312–20320 RSC.
- A. Vlad, A. Stierle, R. Westerström, S. Blomberg, A. Mikkelsen and E. Lundgren, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 035407 CrossRef.
- J. Klikovits, E. Napetschnig, M. Schmid, N. Seriani, O. Dubay, G. Kresse and P. Varga, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 045405 CrossRef.
- S. Blomberg, J. Zetterberg, J. Zhou, L. R. Merte, J. Gustafson, M. Shipilin, A. Trinchero, L. A. Miccio, A. Magaña, M. Ilyn, F. Schiller, J. E. Ortega, F. Bertram, H. Grönbeck and E. Lundgren, ACS Catal., 2017, 7, 110–114 CrossRef CAS.
- C. T. Herbschleb, S. C. Bobaru and J. W. M. Frenken, Catal. Today, 2010, 154, 61–67 CrossRef CAS.
- S. B. Roobol, W. G. Onderwaater, M. A. van Spronsen, F. Carla, O. Balmes, V. Navarro, S. Vendelbo, P. J. Kooyman, C. F. Elkjær, S. Helveg, R. Felici, J. W. M. Frenken and I. M. N. Groot, Phys. Chem. Chem. Phys., 2017, 19, 8485–8495 RSC.
- J. Jelic and R. J. Meyer, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 125410 CrossRef.
- J. Jelic, K. Reuter and R. Meyer, ChemCatChem, 2010, 2, 658–660 CrossRef CAS.
- J. M. Lorenzi, S. Matera and K. Reuter, ACS Catal., 2016, 6, 5191–5197 CrossRef CAS.
- A. D. Smeltz, W. N. Delgass and F. H. Ribeiro, Langmuir, 2010, 26, 16578–16588 CrossRef CAS PubMed.
- J. M. Bray and W. F. Schneider, ACS Catal., 2015, 5, 1087–1099 CrossRef CAS.
- C. T. Herbschleb, PhD thesis, Leiden University, 2011.
- M. Ehrensperger and J. Wintterlin, J. Catal., 2014, 319, 274–282 CrossRef CAS.
- V. Navarro, M. A. van Spronsen and J. W. M. Frenken, Nat. Chem., 2016, 8, 929–934 CrossRef CAS PubMed.
- A. Banerjee, V. Navarro, J. W. M. Frenken, A. P. van Bavel, H. P. C. E. Kuipers and M. Saeys, J. Phys. Chem. Lett., 2016, 7, 1996–2001 CrossRef CAS PubMed.
Footnote |
| † Also abbreviated as IRAS or RAIRS, reflective absorption infrared spectroscopy. |
| This journal is © The Royal Society of Chemistry 2017 |