
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe ENGases: versatile biocatalysts for the production of homogeneous N-linked glycopeptides and glycoproteins
Antony J.
Fairbanks
 ab
ab
aDepartment of Chemistry, University of Canterbury, Private Bag 4800, Christchurch 8140, New Zealand. E-mail: antony.fairbanks@canterbury.ac.nz
bBiomolecular Interaction Centre, University of Canterbury, Private Bag 4800, Christchurch 8140, New Zealand
First published on 6th July 2017
Abstract
The endo-β-N-acetylglucosaminidases (ENGases) are an enzyme class (EC 3.2.1.96) produced by a range of organisms, ranging from bacteria, through fungi, to higher order species, including humans, comprising two-sub families of glycosidases which all cleave the chitobiose core of N-linked glycans. Synthetic applications of these enzymes, i.e. to catalyse the reverse of their natural hydrolytic mode of action, allow the attachment of N-glycans to a wide variety of substrates which contain an N-acetylglucosamine (GlcNAc) residue to act as an ‘acceptor’ handle. The use of N-glycan oxazolines, high energy intermediates on the hydrolytic pathway, as activated donors allows their high yielding attachment to almost any amino acid, peptide or protein that contains a GlcNAc residue as an acceptor. The synthetic effectiveness of these biocatalysts has been significantly increased by the production of mutant glycosynthases; enzymes which can still catalyse synthetic processes using oxazolines as donors, but which do not hydrolyse the reaction products. ENGase biocatalysts are now finding burgeoning application for the production of biologically active glycopeptides and glycoproteins, including therapeutic monoclonal antibodies (mAbs) for which the oligosaccharides have been remodelled to optimise effector functions.
 Antony J. Fairbanks | Antony Fairbanks studied Chemistry at the University of Oxford (Wadham College), receiving his BA in 1990, and his DPhil in 1993 (Prof. George Fleet). After Postdocs with Profs Pierre Sinaÿ (ENS, Paris) and Steve Ley (Cambridge) he returned to Oxford as an independent academic at the end of 1996. In 2009 he moved to the University of Canterbury (UC) in Christchurch, New Zealand, where he is currently a Professor of Organic Chemistry. He was Head of the Chemistry Department at UC from 2010–2014 throughout the Christchurch earthquakes. He has long-standing interests in synthetic carbohydrate chemistry. |
1. Introduction
Protein glycosylation1 is the most diverse form of post-translational modification, playing a key role in protein folding, and crucially affecting many important protein properties, such as conformation and stability, susceptibility to proteases, and circulatory lifetime. Protein glycosylation is central to many vital biological processes, such as cell–cell signalling, development, and immune response. In N-linked glycosylation,2 the predominant form, the oligosaccharides/glycans are attached to the amide nitrogen of particular protein asparagine residues, whereas in the alternative O-linked glycosylation, glycans are most commonly attached via the side chains of serine or threonine residues.Many important mammalian proteins of clinical and therapeutic significance are N-glycosylated. Amongst these immunoglobulin G (IgG) antibodies, the main antibodies found in blood and extracellular fluid, are of particular therapeutic and commercial importance. Indeed recombinant monoclonal antibodies (mAbs) are the most significant class of new therapeutic agents to be developed in recent times, 2016 marking the 30th anniversary of the first FDA approval of a mAb for human use.3 Human IgGs contain two N-linked glycans attached to the two Asn-297 residues of the Fc region. Numerous studies have demonstrated that Fc-receptor mediated effector functions of IgGs (e.g. complement-dependent cytotoxicity [CDC] and antibody-dependent cellular cytotoxicity, [ADCC]), are highly dependent on the structure of attached glycans.4 Thus correct glycosylation of mAbs is important for therapeutic efficacy, and correspondingly significant efforts are being made to optimise the glycosylation patterns on mAbs.5
N-Linked glycosylation is also of significance for many mammalian enzymes; for example all lysosomal enzymes are N-glycosylated, and their targeting to the lysosome relies on the attachment of mannose-6-phosphate residues and transport mediated by the mannose-6-phosphate receptors (M6PR's).6 Glycosylation therefore plays an important role in the efficacy of treatment of the lysosomal storage disorders (LSDs) using recombinant glycoproteins as enzyme replacement therapies.7
The biosynthesis of the glycan portions of N-linked glycoproteins is not under direct genetic control; rather a complex sequence of enzymatically catalysed events leads to the formation of diverse N-glycan structures. Firstly the enzyme oligosaccharyl transferase (OST) mediates the transfer of a 14-mer dolichol phosphate bound oligosaccharide (Glc3Man9GlcNAc2) to particular asparagine (Asn) residues (Asn-X-Ser/The)8 as the protein is assembled. Then, sequential trimming of the oligosaccharide chain(s) occurs, and subsequently the action of several other glycosidases and glycosyl transferases leads to the production of glycoproteins bearing a variety of N-glycan structures, commonly classified into three groups; high mannose, hybrid, and complex (Fig. 1). Importantly all of these oligosaccharides contain the same core pentasaccharide unit (Man3GlcNAc2) linked to Asn residues. Due the action of numerous enzymes in competing processes, glycoproteins are invariably produced as inseparable heterogeneous mixtures of different ‘glycoforms’; proteins in which different oligosaccharide structures are linked to the same peptide chain. Therefore all recombinant therapeutic glycoproteins, including mAbs, are currently manufactured and administered as mixtures of materials in which different oligosaccharide structures are linked to the same peptide chain.9
 | ||
| Fig. 1 Chemical structures and Glycobiology representations of typical N-glycans showing the ENGase cleavage site: (a) high mannose; (b) hybrid; (c) complex biantennary with core fucose. | ||
Access to pure single glycoforms of glycopeptides and glycoproteins has consequently become a major scientific objective.10 Amongst the numerous approaches11 that have been adopted to access glycopeptides and glycoproteins in homogeneous form, this review will focus on synthetic applications of endo-β-N-acetyl-glucosaminidase (ENGase) enzymes.
2. The endo-β-N-acetylglucosaminidases (ENGases)
2.1 Hydrolytic activity
The endo-β-N-acetylglucosaminidases or ENGases (sometimes also referred to as endohexosaminidases, of which they are a sub-set) are an enzyme class (EC 3.2.1.96), first identified in the early 1970's, that are produced by a range of organisms, ranging from bacteria, through fungi, to higher order species, including humans. Interest in them initially arose because of their ability to act on glycoproteins, hydrolysing attached N-glycans. All of the ENGases so far identified have been classified by sequence analysis into one of two of the Carbohydrate Active enZYme database (CAZY)12 families13 of the glycoside hydrolases, either family GH18 or family GH85, and all have similar (β/α)8-TIM-barrel structures consisting of eight α-helices and eight parallel β-sheets. Fig. 2 shows two views of an X-ray crystal structure14 of Endo A, a family GH85 ENGase. The major domain comprises the eight α-helices (red) and eight β-sheets (yellow) in a TIM-barrel fold; two other domains also comprise β-sheets. | ||
| Fig. 2 Two different views of the X-ray crystal structure14 of WT Endo A (PDB: 3FHA). α-Helices are shown in red, β-sheets in yellow. Domain 1 (residues 2–350) displays a typical (β/α)8-TIM-barrel fold. An arrow shows the position of the catalytic site. | ||
In their action as endo-glycosidases the ENGases specifically cleave the diacetylchitobiose core [GlcNAc β(1–4)GlcNAc] of N-linked glycans between the two N-acetylglucosamine (GlcNAc) residues.15 When applied to an N-linked glycoprotein/glycopeptide the two products of hydrolysis are therefore truncated N-glycan oligosaccharides and a protein/peptide which possesses only a single GlcNAc residue at any N-glycosylation site. Their application as biocatalysts to achieve the reverse process, namely the attachment of N-glycans to GlcNAc residues, represents a highly effective convergent method to access homogenous glycopeptides and glycoproteins, which comprise N-linked glycans of optimal structure for desired biological activity.16Fig. 3 demonstrates the principles of glycoprotein remodelling and convergent glycopeptide synthesis using ENGases.
 | ||
| Fig. 3 The use of ENGases to catalyse: (a) glycoprotein remodelling; (b) convergent glycopeptide synthesis. | ||
The two-step procedure for in vitro glycoprotein remodelling (Fig. 3a) firstly entails ENGase catalysed hydrolysis of the heterogeneous mixture of protein glycoforms that is invariably produced by expression, so that N-glycans are ‘trimmed’ back to a single GlcNAc residue at N-linked glycosylation sites. In a second step, a defined glycan is attached to this GlcNAc ‘handle’, again in a process catalysed by an ENGase, but one which is acting in the reverse sense. This synthetic process may be achieved either by a classical ‘transglycosylation’ reaction employing an oligosaccharide comprising two GlcNAc residues to act as the source of the glycan to be added (X = GlcNAc), or alternatively by a ‘glycosylation’ reaction using an activated donor that possesses only a single GlcNAc residue which is activated at the reducing terminus (X = leaving group). In a similar fashion ENGases can be applied to the glycosylation of synthetic peptides assembled by solid-phase synthesis that contain GlcNAc ‘handles’ attached to particular Asn residues (Fig. 3b). Thus, ENGase-catalysed attachment of an N-glycan, again either by ‘transglycosylation’ or ‘glycosylation’, represents an efficient convergent method to access defined glycopeptides.
Although all of the ENGases carry out the same hydrolytic reaction, they have different tolerances as to the precise structure of the N-glycans that they can hydrolyse. A summary of some of the most well studied ENGases to date is given in Table 1, including their hydrolytic specificities and their active site residues. The ENGases are all retaining glycosidases that hydrolyse substrates via a two-step mechanism involving general acid/base catalysis (Fig. 4). The family GH18 enzymes (Fig. 4a) all contain two carboxylic acid residues in the active site, a glutamic acid and an aspartic acid. In contrast the family GH85 enzymes (Fig. 4b) contain only one carboxylic acid residue, a glutamic acid; the other key catalytic residue is an asparagine. The pH/rate dependency for the hydrolytic activity of the ENGases is typically a bell-shaped curve centred around pH 5.0–8.0, the precise pH optimum depending on the enzyme.17–19 The upper and lower limits of the curve generally correspond with the kinetic pKa values of the two catalytic carboxylic acid residues in the enzyme active site; for example the pH optimum for Endo H is pH 5.5, and its hydrolytic activity drops rapidly as the pH is either increased from pH 6.0 to pH 8.0, or is decreased from pH 5.0 to pH 4.0.18 Although the family GH85 ENGases actually only possess a single catalytic carboxylic acid residue in their active sites, similar pH/rate profiles have generally been observed for their hydrolytic activity.17,19
| Enzyme | Source | GH family | Glycan structure hydrolysed | Glycans with core fucose hydrolysed? | Catalytic residues | Glycosynthase mutants |
|---|---|---|---|---|---|---|
| a Amino acid numbering refers to the peptide chain after cleavage of an N-terminal signal peptide. | ||||||
| Endo-A | Arthrobacter protophormiae | 85 | High mannose/hybrid | No |
E173a
N171a |
N171Aa
E173Qa E173Ha |
| Endo-M | Mucor hiemalis | 85 | High mannose/hybrid and biantennary complex | No |
E177
N175 |
N175A
N175Q N175Q/W251N |
| Endo-D | Streptococcus pneumoniae | 85 | Truncated core structures only | Yes |
E324
N322 |
N322A
N322Q |
| Endo-CC1 | Coprinopsis cinerea | 85 | High mannose/hybrid and biantennary complex | — |
E182
N180 |
N180H
N180Q |
| Endo-CC2 | Coprinopsis cinerea | 85 | High mannose/hybrid and biantennary complex | — |
E188
N186 |
— |
| Endo Om | Ogatea minuta | 85 | High mannose/hybrid and biantennary complex | No |
E196
N194 |
— |
| Endo-CE | Caenorhabditis elegans | 85 | High mannose/hybrid | No |
E154
N152 |
— |
| Endo H | Streptomyces plicatus (griseus) | 18 | High mannose/hybrid | Yes |
E132a
D130a |
— |
| Endo F1 | Elizabethkingia meningoseptica | 18 | High mannose/hybrid | No |
E132a
D130a |
— |
| Endo F2 | Elizabethkingia meningoseptica | 18 | Biantennary complex | Yes |
E126a
D124a |
— |
| Endo F3 | Elizabethkingia meningoseptica | 18 | Bi-/triantennary complex | Yes |
E128a
D126a |
D126Aa
D126Qa |
| Endo S | Streptococcus pyogenes | 18 | Complex (IgG Fc specific) | Yes |
E235
D233 |
D233A
D233Q |
| Endo S2 | Streptococcus pyogenes (M49) | 18 | High mannose/hybrid and biantennary complex | Yes |
E186
D184 |
D184M
D184Q |
 | ||
| Fig. 4 Hydrolytic mechanism of the ENGases showing key catalytic residues for some enzymes from: (a) family GH18; (b) family GH85. | ||
Regardless of whether the active site contains one or two carboxylic acids, the hydrolytic mechanism20 catalysed by the ENGases involves neighbouring group participation of the 2-acetamide of the second GlcNAc residue. A glycosyl oxazoline (or oxazolinium ion) is therefore implicated as a high-energy intermediate in the ENGase mediated hydrolytic process. A key breakthrough in the field was the realisation that N-glycan oxazolines may act as substrates for these enzymes; in particular that they may be intercepted by a GlcNAc residue attached to a peptide or protein in a synthetic reaction that is formally the reverse of hydrolysis.
In 2001 Shoda and co-workers published their first seminal report in the field21 in which they demonstrated that disaccharide oxazoline 1 could be used as an activated glycosyl donor substrate for some of the ENGases (Scheme 1). In particular Endo A and Endo M (both family GH85 enzymes) catalysed the glycosylation of GlcNAcOpNP 2 by the disaccharide oxazoline 1, to produce the corresponding trisaccharide 3. However neither Endo H nor Endo F1 (both family GH18 enzymes), were found to catalyse this synthetic reaction. Besides paving the way for the use of more extended oxazolines as privileged donor substrates for ENGase catalysed glycosylation, these initial results also seemed to imply that perhaps only family GH85 enzymes would be useful synthetically; subsequently this led to an initial focus on the family GH85 enzymes. An additional advantage of the use of oxazolines as donors is that in many cases wild-type (WT) ENGases are able to catalyse synthetic reactions using either truncated or ‘non-natural’ oxazolines, and often the products of these reactions are not hydrolytic substrates for that enzyme. This was indeed the case in the first example by Shoda (Scheme 1), i.e. trisaccharide 3 was not a hydrolytic substrate for either Endo A or Endo M. Thus, in these cases WT ENGases can catalyse irreversible synthetic reactions using oxazoline donors.
 | ||
| Scheme 1 Shoda's demonstration of the first use of oxazolines as glycosyl donors for ENGase catalysed glycosylation.21 | ||
Since this first report oxazolines have become the donors of choice for the ENGase-catalysed synthesis.22 Moreover, following further important developments made by Shoda,23 they may now be readily synthesised directly at the reducing end of complex oligosaccharides without the need for any protecting groups.24 Besides the use of oxazolines, a variety of other factors can increase glycosylation efficiency, for example the use of variable amounts of organic co-solvents25,26 and/or control of reaction pH;27 modulation of both of these can decrease the rates of competitive hydrolytic reactions.
2.2 Mutant enzymes
The use of glycosidases for synthetic purposes, either in ‘true’ transglycosylation reactions, or when catalysing ‘glycosylation’ using activated glycosyl donors, such as oxazolines, is inevitably limited by the rate at which the enzyme hydrolyses the product. Although in certain cases when using truncated or ‘non-natural’ oxazolines as donors, the products of glycosylation are not hydrolysed by the ENGase, in general they are. Indeed when using full-length glycans the rate of product hydrolysis can sometimes far exceed the rate of the synthetic reaction, meaning that synthesis is unachievable. A solution to this problem is to access mutant ‘glycosynthase’ enzymes,28 as originally developed by Withers29 and Planas,30 by site-directed mutagenesis. In this original work, the catalytic nucleophile of retaining glycosidases was replaced with a non-participating residue, for example alanine; these mutant enzymes could process activated donors (such as glycosyl fluorides) of opposite anomeric configuration to the natural substrate, but were incapable of hydrolysing the reaction product. In the case of the ENGases, the catalytic nucleophile is the 2-acetamido group on the GlcNAc residue undergoing hydrolysis; therefore a direct analogy is not possible. Two approaches have been developed to accessing glycosynthase mutants of ENGases, namely mutation of one of the two key catalytic site residues. By far the most common approach has been to mutate either the asparagine (family GH85) or aspartate (family GH18) residue that stabilises/deprotonates the oxazolinium intermediate during the hydrolytic reaction; i.e. the catalytic residue that acts as the base. The first glycosynthase mutant of any ENGase, reported by Wang and Yamamoto,31 was a mutant of Endo M in which residue N175 was replaced by alanine (Fig. 5a), and this was a reasonably effective synthase enzyme. Further studies in which all other possible mutations were made at this site revealed that the asparagine to glutamine mutation was the most effective in terms of synthetic efficiency (Fig. 5b); this E175Q Endo M mutant32 is now available commercially. Wang has subsequently performed the analogous mutations on a variety of other family GH85 enzymes including Endo A and Endo D, to produce a variety of synthetically useful synthase mutants. Moreover Wang extended this approach to some of the family GH18 enzymes, and has shown that replacement of the aspartic acid active site residue of both Endo F3 and Endo S by either alanine (Fig. 4a) or glutamine (Fig. 5b), also produces extremely useful glycosynthase mutants. Whilst this N/D to A or Q mutation is the most reliable in terms of the production of an active glycosynthase enzyme, very recently two other mutations of the catalytic base residue have been reported (Endo CC1 N180H33 and Endo S2 D184M34). | ||
| Fig. 5 Design of mutant glycosynthases of the ENGases: (a) mutations of the catalytic base (family GH18: D, family GH85: N) to alanine; (b) mutations of the catalytic base (family GH18: D, family GH85: N) to glutamine; (c) mutation of the catalytic acid (E) to glutamine; (d) mutation of the catalytic acid (E) to histidine. | ||
An alternative approach, developed by Fairbanks, is to mutate the catalytic glutamic acid residue (conserved for GH18 and GH85 enzymes) that acts as a general acid to protonate the anomeric oxygen during the hydrolytic process.35 Adopting this approach Fairbanks made the first two glycosynthase mutants of Endo A, E173Q (Fig. 5c) and E173H (Fig. 5d); potential hydrogen bond acceptors were chosen as the replacements for the glutamic acid as this residue also normally acts as a general base to direct the incoming nucleophile during the second step of the hydrolytic reaction. The E173Q and E173H Endo A mutants35 were both capable of irreversibly catalysing glycosylation of GlcNAc acceptors, including de-glycosylated RNase B, with a tetrasaccharide oxazoline. However Yamamoto reported that the analogous Endo M mutants (E177Q and E177H) were not synthetically active.32
Further site directed mutagenesis studies have also revealed that mutations to other active residues can increase the synthetic efficiency of these enzymes, particularly by decreasing the rate of direct competitive oxazoline hydrolysis. For example the Y217F mutant32 of Endo M and the Y205F mutant14 of Endo A both showed increased rates of synthetic glycosylation; however such mutants are actually still capable of hydrolysing the product.
The total number of mutant ENGases that have now been made is increasing rapidly (vide infra), as more and more enzymes are cloned and expressed in a drive to expand synthetic capability. A summary of some of the most useful synthetic mutants produced so far is provided in Table 2. Finally a key point worth re-iterating is that these mutant enzymes will not catalyse transglycosylation using full-length N-glycans as substrates, because the first step of ‘transglycosylation’ necessarily involves hydrolysis of the full length N-glycan. In many previous papers the term transglycosylation has also (rather erroneously) been used to refer to ENGase-catalysed reactions using oxazolines as donors. The ability of mutant ENGases to catalyse one and not the other highlights the important, yet mostly overlooked, difference between the two.
| Mutant enzyme | Donor oxazolines structures processed | Fucosylation of the acceptor tolerated (or required)? | Protein and IgG applicability | Comments |
|---|---|---|---|---|
| Endo A N171A | High mannose/hybrid | No |
Protein: yes
IgG: Fc only |
Will also transfer truncated glycans with bisecting GlcNAc
Tolerates glycan phosphorylation |
| Endo A E173H | High mannose/hybrid | No |
Protein: yes
IgG: not known |
High mannose only up to Man6 structures |
| Endo M N175Q |
High mannose/hybrid
Biantennary complex |
No |
Protein: yes
IgG: no |
Commercially available
Will not glycosylate the Fc region of IgGs |
| Endo M N175Q/W251N |
High mannose/hybrid
Biantennary complex |
Yes |
Protein: not known
IgG: not known |
Will act on IgG Fc-derived peptides |
| Endo D N322Q | Core structure only | Yes |
Protein: yes
IgG: yes |
Only up to pentasaccharide |
| Endo F3 D126A |
Biantennary complex
Triantennary complex |
Required |
Protein: not known
IgG: yes |
Will transfer triantennary structures |
|
Endo S D233A
Endo S D233Q |
Core structure
Biantennary complex |
Yes |
Protein: no
IgG: yes |
Endo S is highly IgG specific
Will not transfer high mannose oxazolines |
| Endo S2 D184M |
High mannose/hybrid
Biantennary complex |
Yes |
Protein: not known
IgG: yes |
Transfers all three classes of glycan to IgGs, both with and without core fucose |
| Endo CC1 N180H | Biantennary complex | Not known |
Protein: yes
IgG: not known |
Reportedly more thermostable than Endo M
Not yet tested with high mannose oxazolines |
2.3 Family GH85 enzymes
According to the CAZy database,12 family GH85 currently contains 353 members, the vast majority of which come from bacterial sources. So far 11 enzymes have been characterised, and crystal structures for two of them have been published and deposited in the Protein Data Bank (PDB), confirming their (β/α)8-TIM-barrel structures; for Endo A (PDB accession codes WT: 3FHQ, and an E173Q mutant: 2VTF), and Endo D (PDB: 2W91 and 2W92). An amino acid alignment of the catalytic region of some of the most well-studied GH85 enzymes is shown in Fig. 6, showing conserved and key active site residues. Note there is some inconsistency in the amino acid numbering used in the literature; for GH85 enzymes uniquely for Endo A the numbering refers to the amino acid position after cleavage of an N-terminal signal peptide. | ||
| Fig. 6 Amino acid alignment of the catalytic region of some family GH85 ENGases, showing conserved and key active site residues. In the case of Endo A the amino acid numbering refers to the peptide chain after signal peptide cleavage. Catalytic site residues are highlighted in pink; conserved residues across at least three enzymes are highlighted in grey. The symbol † shows the position of aromatic amino acids demonstrated as important for (trans)glycosylation activity for some enzymes. An asterisk indicates positions which have a single, fully conserved residue; a colon or a full stop indicates conservation between groups of strongly, or weakly, similar properties respectively. | ||
A crystal structure14 showing the active site of Endo A in complex with a tetrasaccharide thiazoline, highlighting the key catalytic residues (N171 and E173) and aromatic residues (Y205, W216 and W244) important for (trans)glycosylation activity is shown in Fig. 7.
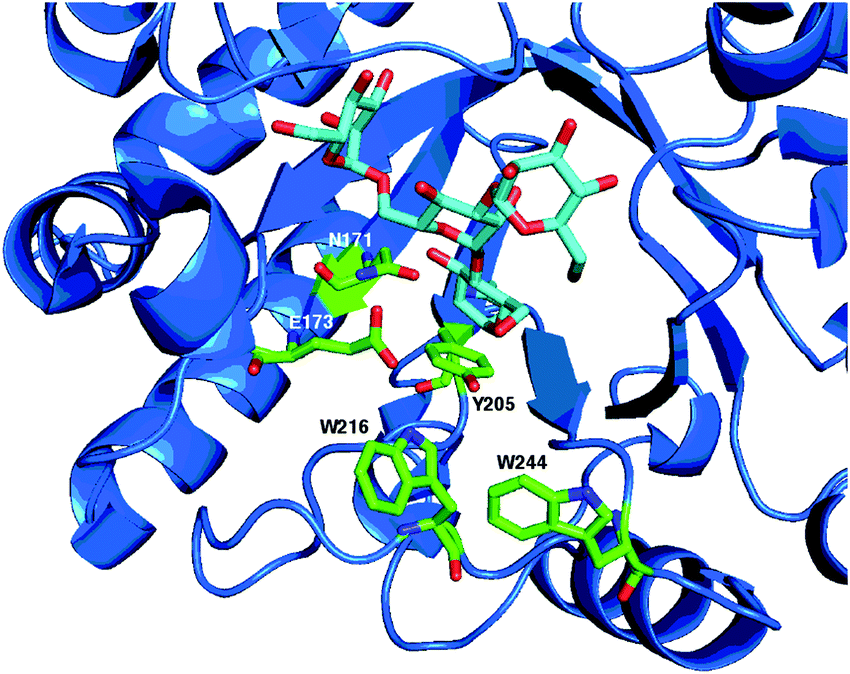 | ||
| Fig. 7 X-Ray crystal structure14 of WT Endo A (PDB: 3FHQ) in complex with a tetrasaccharide (Man3GlcNAc) thiazoline, showing catalytic active site residues (N171 and E173) and aromatic residues (Y205, W216 and W244) important for (trans)glycosylation activity. | ||
As for Endo A, in their seminal paper Shoda and collaborators21 showed that a disaccharide oxazoline was a good donor substrate for Endo M. Fairbanks60 then went on to show that larger N-glycan oxazolines61,62 were also donor substrates for Endo M, and additionally reported that non-natural oxazolines,63 for example those containing glucose rather than mannose attached to the GlcNAc, could act as donors for Endo M, to give products that were not hydrolytic substrates for the enzyme. The issue of competitive product hydrolysis was completely overcome by the development of the first glycosynthase mutant of any ENGase, namely the Endo M N175A mutant by Wang and Yamamoto.31 Additionally an Endo M Y217F mutant also showed enhanced glycosylation activity, though it was still capable of catalysing product hydrolysis. Subsequent studies in which all other possible mutations were made at position 175 revealed that the N175Q mutant was the most effective in terms of synthetic efficiency; a double N175Q/Y217F mutant was not more effective. This Endo M N175Q mutant32 has so far proven to be the most widely applicable and useful glycosynthase of an ENGase, and is now commercially available from Tokyo Chemical Industry Co. Ltd (TCI). Its applications in synthesis will be discussed later.
Inazu mapped out the acceptor requirements64 for transglycosylation catalysed by WT Endo M, confirming that free hydroxyls at positions-4 and 6 are needed on a GlcNAc acceptor (mannose and glucose also acted as acceptors), but that neither the ring oxygen nor the substituents at positions-1 or 2 were important. This minimum required structure corresponds to a 1,3-diol structure with secondary and primary hydroxyl groups; Inazu additionally showed that an acyclic diol would also act as an acceptor, but not glycerol, which possesses two primary hydroxyl groups.
This requirement for a free primary hydroxyl group at position-6 of GlcNAc means Endo M will not act, either synthetically or hydrolytically, on glycans that have core fucose (i.e. with fucose attached to position-6 of the innermost GlcNAc), limiting the synthetic potential of its glycosynthases. Very recently Yamamoto65 performed mutagenic studies to find a mutant enzyme that would cleave fucosylated glycans and discovered that the W251N mutation, asparagine being the amino acid at the corresponding position (N413) in Endo D, a family GH85 ENGase which can cleave fucosylated glycans, increased hydrolytic activity on fucosylated glycans. The incorporation of the double mutation N175Q/W251N65 then produced a glycosynthase Endo M which could glycosylate (∼20% yield) a Fucα(1–6)GlcNAc-biotin conjugate using a complex biantennary oxazoline.
Particular interest in the enzyme arose from its ability to hydrolyse substrates that are core fucosylated, a common modification of N-glycans attached to IgG's. The first demonstration of the ability of Endo D to process oxazolines as donors was reported by Fairbanks,70 although significant limitations of utility of the wild type enzyme were revealed due to the high rates of direct oxazoline hydrolysis. Additionally the fact that Endo D only hydrolyses very truncated N-glycans precludes its application to larger structures.
Subsequently Wang produced two glycosynthase mutants of Endo D (N322A and N322Q),71 which, although they displayed enhanced synthetic efficiency including the ability to transfer a tetrasaccharide oxazoline to an IgG-Fc fragment carrying a Fucα(1,6)GlcNAc moiety, were unable to accept more extended oxazolines as substrates. Mutations of other residues (E324Q, Y360F and H371W) were also made by analogy with the Endo A/M sequences and corresponding mutations that had improved the glycosylation activity of those enzymes. However, for Endo D these were all found to be less effective than the N322A and N322Q mutants.
2.4 Family GH18 enzymes
Family GH18 of the glycoside hydrolases, which currently contains more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 enzymes, also comprises many chitinases of classes II and IV (EC 3.2.1.14) in addition to numerous ENGase enzymes (EC 3.2.1.96). All of the GH18 enzymes have similar (β/α)8-barrel folds, and the catalytic residues are located at the carboxyl end of β-strand 4. An amino acid alignment of the catalytic region of a selection of the family GH18 ENGases is shown in Fig. 8, highlighting the key catalytic acid (E) and base (D) residues. Again there is inconsistency in the amino acid numbering used in the literature, as all of the GH18 enzymes have N-terminal signal peptides; initially the numbering used referred to the amino acid position after signal peptide cleavage, but this has not been adopted for Endo S and Endo S2. Although crystallographic and mutagenic studies were performed on the GH18 enzymes before the GH85 enzymes, it is only more recently that the synthetic potential of this family of the ENGases has begun to be realised. An X-ray crystal structure79 of WT Endo F3 (PDB: 1EOM) in complex with a biantennary complex oligosaccharide is shown in Fig. 9. Catalytic site base (D126) and acid (E128) residues are shown.
000 enzymes, also comprises many chitinases of classes II and IV (EC 3.2.1.14) in addition to numerous ENGase enzymes (EC 3.2.1.96). All of the GH18 enzymes have similar (β/α)8-barrel folds, and the catalytic residues are located at the carboxyl end of β-strand 4. An amino acid alignment of the catalytic region of a selection of the family GH18 ENGases is shown in Fig. 8, highlighting the key catalytic acid (E) and base (D) residues. Again there is inconsistency in the amino acid numbering used in the literature, as all of the GH18 enzymes have N-terminal signal peptides; initially the numbering used referred to the amino acid position after signal peptide cleavage, but this has not been adopted for Endo S and Endo S2. Although crystallographic and mutagenic studies were performed on the GH18 enzymes before the GH85 enzymes, it is only more recently that the synthetic potential of this family of the ENGases has begun to be realised. An X-ray crystal structure79 of WT Endo F3 (PDB: 1EOM) in complex with a biantennary complex oligosaccharide is shown in Fig. 9. Catalytic site base (D126) and acid (E128) residues are shown.
 | ||
| Fig. 8 Amino acid alignment of the catalytic region of some family GH18 ENGases, showing conserved and key active site residues. Amino acid numbering refers to the peptide chain after signal peptide cleavage, except in the cases of Endo S and Endo S2. Catalytic site residues are highlighted in pink. Conserved residues between at least three enzymes are highlighted in grey. An asterisk indicates positions which have a single, fully conserved residue; a colon or a full stop indicates conservation between groups of strongly, or weakly, similar properties respectively. | ||
 | ||
| Fig. 9 X-Ray crystal structure79 of WT Endo F3 (PDB: 1EOM) in complex with a biantennary complex octasaccharide, showing catalytic active site residues (D126 and E128). | ||
 ) is absolutely required for processing. Additionally it can hydrolyse hybrid N-glycans that contain core fucose. Following its cloning and expression in E. coli,83 and identification of active site residues by mutagenesis,18 the crystal structures84 of WT Endo H (PDB: 1EDT) and then a variety of mutants were solved;85 a total of 8 structures are now available (PDB: 1C3F, 1C8X, 1CBY, 1C90, 1C91, 1C92, 1C93). An early example of very low efficacy transglycosylation activity of Endo H (alongside Endo F1) was reported by Trimble86 during the hydrolysis of Asn-linked high mannose N-glycans when transfer to glycerol was observed. However, Shoda later reported that Endo H was not capable of effecting glycosylation using a disaccharide oxazoline,21 and larger N-glycan oxazolines were also found not to be donor substrates by both Fairbanks48 and Wang.87 Therefore further synthetic investigations of Endo H ceased. Indeed it was probably these early negative results with a family GH18 ENGase that caused synthetic attention to be focussed exclusively on the GH85 ENGases for a considerable time.
) is absolutely required for processing. Additionally it can hydrolyse hybrid N-glycans that contain core fucose. Following its cloning and expression in E. coli,83 and identification of active site residues by mutagenesis,18 the crystal structures84 of WT Endo H (PDB: 1EDT) and then a variety of mutants were solved;85 a total of 8 structures are now available (PDB: 1C3F, 1C8X, 1CBY, 1C90, 1C91, 1C92, 1C93). An early example of very low efficacy transglycosylation activity of Endo H (alongside Endo F1) was reported by Trimble86 during the hydrolysis of Asn-linked high mannose N-glycans when transfer to glycerol was observed. However, Shoda later reported that Endo H was not capable of effecting glycosylation using a disaccharide oxazoline,21 and larger N-glycan oxazolines were also found not to be donor substrates by both Fairbanks48 and Wang.87 Therefore further synthetic investigations of Endo H ceased. Indeed it was probably these early negative results with a family GH18 ENGase that caused synthetic attention to be focussed exclusively on the GH85 ENGases for a considerable time.
3. Synthetic applications of ENGase enzymes
ENGase enzymes may be used to transfer N-glycans onto a wide variety of other species providing that they possess a GlcNAc residue to act as an acceptor. An additional requirement is that other functionalization of this GlcNAc residue is tolerated by the enzyme in question; for example the presence of a fucose at position-6, a xylose at position-3, or protecting groups on any of the hydroxyl groups. Below are some recent highlights from three general areas of application of ENGase biocatalysis.3.1 Production of bioactive glycopeptides
Many early applications of ENGases to catalyse transglycosylation using full-length Asn-linked glycans as donors focussed on peptides with a GlcNAc residue attached to asparagine residue as the acceptor. For example Wang reported108 the convergent ENGase (both Endo A and Endo M) mediated synthesis of CD52 glycopeptides, bearing both high mannose and complex biantennary glycans; CD52 is a 12-amino acid GPI-anchored cell-surface glycopeptide found on all human lymphocytes and sperm cells.C34 is a 34-mer peptide derived from the C-terminal ectodomain of HIV-1 envelope glycoprotein, gp41. Synthetic C34 peptides have been shown to possess potent anti-HIV activity (typical IC50's ∼1.1 nM). Wang reported109 the synthesis of a high mannose (Man9GlcNAc2) glycoform of C34 using Endo A to catalyse the transglycosylation of a synthetic 34-mer peptide using a full length Man9GlcNAc2Asn as donor in 11% yield. However, unfortunately glycosylation with the high mannose N-glycan actually decreased the anti-HIV activity of C34 by a factor of ∼7.
The very significant increase in efficiency achieved by using oxazolines as donors was clearly demonstrated by Wang in the Endo A and Endo M catalysed synthesis of glycosylated versions of both 34-mer HIV gp41 and 7-mer gp120 fragments using di- and tetrasaccharide oxazolines as donors (yields 73–82%).44 Similarly Wang also produced di-glycosylated 47-mer cyclic glycopeptides corresponding to the third variable domain (V3) of HIV gp120 domain, in 60% and 86% yields (Scheme 2a) respectively.45
 | ||
| Scheme 2 Selected examples of the convergent synthesis of glycopeptides using ENGases: (a) di-glycosylated 47-mer V3 domain of HIV gp120 made with WT Endo A; (b) high mannose (Man9GlcNAc2) glycosylated pentapeptide made using the Endo A N171A mutant; (c) high mannose (Man9GlcNAc2) glycosylated CMV tegument protein made using the Endo M N175Q mutant; (d) glycosylated variants of the 37-mer anti-diabetes drug pramlintide made using the Endo A E173H and Endo M N175Q mutants. | ||
With larger oligosaccharide oxazolines the rate of product hydrolysis frequently outcompetes the rate of the synthetic reaction. Therefore it was only by the use of glycosynthase mutants, for example Endo A N171A, that larger structures, such as a Man9GlcNAc oxazoline could be added to a pentapeptide (Scheme 2b). In the same report the N175A Endo M mutant was similarly used to attach complex biantennary glycans to the HIV C34 peptide.50
A combination of solid phase peptide synthesis (installing GlcNAc and GalNAc at the pre-determined N- and O-glycosylation sites), and the use of an ENGase (either WT Endo A, or the Endo M N175A or Y217F mutants) to attach N-glycans to GlcNAc, and a galactosyl transferase to attach galactose to GalNAc, was developed by Wang110 to produce CD52 glycopeptides bearing both N- and O-glycan structures. Wang111 has also used the ENGases (WT Endo A and N175A Endo M) in combination with an N-glycosyl transferase (NGT), which catalyses the addition of a glucose residue to asparagine residues in peptides, for the production of other glycosylated versions of the HIV-1 gp41 polypeptide C34. Similarly, Nakahara112 and co-workers used the Endo M N175Q mutant to glycosylate Saposin C with a truncated complex biantennary glycan; the Saposin C 80-mer peptide was assembled by the combined use of solid phase peptide synthesis and native chemical ligation (NCL).
Neither Endo A nor Endo M (nor their mutants) is capable of catalysing the glycosylation of GlcNAc residues with a core fucose. Wang has therefore developed the use of Endo F2 and Endo F3 for the production of core-fucosylated glycopeptides. Their application, together with a complex bi-antennary N-glycan oxazoline donor, was exemplified by the synthesis of a complex core-fucosylated CD52 antigen (52% yield with Endo F3, 25% yield with Endo F2).87
Fairbanks has applied mutant ENGases for the synthesis of putative glycopeptide vaccine candidates (Scheme 2c).113 In particular it was shown that the conjugation of high mannose glycans to a human cytomegalovirus (CMV) tegument 19-mer peptide increased its binding to antigen presenting cells (APCs), presumably via interaction with the mannose receptor (MR) which is widely expressed on APCs. Furthermore an embedded 9-mer peptide epitope was cross-presented by the APCs to a cytotoxic T-lymphocyte clone (CTL, CD8+) provided that the site of glycosylation was outside the 9-mer epitope.
ENGases have also been used to access glycosylated variants of therapeutic peptides. Fairbanks used both the commercially available N175Q Endo M mutant, and the E173H Endo A mutant to access a variety of glycosylated variants of the anti-diabetes drug pramlintide,114 which is a non-fibril forming analogue of human amylin (Scheme 2d). The rationale for this study was an attempt to improve the pharmacokinetic properties of the current drug, which is itself of low solubility and has a low in vivo half-life. The glycopeptides produced displayed both in vitro and in vivo activity, the latter demonstrated by a blood glucose tolerance test in rats. Further in vitro studies also correlated the position of glycosylation with the ability of the glycopeptide to agonise amylin receptors.115
3.2 Production of remodelled glycoproteins
RNase B has been the glycoprotein of choice for remodelling using various ENGases and different oxazolines. In the first step commercially available bovine RNase B (an inseparable mixture of high mannose glycoforms) is typically treated with Endo H, which cleaves the N-glycan at Asn-34 back to a single GlcNAc, giving de-glycosylated RNase B (dRNase B). This protein is then used as a substrate for the ENGase catalysed attachment of N-glycans. Transglycosylation using full length N-glycans is not an effective method for glycoprotein production; even when using very large excesses of glycan yields are typically less than 5%.38,116 Wang46 was the first to demonstrate that the use of oxazolines was significantly more efficient than transglycosylation for the remodelling of RNase B (Scheme 3a). Using Endo A and a variety of different oxazolines he went on to produce a diverse array of RNase glycoforms,117 including the attachment of azido-substituted glycans; these re-modelled glycoproteins could then be used as substrates for further modification by click chemistry. Building on the synthesis of glycopeptides, Wang showed that the glycosynthase mutants Endo A N171A and Endo M 175A were capable of producing re-modelled RNase B, bearing high mannose (Scheme 3b) or truncated complex biantennary glycans respectively, in excellent yield.50 In an elegant example of ‘one-pot remodelling’ (Scheme 3c) Wang and Yamamoto showed that the combined use of WT Endo A (which cleaves high mannose glycans) and the N175A Endo M mutant (which is capable of catalysing the synthesis of complex biantennary glycans) together with a complex biantennary oxazoline derived from egg yolks allowed the direct conversion of RNase B (a mixture of high mannose glycoforms) into RNase C (a single sialylated complex biantennary glycoform).118 | ||
| Scheme 3 Selected examples of the synthesis of remodelled RNase B using ENGases: (a) truncated glycoforms produced using Endo A; (b) high mannose (Man9GlcNAc2) glycoform produced using the Endo A N171A mutant; (c) one-pot remodelling of a mixture of RNase B glycoforms into RNase C using Endo A and the Endo M N175A mutant; (d) glucosylated high mannose (GlcMan9GlcNAc2) glycoform produced using the Endo A N171A mutant; (e) mannose-6-phosphate containing glycoform produced using Endo A. | ||
Wang also produced119 further high mannose glycoforms of RNase which additionally contained glucose as ligands for the lectin-like molecular chaperones calnexin and calreticulin using the Endo A N171A mutant (Scheme 3d). A notable point of this work was that galactose, which will not act as an acceptor for the ENGases, was used to ‘protect’ the glucose residue (which itself may be glycosylated by ENGases) at the non-reducing end of the oligosaccharide; following glycoprotein remodelling using the ENGase mutant, the galactose was removed with a β-galactosidase.
Protein transport to the lysosome is mediated by the mannose-6-phosphate receptors (M6PRs), which recognise high mannose glycans comprising mannose-6-phosphate (M6P) residues. The production of glycoproteins that possess optimal phosphorylation of mannose residues is therefore of particular significance for improving the efficacy of enzyme replacement therapies for the treatment of lysosomal storage disorders.7 Fairbanks and co-workers recently reported120 the first production of a phosphorylated glycoprotein using ENGase catalysis. In this work a synthetic glycan bearing M6P residues was converted to an oxazoline and transferred to both a glycosyl amino acid and de-glycosylated RNase B (dRNase B) using WT Endo A (Scheme 3e). Shortly afterwards Wang and co-workers published121 a similar approach, using both WT Endo A and the N171A Endo A mutant, and additionally reported surface plasmon resonance (SPR) studies of glycopeptide/protein binding to the immobilised cation independent mannose-6-phosphate receptor (CI-M6PR).
3.3 Production of remodelled mABs
A significant focus of recent attention has been application of ENGase-mediated remodelling of the glycans attached to the Fc region of IgGs, with the objective of maximising the effector functions of clinically administered mAbs. One key aspect of this work is that the heterogeneous N-glycans attached to Asn-297 of the Fc region of IgGs expressed in mammalian cell lines are frequently fucosylated, precluding their remodelling by many of the ENGases.In the first example (Scheme 4a) WT Endo A was used to remodel the Fc region of human IgG1, which was expressed in Pichia pastoris. The heterogeneous mixture of non-fucosylated yeast glycoforms was first trimmed back to a single GlcNAc at the Asn-297 residues of the homodimer by treatment with Endo H, and then extended with tetra- and hexasaccharide oxazolines.122 Alternatively high mannose glycoforms of IgG1 could be produced by expression in a mammalian cell line (CHO cells) in the presence of the glycosidase inhibitor kifunensine; again treatment with Endo H then trimmed back these glycans to a single GlcNAc at Asn-297. However, remodelling of the Fc region of IgG1 with the Endo M N175A and N175Q mutants was unsuccessful.123 Similarly WT Endo A, which is high mannose specific, was also unable to transfer full-length complex biantennary glycans to the Fc region, though it could attach a variety of truncated structures, including those with a ‘bisecting GlcNAc’.
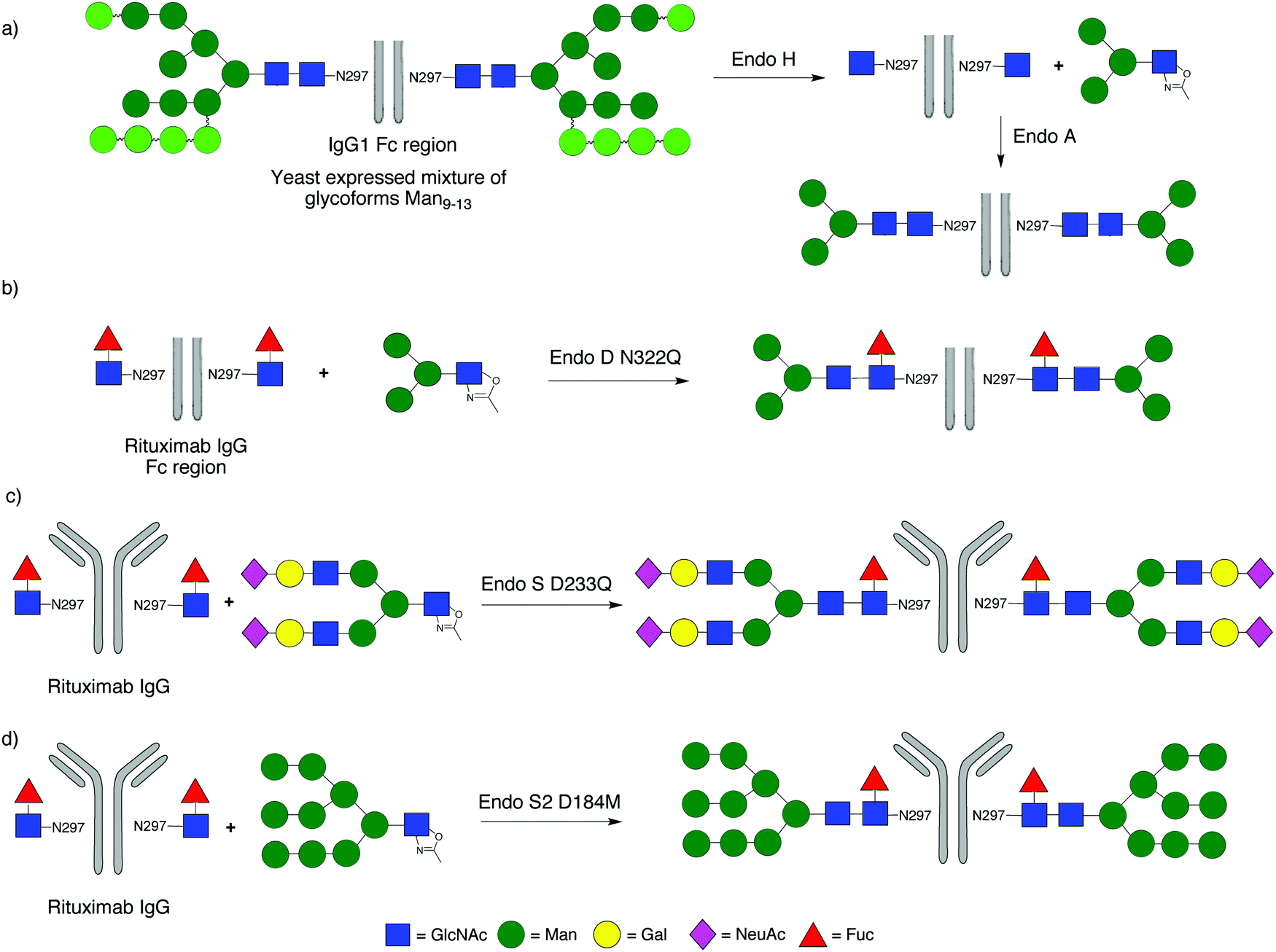 | ||
| Scheme 4 Selected examples of the remodelling of IgGs using ENGases; (a) Endo A catalysed attachment of a truncated core glycan to the non-fucosylated Fc region of human IgG; (b) Endo D N322Q mutant catalysed attachment of a truncated core glycan to the fucosylated Fc region of the mAb rituximab; (c) Endo S D233Q mutant catalysed attachment of a complex biantennary glycan to fucosylated intact rituximab; (d) Endo S2 D184M mutant catalysed attachment of a high mannose glycan to fucosylated intact rituximab. | ||
In subsequent studies Wang turned attention to glycosylation of IgG fragments that were core fucosylated. Firstly the IgG Fc region of the therapeutic mAb rituximab (used for the treatment of leukaemia and lymphoma) was produced by papain digestion, and the heterogeneous N-glycans at Asn-297 of commercial rituximab, which are core-fucosylated biantennary complex type oligosaccharides, were trimmed back to Fucα(1–6)GlcNAc using Endo S. Wang then demonstrated that the N322Q mutant of Endo D was capable of attaching the Man3GlcNAc tetrasaccharide oxazoline (Scheme 4b).71 However the narrow substrate specificity of Endo D precluded its application to larger oligosaccharide structures. Given these synthetic limitations attention turned to the family GH18 ENGase Endo S; an ENGase which preferentially acts hydrolytically on complex type N-glycans attached to folded IgG's. The first report of the synthetic use of WT Endo S by Davis98 demonstrated its ability to transfer a tetrasaccharide oxazoline to the fucosylated Fc region of human IgG. Shortly afterwards Wang99 developed D233A and D233Q glycosynthase mutants of Endo S, and applied them for remodelling of rituximab with full-length complex biantennary structures (Scheme 4c). Firstly the heterogeneous N-glycans at Asn-297 were trimmed back to Fucα(1–6)GlcNAc using WT Endo S. Then the D233A and D233Q Endo S mutants were shown to be capable of catalysing the attachment of a variety of glycans, including a full length complex biantennary glycan, as well as asialylated complex and core structures. In this study Wang also showed that the core fucose attached to the GlcNAc at Asn-297 of rituximab could be removed by a fucosidase after the hydrolytic action of Endo S, and that subsequently the D233A and D233Q Endo S mutants could also transfer glycans to the non-fucosylated GlcNAc. However, these Endo S mutants would not transfer high mannose structures.
The Wang Endo S D233Q mutant has subsequently been applied by others124 for the production of a variety of defined versions of IgGs, including the therapeutic mAbs herceptin125 and rituximab.126 Wang has also demonstrated127 the ability of the D126A mutant of Endo F3 (numbering after signal peptide cleavage) for the glycosylation of the fucosylated Fc region of rituximab with an asialo triantennary complex type oxazoline to produce homogeneous intact antibody glycoforms; a transformation that mutants of Endo S were incapable of performing. Most recently Wang34 has developed D184M and D184Q glycosynthase mutants of the new enzyme Endo S2, and shown that these have more relaxed substrate specificity than the corresponding Endo S mutants. For example they were capable of transferring all three major types of N-glycan (high mannose, hybrid and complex) to a range of IgGs, both with (Scheme 4d) and without core fucose in a highly efficient manner.
3.4 Scope and limitations
Although the use of ENGases is a powerful approach to access defined homogeneous glycoconjugates, there are currently some clear limitations. Firstly, and perhaps most obviously, their use only allows remodelling of N-glycan structures. As many mammalian glycoproteins, for example the therapeutic glycoprotein erythropoietin (EPO), also contain O-linked oligosaccharides, it is clear that ENGases cannot on their own be used to access versions comprising defined O-glycan structures. One possible solution is the use of glycosyl transferases to build more extended O-glycan structures from GalNAc-Ser/Thr, as has already been demonstrated by Wang for the production of glycopeptides bearing both defined N- and O-glycan structures.110 Alternatively one may in the future envisage the further128 synthetic development of endo-α-N-acetylgalactosaminidases, for example from family GH101, the use of which may perhaps allow the direct attachment of extended O-glycan structures to protein serine and threonine residues.Another issue is that many glycoproteins possess several different N-glycans at different glycosylation sites. Possible strategies to overcome this hurdle are already evident. For example in the very first ENGase-mediated synthesis of a glycopeptide bearing different N-glycans at two different sites Haneda and Toma used the differential hydrolytic and transglycosylation activities of Endo M and Endo A to transfer first a complex and then a high mannose glycan to different positions on the same peptide.129 Although ENGases have not yet been applied for the fabrication of glycoproteins bearing multiple different N-glycan structures, similar strategies may be envisaged. A caveat is that most ENGases have only rather broad hydrolytic specificity and so differentiating between different N-linked glycosylation sites may not always be possible. This is particularly true from a synthetic perspective where one may expect limited control over the relative rates of ENGase-catalysed glycosylation of different GlcNAc residues at different N-linked glycosylation sites; in the Haneda/Toma case129 it was perhaps fortuitous that one GlcNAc was considerably more reactive than the other, and so selectively was achievable.
Finally it is important to note that the use of ENGases together with oxazolines as donors is not without competing side reactions. For example, Davis125 and co-workers recently reported that a non-ENGase catalysed non-specific glycation of the IgG herceptin occurred during attempted glycosylation when using a large excess of oxazoline. In this instance Davis and colleagues were able to find conditions which suppressed this competing glycation; namely the use of the Endo S D233Q mutant at higher enzyme loading (10% w/w) and by performing multiple additions of oxazoline (e.g. 7 additions of 15 equivalents at 15 min intervals) to reduce its peak concentration.125 However, since analysis of the products of many ENGase-catalysed IgG and protein glycosylations has typically been performed solely on the basis of mass spectrometry, this type of glycation may have been previously overlooked (or at least underestimated), particularly in cases where large excesses of oxazoline have been used.
4. Conclusions
ENGase enzymes are potent biocatalysts that may be applied for the convergent synthesis of glycopeptides, and the re-modelling of the heterogeneous mixtures of glycoprotein glycoforms, including IgGs, that are invariably produced during expression. Two key developments have transformed the ENGases from being tools primarily for the Glycobiologist into highly effective and practical synthetic biocatalysts; namely the development of oxazolines as activated donors, and the production of glycosynthase mutants. An ever-increasing number of glycosynthase ENGase mutants is available. A summary of some of the most synthetically useful mutant enzymes produced so far is given in Table 2. At this point in time it is fair to say that there is no ‘best enzyme for every job’; rather each mutant has a different substrate tolerance, both with respect to donor (i.e. oxazoline structure) and acceptor (i.e. functionalization of the GlcNAc, and what is it attached to). The situation is therefore rather complex, and the precise specificities of each enzyme need to be considered in detail before a catalyst is selected.With respect to the cloning and expression of new enzymes, then considering the total number of ENGase genes already identified to date, it is clear that so far only the very surface has been scratched. Given the broad diversity of organisms that produce ENGases, and the wide variety of N-linked glycoconjugates on which they act, it is certain that new enzymes with useful action will continue to be identified, expanding the synthetic toolkit that is available. In terms of future applications the use of ENGase remodelling of therapeutic mABs in order to optimise glycan structure and maximise effector functions and subsequent clinical investigations of these materials appears to be only a matter of time. Moreover other significant applications of ENGases, for example for the production of optimised glycoprotein therapeutics and the convergent synthesis of therapeutic glycopeptides with improved pharmacokinetic properties, all beckon.
References
- A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, Essentials of Glycobiology, Cold Spring Harbour Laboratory Press, Cold Spring Harbour, NY, 2009 Search PubMed.
- A. Helenius and M. Aebi, Science, 2001, 291, 2364–2369 CrossRef CAS PubMed.
- K. R. Rodgers and R. C. Chou, Biotechnol. Adv., 2016, 34, 1149–1158 CrossRef CAS PubMed.
- R. Jefferis, Biotechnol. Prog., 2005, 21, 11–16 CrossRef CAS PubMed.
- R. Jefferis, Nat. Rev. Drug Discovery, 2009, 8, 226–234 CrossRef CAS PubMed.
- M. L. Reitman and S. Kornfeld, J. Biol. Chem., 1981, 256, 11977–11980 CAS.
- Y. Zhu, X. Li, J. Kyazike, Q. Zhou, B. L. Thurberg, N. Raben, R. J. Mattaliano and S. H. Cheng, J. Biol. Chem., 2004, 279, 50336–50341 CrossRef CAS PubMed.
- R. Kornfield and S. Kornfield, Annu. Rev. Biochem., 1985, 54, 631–664 CrossRef PubMed.
- N. Jenkins, R. B. Parekh and D. C. James, Nat. Biotechnol., 1996, 14, 975–981 CrossRef CAS PubMed.
- N. Sethuraman and T. A. Stadheim, Curr. Opin. Biotechnol., 2006, 17, 341–346 CrossRef CAS PubMed.
- (a) C. Unverzagt and Y. Kajihara, Chem. Soc. Rev., 2013, 42, 4408–4420 RSC; (b) D. P. Gamblin, E. M. Scanlan and B. G. Davis, Chem. Rev., 2009, 109, 131–163 CrossRef CAS PubMed.
- http://www.cazy.org. See: V. Lombard, H. Golaconda Ramulu, E. Drula, P. M. Coutinho and B. Henrissat, Nucleic Acids Res., 2013, 42, D490–D495 CrossRef PubMed.
- B. Henrissat and G. Davies, Curr. Opin. Struct. Biol., 1997, 7, 637–644 CrossRef CAS PubMed.
- J. Yin, L. Li, N. Shaw, Y. Li, J. K. Song, W. Zhang, C. Xia, R. Zhang, A. Joachimiak, H.-C. Zhang, L.-X. Wang, Z.-J. Liu and P. Wang, PLoS One, 2009, 4, e4658 Search PubMed.
- (a) F. Maley, R. B. Trimble, A. L. Tarentino and T. H. Plummer, Anal. Biochem., 1989, 180, 195–204 CrossRef CAS PubMed; (b) Y. Karamanos, S. Bourgerie, J. P. Barreaud and R. Julien, Res. Microbiol., 1995, 146, 437–443 CrossRef CAS PubMed.
- For some previous reviews see: (a) L.-X. Wang, Carbohydr. Res., 2008, 343, 1509–1522 CrossRef CAS PubMed; (b) L.-X. Wang and W. Huang, Curr. Opin. Chem. Biol., 2009, 13, 592–600 CrossRef CAS PubMed; (c) A. J. Fairbanks, C. R. Chim., 2011, 14, 44–58 CrossRef CAS.
- S. Kadowaki, K. Yamamoto, M. Fujisaki, K. Izumi, T. Tochikura and T. Yokoyama, Agric. Biol. Chem., 1990, 54, 97–106 CAS.
- B. F. Schmidt, E. Ashizawa, A. S. Jarnagin, S. Lynn, G. Noto, L. Woodhouse, D. A. Estell and P. Lad, Arch. Biochem. Biophys., 1994, 311, 350–353 CrossRef CAS PubMed.
- D. W. Abbott, M. S. Macauley, D. J. Vocadlo and A. B. Boraston, J. Biol. Chem., 2009, 284, 11676–11689 CrossRef CAS PubMed.
- I. Tews, A. van Scheltinga, A. Perrakis, K. Wilson and B. Dijkstra, J. Am. Chem. Soc., 1997, 119, 7954–7959 CrossRef CAS.
- M. Fujita, S.-i. Shoda, K. Haneda, T. Inazu, K. Takegawa and K. Yamamoto, Biochim. Biophys. Acta, 2001, 1528, 9–14 CrossRef CAS.
- For two personal perspectives on the parallel development of more extended oxazolines as donors for ENGase enzymes see: (a) L.-X. Wang, Trends Glycosci. Glycotechnol., 2011, 23, 33–52 CrossRef CAS PubMed; (b) A. J. Fairbanks, Pure Appl. Chem., 2013, 85, 1847–1863 CrossRef CAS.
- M. Noguchi, T. Tanaka, H. Gyakushi, A. Kobayashi and S.-I. Shoda, J. Org. Chem., 2009, 74, 2210–2212 CrossRef CAS PubMed.
- M. Umekawa, T. Higashiyama, Y. Koga, T. Tanaka, M. Noguchi, A. Kobayashi, S.-I. Shoda, W. Huang, L.-X. Wang, H. Ashida and K. Yamamoto, Biochim. Biophys. Acta, 2010, 1800, 1203–1209 CrossRef CAS PubMed.
- J. Q. Fan, K. Takegawa, S. Iwahara, A. Kondo, I. Kato, C. Abeygunawardana and Y. C. Lee, J. Biol. Chem., 1995, 270, 17723–17729 CrossRef CAS PubMed.
- E. Akaike, M. Tsutsumida, K. Osumi, M. Fujita, T. Yamanoi, K. Yamamoto and K. Fujita, Carbohydr. Res., 2004, 339, 719–722 CrossRef CAS PubMed.
- C. D. Heidecke, T. B. Parsons and A. J. Fairbanks, Carbohydr. Res., 2009, 344, 2433–2438 CrossRef CAS PubMed.
- For a recent review which also includes a discussion of ENGase glycosynthases see: P. M. Danby and S. G. Withers, ACS Chem. Biol., 2016, 11, 1784–1794 CrossRef CAS PubMed.
- L. F. Mackenzie, Q. R. Wang, R. A. J. Warren and S. G. Withers, J. Am. Chem. Soc., 1998, 120, 5583–5584 CrossRef CAS.
- C. Malet and A. Planas, FEBS Lett., 1998, 440, 208–212 CrossRef CAS PubMed.
- M. Umekawa, W. Huang, B. Li, K. Fujita, H. Ashida, L.-X. Wang and K. Yamamoto, J. Biol. Chem., 2008, 283, 4469–4479 CrossRef CAS PubMed.
- M. Umekawa, C. Li, T. Higashiyama, W. Huang, H. Ashida, K. Yamamoto and L.-X. Wang, J. Biol. Chem., 2010, 285, 511–521 CrossRef CAS PubMed.
- Y. Higuchi, Y. Eshima, Y. Huang, T. Kinoshita, W. Sumiyoshi, S.-I. Nakakita and K. Takegawa, Biotechnol. Lett., 2017, 39, 157–162 CrossRef CAS PubMed.
- T. Li, X. Tong, Q. Yang, J. P. Giddens and L.-X. Wang, J. Biol. Chem., 2016, 291, 16508–16518 CrossRef CAS PubMed.
- C. D. Heidecke, Z. Ling, N. C. Bruce, J. W. B. Moir, T. B. Parsons and A. J. Fairbanks, ChemBioChem, 2008, 9, 2045–2051 CrossRef CAS PubMed.
- K. Takegawa, M. Nakoshi, S. Iwahara, K. Yamamoto and T. Tochikura, Appl. Environ. Microbiol., 1989, 55, 3107–3112 CAS.
- J. Q. Fan, M. S. Quesenberry, K. Takegawa, S. Iwahara, A. Kondo, I. Kato and Y. C. Lee, J. Biol. Chem., 1995, 270, 17730–17735 CrossRef CAS PubMed.
- K. Takegawa, M. Tabuchi, S. Yamaguchi, A. Kondo, I. Kato and S. Iwahara, J. Biol. Chem., 1995, 270, 3094–3099 CrossRef CAS PubMed.
- K. Fujita and K. Takegawa, Biochem. Biophys. Res. Commun., 2001, 282, 678–682 CrossRef CAS PubMed.
- K. Takegawa, K. Yamabe, K. Fujita, M. Tabuchi, M. Mita, H. Izu, A. Watanabe, Y. Asada, M. Sano, A. Kondo, I. Kato and S. Iwahara, Arch. Biochem. Biophys., 1997, 338, 22–28 CrossRef CAS PubMed.
- K. Fujita and K. Takegawa, Biochem. Biophys. Res. Commun., 2001, 283, 680–686 CrossRef CAS PubMed.
- J. Q. Fan, L. H. Huynh, B. B. Reinhold, V. N. Reinhold, K. Takegawa, S. Iwahara, A. Kondo, I. Kato and Y. C. Lee, Glycoconjugate J., 1996, 13, 643–652 CrossRef CAS PubMed.
- Y. Tomabechi, M. A. Squire and A. J. Fairbanks, Org. Biomol. Chem., 2014, 12, 942–955 CAS.
- B. Li, Y. Zeng, S. Hauser, H. Song and L.-X. Wang, J. Am. Chem. Soc., 2005, 127, 9692–9693 CrossRef CAS PubMed.
- H. Li, B. Li, H. Song, L. Breydo, I. V. Baskakov and L.-X. Wang, J. Org. Chem., 2005, 70, 9990–9996 CrossRef CAS PubMed.
- B. Li, H. Song, S. Hauser and L.-X. Wang, Org. Lett., 2006, 8, 3081–3084 CrossRef CAS PubMed.
- Y. Zeng, J. Wang, B. Li, S. Hauser, H. Li and L.-X. Wang, Chem. – Eur. J., 2006, 12, 3355–3364 CrossRef CAS PubMed.
- T. W. D. F. Rising, C. D. Heidecke, J. W. B. Moir, Z. Ling and A. J. Fairbanks, Chem. – Eur. J., 2008, 14, 6444–6464 CrossRef CAS PubMed.
- M. D. L. Suits, Z. Ling, R. J. Bingham, N. C. Bruce, G. J. Davies, A. J. Fairbanks, J. W. B. Moir and E. J. Taylor, J. Mol. Biol., 2009, 389, 1–9 CrossRef PubMed.
- W. Huang, C. Li, B. Li, M. Umekawa, K. Yamamoto, X. Zhang and L.-X. Wang, J. Am. Chem. Soc., 2009, 131, 2214–2223 CrossRef CAS PubMed.
- S. Kadowaki, K. Yamamoto, M. Fujisaki, H. Kumagai and T. Tochikura, Agric. Biol. Chem., 1988, 52, 2387–2389 CAS.
- K. Yamamoto, S. Kadowaki, M. Fujisaki, H. Kumagai and T. Tochikura, Biosci., Biotechnol., Biochem., 1994, 58, 72–77 CrossRef CAS PubMed.
- K. J. Yamamoto, S. Kadowaki, J. Watanabe and H. Kumagai, Biochem. Biophys. Res. Commun., 1994, 203, 244–252 CrossRef CAS PubMed.
- K. Haneda, T. Inazu, K. Yamamoto, H. Kumagai, Y. Nakahara and A. Kobata, Carbohydr. Res., 1996, 292, 61–70 CrossRef CAS PubMed.
- K. Yamamoto, K. Fujimori, K. Haneda, M. Mizuno, T. Inazu and H. Kumagai, Carbohydr. Res., 1997, 305, 415–422 CrossRef CAS PubMed.
- K. Haneda, T. Inazu, M. Mizuno, R. Iguchi, K. Yamamoto, H. Kumagai, S. Aimoto, H. Suzuki and T. Noda, Bioorg. Med. Chem. Lett., 1998, 8, 1303–1306 CrossRef CAS PubMed.
- M. Mizuno, K. Haneda, R. Iguchi, I. Muramoto, T. Kawakami, S. Aimoto, K. Yamamoto and T. Inazu, J. Am. Chem. Soc., 1999, 121, 284–290 CrossRef CAS.
- K. Fujita, K. Kobayashi, A. Iwamatsu, M. Takeuchi, H. Kumagai and K. Yamamoto, Arch. Biochem. Biophys., 2004, 432, 41–49 CrossRef CAS PubMed.
- K. Osumi, Y. Makino, E. Akaike, T. Yamanoi, M. Mizuno, M. Noguchi, T. Inazu, K. Yamamoto and K. Fujita, Carbohydr. Res., 2004, 339, 2633–2635 CrossRef CAS PubMed.
- T. W. D. F. Rising, T. D. W. Claridge, N. Davies, D. P. Gamblin, J. W. B. Moir and A. J. Fairbanks, Carbohydr. Res., 2006, 341, 1574–1596 CrossRef CAS PubMed.
- T. W. D. F. Rising, C. D. Heidecke, J. W. B. Moir, Z. Ling and A. J. Fairbanks, Chem. – Eur. J., 2008, 14, 6444–6464 CrossRef CAS PubMed.
- T. B. Parsons, J. W. B. Moir and A. J. Fairbanks, Org. Biomol. Chem., 2009, 7, 3128–3140 CAS.
- T. W. D. F. Rising, T. D. W. Claridge, J. W. B. Moir and A. J. Fairbanks, ChemBioChem, 2006, 7, 1177–1180 CrossRef CAS PubMed.
- Y. Tomabechi, Y. Odate, R. Izumi, K. Haneda and T. Inazu, Carbohydr. Res., 2010, 345, 2458–2463 CrossRef CAS PubMed.
- T. Katoh, T. Katayama, Y. Tomabechi, Y. Nishikawa, J. Kumada, Y. Matsuzaki and K. Yamamoto, J. Biol. Chem., 2016, 291, 23305–23317 CrossRef CAS PubMed.
- T. Muramatsu, J. Biol. Chem., 1971, 246, 5535–5537 CAS.
- (a) N. Koide and T. Muramatsu, J. Biol. Chem., 1974, 249, 4897–4904 CAS; (b) T. Tai, K. Yamashita, M. Ogata-Arakawa, N. Koide, T. Muramatsu, S. Iwashita, Y. Inoue and A. Kobata, J. Biol. Chem., 1975, 250, 8569–8575 CAS; (c) N. Koide, M. Nose and T. Muramatsu, Biochem. Biophys. Res. Commun., 1977, 75, 838–844 CrossRef CAS PubMed; (d) T. Muramatsu, N. Koide and K.-i. Maeyama, J. Biochem., 1978, 83, 363–370 CrossRef CAS PubMed; (e) T. Muramatsu, Methods Enzymol., 1978, 50, 555–559 CAS.
- H. Muramatsu, H. Tachikui, H. Ushida, X.-j. Song, Y. Qiu, S. Yamamoto and T. Muramatsu, J. Biochem., 2001, 129, 923–928 CrossRef CAS PubMed.
- S. Yamamoto, H. Muramatsu and T. Muramatsu, Glycoconjugate J., 2005, 22, 35–42 CrossRef CAS PubMed.
- T. B. Parsons, M. K. Patel, A. B. Boraston, D. J. Vocadlo and A. J. Fairbanks, Org. Biomol. Chem., 2010, 8, 1861–1869 CAS.
- S.-Q. Fan, W. Huang and L.-X. Wang, J. Biol. Chem., 2012, 287, 11272–11281 CrossRef CAS PubMed.
- Y. Eshima, Y. Higuchi, T. Kinoshita, S. I. Nakakita and K. Takegawa, PLoS One, 2015, 10, e0132859 Search PubMed.
- S. Murakami, Y. Takaoka, H. Ashida, K. Yamamoto, H. Narimatsu and Y. Chiba, Glycobiology, 2013, 23, 736–744 CrossRef CAS PubMed.
- T. Kato, K. Fujita, M. Takeuchi, K. Kobayashi, S. Natsuka, K. Ikura, H. Kumagai and K. Yamamoto, Glycobiology, 2002, 12, 581–587 CrossRef CAS PubMed.
- T. Suzuki, K. Yano, S. Sugimoto, K. Kitajima, W. Lennarz, S. Inoue, Y. Inoue and Y. Emori, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9691–9696 CrossRef CAS PubMed.
- K. Fujita, H. Takami, K. Yamamoto and K. Takegawa, Biosci., Biotechnol., Biochem., 2004, 68, 1059–1066 CrossRef CAS PubMed.
- R. M. Fischl, J. Stadlmann, J. Grass, F. Altmann and R. Léonard, Plant Mol. Biol., 2011, 77, 275–284 CrossRef CAS PubMed.
- Y. Kimura, Y. Takeoka, M. Inoue, M. Maed and K. Fujiyama, Biosci., Biotechnol., Biochem., 2014, 75, 1019–1021 CrossRef PubMed.
- C. A. Waddling, T. H. Plummer, A. L. Tarentino and P. Van Roey, Biochemistry, 2000, 39, 7878–7885 CrossRef CAS PubMed.
- (a) A. L. Tarentino and F. Maley, J. Biol. Chem., 1974, 249, 811–817 CAS; (b) A. L. Tarentino, J. Thomas, H. Plummer and F. Maley, J. Biol. Chem., 1974, 249, 818–824 CAS.
- R. B. Trimble and F. Maley, Anal. Biochem., 1984, 141, 515–522 CrossRef CAS PubMed.
- R. A. O'Neill, J. Chromatogr. A, 1996, 720, 201–215 CrossRef.
- R. J. Trumbly, P. W. Robbins, M. Belfort, F. D. Ziegler, F. Maley and R. B. Trimble, J. Biol. Chem., 1985, 260, 5683–5690 CAS.
- V. Rao, C. Guan and P. V. Roey, Structure, 1995, 3, 449–457 CrossRef CAS PubMed.
- V. Rao, T. Cui, C. Guan and P. Van Roey, Protein Sci., 1999, 8, 2338–2346 CrossRef CAS PubMed.
- R. B. Trimble, P. H. Atkinson, A. L. Tarentino, T. H. Plummer, F. Maley and K. B. Tomer, J. Biol. Chem., 1986, 261, 12000–12005 CAS.
- W. Huang, J. Li and L.-X. Wang, ChemBioChem, 2011, 12, 932–941 CrossRef CAS PubMed.
- J. H. Elder and S. Alexander, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 4540–4544 CrossRef CAS.
- A. L. Tarentino, G. Quinones, W. P. Schrader, L. M. Changchien and J. T. H. Plummer, J. Biol. Chem., 1992, 267, 3868–3872 CAS.
- A. L. Tarentino, G. Quinones, L. M. Changchien and T. H. Plummer, J. Biol. Chem., 1993, 268, 9702–9708 CAS.
- P. Van Roey, V. Rao, T. H. Plummer and A. L. Tarentino, Biochemistry, 1994, 33, 13989–13996 CrossRef CAS PubMed.
- R. B. Trimble and A. L. Tarentino, J. Biol. Chem., 1991, 266, 1646–1651 CAS.
- B. Grimwood, T. Plummer and A. Tarentino, Glycobiology, 1998, 8, 633–636 CrossRef.
- J. P. Giddens, J. V. Lomino, M. N. Amin and L.-X. Wang, J. Biol. Chem., 2016, 291, 9356–9370 CrossRef CAS PubMed.
- M. Collin and A. Olsen, EMBO J., 2001, 20, 3046–3055 CrossRef CAS PubMed.
- M. Allhorn, A. Olsén and M. Collin, BMC Microbiol., 2008, 8, 3 CrossRef PubMed.
- M. Collin and A. Olsen, Infect. Immun., 2001, 69, 7187–7189 CrossRef CAS PubMed.
- J. J. Goodfellow, K. Baruah, K. Yamamoto, C. Bonomelli, B. Krishna, D. J. Harvey, M. Crispin, C. N. Scanlan and B. G. Davis, J. Am. Chem. Soc., 2012, 134, 8030–8033 CrossRef CAS PubMed.
- W. Huang, J. Giddens, S.-Q. Fan, C. Toonstra and L.-X. Wang, J. Am. Chem. Soc., 2012, 134, 12308–12318 CrossRef CAS PubMed.
- B. Trastoy, J. V. Lomino, B. G. Pierce, L. G. Carter, S. Günther, J. P. Giddens, G. A. Snyder, T. M. Weiss, Z. Weng, L.-X. Wang and E. J. Sundberg, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6714–6719 CrossRef CAS PubMed.
- J. Sjögren, W. B. Struwe, E. F. J. Cosgrave, P. M. Rudd, M. Stervander, M. Allhorn, A. Hollands, V. Nizet and M. Collin, Biochem. J., 2013, 455, 107–118 CrossRef PubMed.
- J. Sjögren, E. F. J. Cosgrave, M. Allhorn, M. Nordgren, S. Björk, F. Olsson, S. Fredriksson and M. Collin, Glycobiology, 2015, 25, 1053–1063 CrossRef PubMed.
- I. Stals, S. Karkehabadi, S. Kim, M. Ward, A. Van Landschoot, B. Devreese and M. Sandgren, PLoS One, 2012, 7, e40854 CAS.
- I. Stals, B. Samyn, K. Sergeant, T. White, K. Hoorelbeke, A. Coorevits, B. Devreese, M. Claeyssens and K. Piens, FEMS Microbiol. Lett., 2010, 303, 9–17 CrossRef CAS PubMed.
- T. Hamaguchi, T. Ito, Y. Inoue, T. Limpaseni, P. Pongsawasdi and K. Ito, Glycobiology, 2010, 20, 420–432 CrossRef CAS PubMed.
- J. Garbe, J. Sjögren, E. F. J. Cosgrave, W. B. Struwe, M. Bober, A. I. Olin, P. M. Rudd and M. Collin, PLoS One, 2014, 9, e91035 Search PubMed.
- M. Collin and V. A. Fischetti, J. Biol. Chem., 2004, 279, 22558–22570 CrossRef CAS PubMed.
- H. Li, S. Singh, Y. Zeng, H. Song and L. Wang, Bioorg. Med. Chem. Lett., 2005, 15, 895–898 CrossRef CAS PubMed.
- L.-X. Wang, H. Song, S. Liu, H. Lu, S. Jiang, J. Ni and H. Li, ChemBioChem, 2005, 6, 1068–1074 CrossRef CAS PubMed.
- W. Huang, X. Zhang, T. Ju, R. D. Cummings and L.-X. Wang, Org. Biomol. Chem., 2010, 8, 5224–5233 CAS.
- J. V. Lomino, A. Naegeli, J. Orwenyo, M. N. Amin, M. Aebi and L.-X. Wang, Bioorg. Med. Chem., 2013, 21, 2262–2270 CrossRef CAS PubMed.
- H. Hojo, H. Tanaka, M. Hagiwara, Y. Asahina, A. Ueki, H. Katayama, Y. Nakahara, A. Yoneshige, J. Matsuda, Y. Ito and Y. Nakahara, J. Org. Chem., 2012, 77, 9437–9446 CrossRef CAS PubMed.
- J. D. McIntosh, M. A. Brimble, A. E. S. Brooks, P. R. Dunbar, R. Kowalczyk, Y. Tomabechi and A. J. Fairbanks, Chem. Sci., 2015, 6, 4636–4642 RSC.
- Y. Tomabechi, G. Krippner, P. M. Rendle, M. A. Squire and A. J. Fairbanks, Chem. – Eur. J., 2013, 19, 15084–15088 CrossRef CAS PubMed.
- R. Kowalczyk, M. A. Brimble, Y. Tomabechi, A. J. Fairbanks, M. Fletcher and D. L. Hay, Org. Biomol. Chem., 2014, 12, 8142–8151 CAS.
- K. Fujita and K. Yamamoto, Biochim. Biophys. Acta, 2006, 1760, 1631–1635 CrossRef CAS PubMed.
- H. Ochiai, W. Huang and L.-X. Wang, J. Am. Chem. Soc., 2008, 130, 13790–13803 CrossRef CAS PubMed.
- W. Huang, Q. Yang, M. Umekawa, K. Yamamoto and L.-X. Wang, ChemBioChem, 2010, 11, 1350–1355 CrossRef CAS PubMed.
- M. N. Amin, W. Huang, R. M. Mizanur and L.-X. Wang, J. Am. Chem. Soc., 2011, 133, 14404–14417 CrossRef CAS PubMed.
- P. Priyanka, T. B. Parsons, A. Miller, F. M. Platt and A. J. Fairbanks, Angew. Chem., Int. Ed., 2016, 55, 5058–5061 CrossRef CAS PubMed.
- T. Yamaguchi, M. N. Amin, C. Toonstra and L.-X. Wang, J. Am. Chem. Soc., 2016, 138, 12472–12485 CrossRef PubMed.
- Y. Wei, C. Li, W. Huang, B. Li, S. Strome and L.-X. Wang, Biochemistry, 2008, 47, 10294–10304 CrossRef CAS PubMed.
- G. Zou, H. Ochiai, W. Huang, Q. Yang, C. Li and L.-X. Wang, J. Am. Chem. Soc., 2011, 133, 18975–18991 CrossRef CAS PubMed.
- M. Kurogochi, M. Mori, K. Osumi, M. Tojino, S.-I. Sugawara, S. Takashima, Y. Hirose, W. Tsukimura, M. Mizuno, J. Amano, A. Matsuda, M. Tomita, A. Takayanagi, S.-I. Shoda and T. Shirai, PLoS One, 2015, 10, e0132848 Search PubMed.
- T. B. Parsons, W. B. Struwe, J. Gault, K. Yamamoto, T. A. Taylor, R. Raj, K. Wals, S. Mohammed, C. V. Robinson, J. L. P. Benesch and B. G. Davis, Angew. Chem., Int. Ed., 2016, 55, 2361–2367 CrossRef CAS PubMed.
- C.-W. Lin, M.-H. Tsai, S.-T. Li, T.-I. Tsai, K.-C. Chu, Y.-C. Liu, M.-Y. Lai, C.-Y. Wu, Y.-C. Tseng, S. S. Shivatare, C.-H. Wang, P. Chao, S.-Y. Wang, H.-W. Shih, Y.-F. Zeng, T.-H. You, J.-Y. Liao, Y.-C. Tu, Y.-S. Lin, H.-Y. Chuang, C.-L. Chen, C.-S. Tsai, C.-C. Huang, N.-H. Lin, C. Ma, C.-Y. Wu and C.-H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10611–10616 CrossRef CAS PubMed.
- J. P. Giddens, J. V. Lomino, M. N. Amin and L.-X. Wang, J. Biol. Chem., 2016, 291, 9356–9370 CrossRef CAS PubMed.
- (a) H. Ashida, H. Ozawa, K. Fujita, S. Suzuki and K. Yamamoto, Glycoconjugate J., 2010, 27, 125–132 CrossRef CAS PubMed; (b) H. Ashida, K. Yamamoto, T. Murata, T. Usui and H. Kumagai, Arch. Biochem. Biophys., 2000, 373, 394–400 CrossRef CAS PubMed.
- K. Haneda, M. Takeuchi, M. Tagashira, T. Inazu, K. Toma, Y. Isogai, M. Hori, K. Kobayashi, M. Takeuchi, K. Takegawa and K. Yamamoto, Carbohydr. Res., 2006, 341, 181–190 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |