
Multifunctional metal–organic framework catalysts: synergistic catalysis and tandem reactions
Yuan-Biao
Huang
,
Jun
Liang
,
Xu-Sheng
Wang
and
Rong
Cao
*
State Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, China. E-mail: rcao@fjirsm.ac.cn
First published on 14th November 2016
Abstract
Metal–organic frameworks (MOFs) are porous crystalline materials constructed from metal ions or clusters and multidentate organic ligands. Recently, the use of MOFs or MOF composites as catalysts for synergistic catalysis and tandem reactions has attracted increasing attention due to their tunable open metal centres, functional organic linkers, and active guest species in their pores. In this review, the applications of MOFs with multiple active sites in synergistic organic catalysis, photocatalysis and tandem reactions are discussed. These multifunctional MOFs can be categorized by the type of active centre as follows: (i) open metal centres and functional organic linkers in the MOF structure, (ii) active guest sites in the pores and active sites in the MOF structure, and (iii) bimetallic nanoparticles (NPs) on MOF supports. The types of synergistic catalysis and tandem reactions promoted by multifunctional MOFs and their proposed mechanisms are presented in detail. Here, catalytic MOFs with a single type of active site and MOFs that only serve as supports to enhance substrate adsorption are not discussed.
 Yuan-Biao Huang | Dr Yuan-Biao Huang obtained his PhD in 2009 under the supervision of Prof. G.-X. Jin from Fudan University, where he focused on olefin polymerization catalysis. In the same year, he joined Prof. Rong Cao's group at Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences. In 2014, he joined Prof. Qiang Xu's group at National Institute of Advanced Industrial Science and Technology as a JSPS (Japan Society for the Promotion of Science) fellow. In 2015, he moved back to FJIRSM Cao's research group at FJIRSM. His research interests are focused on porous MOF materials for heterogeneous catalysis. |
 Jun Liang | Jun Liang studied chemistry at Northeast Normal University, where he obtained his MSc working on the synthesis of metal–organic rotaxane frameworks in June 2014. In the same year, he joined the group of Prof. Cao at the Fujian Institute of Research on Structure of Matter (FJIRSM), Chinese Academy of Sciences, as a PhD student of Xiamen University. His research interests are focused on porous MOFs for sustainable conversion of CO2 into value-added chemicals. |
 Xu-sheng Wang | Xu-Sheng Wang was born in Hebei, P. R. China, in 1988. He received his Master’s degree from the Fujian Institute of Research on the Structure of Matter (FJIRSM), Chinese Academy of Sciences (CAS), in 2014 under the supervision of Prof. Rong Cao. After one year as a Research Assistant at FJIRSM, he started as a PhD student in the same research group. His research currently focuses on porous MOF composites for catalysis and clean energy transformation. |
 Rong Cao | Rong Cao was born in Fujian province, China. He obtained his PhD from Fujian Institute of Research on the Structure of Matter (FJIRSM), Chinese Academy of Sciences (CAS), in 1993. Following post-doctoral experience in the Hong Kong Polytechnic University and JSPS Fellowship in Nagoya University, he became a professor at FJIRSM in 1998. Now, he is the director of FJIRSM. His main research interests include supramolecular chemistry, inorganic–organic hybrid materials and nanocatalysis. |
1. Introduction and scope of this review
Currently, people are becoming increasingly concerned with the growing energy shortage and environmental pollution. A sustainable development model that integrates considerations of economic viability and environmental integrity must be developed. In particular, the development of sustainable chemical science involves new technologies and efficient processes, which are considered to be effective means for addressing environmental issues. Improving catalytic efficiencies, reducing waste, and using environmentally friendly reagents are the key factors of green chemistry.1 From a sustainable chemistry viewpoint, synergistic catalysis and tandem reactions are efficient, environmentally friendly chemical synthesis methods.Synergistic catalysis is a powerful strategy in which the reactants are simultaneously activated by two or more distinct active sites and the reaction energy barrier is significantly lowered.1 As a result, the efficiency and selectivity of existing reactions can be improved, and new chemical transformations can be realized. Synergistic catalysts are composed of two catalysts or one catalyst with two or more active sites and have been used for organic catalysis,2 photocatalysis,3 nanocatalysis,4 electrocatalysis,5 and enzyme catalysis.6 In synergistic catalysis, the cooperative action of the active sites leads to an increase in the HOMO (highest occupied molecular orbital) energy level of one reactant and a decrease in the LUMO (lowest unoccupied molecular orbital) energy level of the other reactant. Due to the judicious combination of multifunctional active species, the resulting catalysts can synergistically promote many chemical reactions that cannot be easily or efficiently catalysed by a single active species.
Tandem or domino (cascade) reactions consist of two or more successive independent reactions performed in a one pot system without separating and purifying the intermediates.1,7 The reactions can be catalysed by a single active site (catalyst) sequentially, but this approach is usually limited to reactions that have similar mechanisms (Scheme 1a). More frequently, tandem reactions are promoted by two catalysts or a bifunctional catalyst with two or more types of active sites. The reactants are activated by the first type of active site (catalyst) to produce an intermediate, which is further catalysed by the second type of active site (catalyst) to give the final product (Scheme 1b). Consequently, tandem reactions not only reduce the number of reaction steps, energy consumption and waste but also minimize the use of solvents and reagents.8 These advantages make tandem reactions sustainable green processes that illustrate the concepts of efficiency and atom economy. Tandem reactions offer many opportunities for improving chemical transformations such as Lewis acid–base catalysis, hydrogenation reactions and alkene metathesis.8
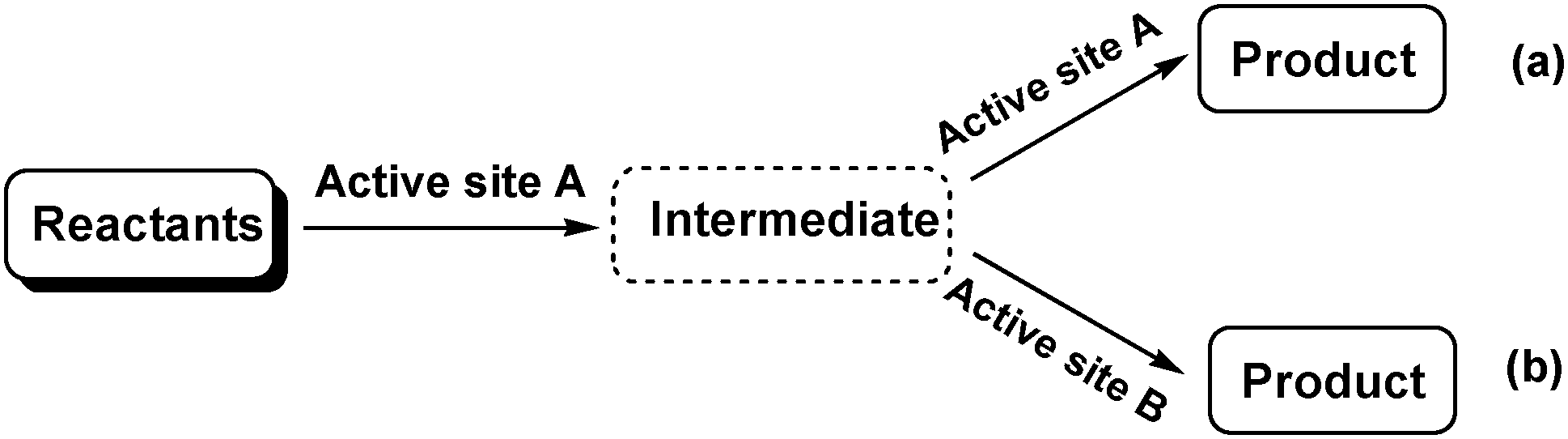 | ||
| Scheme 1 Tandem reactions catalysed by (a) one active site or (b) two types of active sites. | ||
Metal–organic frameworks (MOFs), which are also known as porous coordination polymers (PCPs), are porous crystalline materials consisting of metal ions or clusters coordinated to multidentate organic ligands to form two- or three-dimensional infinite structures.9–13 The structures, topologies, pores and functionalities of MOFs can be easily designed and tuned by changing the metal nodes and ligands and by post-synthetic modification (PSM).14–18 Over the past several years, MOFs have become one of the most popular research areas in the chemical and materials sciences due to their large surface areas, tunable pore sizes and versatile architectures. These outstanding features make MOFs promising materials for use in applications such as gas adsorption and separation,19,20 heterogeneous catalysis,21–26 sensing,27 luminescence,28 proton conduction,29 and drug delivery.30 In particular, MOFs have attracted much attention in organic catalysis,22 asymmetric catalysis,25 and photocatalysis.23 The possible compositions and structures of MOFs are nearly infinite, as demonstrated by the large number of metal nodes and functional ligands that can be used in their fabrication. These MOF building blocks can be employed as active centres for organic catalysis and photocatalysis. In particular, compared with other porous materials, homochiral MOFs for asymmetric catalysis can be synthesized relatively easily using enantiotropic ligands. Other main features of MOFs include their large surface areas and pore volumes, which enable active guest species to be introduced into the pores/cages/channels and allow substrates to access the internal active sites. Thus, the combination of different types of MOF active sites, such as metal nodes, functional organic linkers, and guest species in the pores, makes MOFs promising multifunctional materials for synergistic catalysis and tandem reactions (Fig. 1).
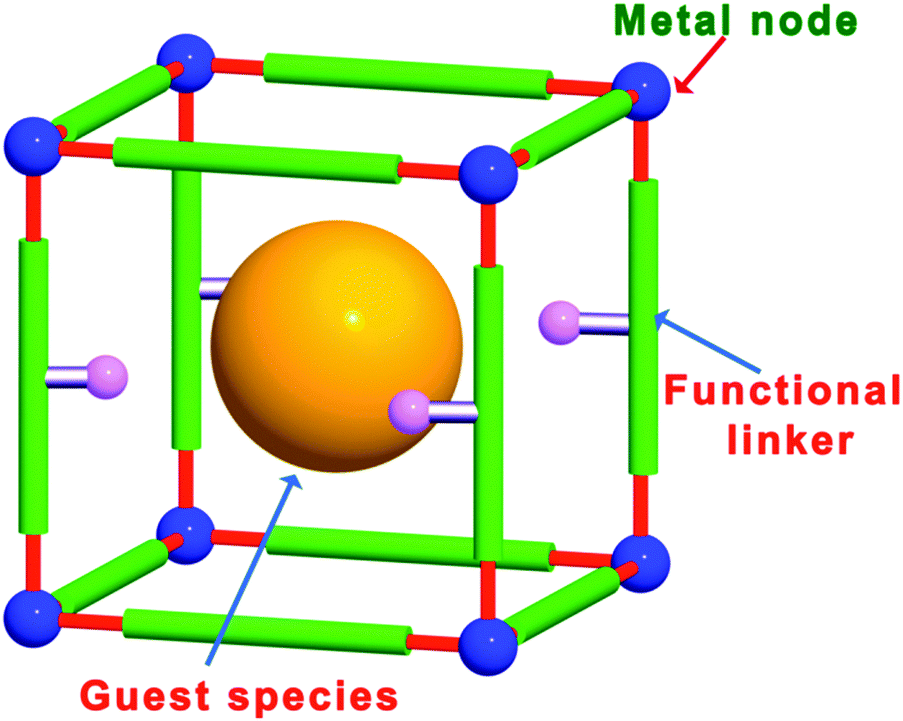 | ||
| Fig. 1 Different types of MOF active sites, including metal nodes, functional organic linkers, and guest species in the pores. | ||
In this review, the reports of the use of MOFs with multiple types of active sites as multifunctional catalysts for synergistic catalysis and/or tandem reactions are summarized. The active sites of MOF catalysts can be categorized as follows: (i) coordinatively unsaturated metal (CUM) centres and functional linkers, (ii) functional groups attached to the linkers and/or metal centres by direct synthesis or PSM, and (iii) active guest species such as metal nanoparticles (MNPs), complexes, and polyoxometallates (POMs) encapsulated in the pores. The comprehensive discussion of the use of MOFs in synergistic catalysis and tandem reactions is mainly divided into the following sections based on the type of active sites: (i) two or more active sites on the MOF backbone, (ii) active site@MOF composites, (iii) bimetallic NPs on MOF supports, and (iv) multifunctional MOF catalysts for synergistic photocatalysis. It should be noted that reports of tandem reactions catalysed by a single active site and MOFs acting only as supports to enhance substrate adsorption are not discussed.22,24 The design and preparation of multifunctional MOFs, as well as their use in synergistic catalysis and tandem reactions and their catalytic mechanisms, are described. Hopefully, this review will attract the attention of chemists and stimulate more research on the design of multifunctional MOF-based materials based on the synergistic catalysis and tandem reaction mechanisms discussed below.
2. Bifunctional active sites on MOF backbones
The many different metal ions or clusters and organic linkers with pendant functional groups that can be used to fabricate MOFs make MOFs versatile heterogeneous catalysts for various organic transformations, such as cyanosilylation, oxidation, aldol condensation, and transesterification reactions. In particular, the different combinations of mixed metal centres, mixed ligands, metal centres and ligands in MOFs can act as bifunctional active sites to synergistically enhance the catalytic performance and/or promote tandem reactions. Examples of bifunctional MOFs and the reactions that they catalyse are summarized in Table 1.| MOF | Reaction(s) | Active sites | Ref. |
|---|---|---|---|
| InGaPF | Strecker reaction | Acid, base | 31 |
| MIL-100(Sc,Fe) | Friedel–Crafts addition | Lewis acid | 32 |
| Cu/ZIF-8 |
Friedländer reaction
Combes condensation |
Acid sites | 33 |
| Cu/ZIF-8 | [3+2] Huisgen dipolar cycloaddition | Acid sites | 33 |
| Cu/ZIF-67 | Photodegradation of methyl orange | Redox sites | 35 |
| Ag/HKUST-1 | Toluene selective oxidation | Redox sites | 36 |
| MIL-101–SO3H–NH2 | Deacetalization–Henry reaction | Acid, base | 38 and 68 |
| IRMOF-9-Irdcppy-NH2 | Knoevenagel condensation and allylic N-alkylation | Base, metal complex | 39 |
| USTC-253–TFA | CO2 cycloaddition | Lewis and Brønsted acid | 41 |
| MIL-101–Cr–SO3H·Al(III) | Benzylation | Lewis and Brønsted acid | 44 |
| UiO-66-NH2 | Cross-aldol condensation | Acid, base | 45 |
| UiO-66-NH2 | Knoevenagel condensation | Acid, base | 46 and 47 |
| IRMOF-3 | Knoevenagel condensation | Acid, base | 48 and 49 |
| Tb–TCA | Knoevenagel condensation | Acid, base | 50 |
| MOF-74 | Knoevenagel condensation and Michael addition | Acid, base | 51 |
| UiO-66-NH2 | CO2 cycloaddition | Acid, base | 52 |
| ZIF-8-H | [3+3] cycloaddition | Acid, base | 53 |
| ZIF-8 | Transesterification | Acid, base | 54 |
| PCN-124 | Deacetalization–Knoevenagel condensation | Acid, base | 55 |
| MIL-101(Al)-NH2 | Deacetalization–Knoevenagel condensation | Acid, base | 56 |
| MIL-101(Al)-NH2 | Meinwald rearrangement–Knoevenagel condensation | Acid, base | 60 |
|
UiO-66-NH2LIr
IRMOF-3-LIr |
Nitroarene reductive N-alkylation with aldehydes | Acid, metal complex | 62 |
|
(NNN)–M–Zr-MOF
M = Rh, Ir |
Olefination–hydrogenation | Base, metal complex | 63 |
| Fe@Hf-2 | Epoxidation–epoxide ring-opening | Acid, metal complex | 64 |
| Cu3(BTC)2–[Pd] | Sonogashira–click | Acid, metal complex | 69 |
| (salen)MnCl-based CMOF-1 | Epoxidation–epoxide ring-opening | Acid, metal complex | 71 |
2.1 Mixed metal centres as active sites for synergistic catalysis and tandem reactions
Solid-solution MOFs in which the mixed metal centres occupy equivalent crystallographic framework sites exhibit enhanced catalytic activities and/or yield different products. The solid-solution InGaPF MOFs with the formula [InxGa1−x(O2C2H4)0.5(hfipbb)] (H2hfipbb = 4,4′-(hexafluoroisopropylidene)bis(benzoic acid)) contain both Lewis acid (metal centres) and Lewis base (bridging oxygens) sites. These MOFs were synthesized and were found to exhibit high activity and selectivity in a three-component, one-pot Strecker reaction to produce α-aminonitrile.31 InGaPF MOFs are isostructural with their monometal counterparts, i.e., AlPF-1 (Al(OH)(hfipbb)), GaPF-1 (Ga(OH)(hfipbb)), and InPF-11β (In(O2C2H4)0.5(hfipbb)). Interestingly, when these materials were used in the one-pot Strecker reaction between benzaldehyde, trimethylsilyl cyanide (TMSCN) and aniline, different products were obtained (Scheme 2). The expected Strecker reaction product α-aminonitrile was obtained when AlPF-1 was used. However, in the case of GaPF-1, the aldehyde cyanosilylation product was obtained, suggesting that both the silyl and carbonyl groups were quickly activated and TMSCN had completely reacted before imine formation occurred. When InPF-11β was used as the catalyst, the main final product was the imine, indicating that the silyl group was activated by the Lewis base site and the cyano group addition was inhibited. Surprisingly, the α-aminonitrile product was formed quantitatively within 0.33 h when the mixed metal solid–solution In0.28Ga0.72PF MOF was employed. In this reaction, benzaldehyde was firstly activated by the Lewis acid sites, and the imine was formed. Then, the Lewis base-activated TMSCN attacked the imine group, resulting in the formation of α-aminonitrile, which could be hydrolysed to obtain an α-amino acid. More importantly, although ketones are more difficult to activate than aldehydes, In0.28Ga0.72PF could catalyse their conversion to Strecker products with high yields.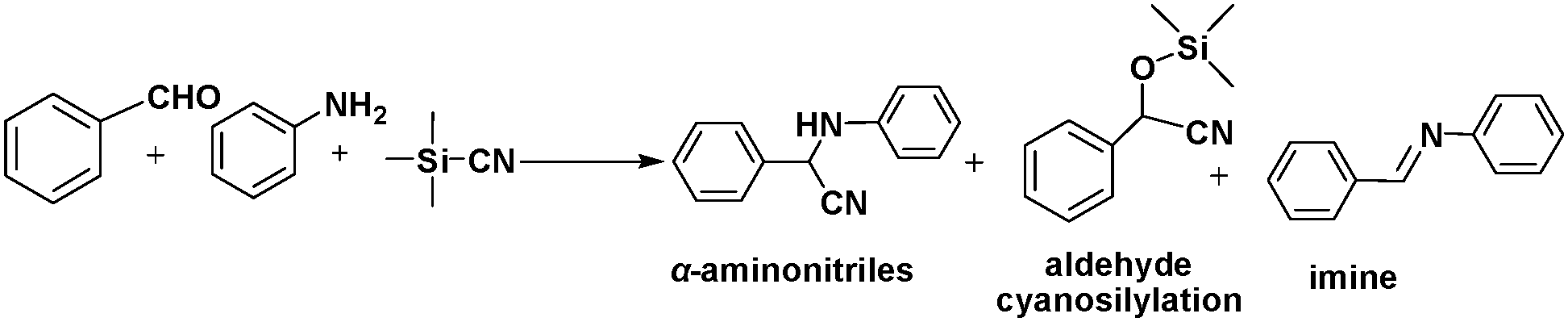 | ||
| Scheme 2 Strecker reaction of benzaldehyde, aniline, and TMSCN. | ||
In one study, Fe(III) was incorporated into MIL-100(Sc) to form a solid–solution MIL-100(Sc,Fe) MOF that could catalyse oxidation reactions.32 In particular, the MIL-100(Sc60Fe40) catalyst contained CUMs and could act as a bifunctional Lewis acid-oxidation catalyst for the tandem reaction consisting of the Friedel–Crafts addition of 2-methylindole to trifluoroacetaldehyde ethyl hemiacetal, followed by the oxidation of the product in the presence of t-butyl hydroperoxide (Scheme 3). Moreover, other substrates, such as indole, N-methylindole, pyrrole, and dimethoxybenzene, could also be converted by the MOF. In contrast, a simple physical mixture of MIL-100(Sc) and MIL-100(Fe) with the same Sc/Fe ratio exhibited lower conversions in the tandem reaction, providing evidence of the synergistic effect of the Lewis acid centres and oxidation sites in the mixed-metal MOF.
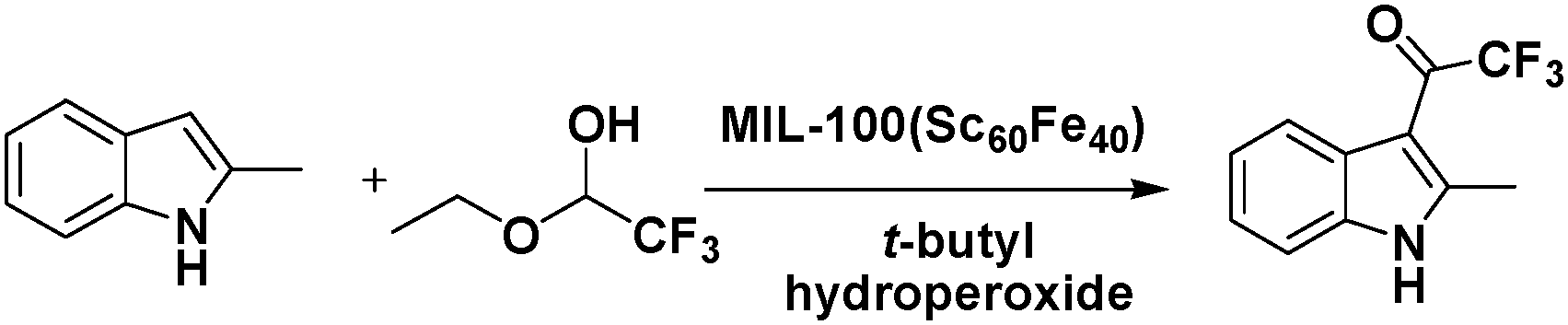 | ||
| Scheme 3 Tandem Friedel–Crafts addition of 2-methylindole to trifluoroacetaldehyde ethyl hemiacetal. | ||
Cu(II) was doped into the nodes of zeolitic imidazolate framework ZIF-8 to give Cu/ZIF-8 by reacting Cu(NO3)2, Zn(NO3)2, and 2-methylimidazole.33 The Cu(II) doping significantly increased the Lewis acidity of the ZIF. Therefore, quantitative product yields were obtained in the Friedländer reaction of 2-aminobenzophenone with an active methylene compound catalysed by the Cu/ZIF-8 material (Scheme 4a). Furthermore, the Combes condensation of aniline with acetylacetone also proceeded smoothly to give 2,4-dimethylquinoline when Cu5%/ZIF-8 was used (Scheme 4b). More importantly, the Cu/ZIF-8 material doped with 25% Cu(II) exhibited excellent activity for the [3+2] Huisgen dipolar cycloaddition of organic azides to alkynes to give 1,2,3-triazoles with very high regioselectivity (Scheme 4c). Notably, this reaction is typically catalysed by Cu(I) catalysts.34 Copper was doped into the metal nodes of cobalt zeolitic imidazolate framework ZIF-67 by mixing Cu(COO)2, Co(COO)2 and 2-methylimidazole, and the resulting material exhibited enhanced photocatalytic activity.35 Although Cu(COO)2 was added as a metal salt, the authors proposed that the Cu(II) ion was partially reduced to Cu(I) in the presence of ethyleneurea hemihydrate under the solvothermal conditions. Although the copper location and valence state were unknown, the copper doping enhanced the photocatalytic degradation of methyl orange. In another study, the copper ions in the nodes of Cu3(BTC)2 were partially replaced by Ag(I) using a post-synthetic exchange (PSE) method.36 The resulting material was used to catalyse the selective oxidation of toluene to benzaldehyde. Although doping Cu3(BTC)2 with Ag led to an increase in the conversion from 6.5% to 12.7%, the Ag ion distribution and the valence state of the compound were unknown. Nevertheless, introducing another metal ion by direct synthesis or PSE can enhance the activity and selectivity of a MOF catalyst because it alters the Lewis acidity and electronic structure of the parent MOF.
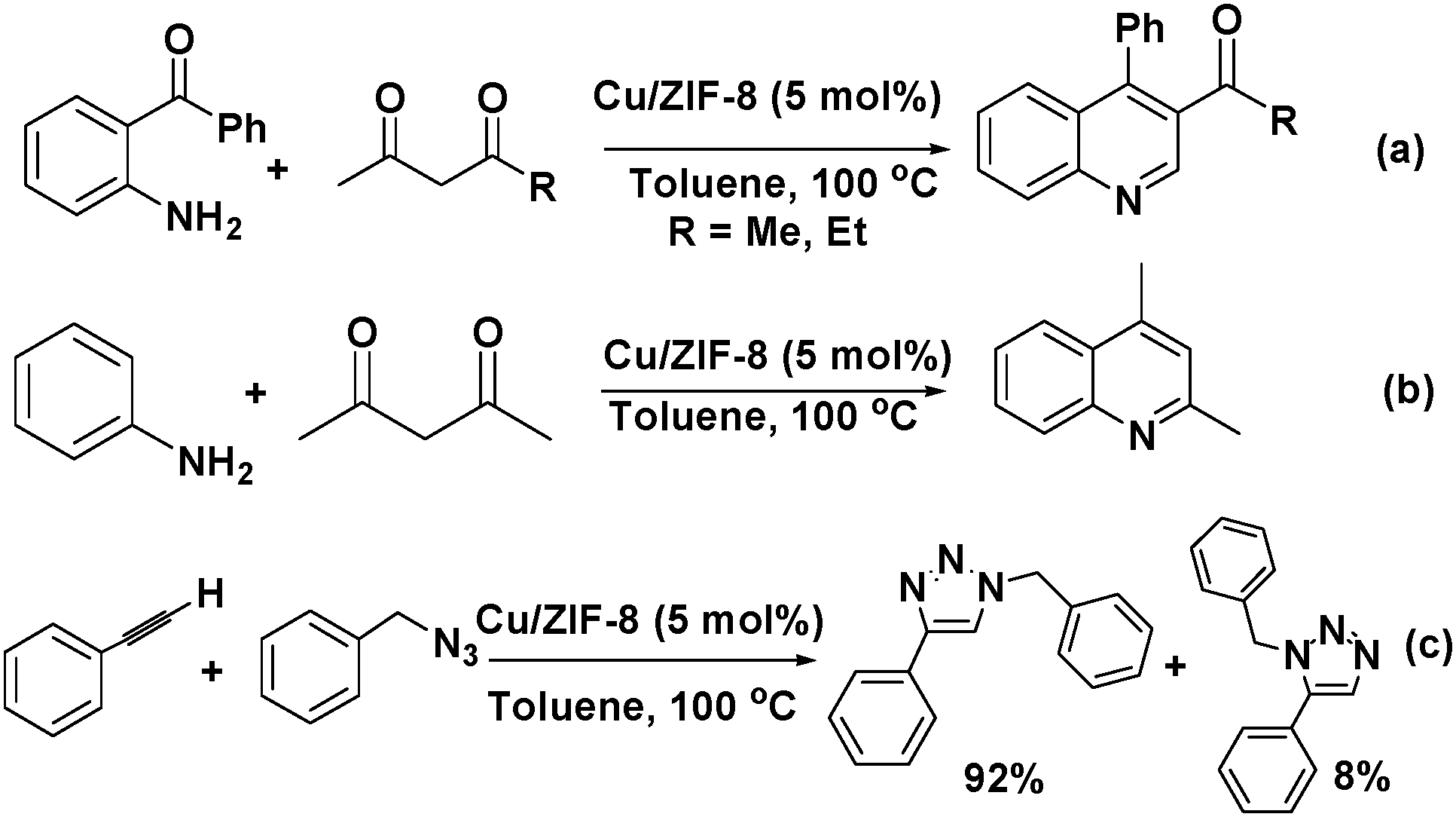 | ||
| Scheme 4 Reactions catalysed by Cu-doped ZIF-8: (a) Friedländer reaction of 2-aminobenzophenone and an active methylene compound, (b) Combes condensation of aniline with acetylacetone, and (c) [3+2] Huisgen dipolar cycloaddition of organic azides to alkynes. | ||
2.2 Mixed linkers as bifunctional active sites for tandem reactions
Incorporating different active sites onto the same linker or using mixed linkers in one MOF could provide a multifunctional platform for synergistic catalysis and/or tandem reactions.37 To circumvent the weak Lewis acidity of CUMs, Ahn and coworkers prepared the site-isolated, bifunctional Brønsted acid–base catalyst MIL-101(Cr)–NH2–SO3H by using two different (–NO2, –SO3H) terephthalate linkers in the synthesis and then reducing the resulting MOF (Scheme 5).38 The bifunctional Brønsted acid–base catalyst achieved a very high conversion and selectivity in the one-pot tandem deacetalization–nitroaldol reaction converting benzaldehyde dimethyl acetal to trans-1-nitro-2-phenylethylene (Scheme 6). Notably, the coexistence of acid and base sites, which was achieved in this MOF, cannot be achieved simultaneously in homogeneous catalysts, making this unique MOF a promising catalyst for tandem reactions.7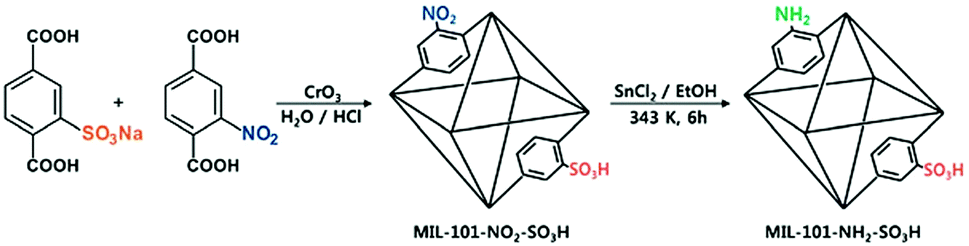 | ||
| Scheme 5 Preparation of the site-isolated, bifunctional Brønsted acid–base catalyst MIL-101(Cr)–NH2–SO3H. Reproduced from ref. 38 with permission from Royal Society of Chemistry. | ||
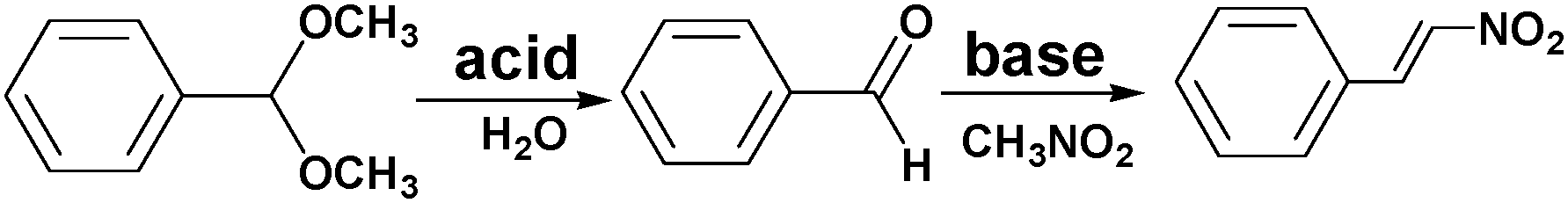 | ||
| Scheme 6 One-pot tandem deacetalization–nitroaldol reaction of benzaldehyde dimethyl acetal to trans-1-nitro-2-phenylethylene. | ||
Linker active sites can be introduced into MOFs by direct synthesis or by grafting them onto the linkers by PSM, which would prevent their coordination to the metal centres and subsequent deactivation during the MOF synthesis.15 An organometallic Ir(I) complex was incorporated into the amino-functionalized Zn(II)-based IRMOF-9-Irdcppy-NH2 (IRMOF = isoreticular metal–organic framework) by PSM (Scheme 7), and the resulting material was used to catalyse a one-pot tandem Knoevenagel condensation–allylic N-alkylation reaction (Scheme 8).39 In this reaction, the Knoevenagel condensation was catalysed by the basic amino sites to yield 2-(indolin-7-ylmethylene)malononitrile. The Ir(I) active sites subsequently catalysed the allylic N-alkylation of this intermediate. Although a high yield was obtained after 36 h, the catalyst was unstable due to the acidic malononitrile substrate. As a result, the yield decreased from 95% for the first run to ∼24% for the second run and 0% for the third run.
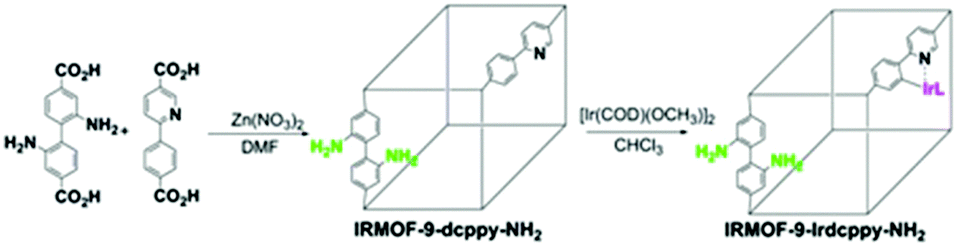 | ||
| Scheme 7 Synthesis of IRMOF-9-Irdcppy-NH2 by PSM. Reproduced from ref. 39 with permission from American Chemical Society. | ||
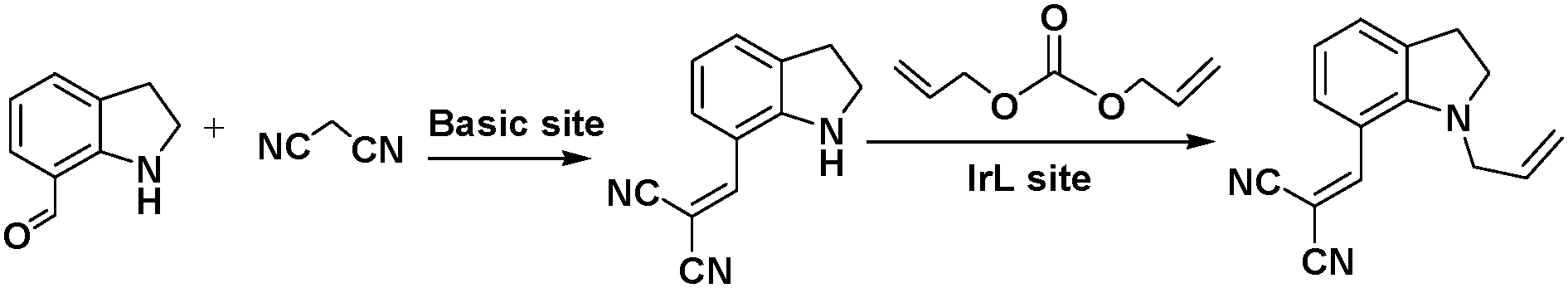 | ||
| Scheme 8 Tandem Knoevenagel condensation–allylic N-alkylation reaction. | ||
2.3 Metal centres and linkers as bifunctional active sites for synergistic catalysis and tandem reactions
Metal nodes of MOF can act as redox active sites and/or Lewis acid sites after coordinated guest molecules are removed, and functionalities can be introduced into the organic linkers. Bifunctional MOFs with open metal centres and functional linkers that can act as active sites are suitable for use in synergistic catalysis and tandem reactions.It is well known that the synergy between Lewis acid and Brønsted acid sites can improve the catalytic performance of a material considerably.40 The sulfone-functionalized USTC-253–TFA MOF has defects and thus contains Lewis and Brønsted acid sites. This MOF exhibited enhanced activity for CO2 cycloaddition to epoxides in the presence of nBu4NBr (TBAB) at room temperature under 1 bar CO2.41 USTC-253–TFA is similar to MOF-253,42 in which the Al centres act as Lewis acid sites and are connected by OH groups that act as Brønsted acid sites. Similar to the defect-containing UiO-66 MOF,43 trifluoroacetic acid (TFA) is partially coordinated to the Al centres in USTC-253–TFA, and removal of these TFA molecules results in the formation of Lewis acid CUMs and missing-linker defects. In the CO2 addition reaction catalysed by USTC-253–TFA, the epoxy ring was activated by the Lewis or Brønsted acid sites. The less-hindered C atom of the activated epoxide was subsequently attacked by the TBAB Br− anion. Then, CO2 interacted with the oxygen anion of the opened epoxy ring to produce an alkylcarbonate anion, which was then converted to the cyclic carbonate. Thus, the synergistic effect of the Brønsted acid sites and Lewis acid CUMs in USTC-253–TFA was responsible for its enhanced activity for the CO2 cycloaddition reaction. To exploit the possible synergy between different types of acid sites, Ma et al. introduced strong Al(III) Lewis acid centres into a Brønsted acid MOF by reacting AlCl3 with MIL-101–Cr–SO3H and then treating the material with water to obtain MIL-101–Cr–SO3H·Al(III).44 Nearly all of the chloride atoms were converted to hydroxyl groups, and the Al(III) ion was octahedrally coordinated to one sulfonate O atom, two OH groups, two μ-OH groups, and one water molecule. The catalyst exhibited high catalytic activity and selectivity in a series of fixed-bed reactions, such as the benzylation of aromatic hydrocarbons and benzyl alcohol. Surprisingly, this catalyst outperformed benchmark zeolite catalysts and several other MOFs. Much lower yields were obtained when Al2O3, MIL-101–Cr, MIL-101–Cr–SO3H and AlCl3 were used as catalysts instead of MIL-101–Cr–SO3H·Al(III), highlighting the synergistic effect of the Lewis acid (Al(III) and Cr(III) centres) and Brønsted acid sites in this MOF on aromatic hydrocarbon bond activation.
Lewis acid CUMs and basic active sites integrated into one MOF are suitable for catalytic transformations such as cross-aldol and Knoevenagel condensation reactions. Vos and coworkers reported the use of the amino-functionalized UiO-66-NH2 as a bifunctional acid–base catalyst for a cross-aldol condensation reaction.45 The amino-functionalized UiO-66-NH2 exhibited a higher conversion and selectivity than UiO-66 in the cross-aldol condensation of benzaldehyde with heptanal. In this reaction, heptanal was activated by a basic amino site inside the cages. The benzaldehyde carbonyl group interacted with a Zr Lewis acid site, which increased its polarization, thus facilitating the attack of heptanal. Thus, the synergistic effect between the Zr Lewis acid sites and the basic amino groups suppressed byproduct formation and promoted the cross-aldol reaction. UiO-66-NH2 also exhibited synergistically enhanced activity for the Knoevenagel condensation of benzaldehyde with ethyl cyanoacetate or malononitrile.46,47 It was proposed that the aldehyde was activated by Zr sites in close proximity to the amino groups, which favoured the formation of an aldimine intermediate. Then, this intermediate reacted smoothly with a methylene compound to form the final product. A similar enhancement was also observed for a Knoevenagel condensation catalysed by the amino-functionalized IRMOF-3.46 However, it should be noted that the active acid sites might originate from the presence of defects in the Zn4O clusters and/or occluded ZnO species produced by partial framework decomposition.48,49 Duan et al. studied the reactant activation processes in a Knoevenagel condensation catalysed by a bifunctional MOF containing Lewis acid and base sites.50 In particular, they used the lanthanide–organic framework Tb–TCA (H3-TCA = tricarboxytriphenylamine), which contains Tb(III) Lewis acid and base (triphenylamine) sites, as a bifunctional catalyst in a Knoevenagel condensation reaction. The luminescence responses and IR analysis confirmed that the aldehyde was activated by the interaction between its C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group and Tb(III). At the same time, the methylenic group of the malononitrile reactant was activated by a triphenylamine Lewis base site. Thus, the synergistic effect of the Lewis acid and base sites led to the high catalytic performance of Tb–TCA in the studied Knoevenagel condensation reaction. In addition to the amino-functionalized MOFs, the M2dobdc MOFs (dobdc4− = 2,5-dioxidoterephthalate; M2+ = Mg2+, Co2+, Ni2+, Cu2+ and Zn2+), denoted MOF-74 or CPO-27, were also active in Knoevenagel condensations and Michael additions due to their intrinsic basic sites.51 The intrinsic basicity of MOF-74 derives from the phenolate oxygen atoms coordinated to the metal ions. The close proximity of these basic active sites to CUMs in MOF-74 led to the synergistic activation of the substrates in Knoevenagel condensations and Michael additions. More specifically, the basic phenolate oxygen atoms deprotonated the acidic reactants, e.g., malononitrile or ethyl cyanoacetate, and the adjacent CUMs acted as docking sites for the deprotonated reactant molecules. Then, the second reactant of the Knoevenagel condensation, e.g., benzaldehyde, adsorbed on the protonated phenolate oxygen, resulting in smooth product formation. It should be noted that Ni2dobdc was the most active catalyst in this MOF series due to its strong basicity.
O group and Tb(III). At the same time, the methylenic group of the malononitrile reactant was activated by a triphenylamine Lewis base site. Thus, the synergistic effect of the Lewis acid and base sites led to the high catalytic performance of Tb–TCA in the studied Knoevenagel condensation reaction. In addition to the amino-functionalized MOFs, the M2dobdc MOFs (dobdc4− = 2,5-dioxidoterephthalate; M2+ = Mg2+, Co2+, Ni2+, Cu2+ and Zn2+), denoted MOF-74 or CPO-27, were also active in Knoevenagel condensations and Michael additions due to their intrinsic basic sites.51 The intrinsic basicity of MOF-74 derives from the phenolate oxygen atoms coordinated to the metal ions. The close proximity of these basic active sites to CUMs in MOF-74 led to the synergistic activation of the substrates in Knoevenagel condensations and Michael additions. More specifically, the basic phenolate oxygen atoms deprotonated the acidic reactants, e.g., malononitrile or ethyl cyanoacetate, and the adjacent CUMs acted as docking sites for the deprotonated reactant molecules. Then, the second reactant of the Knoevenagel condensation, e.g., benzaldehyde, adsorbed on the protonated phenolate oxygen, resulting in smooth product formation. It should be noted that Ni2dobdc was the most active catalyst in this MOF series due to its strong basicity.
The coexistence of Lewis acid and base sites in MOFs is desirable for achieving high catalytic activity for CO2 cycloaddition to styrene epoxide.52 The acidity and basicity of MOF materials can be determined by NH3- and CO2-TPD, respectively. CO2-TPD results have indicated that the basicity sequence of some common MOFs is IRMOF-3 > UiO-66-NH2 > ZIF-8 > UiO-66, Mg-MOF-74 ≫ MIL-101 > Cu3(BTC)2, MOF-5. The basicities of UiO-66-NH2 and IRMOF-3 are due to their amino functionalities, whereas imidazole N atoms contribute to the basicity of ZIF-8. UiO-66, Mg-MOF-74, and MIL-101 also exhibit some basicity, probably due to the O atoms in the metal clusters connecting the organic ligands. NH3-TPD results have shown that UiO-66-NH2, UiO-66, and Mg-MOF-74 are weakly to moderately acidic, whereas Cu3(BTC)2 and MIL-101 exhibit moderate to strong Lewis acidity. ZIF-8, IRMOF-3, and MOF-5 are very weakly acidic. In CO2 cycloaddition to styrene epoxide catalysed by these MOFs, CO2 was activated by the Lewis base site, and the epoxide was polarized on the adjacent Lewis acid site, resulting in a smooth reaction. Of the MOF catalysts discussed above, the acidity and basicity of UiO-66-NH2 are suitable for catalysing CO2 cycloaddition to styrene epoxide, and therefore, this MOF exhibited the highest catalytic activity and nearly 100% selectivity to carbonate in chlorobenzene at 373 K and 2.0 M Pa.
Li and coworkers prepared a hollow zeolitic imidazolate framework (ZIF-8-H) using carboxylate-terminated polystyrene (PS) nanospheres as a template.53 The close proximity of weakly acidic Zn(II) sites and basic imidazolate species in ZIF-8-H led to a synergistic effect on the [3+3] cycloaddition of 1,3-cyclohexanediones to enals. ZIF-8-H catalysed these reactions with a high conversion and selectivity. In fact, the acido-basicity at the external surface of ZIF-8 was probed by FTIR and confirmed by ab initio calculations in another study.54 Active sites including OH and NH groups, hydrogenocarbonates, low-coordinated Zn atoms, and free linker N− moieties were detected on the surface. It was proposed that the Zn(II) Lewis acid sites and Brønsted acid sites (NH groups) as well as the base sites (OH groups and N− moieties) in ZIF-8 were responsible for its significant activity for the transesterification of vegetable oil with various alcohols (methanol, ethanol, 1-propanol, 1-butanol, isopropanol, and t-butanol).
MOFs containing Lewis acid and base active sites not only perform synergistic catalysis but also promote tandem reactions, such as deacetalization–Knoevenagel reactions. Accordingly, Zhou's group designed and constructed the porous coordination network PCN-124 from Cu paddlewheel motifs and the 5,5′-((pyridine-3,5-dicarbonyl)bis(azanediyl))diisophthalate ligand, which bears carboxylate, pyridine, and amide groups.55 Interestingly, the cooperative action of the Cu(II) Lewis acid centres and the Lewis basic pyridine and amide groups makes PCN-124 an efficient catalyst for one-pot tandem deacetalization–Knoevenagel condensation reactions (Scheme 9). The amino-functionalized mesoporous MIL-101(Al)-NH2 was also used as an excellent bifunctional acid–base catalyst in a one-pot tandem deacetalization–Knoevenagel condensation reaction (Scheme 9).56 The Brønsted acidity of MIL-101(Al)-NH2 is due to the carboxylic acid moieties (COOH groups) of the bridging linker on the particle external surfaces.57 These Brønsted acid sites and the coordinately unsaturated Al Lewis acid sites were responsible for benzaldehyde dimethylacetal deacetalization,58 whereas the subsequent Knoevenagel condensation of benzaldehyde with malononitrile was catalysed by the amine basic sites and coordinatively unsaturated Al Lewis acid sites in the MOF.59 MIL-101(Al)-NH2 was also employed as a bifunctional Lewis acid–Brønsted base catalyst in a tandem Meinwald rearrangement–Knoevenagel condensation reaction and was found to exhibit significant substrate selectivity (Fig. 2).60 The Meinwald rearrangement proceeded via a benzylic 3° carbocation intermediate to form the aldehyde with high yield, and the Al(III) Lewis acid sites catalysed this epoxide ring-opening reaction with exceptional substrate selectivity. The basic MOF amino groups subsequently catalysed the Knoevenagel condensation of the aldehyde with an active methylene nucleophile (Scheme 10).
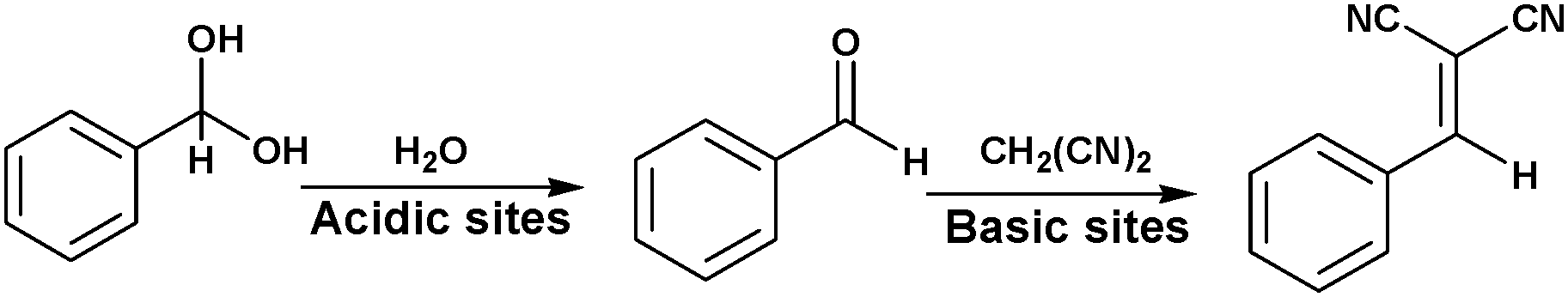 | ||
| Scheme 9 Tandem deacetalization–Knoevenagel condensation reaction. | ||
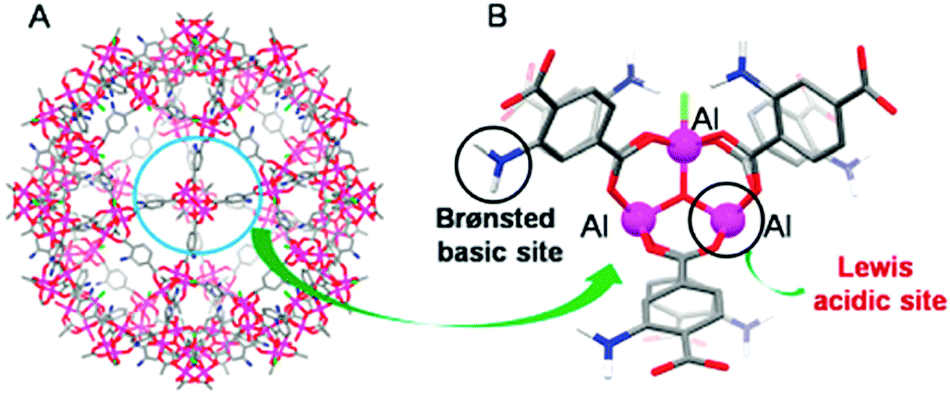 | ||
| Fig. 2 (A) Large cage in MIL-101(Al)-NH2 and (B) its Lewis acid sites (Al(III) centres) and Brønsted base sites (aminoterephthalate linkers). Reproduced from ref. 60 with permission from Royal Society of Chemistry. | ||
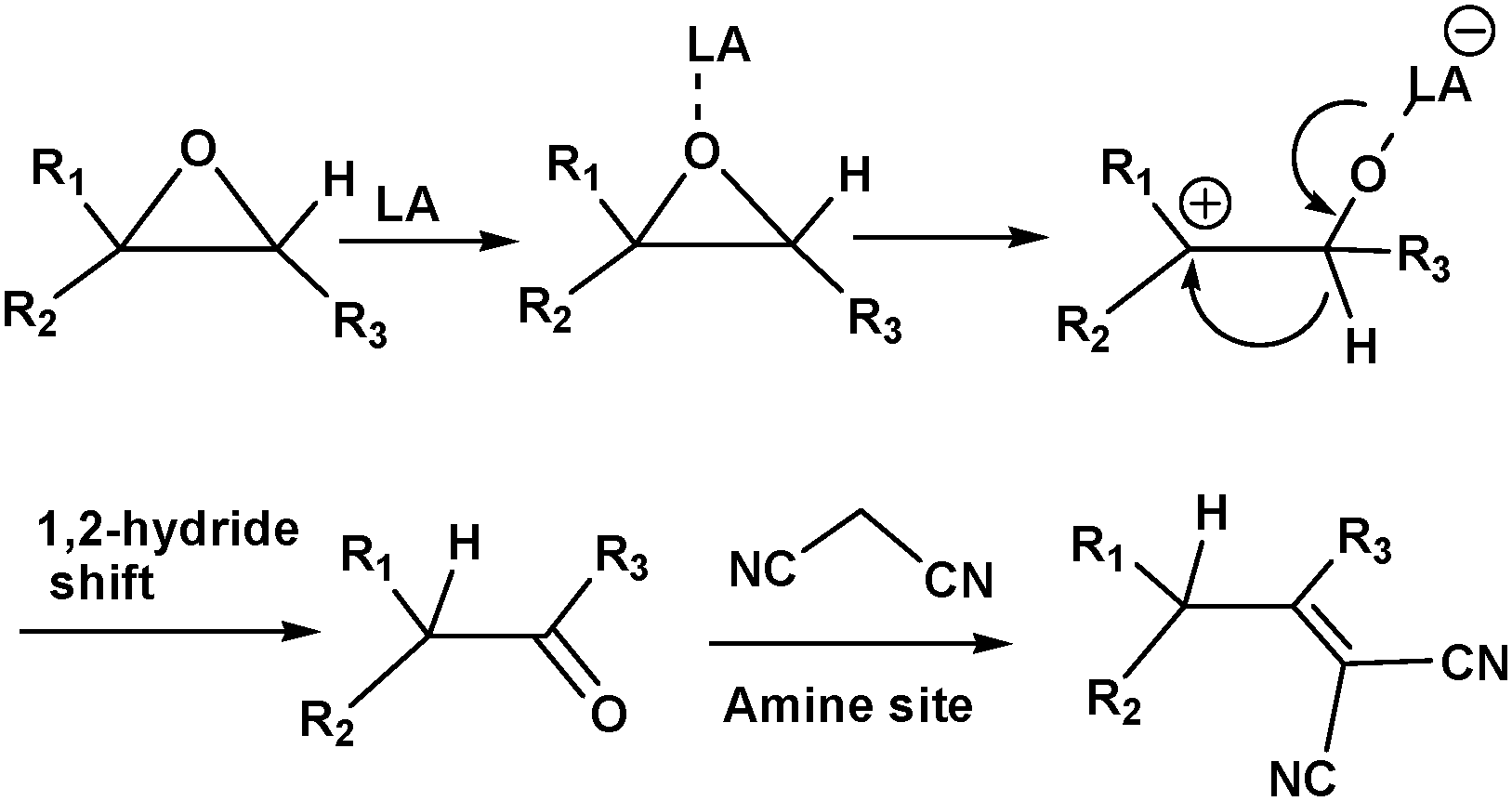 | ||
| Scheme 10 Tandem Meinwald rearrangement–Knoevenagel condensation reaction catalysed by MIL-101(Al)-NH2. | ||
Alternatively, MOF catalysts can be prepared by introducing metalloligand active sites by direct synthesis and/or PSM.61 Corma and coworkers introduced iridium complexes into UiO-66-NH2via PSM and used the resulting material (UiO-66-NH2LIr) to catalyse a one-pot, three-step tandem reaction (Scheme 11).62 UiO-66-NH2LIr exhibited higher selectivity than the corresponding homogeneous catalyst in the synthesis of mono-N-alkylamine by nitroarene reductive N-alkylation with aldehydes in the presence of H2 (Scheme 12). In this reaction, the nitro compound was reduced to the corresponding amine at an iridium site in the presence of hydrogen. Then, the aromatic amine and the carbonyl compound formed an imine via a condensation reaction catalysed by a MOF Zr acid site. Finally, the imine intermediate was hydrogenated to the secondary amine at the Ir active site. The successful introduction of Ir active sites into a MOF for tandem catalysis motivated the same group to introduce chiral NNN-pincer aminopyridineimine ligands into UiO-66-NH2 and UiO-67-NH2via a condensation reaction and subsequently add metal precursors to form (NNN)–M–Zr-MOF complexes (M = Rh, Ir; Scheme 13).63 The (NNN)–Rh–Zr-MOF compound exhibited excellent catalytic activity in tandem aldehyde olefination–hydrogenation reactions (Scheme 14). First, the (NNN)–Rh–Zr-MOF basic amine groups catalysed the Knoevenagel condensation of the aldehyde with ethyl nitroacetate to give ethyl 2-nitro-3-phenylacrylate. The CC double bond of this intermediate was subsequently reduced by the Rh active sites in the presence of hydrogen to give ethyl 2-nitro-3-phenylpropanoate, which could be converted to ethyl 2-amino-3-phenylpropanoate at longer reaction times. Under the same conditions, the corresponding Ir compound (NNN)–Ir–Zr-MOF exhibited a lower yield (30%), selectivity and stability. Notably, although a chiral amino group was introduced into the catalyst, no enantioselectivity was observed in the tandem reaction, even in the presence of a chiral diphosphine, such as 2,3-(isopropylidenedioxy)-2,3-dihydroxy-1,4-bis(diphenylphosphanyl)butane.
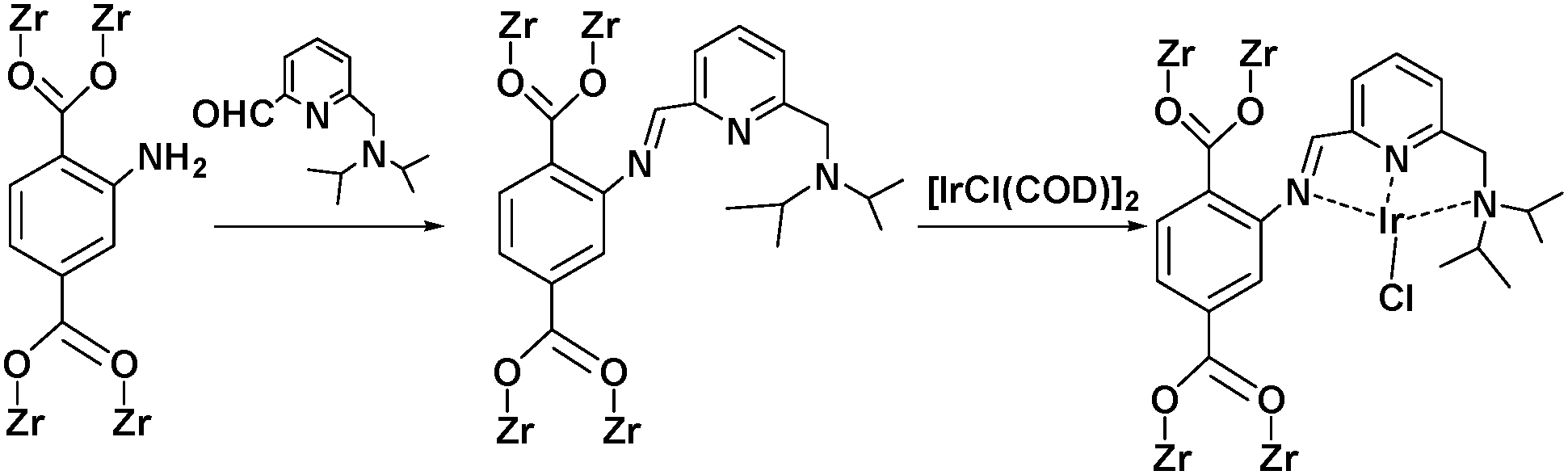 | ||
| Scheme 11 Synthesis of UiO-66-NH2LIr by PSM. | ||
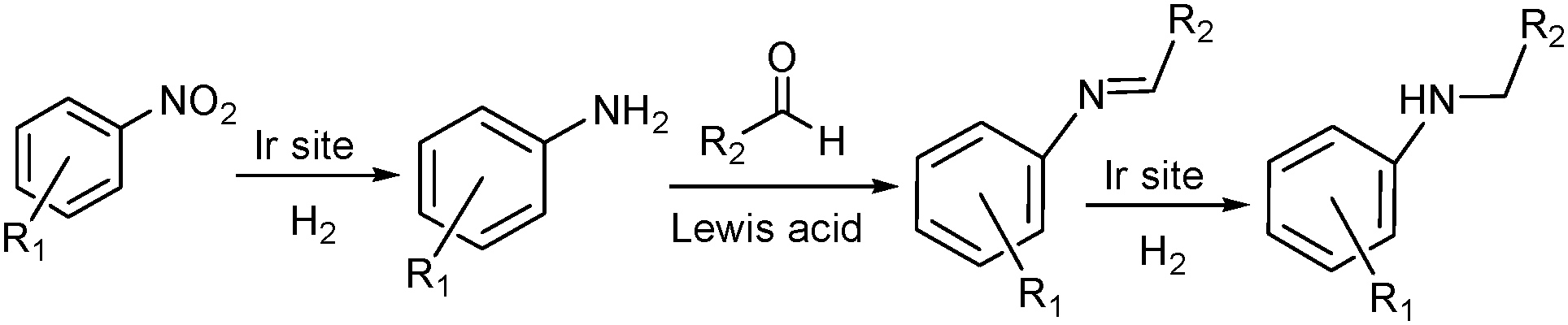 | ||
| Scheme 12 Mono-N-alkylamine synthesis by nitroarene reductive N-alkylation with aldehydes in the presence of H2. | ||
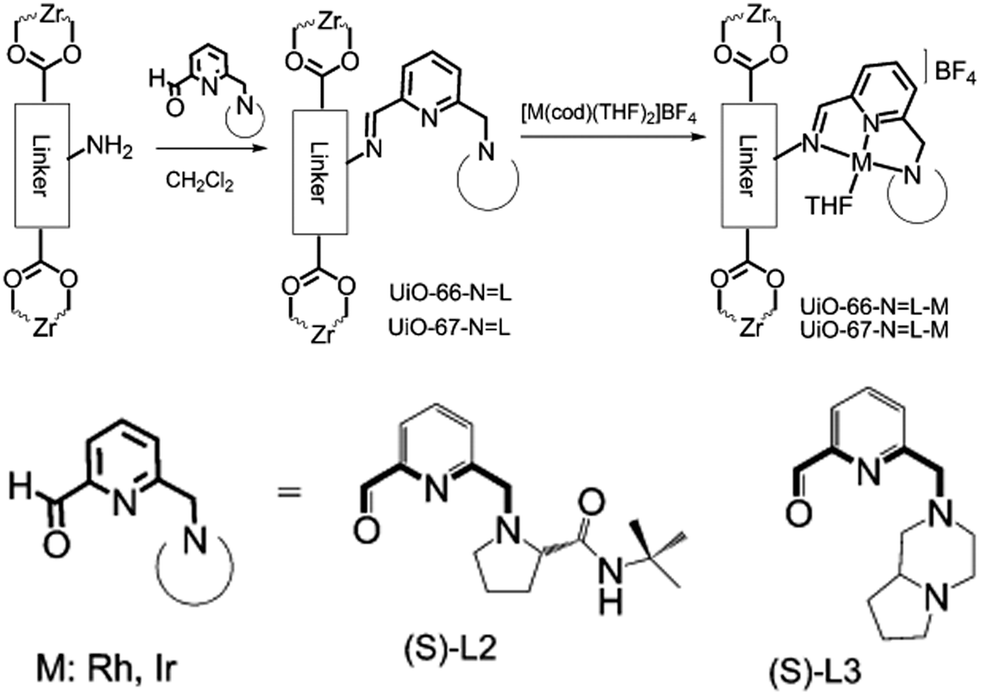 | ||
| Scheme 13 Introduction of NNN-pincer Rh and Ir complexes into UiO-66-NH2 and UiO-67-NH2 by PSM. Reproduced from ref. 63 with permission from Wiley-VCH. | ||
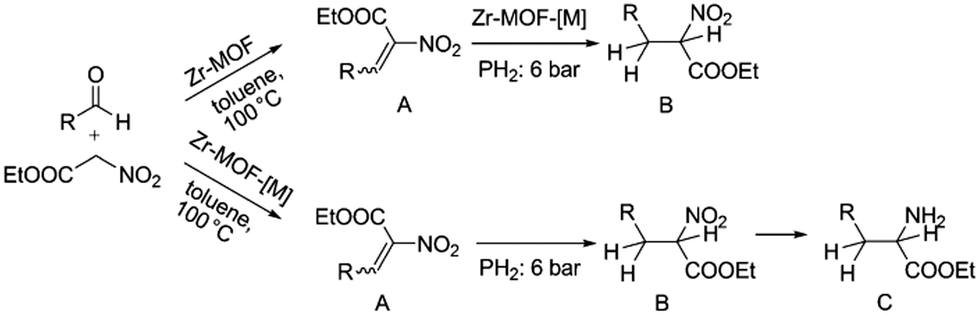 | ||
| Scheme 14 M–Zr-MOF-catalysed tandem olefination–hydrogenation reaction. R = Ar; M = Rh, Ir. Reproduced from ref. 63 with permission from Wiley-VCH. | ||
Farha and coworkers prepared an Hf-NU-1000 analogue using Fe-porphyrin struts (denoted Hf-2) and used it to catalyse a one-pot tandem epoxidation–epoxide ring-opening reaction.64 In particular, the bifunctional mesoporous Hf-2, which has the same topology as PCN-222 (MOF-545), was constructed from Hf6-oxo clusters and meso-tetra(4-carboxyphenyl)porphyrin-Fe(III) chloride.65,66 To ensure that every porphyrin was metallated, anhydrous FeCl3 was added to a DMF suspension of Hf-2. Interestingly, single-crystal X-ray analysis and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) indicated that the Fe atoms in the resulting material, denoted Fe@Hf-2, were not only incorporated into the porphyrin struts but also coordinated to the –OH and –OH2 ligands of the Hf nodes. Fe@Hf-2 was found to catalyse the tandem styrene epoxidation–epoxide ring-opening reaction (Scheme 15) with complete conversion within 10 h. Surprisingly, the azide reactant attacked the terminal carbon to give the protected 1,2-aminoalcohol product shown in Scheme 15c. When the reaction of styrene epoxide with TMS-N3 was catalysed by the Lewis acid sites in Hf-NU-1000, the other regioisomer of the product was obtained (Scheme 15a).67 In this catalytic system, the 1,2-hydroxyl amine precursor was obtained by styrene oxide ring opening at the benzylic carbon atom. The regioselectivity of the product appeared to be controlled by the styrene epoxidation catalytic mechanism. The authors proposed that the bis-μ-oxo di-iron functionality on the Fe@Hf-2 Hf6 nodes contributed to the inverse selectivity.
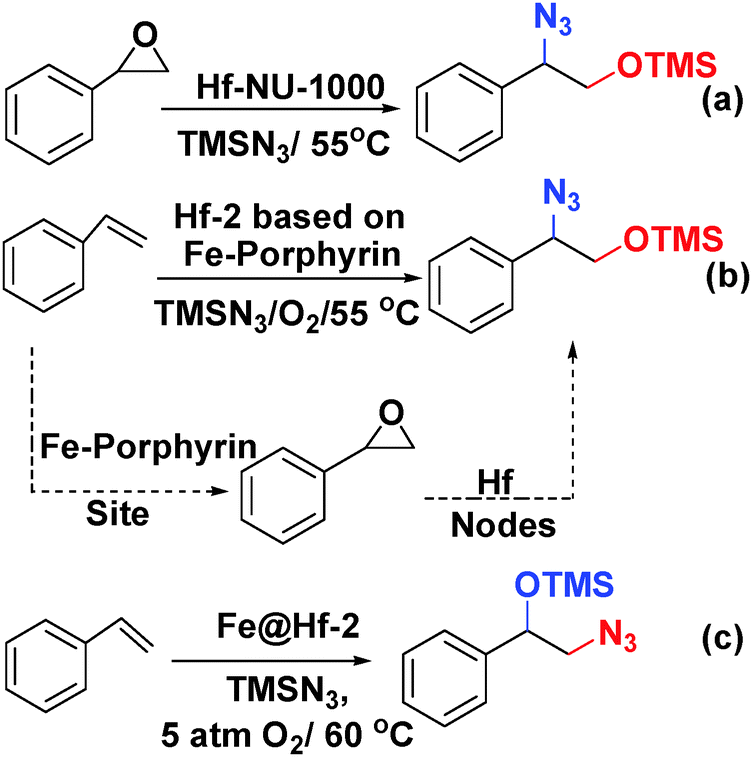 | ||
| Scheme 15 (a) Epoxide ring-opening reaction catalysed by Hf-NU-1000. One-pot tandem epoxidation–epoxide ring-opening reaction catalysed by Hf-2 (b) and Fe@Hf (c). | ||
2.4 Functional active sites coordinated to metal nodes for tandem reactions
Active sites can be introduced into MOFs by coordinating functional groups or molecules to the metal nodes after the removal of any coordinated solvent molecules. These coordinated active sites can then act cooperatively with the open metal nodes or functional ligands in the MOF to facilitate synergistic catalysis and tandem reactions. To overcome the weak Lewis acidity of CUMs, Li et al. introduced free –SO3H functionalities into the organic ligands of MIL-101 and coordinated –NH2 groups to the CUMs by post-synthesis modification.68 More specifically, they first coordinated mono-BOC-ethylenediamine to the Cr(III) centres to form BOC-protected MIL-101-NHBOC. The phenylene linkers were subsequently sulfonated by treatment with chlorosulfonic acid, giving MIL-101-SO3H-NHBOC. Finally, the bifunctional MIL-101–SO3H–NH2 catalyst was obtained by deprotecting the amino groups through thermal treatment. Remarkably, this catalyst exhibited enhanced activity for the tandem dimethyl acetal hydrolysis–Henry reaction (Scheme 6). However, the catalyst preparation process was laborious and time consuming, limiting its practical application.In another study, Corma and coworkers introduced a Pd complex into the cages of Cu3(BTC)2 to obtain a bifunctional Pd–Cu MOF catalyst denoted MOF-Cu3(BTC)2–[Pd] (Scheme 16), which exhibited high activity for one-pot tandem Sonogashira–click reactions.69 First, 4-substituted pyridine ligands were grafted onto the CUMs in the MOF cages, and the Pd complex PdCl2(PhCN)2 was then added to obtain the desired Cu–Pd MOF. The resulting MOF-Cu3(BTC)2–[Pd] catalyst, which contained Cu(II) and Pd(II) sites, acted as a bifunctional catalyst in the one-pot tandem Sonogashira C–C coupling–intramolecular cycloaddition click reactions of 2-iodobenzyl bromide, NaN3, and different alkynes to give triazolo[5,1-a]isoindoles with high yields. The formation of the Sonogashira C–C coupling product was catalysed by the Pd sites, whereas the Cu(II) sites catalysed the intramolecular cycloaddition click reaction (Scheme 17),70 which is usually promoted by Cu(I) species.34
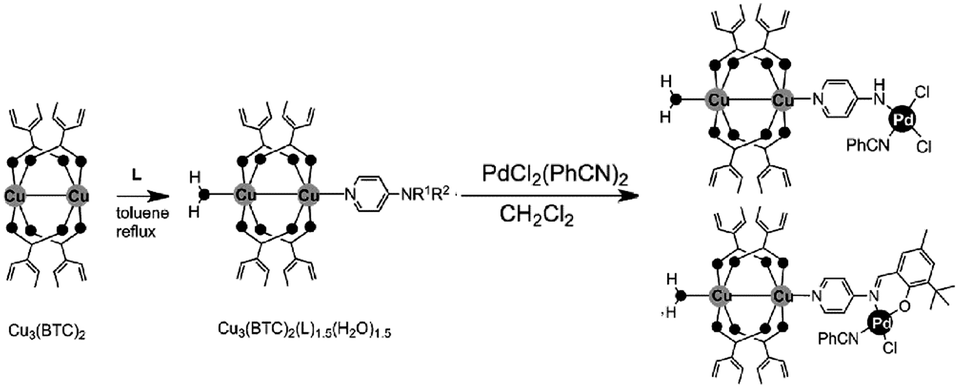 | ||
| Scheme 16 Functionalization of Cu3(BTC)2 with palladium complexes. Reproduced from ref. 69 with permission from Wiley-VCH. | ||
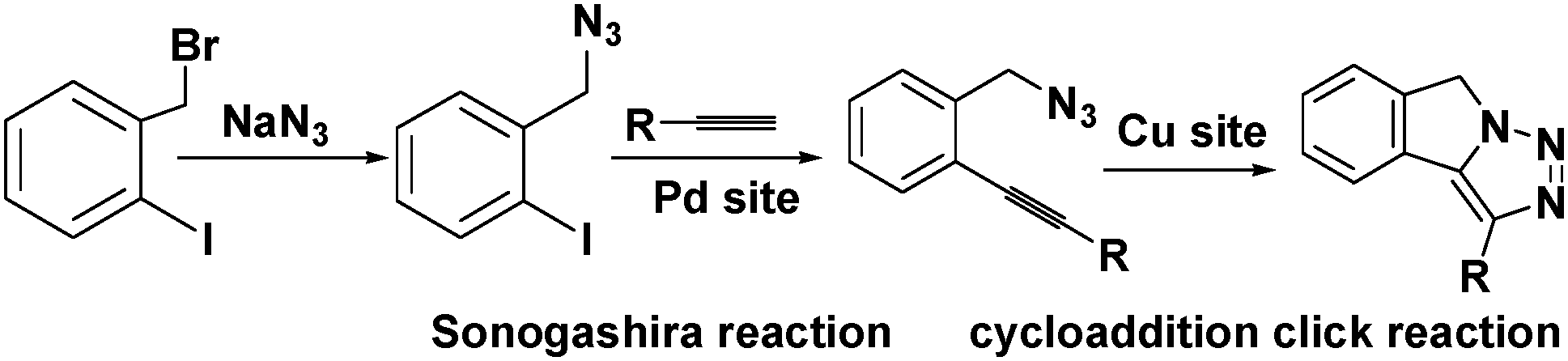 | ||
| Scheme 17 Tandem Sonogashira C–C coupling–cycloaddition click reaction catalysed by MOF-Cu3(BTC)2–[Pd]. | ||
2.5 Bifunctional homochiral MOFs for asymmetric tandem reactions
Compared with traditional porous materials such as zeolites and carbon materials, homochiral MOFs can be easily designed and fabricated by direct synthesis using chiral ligands or by introducing chiral groups via a PSM method.26 Chiral (salen)MnCl complexes constitute a family of classic asymmetric catalysts for alkene epoxidation,71 and the Zn4O(O2CR)6 secondary building units (SBUs) in MOFs can be utilized as weak Lewis acid sites for catalysis.51 Lin and coworkers used a chiral (salen)MnCl ligand and the [Zn4(μ4-O)(O2CR)6] SBU to synthesize the homochiral CMOF-1, which exhibited high regioselectivity and stereoselectivity in tandem alkene epoxidation/epoxide ring-opening reactions (Fig. 3).72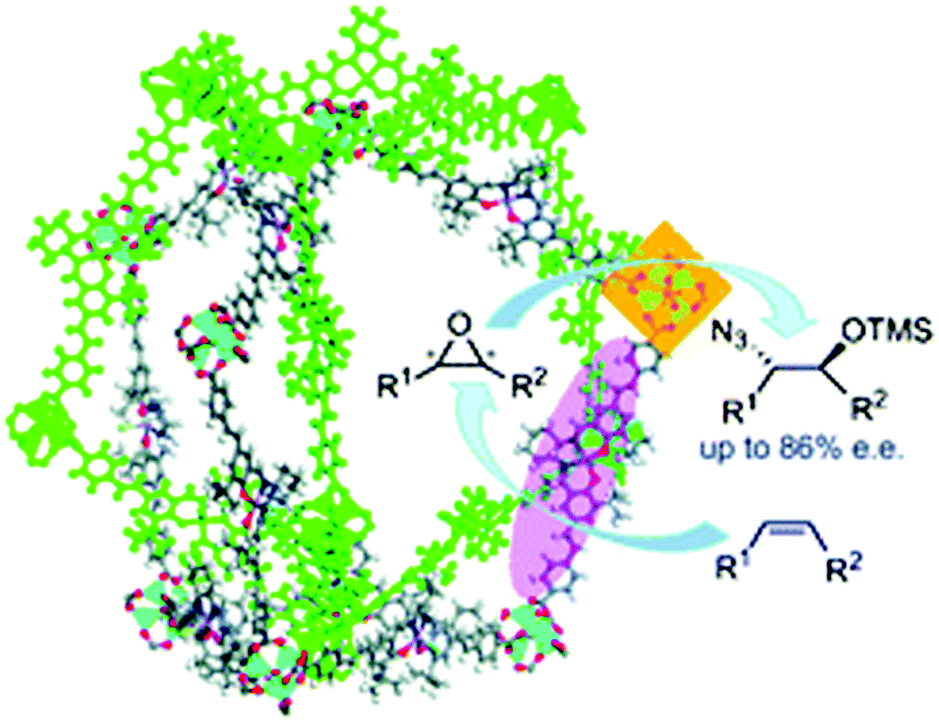 | ||
| Fig. 3 Tandem asymmetric alkene epoxidation–epoxide ring-opening reaction catalysed by (salen)MnCl-based CMOF-1. Reproduced from ref. 72 with permission from Royal Society of Chemistry. | ||
Although CMOF-1 could not adsorb N2 due to significant framework distortion upon solvent removal from the pores, it adsorbed 34.0 wt% Brilliant Blue R-250, indicating that the pores are accessible to large molecules. Interestingly, the isolated chiral (salen)MnCl sites and Zn Lewis acid sites in CMOF-1 could effectively catalyse the tandem asymmetric epoxidation–epoxide ring-opening reaction of unactivated alkenes with TMSN3 using 2-(t-butylsulfonyl)iodosylbenzene as the oxidant.
3. Guest species and MOFs as active sites for synergistic catalysis and tandem reactions
The well-defined pore structures of MOFs make them excellent candidates for hosting catalytically active guest species.13,73–77 In particular, the pore confinement effect can limit metal nanoparticle (MNP) growth, and the functional linkers can stabilize the NPs, resulting in the formation of uniform surfactant-free MNPs that are of the same size as the pore width.74 Then, the active guest species can act cooperatively with nearby open metal sites and/or functional groups in the MOF to achieve synergistic catalysis and catalyse tandem reactions (Table 2).| Catalyst | Reaction | Active sites | Ref. |
|---|---|---|---|
| Pd@MIL-101 | Methyl isobutyl ketone (MIBK) synthesis from acetone | Lewis acid, Pd NPs | 78 |
| Pd/MIL-101 | Selective phenol hydrogenation to cyclohexanone | Lewis acid, Pd NPs | 79 |
| Rh@SMIL-101 | Phenol hydrogenation to cyclohexanone | Lewis acid, Ru NPs | 81 |
| Pd@MIL-101 | Citronellal cyclization to isopulegol | Lewis acid, Pd NPs | 82 |
| Pd@MIL-101 and Pt@MIL-101 | Tandem nitroarene reduction and reductive amination of carbonyl compounds | Lewis acid, Pd or Pt NPs | 83 |
| Pd–Ag@MIL-101 | Tandem nitrobenzene hydrogenation and acetophenone reductive amination | Lewis acid, PdAg NPs | 84 |
| Au–Pd/MIL-101 | Aerobic oxidation of the primary C–H bonds in toluene and its derivatives | Lewis acid, AuPd NPs | 86 |
| Pd@UiO-66-NH2 | Oxidation–acetalization | Acid, Pd NPs | 88 |
| Pt–Pd/MM-MIL-53 | Oxidant-free catalytic alcohol dehydrogenation | Lewis acid, base, PtPd NPs | 90 |
| Pd/NH2-MIL-125 | Formic acid dehydrogenation | Base, Pd NPs | 95 |
| Au/NH2-MIL-53 | One-pot aerobic benzyl alcohol oxidation/Knoevenagel condensation reaction | Base and Au NPs | 98 |
| Pd@IRMOF-3 | Knoevenagel–hydrogenation | Base, Pd NPs | 99 |
| Pt⊂UiO-66-S and Pt⊂UiO-66-N | Methylcyclopentane (MCP) conversion to benzene, olefins, cyclohexane, and acyclic isomers | Pd, (–SO3H, –NH3+) | 100 |
| Pd@UiO-66-X, X = H, NH2, OMe | Aerobic benzaldehyde reaction with ethylene glycol | Pd, (OMe, NH2) | 101 |
| Pt/MIL-101–cinchona alkaloid | Asymmetric hydrogenation of ethyl pyruvate and ethyl 2-oxo-4-phenylbutyrate | Pt, chiral cinchona alkaloid | 102 |
3.1 MNP@MOF composites for synergistic catalysis and tandem reactions
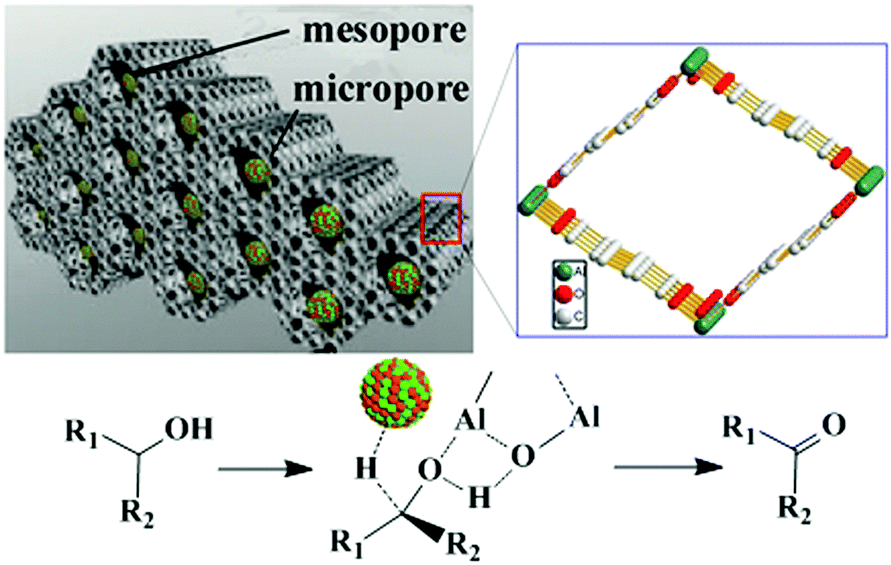
MNPs and open metal centres in MOFs could provide multiple active sites for synergistic catalysis and tandem reactions. In particular, MIL-101 is a popular mesoporous material with large pores of 2.9 and 3.4 nm and a high BET surface area of ca. 4000 m2 g−1. It has been widely employed as a support for small catalytically active MNPs.77 More importantly, it has a high density of open chromium centres, which can act as Lewis acid sites for catalysis after the terminal water molecules have been removed. Accordingly, MNPs embedded in MIL-101 can act cooperatively with the Cr(III) Lewis acid sites to achieve synergistic catalysis or promote tandem reactions.78,79,81–84,86 Pd NPs on the MIL-101 support were first employed as efficient bifunctional catalysts for the one-step synthesis of methyl isobutyl ketone (MIBK) from acetone in the presence of H2 (Scheme 18).78 In this reaction, acetone was firstly converted to mesityl oxide (MO) with high selectivity due to the high density and improved accessibility of the Cr(III) Lewis acid sites, which can catalyse acetone condensation and dehydration. Then, the Pd NPs catalysed MO hydrogenation to methyl isobutyl ketone (MIBK) with 90.2% selectivity at 60% conversion, which is suitable for industrial applications. The same group also reported that Pd/MIL-101 efficiently catalysed selective phenol hydrogenation to cyclohexanone in water at atmospheric pressure and room temperature.79 This bifunctional catalyst exploited the synergy between the Lewis acid and Pd NP sites to convert >99.9% of the phenol reactant to cyclohexanone with a selectivity of >99.9%. The observed high selectivity was because the cyclohexanone carbonyl group was coordinated to the Cr(III) Lewis acid site, thus inhibiting further hydrogenation to cyclohexanol.80 However, further research must be performed to verify the mechanism because the solvent water molecules might coordinate to the Cr(III) nodes, which could affect the acidity of the material. At a high initial H2 pressure (5 bar), Rh NPs encapsulated in sulfonic acid-functionalized MIL-101 (Rh@SMIL-101) also exhibited high selectivity in the hydrogenation of phenol to cyclohexanone in water.81 Although the catalyst contains sulfonic acid sites, the authors proposed the same mechanism as that proposed for Pd/MIL-101.79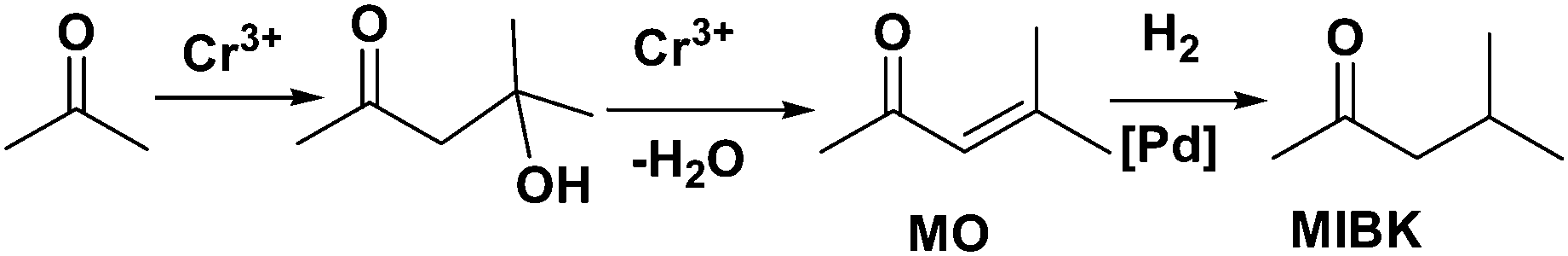 | ||
| Scheme 18 MIBK synthesis from acetone and H2 catalysed by Pd/MIL-101. | ||
Corma and coworkers used Pd@MIL-101 as a bifunctional catalyst for citronellal cyclization to isopulegol, which was subsequently transformed into menthol (Scheme 19).82 Nearly complete citronellal conversion was achieved by the Cr(III) Lewis acid sites with a diastereoselectivity to rac-isopulegol of 74% after 12 h. After completion of the cyclization step, isopulegol hydrogenation was catalysed by the Pd active sites in 6 h, yielding 70% of the desired (−)-menthol product.
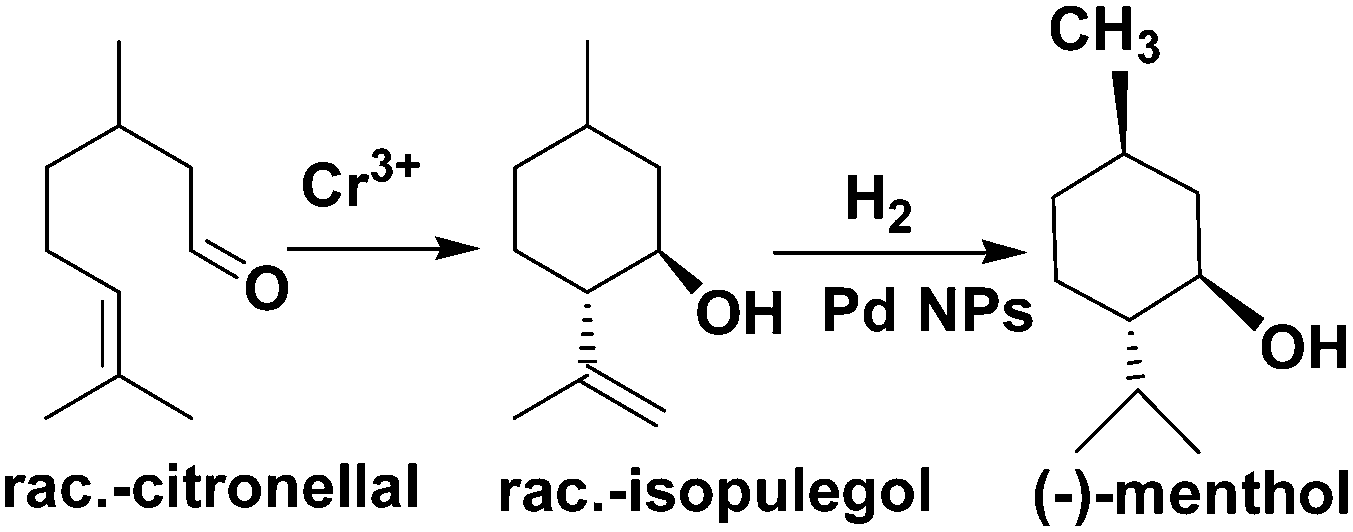 | ||
| Scheme 19 Tandem reaction for citronellal conversion to (−)-menthol catalysed by Pd/MIL-101. | ||
Tandem reactions consisting of nitroarene reduction and the reductive amination of carbonyl compounds could be catalysed by the cooperative action of Pd,83 Pt83 and Pd–Ag alloy84 NPs and MIL-101 Cr(III) Lewis acid sites. Secondary arylamines were obtained by tandem nitroarene hydrogenation–aldehyde or ketone reductive amination reactions with high activity and selectivity (Scheme 20a). In this class of reactions, a nitroarene was first hydrogenated to an aromatic amine at a Pd or Pt active site. The amine then reacted with an aldehyde or ketone over a Cr(III) Lewis acid site to form an imine intermediate, which was hydrogenated to give the secondary arylamine product (Scheme 20a). The tandem reaction mechanism was verified using the Pd–Ag alloy NPs encapsulated in MIL-101 (Fig. 4).84 Although complete conversion could be achieved faster with Pd/MIL-101 than with Pd–Ag/MIL-101, Ag doping improved the selectivity to the desired secondary arylamine product. This result was ascribed to the fact that Ag addition slowed down the rate of benzaldehyde reduction to the alcohol and thus enhanced the benzaldehyde conversion to the secondary arylamine.85 Interestingly, the bifunctional Pd/MIL-101 and Pt/MIL-101 catalysts could also be employed in the syntheses of quinolines, pyrroles and 3-arylpyrrolidines, which involve similar tandem hydrogenation–reductive amination reactions.83 More specifically, tetrahydroquinoline was obtained by nitroarene hydrogenation, followed by 2-nitrocinnamaldehyde intramolecular hydrogenation/reductive amination (Scheme 20b), whereas pyrroles were synthesized by nitroarene hydrogenation, followed by Paal–Knorr condensation with 2,5-hexanedione (Scheme 20c). Remarkably, N-substituted 3-arylpyrrolidines were obtained by tandem Michael addition, nitroalkane hydrogenation, reductive amination and heterocyclic hydrogenation processes (Scheme 20d).
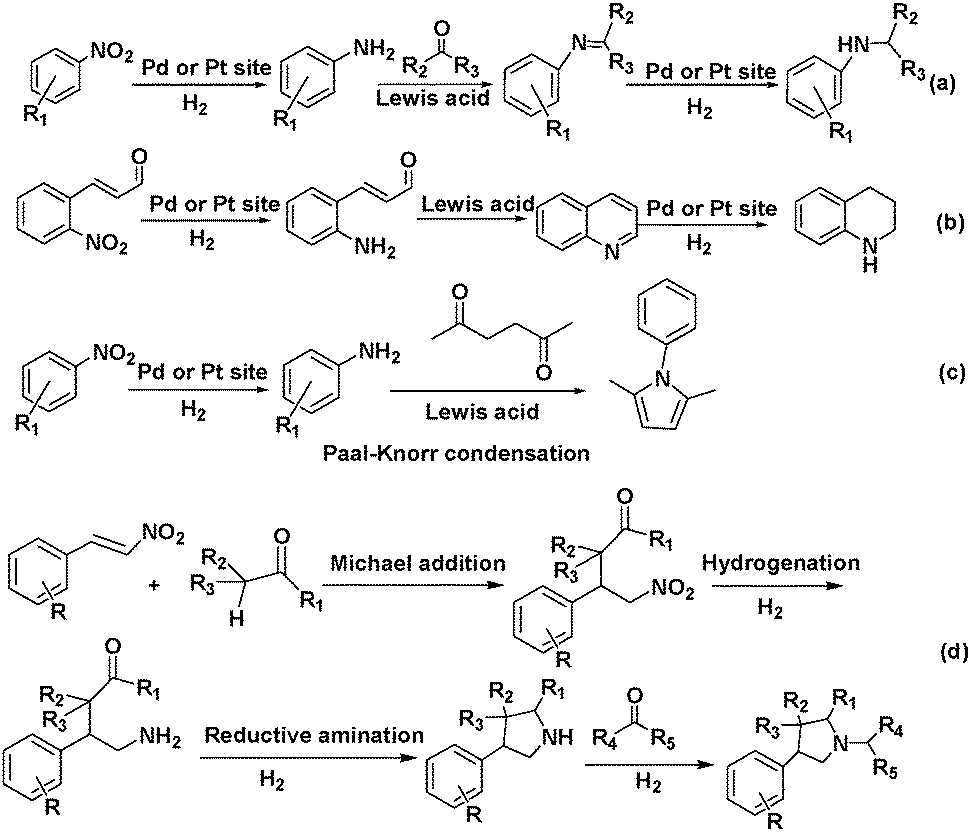 | ||
| Scheme 20 Tandem reactions catalysed by MIL-101-supported Pd or Pt NPs or their corresponding complexes: (a) secondary arylamine synthesis by nitroarene hydrogenation and reductive amination of aldehydes or ketones; (b) tetrahydroquinoline synthesis by nitroarene hydrogenation and 2-nitrocinnamaldehyde intramolecular hydrogenation/reductive amination; (c) pyrrole synthesis via nitroarene hydrogenation and Paal–Knorr condensation with 2,5-hexanedione; (d) N-substituted 3-arylpyrrolidine synthesis by Michael addition, nitroalkane hydrogenation, reductive amination and heterocyclic hydrogenation. | ||
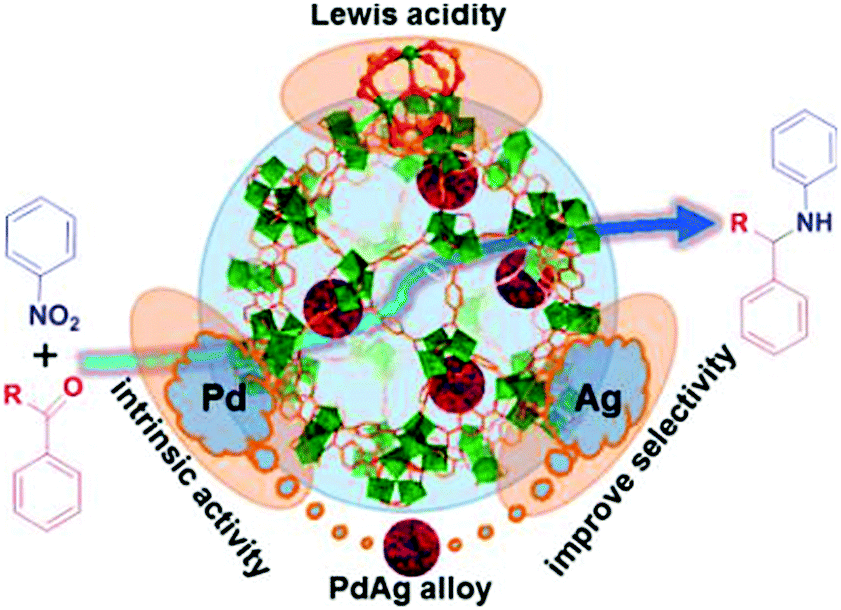 | ||
| Fig. 4 Secondary arylamine synthesis by a tandem reaction consisting of nitroarene hydrogenation and reductive amination of aldehydes or ketones catalysed by Pd–Ag/MIL-101. Reproduced from ref. 84 with permission from American Chemical Society. | ||
Au–Pd NPs on a MIL-101 support and the MIL-101 Cr(III) Lewis acid sites could cooperatively catalyse the aerobic oxidation of the primary C–H bonds in toluene and its derivatives.86 This Au–Pd/MIL-101 catalyst oxidized toluene to the major benzyl benzoate product with 98.6% conversion. Interestingly, neither benzyl alcohol nor benzoic acid was detected, in contrast to the results obtained for AuPd NPs on carbon or TiO2 supports.87 Because the interaction between the toluene aromatic ring and the Cr(III) Lewis acid site made the methyl carbon atom of the methyl group relatively electron deficient, the activated oxygen species adsorbed on the Au–Pd NP surface could easily attack it. Furthermore, the CO group of the benzaldehyde intermediate could coordinate to the Cr(III) Lewis acid sites, preventing its further oxidation to benzoic acid. Thus, the nucleophilic attack of benzyl alcohol on the benzaldehyde carbonyl carbon atom led to the formation of the corresponding hemiacetal, which was further oxidized to benzyl benzoate.
The strategy of combining Lewis acid sites and MNPs to improve the catalytic performance can be expanded to other MOF composites. For example, Pd@UiO-66-NH2 was utilized in a one-pot tandem oxidation–acetalization reaction with excellent activity and selectivity.88 In this tandem reaction, benzyl alcohol was firstly oxidized to benzaldehyde by the Pd NPs, and benzaldehyde acetalization with ethylene glycol occurred at the UiO-66-NH2 Lewis acid sites to give the 2-phenyl-1,3-dioxolane product with 99.9% selectivity.89 More recently, Huang et al. reported that Pt–Pd alloy NPs and a bifunctional MOF with acid and base sites synergistically catalysed oxidant-free alcohol dehydrogenation.90 It is well known that the oxidation of alcohols to carbonyl compounds generally requires the use of oxygen as the oxidant, which is dangerous and costly.91 Therefore, this O2-free strategy is an atom-efficient, safe method for performing this catalytic reaction. The MM-MIL-53 MOF containing Al(III) centres has mesopores that aid the diffusion of the substrates and products and provide access to the internal active sites and micropores that provide large surface areas with many active sites to enhance the catalytic activity. More importantly, the interconnected chains of corner-sharing AlO4(OH)2 octahedra in this MOF can act as acid–base active sites to enhance the oxidant-free alcohol dehydrogenation reaction (Fig. 5). Indeed, several secondary alcohols, such as 2-octanol, were converted to their corresponding ketones with moderate yields under a N2 flow. Moreover, the Pt–Pd/MM-MIL-53(Al) catalyst with PtPd alloy NPs exhibited higher conversions than the corresponding monometallic NP catalysts, indicating that the alloy had a synergistic effect on the catalysis. The proposed mechanism involves proton abstraction from the alcohol by a basic site of MM-MIL-53 to yield an alkoxide group on the Al(III) Lewis acid site. Then, an alkoxide C–H bond was dissociated at a PtPd site to form PtPd-H, and the final ketone product was obtained with the release of H2 gas.
 | ||
| Fig. 5 Oxidant-free alcohol dehydrogenation catalysed by Pt–Pd alloy NPs on the MM-MIL-53 support. Reproduced from ref. 90 with permission from Elsevier Inc. | ||
In addition to the open metal sites, the various organic functional groups in MOFs can be employed as active sites along with MNPs to promote synergistic catalysis and tandem reactions. A typical example is amino-decorated MOFs, in which the amino groups not only stabilize MNPs92–94 but also serve as active sites for organic catalysis. Compared with Pd/MIL-125, Pd supported on NH2-MIL-125 exhibited higher activity in formic acid dehydrogenation with a TOF of 214 h−1, producing 48.1 μmol H2 at 305 K.95 The amino group served as a proton scavenger to form +HNH2, which had a positive effect on the O–H bond dissociation.96,97 Then, the intermediate Pd-formate species underwent β-hydride elimination to produce CO2 and a Pd hydride species, which subsequently decomposed to produce H2.
In addition, amino groups are typically used in base catalysis such as Knoevenagel condensation.98,99 For example, Au/NH2-MIL-53 catalyst exhibited efficient catalytic performance in a one-pot tandem aerobic benzyl alcohol oxidation/Knoevenagel condensation reaction.98 The Au NPs catalysed the benzyl alcohol oxidation, whereas the amino groups catalysed the Knoevenagel condensation. Specifically, benzyl alcohol adsorbed on the Au NP surfaces to form a Au–alcoholate species. Then, β-hydride elimination occurred at the gold surface to produce an aldehyde, which further reacted with malononitrile to produce 2-benzylidenemalononitrile by an amino-catalysed Knoevenagel condensation. Importantly, core–shell structures could improve the efficiency of tandem reactions due to the close proximity of the active sites. Tang and coworkers reported the fabrication and use of a uniform core–shell Pd@IRMOF-3 composite in an efficient tandem Knoevenagel condensation–hydrogenation reaction.99 Pre-synthesized Pd NPs (∼35 nm) were added to an N,N-dimethylformamide (DMF)–ethanol mixture that contained the precursors of IRMOF-3 Zn(NO3)2, 2-aminoterephthalic acid (NH2-H2bdc) and polyvinylpyrrolidone (PVP), and a solvothermal treatment was subsequently applied to obtain the core–shell nanostructure (Scheme 21a). The Knoevenagel condensation of 4-nitrobenzaldehyde (A) with malononitrile was catalysed by the amino groups on the IRMOF-3 shell to give 2-(4-nitrobenzylidene)malononitrile (B). The –NO2 group of this intermediate was then selectively reduced to an amine by the Pd NP cores in the presence of H2 (Scheme 21b).
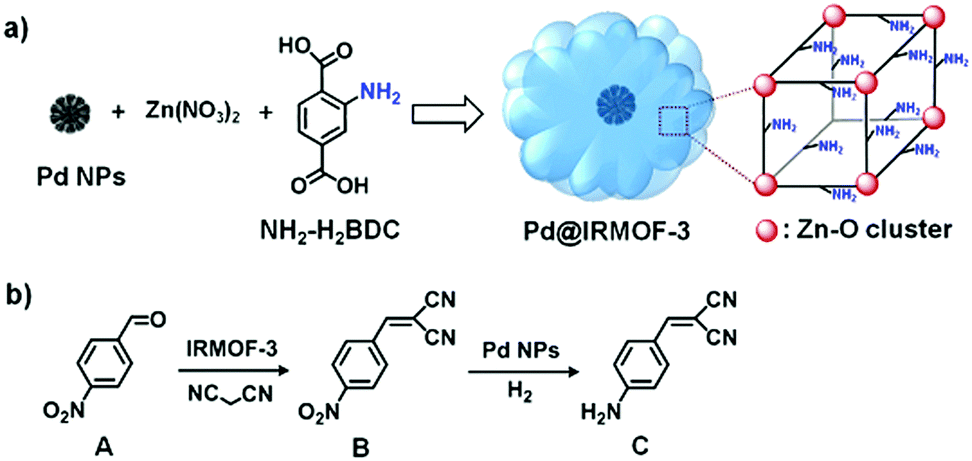 | ||
| Scheme 21 (a) Synthesis of the core–shell Pd@IRMOF-3 composite via a mixed solvothermal method and (b) model tandem reaction involving Knoevenagel condensation of A with malononitrile catalysed by the IRMOF-3 shell and subsequent selective hydrogenation of intermediate B to C catalysed by the Pd NP cores. Reproduced from ref. 99 with permission from American Chemical Society. | ||
The different organic functional groups in MOFs could significantly affect their activity and selectivity in catalytic reactions.100,101 Yaghi et al. introduced 2.5 nm Pt NPs into the crystals of UiO-66 MOF analogues containing sulfonic acid (–SO3H, S) and ammonium (–NH3+, N) functional groups, denoted Pt⊂UiO-66-S and Pt⊂UiO-66-N, respectively.100 They found that these different acidic functionalities play an important role in product selectivity in methylcyclopentane (MCP) conversion to benzene, olefins, cyclohexane, and acyclic isomers. Pt⊂UiO-66-S with strong acid groups exhibited the highest selectivity to C6 cyclic products (62.4% and 28.6% for cyclohexane and benzene, respectively) and did not produce any acyclic isomers. In contrast, Pt⊂UiO-66-N with weak acid sites had selectivities to the acyclic isomer product and C6 cyclic products of 38.6% and <50%, respectively. Moreover, when Pt⊂UiO-66-SN with both functional groups was used as the catalyst, the major product was benzene, olefins and acyclic isomers were formed as minor products, and no cyclohexane was obtained. The different catalytic behaviours of these composites might be attributed to the synergistic interplay between the Pt NPs and the different MOF acid sites. When Pt⊂UiO-66-S and Pt⊂UiO-66-N were used as catalysts, higher selectivity to benzene and cyclohexane was observed than when the Pt⊂UiO-66 catalyst with no acid functional groups was employed due to the lower activation energy for the formation of these two products in the presence of acid sites. Interestingly, the activity of the Pt⊂UiO-66-S catalyst was nearly two times higher than that of Pt⊂UiO-66-N, possibly due to the stronger acidity of –SO3H than –NH3+. More recently, Huang et al. observed a similar phenomenon when Pd NPs encapsulated in isoreticular UiO-66-X MOFs (X = H, NH2, OMe) were employed in the aerobic reaction of benzaldehyde with ethylene glycol and different products were obtained.101 Two reaction mechanisms were proposed for this reaction: (i) benzaldehyde condensation with ethylene glycol to give the hemiacetal and (ii) further condensation to obtain the acetal or oxidation to the ester (Scheme 22). Pd@UiO-66-NH2 exhibited a selectivity to benzaldehyde ethylene acetal of 94%, whereas both Pd@UiO-66-H and Pd@UiO-66-OMe exhibited high selectivity (90% and 97%, respectively) to the ester 2-hydroxyethyl benzoate. Notably, although Pd@UiO-66-NH2 was active for benzyl alcohol oxidation to benzaldehyde, it did not catalyse the oxidation of the hemiacetal to the ester. Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and density functional theory (DFT) calculations revealed that the Pd NPs were coordinated to the –NH2 groups in Pd@UiO-66-NH2 and therefore had a higher chemical potential and were weaker oxidants than those in Pd@UiO-66-OMe. Thus, the interactions between Pd and the –NH2 groups led to the formation of benzaldehyde ethylene acetal. This report demonstrated the potential for changing the MNP chemical microenvironment by changing the MOF linkers, thus opening a new avenue for developing selective MNP–MOF catalysts.
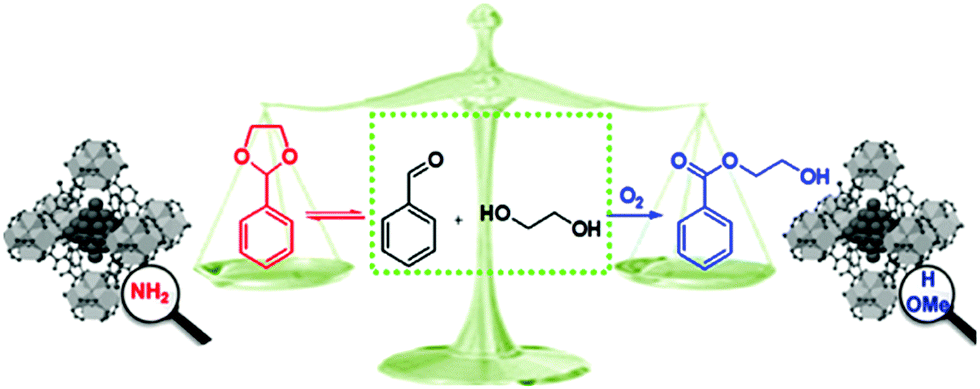 | ||
| Scheme 22 Different products of the reaction of benzaldehyde with ethylene glycol catalysed by Pd@UiO-66-X, X = H, NH2, OMe. Reproduced from ref. 101 with permission from American Chemical Society. | ||
Introducing enantiotropic functional groups into MOFs might lead to asymmetric catalysis systems. A Pt/MIL-101 catalyst modified with a chiral cinchona alkaloid, namely, cinchonidine, exhibited high activity and moderate enantioselectivity in the asymmetric hydrogenation of ethyl pyruvate and ethyl 2-oxo-4-phenylbutyrate (Scheme 23).102 In particular, a TOF of up to 4469 h−1 and a 76.5% enantiomeric excess (ee) of (R)-(+)-ethyl lactate were achieved for ethyl pyruvate, and a TOF of 2000 h−1 and a 76.8% ee of (R)-(+)-ethyl 2-hydroxy-4-phenylbutyrate were achieved for ethyl 2-oxo-4-phenylbutyrate. Although the mechanism was not well understood, the synergistic effect of the Pt NPs and chiral cinchonidine led to the observed moderate enantioselectivity.
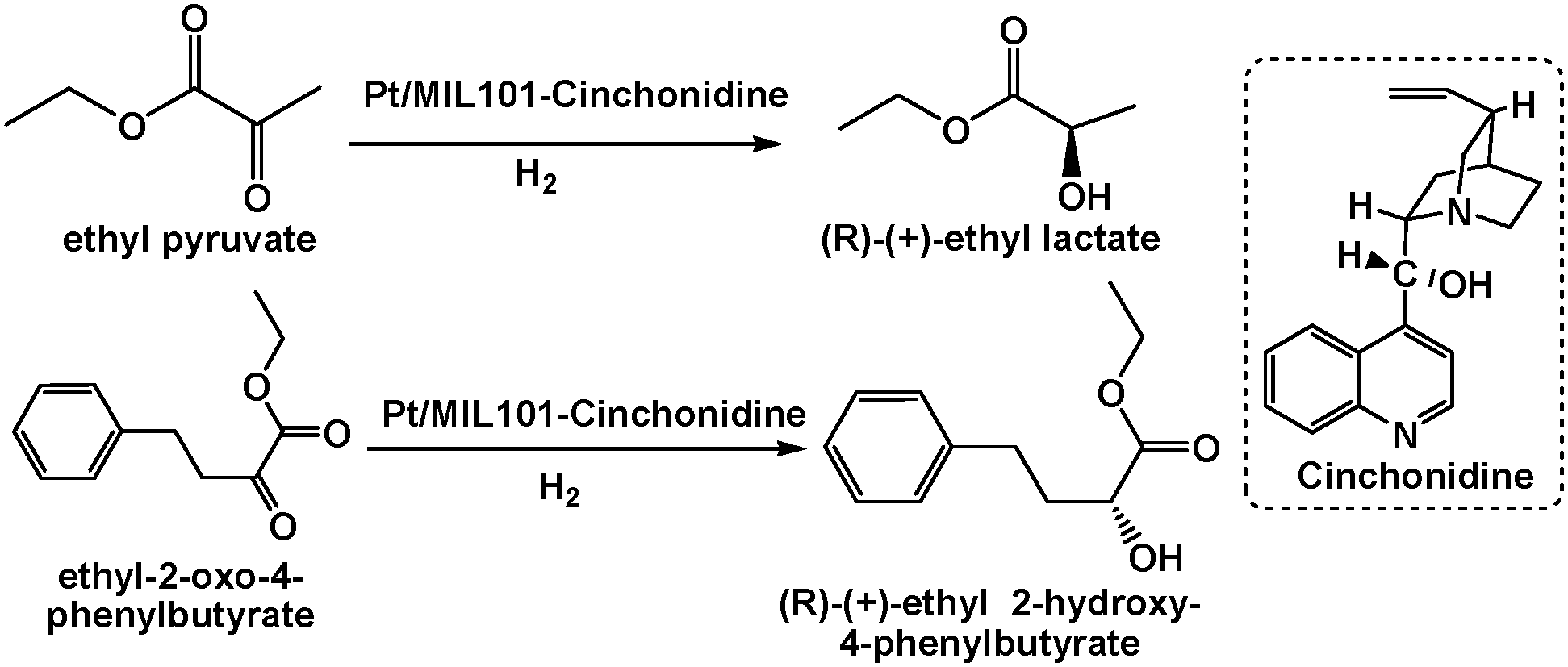 | ||
| Scheme 23 Asymmetric hydrogenation of ethyl pyruvate and ethyl 2-oxo-4-phenylbutyrate by a cinchonidine-modified Pt/MIL-101 catalyst. | ||
3.2 POM@MOF composites for synergistic catalysis and tandem reactions
Polyoxometallates (POMs) are composed of polyanion clusters and counter cations and have diverse structures and properties. They can be encapsulated in MOF pores to serve as different types of active sites, including protons, oxygen atoms, and metal sites, for synergistic catalysis and tandem reactions.103,104 The Keggin-type [BW12O40]5− oxidation catalyst and the chiral L- and D-pyrrolidin-2-ylimidazole (PYI) ligands were used to synthesize the chiral POM-based MOFs denoted Ni–PYI1 and Ni–PYI2, respectively (Fig. 6).105 Both catalysts exhibited moderate activity and very high ee values (>95%) for asymmetric aryl olefin dihydroxylation in the presence of H2O2 (Fig. 6). The authors proposed that the [BW12O40]5− anion was responsible for the catalytic dihydroxylation, whereas the enantioselectivity was controlled by the chiral PYI moiety and the chiral channel environment. Using a similar approach, the same group also prepared the homochiral POM-based ZnW-PYI1 MOF, which was utilized in the tandem asymmetric transformation of CO2 and olefins into value-added enantiomerically pure cyclic carbonates.106 The ZnW12O406− Keggin anion and the chiral PYI organocatalyst were used to fabricate the amino-functionalized homochiral ZnW–PYI1 MOF (Fig. 7). The tandem reaction catalysed by this MOF proceeded via the asymmetric epoxidation of the styrene derivative by the synergistic action of the ZnW12O406− Keggin anion and PYI in the presence of t-butylhydroperoxide (TBHP). The epoxide intermediate and adsorbed CO2 were subsequently activated by the uncoordinated Zn Lewis acid sites in ZnW–PYI1 and by TBABr. The ZnW–PYI1 crystal structure indicates that the high ee values could be attributed to the hydrogen-bonding interactions between the spatially matched ZnW12O406− oxidation catalyst and PYI organocatalyst and to the π–π interactions between the styrene oxide benzene ring and the PYI imidazole ring. The electrophilic ZnW12O406− oxidant and the chiral directing agents in the ZnW–PYI pores also controlled the orientation of the substrates, resulting in stereoselective catalysis. Although complete conversion was only achieved after four days for this one-pot asymmetric catalytic reaction, the general strategy of catalysing a tandem epoxidation–cycloaddition reaction by a chiral MOF is an atom-efficient, environmentally friendly synthesis method for producing useful chiral compounds from CO2.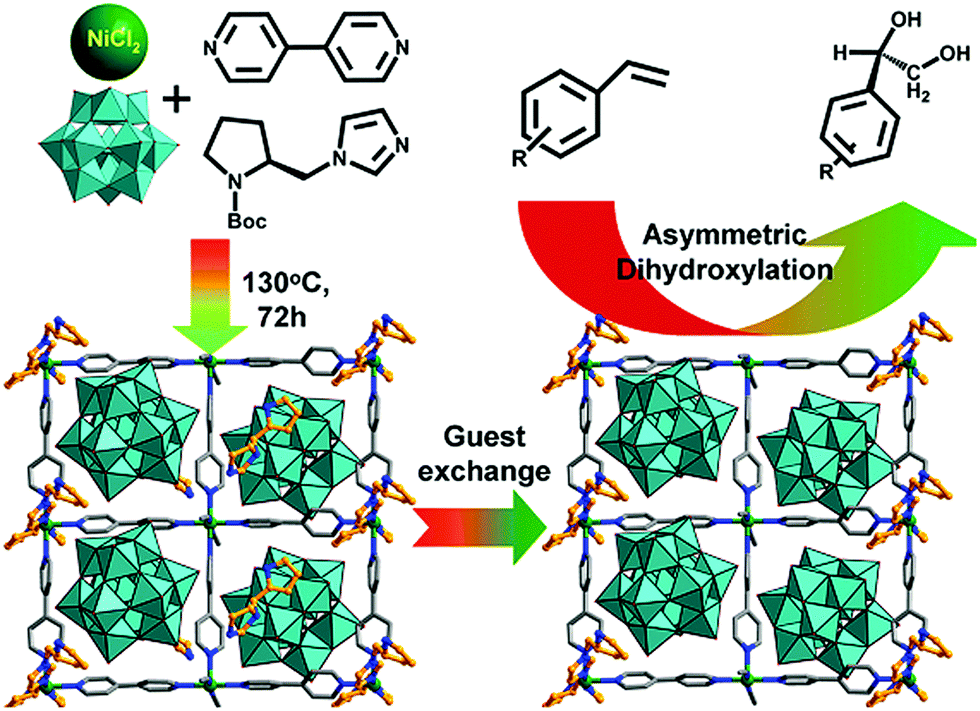 | ||
| Fig. 6 Ni–PYI1 synthesis showing the guest exchange process and the potential amphipathic channel for the asymmetric olefin dihydroxylation catalysis. Reproduced from ref. 105 with permission from American Chemical Society. | ||
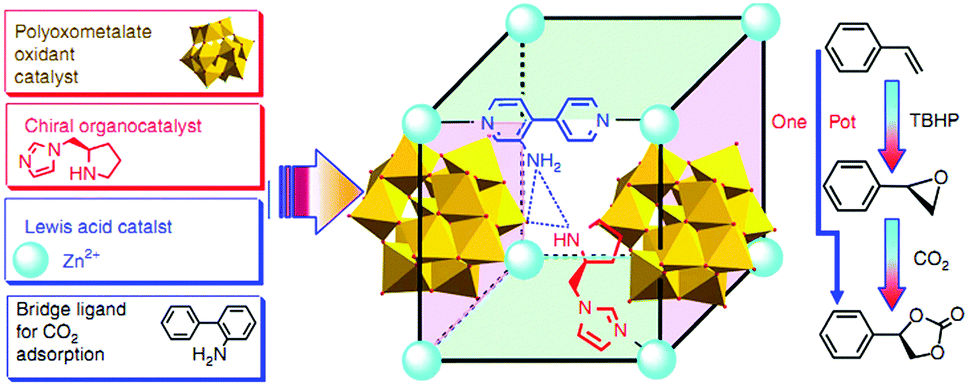 | ||
| Fig. 7 Synthesis route for the homochiral ZnW–PYI1 catalyst and schematic representation of the tandem reaction for asymmetric cyclic carbonate synthesis from olefins and carbon dioxide. Reproduced from ref. 106 with permission from Nature Publishing Group. | ||
4. Bimetallic NPs stabilized on MOFs for synergistic catalysis
It is well known that composite materials consisting of bimetallic alloy or core–shell nanoparticles (NPs) on MOF supports are promising catalysts because they usually exhibit higher catalytic activities than their monometallic NP counterparts. They are expected to exhibit not only a combination of the properties of the two metals but also new outstanding properties due to the synergy between them.107 However, the preparation of bimetallic NPs is much more difficult than that of monometallic NPs. Recently, many groups have made considerable progress in this field. Here, the recent advancements in the chemical synthesis of bimetallic NPs supported on MOFs and their applications in synergistic catalysis and tandem reactions are highlighted.4.1 Bimetallic alloy NPs
Bimetallic alloy NPs consist of homogeneous mixtures of two metals at the atomic level and can be identified by the formation of metal–metal bonds.108 Therefore, bimetallic alloy NP catalysts generally result in synergistic effects with respect to their activity and/or selectivity. Recently, the use of MOF-immobilized bimetallic alloy NPs in synergistic catalysis has gradually increased. In particular, non-precious metals, such as Ni, Cu, and Co, have been used in combination with noble metals to decrease the use of expensive noble metals and thus the cost of these catalysts. The progress in synergistic catalysis by bimetallic alloy NPs on MOF supports is summarized in Table 3, and the discussion of the literature is organized based on the preparation method.| MOF composite | Preparation method | Alloy evidence | Reactions | Ref. |
|---|---|---|---|---|
| Pt–Ru@MOF-5 | MOCVD | PXRD | No test | 109 |
| Ni–Pd@MIL-101 | MOCVD | Molecular dynamics calculations | Reduction of phenol and cyclic and dialkyl ketones | 114 |
| Pd–Cu@MIL-101 | Solution impregnation | No confirmation | CO conversion to CO2 | 115 |
| Pt–Pd@MM-MIL-53 | Solution impregnation | PXRD, EDS line scanning | Oxidant-free alcohol dehydrogenation | 89 |
| Au–Pd/MIL-101 and Au–Pd/ED-MIL-101 | Solution impregnation | PXRD | FA dehydrogenation to H2 | 116 |
| Au–Pd–MnOx/ZIF-8–rGO | Solution impregnation | PXRD, TEM line scanning | FA dehydrogenation to H2 | 118 |
| Ag–Pd@MIL-101 | Solution impregnation | PXRD | FA dehydrogenation to H2 | 120 |
| Ag–Pd/ZIF-8 | Solution impregnation | PXRD | FA dehydrogenation to H2 | 121 |
| Ni–Pt/ZIF-8 | Solution impregnation | Uncertainty | Hydrazine decomposition to hydrogen | 125 |
| Ni–Pt@MIL-101 | Solution impregnation | PXRD | Hydrazine decomposition to hydrogen | 126 |
| Ni–Pt@MIL-96 | Solution impregnation | PXRD | Hydrazine decomposition to hydrogen | 127 |
| Ni–Rh@ZIF-8 | Solution impregnation | HRTEM | Hydrazine decomposition to hydrogen | 128 |
| Au–Pd/MIL-101 | Sol–gel method | XPS | Oxidative esterification of toluene with methanol | 129 |
| Au–Pd/MIL-101 | Sol–gel method | XPS | Selective oxidation of saturated hydrocarbons to ketones and alcohols | 130 |
| Ru–Pt/MIL-101 | Sol–gel method | XPS | Benzene hydrogenation to cyclohexane | 131 |
| Pd–Ag/MIL-101 | DSM | XPS | Tandem nitroarene hydrogenation and reductive amination of aldehydes or ketones | 84 |
| Au–Ni@MIL-101 | DSM | XPS | Ammonia borane hydrolysis to release H2 | 135 |
| Au–Co@MIL-101 | DSM | XPS | Ammonia borane hydrolysis to release H2 | 136 |
| Cu–Co@MIL-101 | DSM | XPS | Ammonia borane hydrolysis to release H2 | 137 |
| Ru–Cu–Co@MIL-101 | Solution impregnation | PXRD | Ammonia borane hydrolysis to release H2 | 138 |
| Pt–Pd@ZIF-8 | Bottle-around-the-ship | PXRD, TEM line scanning | Ethylene photodegradation to CO2 and H2O | 140 |
| Pd–Au@ZIF-8 | Bottle-around-the-ship | PXRD, TEM, EDX elemental mapping | Secondary alcohol oxidation | 141 |
| PdxNiy-in-UiO-67 | Bottle-around-the-ship | XPS | Nitrobenzene hydrogenation | 142 |
| Ag–Pd@MIL-100(Fe) | Bottle-around-the-ship | PXRD | Formic acid dehydrogenation to H2 | 144 |
| Pt–Ni@MOF-74 | Bottle-around-the-ship | TEM line scanning, EDX elemental mapping | Tandem nitroarene reductive imination with carbonyl compounds | 145 |
Liquid impregnation was also used to fabricate Ni–Pt NPs on MOF supports, including ZIF-8,125 MIL-101126 and MIL-96.127 These materials were employed as heterogeneous catalysts for selective hydrazine decomposition in an aqueous solution in the presence of NaOH to produce hydrogen. Compared with the bimetallic catalysts, the monometallic Pt and Ni catalysts and their physical mixtures were either inactive or exhibited very low activity under the same conditions. This observation indicated that the Ni NP surface structure was altered by the presence of a small amount of platinum, resulting in high catalytic performance. Surprisingly, although the average size of Ni–Pt NPs encapsulated in the MIL-101 mesopores was smaller than that dispersed on the MIL-96 surface (1.8 ± 0.4 nm vs. 3.2 nm), the TOF of the latter catalyst was nearly twice that of Ni88Pt12@MIL-101 (114.3 h−1vs. 65.2 h−1) at room temperature. It was suggested that the activity was independent of the NP size. In the Ni–Pt/MIL-96 catalytic system, hydrazine dehydrogenation occurred on the external framework surface, whereas the hydrazine could enter the pores in the MIL-101 system to access the internal active sites. Thus, the lower activity observed for the latter catalyst might have been due to the slow diffusion process in the pores. Using a similar synthesis method, very small Ni–Rh NPs (1.2 ± 0.2 nm) were deposited on ZIF-8 and were found to exhibit high activity for hydrazine dehydrogenation in an alkaline solution with a turnover frequency of 140 h−1 and 100% hydrogen selectivity at 50 °C.128
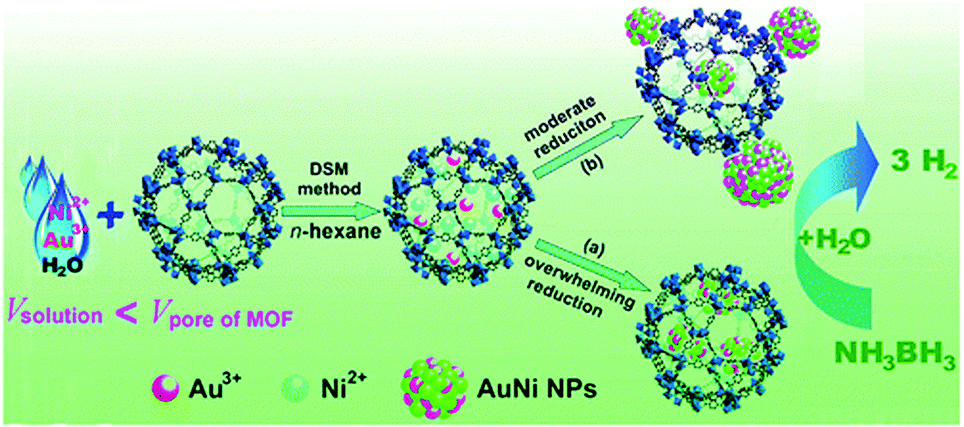 | ||
| Fig. 8 AuNi NPs immobilized on MIL-101 using the DSM and their use in the hydrolytic dehydrogenation of aqueous AB. Reproduced from ref. 135 with permission from American Chemical Society. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of pure Pt@ZIF-8 and pure Pd@ZIF-8, revealing the synergistic effect of the Pt–Pd alloy on the catalysis. Using the same method, Fischer and coworkers prepared composites in which bimetallic Pd–Au alloy NPs were encapsulated in ZIF-8.141 Compared with Pd NPs and Au NPs stabilized with PVP, the unsupported Pd–Au alloy NPs exhibited a synergistically improved activity for secondary alcohol oxidation in an aqueous solution in the presence of K2CO3. However, the ZIF-8-encapsulated Pd–Au catalyst exhibited lower activity for aqueous aerobic alcohol oxidation. This phenomenon resulted from the fact that the small ZIF-8 windows (0.38 nm) and its hydrophobic surface prevented the reactants from accessing the embedded active Pd–Au NPs. A similar phenomenon was also observed for olefin hydrogenation catalysed by ZIF-8-encapsulated Pt NPs.139
:1 mixture of pure Pt@ZIF-8 and pure Pd@ZIF-8, revealing the synergistic effect of the Pt–Pd alloy on the catalysis. Using the same method, Fischer and coworkers prepared composites in which bimetallic Pd–Au alloy NPs were encapsulated in ZIF-8.141 Compared with Pd NPs and Au NPs stabilized with PVP, the unsupported Pd–Au alloy NPs exhibited a synergistically improved activity for secondary alcohol oxidation in an aqueous solution in the presence of K2CO3. However, the ZIF-8-encapsulated Pd–Au catalyst exhibited lower activity for aqueous aerobic alcohol oxidation. This phenomenon resulted from the fact that the small ZIF-8 windows (0.38 nm) and its hydrophobic surface prevented the reactants from accessing the embedded active Pd–Au NPs. A similar phenomenon was also observed for olefin hydrogenation catalysed by ZIF-8-encapsulated Pt NPs.139
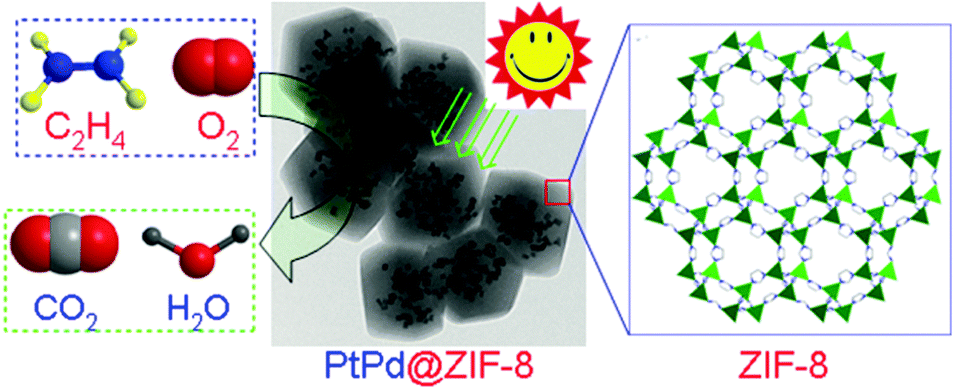 | ||
| Fig. 9 Conversion of adsorbed ethylene into CO2 and H2O photocatalysed by Pt–Pd@ZIF-8 at room temperature. Reproduced from ref. 140 with permission from Royal Society of Chemistry. | ||
Recently, Li and coworkers reported a facile strategy for incorporating a bimetallic precursor into a MOF and subsequently reducing the metal ions to form bimetallic NPs (Scheme 24).142 In particular, a PdCl2(CH3CN)2 and Ni(NO3)2·6H2O precursor mixture was added to a DMF solution of the UiO-67 analogue precursors (2,2′-bipyridine-5,5′-dicarboxylic acid (BPDA) and ZrCl4). During the UiO-67 assembly process, the metal precursors were coordinated to the BPDA ligand, enabling them to be encapsulated in the MOF pores in situ. After reducing the metal ions with NaBH4, highly dispersed Pd–Ni NPs were obtained on the UiO-67 support. However, the particle sizes (3–4 nm) exceeded the diameter of the UiO-67 cavity (1.8 nm). This unusual phenomenon might be explained by local defects/deformations in the host framework caused by the NP growth or by the MNP aggregation caused by the TEM electron beam, which is consistent with the results reported for MNPs/MOF materials in the literature.143 The resulting PdxNiy-in-UiO-67 catalysts were utilized in the model nitrobenzene hydrogenation reaction. The monometallic Ni-in-UiO-67 catalyst was inactive, whereas complete nitrobenzene conversion was achieved in 18 h with Pd-in-UiO-67. Remarkably, the bimetallic Pd7Ni3-in-UiO-67 catalyst achieved a quantitative aniline yield within 2 h, clearly demonstrating the synergistic effect of the bimetallic alloy.
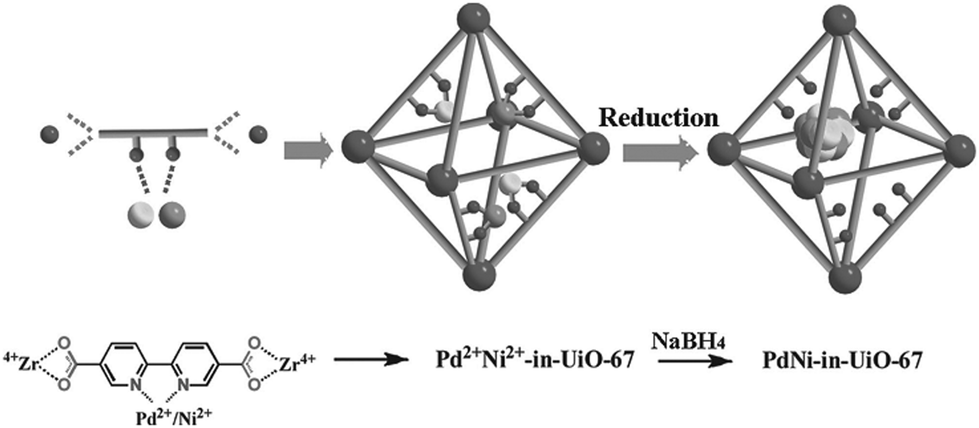 | ||
| Scheme 24 Encapsulation of Pd–Ni NPs in UiO-67 by using a bimetallic precursor as a MOF ligand and post-synthetically reducing the metal ions. Reproduced from ref. 142 with permission from Wiley-VCH. | ||
Bimetallic Ag–Pd alloy NPs were coated with mesoporous MIL-100(Fe) via a facile one-pot method to form Ag–Pd@MIL-100(Fe).144 Uniform particles were obtained by mixing AgNO3, Pd(NO3)2 and the MOF precursors (FeCl3 and H3BTC) in solution in the presence of PVP at 140 °C (Scheme 25). During the synthesis, AgNO3 and Pd(NO3)2 were reduced by DMF to form Ag–Pd alloy NPs. MIL-100(Fe) shells were subsequently grown on the PVP-modified Ag–Pd NP surfaces. The Ag–Pd NP size could be easily controlled from 14 nm to 86 nm, and the shell thickness was varied from 7 nm to 118 nm. The PXRD patterns of the MIL-100(Fe)-encapsulated Ag–Pd NPs exhibited well-defined peaks between the characteristic Ag and Pd peaks, indicating the formation of a Ag–Pd alloy structure. Elemental mapping further demonstrated that the catalysts consisted of a Ag–Pd core, whereas the MIL-100(Fe) C, O, and Fe atoms were distributed around the entire NP core. FA dehydrogenation to H2 was conducted in the presence of the Ag–Pd@MIL-100(Fe) catalyst at room temperature to evaluate its catalytic activity. In the absence of any additive, the Ag–Pd@MIL-100(Fe) catalyst with a shell thickness of 7 nm exhibited higher activity than the monometallic catalysts, with a TOF of 58 h−1 at room temperature.
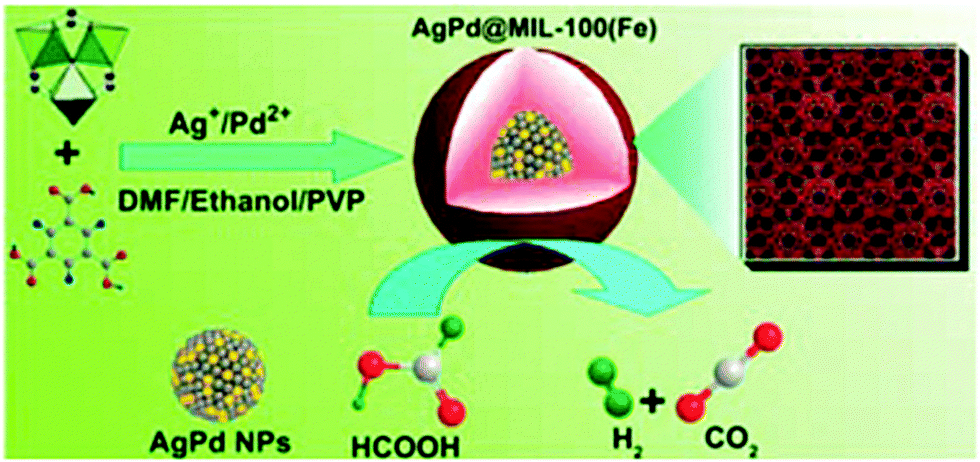 | ||
| Scheme 25 Schematic representation of the synthesis of Ag–Pd@MIL-100(Fe) NPs and their use in FA decomposition at 298 K. Reproduced from ref. 144 with permission from Royal Society of Chemistry. | ||
More recently, Li and coworkers incorporated bimetallic Pt–Ni frames into porous MOF-74 via an in situ etching and coordination synthesis strategy.145 The Pt–Ni frame@MOF-74 catalyst was synthesized by adding the 2,5-dioxidoterephthalate (dobdc) ligand to a DMF solution of the PVP-capped Ni-rich Pt–Ni alloy NPs and then treating the mixture solvothermally (Fig. 10). Here, the Ni metal not only served as the MOF-74 precursor source but also promoted the formation of the Pt–Ni frames after etching. The oxidative etching of the Pt–Ni alloy and the in situ nucleation of MOF-74 occurred simultaneously (eqn (1)–(3)).
| 1/2O2 + H2O + 2e− ⇌ 2OH− | (1) |
| Ni(0) − 2e− ⇌ Ni(II) | (2) |
| 2Ni(II) + dobdc4− ⇌ Ni2(dobdc) | (3) |
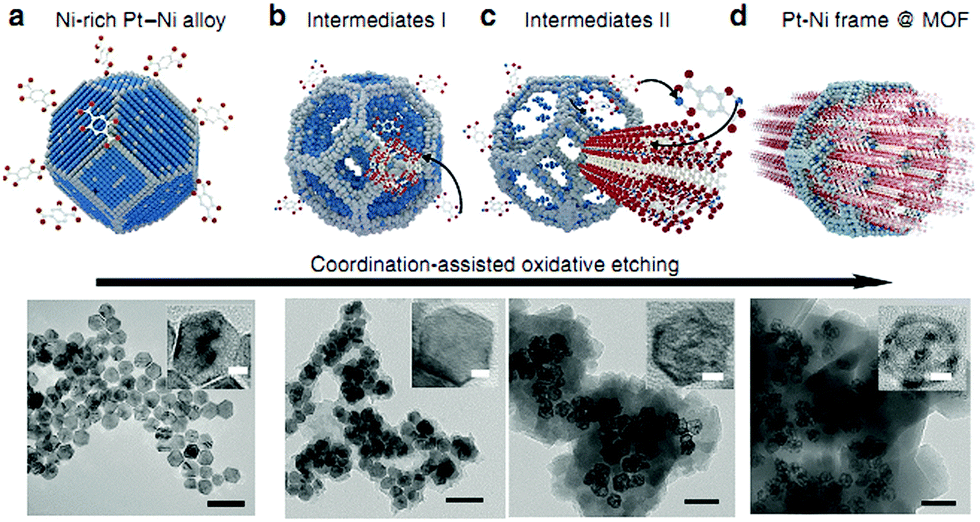 | ||
| Fig. 10 Scheme and corresponding TEM images of the coordination-assisted oxidative etching process. (a) Initial solid Pt–Ni polyhedra. (b) Pt–Ni frame@MOF intermediate I. (c) Pt–Ni frame@MOF intermediate II. (d) Final Pt–Ni frame@MOF. Scale bar: 50 nm (the magnified TEM images are shown in the insets; scale bar: 5 nm). Reproduced from ref. 145 with permission from Nature Publishing Group. | ||
The Ni metal atoms in the Ni-rich Pt–Ni alloy NPs were oxidized to Ni(II) ions, which were then captured by neighbouring dobdc linkers to form the microporous Ni-MOF-74. Compared with Ni, Pt is relatively inert to oxygen, which led to their different diffusion rates during the etching process. Therefore, the formation of the nanoframes might be controlled by the Kirkendall effect.146 When the Pt–Ni alloy polyhedra were etched to their frameworks, Ni-MOF-74 was simultaneously synthesized, encapsulating the alloy framework within its matrix. A 3D tomographic reconstruction indicated that the Pt–Ni frames were fully covered by Ni-MOF-74. Interestingly, the H2 adsorption isotherms at 273 K revealed that the bare Pt–Ni polyhedra only adsorbed 0.03 H per metal atom, whereas Pt–Ni frame@Ni-MOF-74 adsorbed 0.25 H per metal atom. This enhanced H2 adsorption capacity could improve the catalytic efficiency in hydrogenation reactions. Indeed, the Pt–Ni frame@Ni-MOF-74 catalyst exhibited higher hydrogenation activity than the Pt–Ni polyhedra and bare Pt–Ni frames. Furthermore, Pt–Ni frame@Ni-MOF-74 exhibited high selectivity to the imine products in the one-pot tandem reductive imination of nitroarenes with carbonyl compounds due to its outstanding H2 adsorption capacity and molecular sieving properties. The diffusion of the imine towards the interior Pt–Ni frame was inhibited by the small Ni-MOF-74 micropores (0.86 nm), effectively suppressing the over-reduction of the imine to N-phenylbenzylamine (Scheme 26).
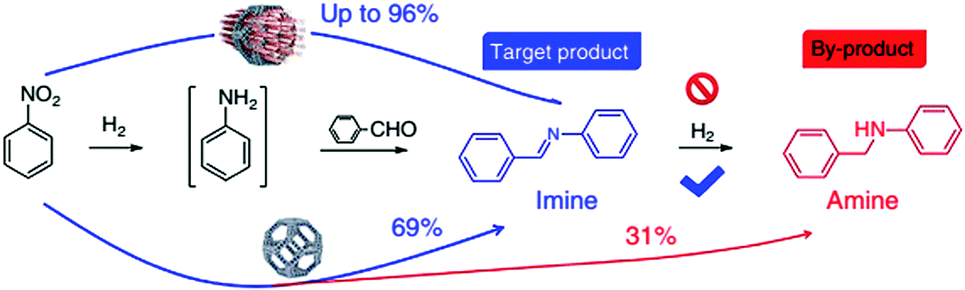 | ||
| Scheme 26 Size-selective catalysis of nitrobenzene reductive imination. Reproduced from ref. 145 with permission from Nature Publishing Group. | ||
4.2 Core–shell MNPs
Core–shell MNPs are another type of widely studied bimetallic particles that usually perform enhanced synergistic catalysis because the metal shell can be significantly affected by the metal core, giving rise to unique physicochemical properties.147 The properties of bimetallic core–shell NPs can be tuned by controlling their size, composition and structure. However, compared with the bimetallic alloy NP syntheses discussed previously, the formation of core–shell structures on MOFs is more difficult because the second type of metal atoms might nucleate separately. Fortunately, a few examples of preparation of bimetallic core–shell NPs on MOF supports have appeared in the literature (Table 4).148,151–154| MOF composite | Preparation method | Reaction | Core–shell evidence | Ref. |
|---|---|---|---|---|
| Au@Ag/ZIF-8 | Sequential deposition–reduction | 4-Nitrophenol reduction | EDS line scanning | 148 |
| Au@Ag/CD-MOF | Reaction–diffusion | No test | UV-Vis spectroscopy and EDS | 151 |
| Pt@Pd/MIL-101 | CO-directed reduction | CO oxidation to CO2 | EDS elemental mapping | 152 |
| Pd@Co@MIL-101 | DSM | AB dehydrogenation | EDS elemental mapping | 153 |
| Pd@Ag-in-UiO-67 | Seed-mediated growth | Phenylacetylene hydrogenation to styrene | EDS mapping and line profile analysis | 154 |
Xu and coworkers prepared Au@Ag core–shell NPs (2–6 nm) supported on ZIF-8 external surfaces by a sequential deposition–reduction method.148 In approach I, the activated ZIF-8 was sequentially soaked in Au and Ag precursor aqueous solutions and reduced after each soak to yield Au@Ag core–shell NPs (Scheme 27). Surprisingly, soaking ZIF-8 in the Ag precursor solution first led to the formation of a similar core–shell structure of Au@AuAg NPs (Scheme 27, approach II). This result was attributed to a galvanic replacement reaction caused by the difference in the reduction potentials of the two soluble metal salts (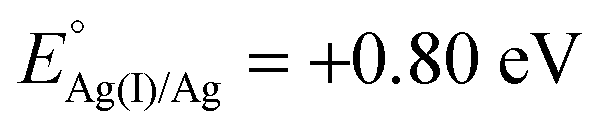 vs. SHE (standard hydrogen electrode);
vs. SHE (standard hydrogen electrode); 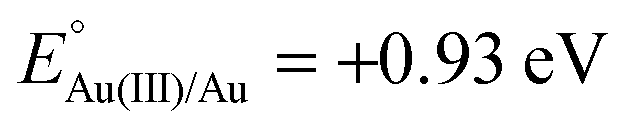 vs. SHE).149 Although the core–shell structure could not be observed in the TEM images, EDS line scanning experiments and point analyses revealed that each particle consisted of a bright Au core coated by a dark Ag shell. The obtained Au@Ag core–shell NPs exhibited higher activity for 4-nitrophenol reduction than the corresponding monometallic and alloyed NPs. This result indicated that the synergistic effect of the Au@Ag core–shell NPs was due to the modified electronic structures of the metals.150 In another study, Au@Ag core–shell NPs were prepared in the crystals of a cyclodextrin-based MOF (CD-MOF) via a reaction–diffusion method.151 Interestingly, the CD-MOF crystals contain homogeneously distributed OH− counterions that could reduce the Ag and Au salts to their respective MNPs. Therefore, Au@Ag core–shell NPs were easily formed in the CD-MOF crystals by successively immersing the MOF crystals in HAuCl4 and AgNO3 solutions (Fig. 11a). Interestingly, when the Ag NPs were uniformly embedded in the CD-MOF crystals first, a galvanic exchange reaction with the Au salt occurred (after ca. 3 h) due to the higher Au(III)/Au redox potential (Fig. 11b). At longer reaction times (ca. 9–10 h), nearly all of the Ag NPs in the CD-MOF were exchanged by Au NPs. Unfortunately, the obtained Au@Ag core–shell NPs were not utilized for catalysis.
vs. SHE).149 Although the core–shell structure could not be observed in the TEM images, EDS line scanning experiments and point analyses revealed that each particle consisted of a bright Au core coated by a dark Ag shell. The obtained Au@Ag core–shell NPs exhibited higher activity for 4-nitrophenol reduction than the corresponding monometallic and alloyed NPs. This result indicated that the synergistic effect of the Au@Ag core–shell NPs was due to the modified electronic structures of the metals.150 In another study, Au@Ag core–shell NPs were prepared in the crystals of a cyclodextrin-based MOF (CD-MOF) via a reaction–diffusion method.151 Interestingly, the CD-MOF crystals contain homogeneously distributed OH− counterions that could reduce the Ag and Au salts to their respective MNPs. Therefore, Au@Ag core–shell NPs were easily formed in the CD-MOF crystals by successively immersing the MOF crystals in HAuCl4 and AgNO3 solutions (Fig. 11a). Interestingly, when the Ag NPs were uniformly embedded in the CD-MOF crystals first, a galvanic exchange reaction with the Au salt occurred (after ca. 3 h) due to the higher Au(III)/Au redox potential (Fig. 11b). At longer reaction times (ca. 9–10 h), nearly all of the Ag NPs in the CD-MOF were exchanged by Au NPs. Unfortunately, the obtained Au@Ag core–shell NPs were not utilized for catalysis.
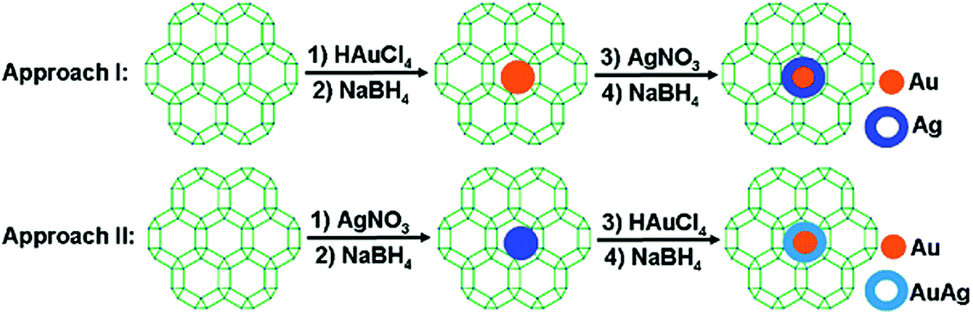 | ||
| Scheme 27 Preparation of Au@Ag core–shell NPs on the ZIF-8 support. Reproduced from ref. 148 with permission from American Chemical Society. | ||
 | ||
| Fig. 11 Au@Ag core–shell NPs prepared on CD-MOF by a reaction–diffusion method. Reproduced from ref. 151 with permission from Wiley-VCH. | ||
Pt@Pd core–shell NPs were immobilized on MIL-101 (Pt@Pd/MIL-101) by a facile CO-directed reduction method at the solid–gas interface.152 Interestingly, the Pt@Pd core–shell NPs mostly formed tetrahedra due to selective CO adsorption on the Pd(111) facets, which slowed down the continuous growth of the minimum surface energy faces. The Pt@Pd/MIL-101 catalyst exhibited higher activity for CO oxidation to CO2 at a lower temperature than Pd/MIL-101 because of the smaller activation energy of this system. However, complete CO conversion to CO2 occurred at a higher temperature when Pt@Pd/MIL-101 was used than when Pt/MIL-101 was employed (200 °C vs. 175 °C). Core–shell bimetallic NPs can also be immobilized in the MIL-101 pores. For example, Jiang and coworkers prepared Pd@Co core–shell NPs within the mesoporous cages of MIL-101, and the resulting catalyst exhibited enhanced activity for hydrolytic AB dehydrogenation and excellent recyclability.153 Pd(II) and Co(II) salts were incorporated into the MIL-101 cages via a DSM that exploited the hydrophilic surfaces of the MIL-101 mesopores. Interestingly, Pd@Co core–shell NPs (Pd@Co@MIL-101) were formed in the presence of the mild reducing agent AB. When the strong reducing agent NaBH4 was used, PdCo alloy NPs were obtained (Scheme 28). The successful formation of a core–shell structure is based on the difference in the reduction potentials of the two metal salts (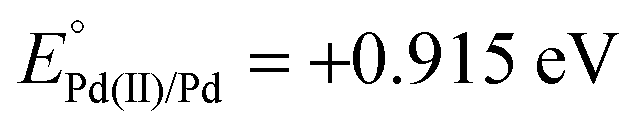 vs. SHE;
vs. SHE; 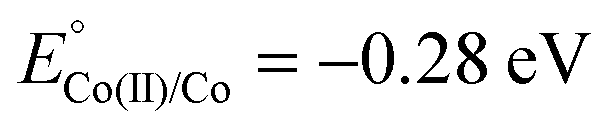 vs. SHE). Therefore, the Pd(II) ions were reduced to Pd NPs by the mild reducing agent AB first, and then, the Co ions were reduced by Pd-H species that were produced during the AB hydrolysis to form shells on the Pd NP cores. Although the core–shell structure could not be observed in the elemental scanning line profile analysis due to the small NP size (2.5 nm), a bright core coated by a dark shell was observed for each particle in the HAADF-STEM images. The core–shell structure was also confirmed by EDS mapping of the Pd and Co NPs. For comparison, samples with Pd@Co NPs located on the MIL-101 external surface (Pd@Co/MIL-101) and with Pd, Co, and PdCo alloy NPs encapsulated in the pores were also prepared. It should be noted that Co@MIL-101 was inactive in hydrolytic AB dehydrogenation, which conflicts with other results reported in the literature.136,137 In contrast, the Pd@Co core–shell NPs exhibited higher activity than the Pd, Co and PdCo alloy NPs in hydrolytic AB dehydrogenation, demonstrating the synergistic effect of Pd and Co and the advantages of core–shell NPs. Notably, although the confined Pd@Co NPs exhibited only slightly higher activity than Pd@Co/MIL-101, they were much more stable. Pd@Co@MIL-101 retained its catalytic activity after five runs without any treatment or activation steps between runs. This result was due to the confinement and stabilization of the NPs in the MOF pores. In contrast, the activity of Pd@Co/MIL-101 decreased sharply during the recycling experiments, probably due to NP aggregation (5–10 nm) on the external surfaces.
vs. SHE). Therefore, the Pd(II) ions were reduced to Pd NPs by the mild reducing agent AB first, and then, the Co ions were reduced by Pd-H species that were produced during the AB hydrolysis to form shells on the Pd NP cores. Although the core–shell structure could not be observed in the elemental scanning line profile analysis due to the small NP size (2.5 nm), a bright core coated by a dark shell was observed for each particle in the HAADF-STEM images. The core–shell structure was also confirmed by EDS mapping of the Pd and Co NPs. For comparison, samples with Pd@Co NPs located on the MIL-101 external surface (Pd@Co/MIL-101) and with Pd, Co, and PdCo alloy NPs encapsulated in the pores were also prepared. It should be noted that Co@MIL-101 was inactive in hydrolytic AB dehydrogenation, which conflicts with other results reported in the literature.136,137 In contrast, the Pd@Co core–shell NPs exhibited higher activity than the Pd, Co and PdCo alloy NPs in hydrolytic AB dehydrogenation, demonstrating the synergistic effect of Pd and Co and the advantages of core–shell NPs. Notably, although the confined Pd@Co NPs exhibited only slightly higher activity than Pd@Co/MIL-101, they were much more stable. Pd@Co@MIL-101 retained its catalytic activity after five runs without any treatment or activation steps between runs. This result was due to the confinement and stabilization of the NPs in the MOF pores. In contrast, the activity of Pd@Co/MIL-101 decreased sharply during the recycling experiments, probably due to NP aggregation (5–10 nm) on the external surfaces.
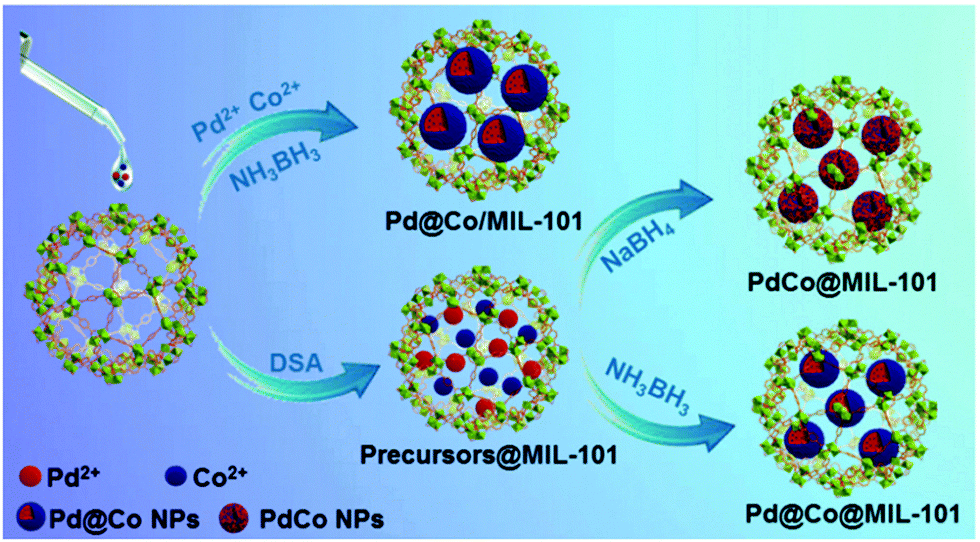 | ||
| Scheme 28 Syntheses of Pd@Co@MIL-101, Pd@Co/MIL-101, and PdCo@MIL-101 catalysts by different procedures using different reducing agents. Reproduced from ref. 153 with permission from Wiley-VCH. | ||
Li and coworkers fabricated tiny Pd@Ag core–shell NPs within the pores of a BPDA-based UiO-67 analogue via a seed-mediated growth method using activated hydrogen atoms as the reducing agent.154 First, Pd NP seeds were encapsulated in the pores of the UiO-67 analogue via a pre-incorporation method. Then, hydrogen molecules were dissociated and activated on the Pd NP surfaces to serve as reducing agents for selective Ag deposition on Pd (Scheme 29). The successful formation of the core–shell structure was attributed to the confinement of the activated hydrogen atoms on the embedded Pd NP surfaces, which led to selective Ag(I) reduction on Pd and suppressed Ag(I) self-nucleation to generate individual Ag NPs. Although it was difficult to observe the Pd@Ag core–shell structure in the HAADF-STEM images because the atomic masses of Pd and Ag are very similar, the structure was unambiguously confirmed by EDS mapping and line profile analysis. However, it should be noted that the average particle size was ca. 2.6–3.1 nm, which is larger than the MOF superoctahedral cage diameter (1.8 nm).155 This unusual phenomenon was explained by the local defects/deformations in the host framework generated during the MNP growth. Although selective alkyne hydrogenation to alkenes is one of the most important chemical transformations in industry, it is still challenging to achieve high alkene selectivity at high conversions. Pd and Pd@Ag NPs supported on UiO-67 (Pd-in-UiO-67 and Pd@Ag-in-UiO-67) were employed in selective phenylacetylene hydrogenation to styrene. Although the styrene selectivity of both catalysts decreased with increasing phenylacetylene conversion, Pd@Ag-in-UiO-67 (Pd/Ag ratio of 30) exhibited higher selectivity (91% selectivity, quantitative conversion) than monometallic Pd-in-UiO-67 (75% selectivity, >99% conversion). This result might be due to surface dilution by Pd atoms and the modified electronic structure of the Ag shell near the Pd surface sites, although Ag-in-UiO-67 was catalytically inactive. In contrast, the adjacent Pd sites on monometallic Pd NPs would favour styrene hydrogenation to ethylbenzene. Therefore, the Ag shell effectively blocked the Pd core sites and isolated Pd sites at the surface, thereby suppressing phenylacetylene hydrogenation to ethylbenzene and enhancing the selectivity to the partially hydrogenated styrene product.
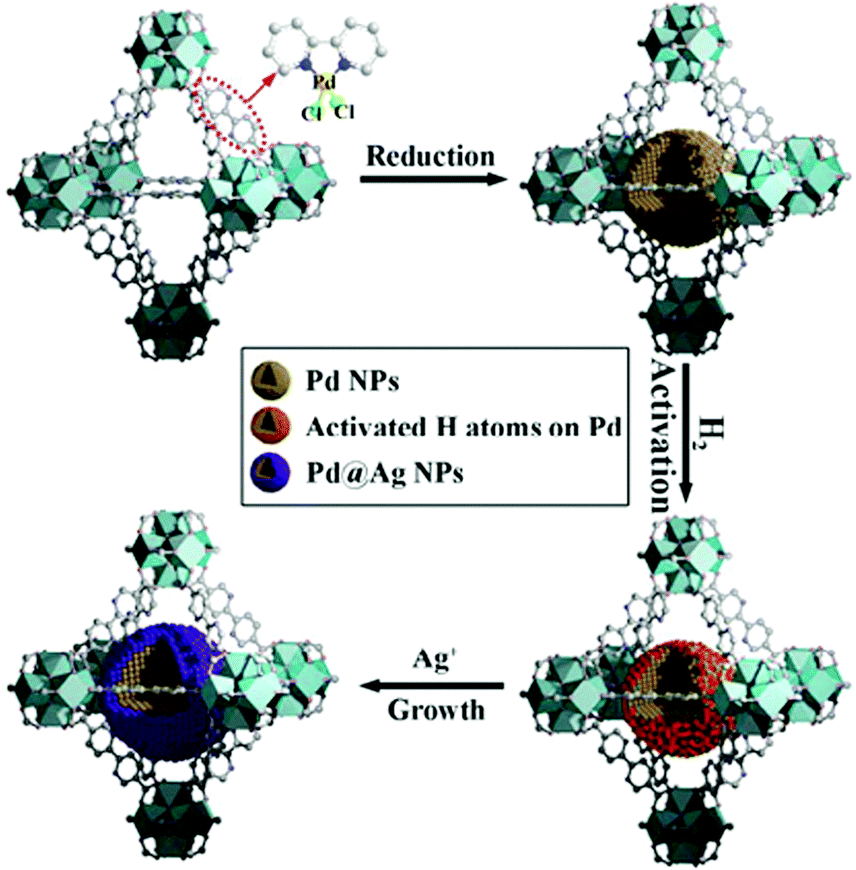 | ||
| Scheme 29 Synthesis of Pd@Ag core–shell NPs encapsulated in the pores of UiO-67. Reproduced from ref. 154 with permission from Royal Society of Chemistry. | ||
4.3 MNPs combined with other active species stabilized on MOFs
The interactions between MNPs and other active species immobilized on MOFs could promote synergistic catalysis. For example, Fischer et al. used the gas-phase loading of CpCuL (L = PMe3, CNtBu) into ZnO@MOF-5 followed by hydrogenolysis at 220 °C to load copper and zinc oxide NPs into MOF-5 (Cu/ZnO@MOF-5).156 The resulting Cu/ZnO@MOF-5 composite was employed in methanol synthesis from a CO/CO2/H2 gas mixture at 1 atm and 220 °C. Interestingly, a high TOF of 212 μmolMeOH gcat−1 h−1 was achieved after 1 h. However, the activity decreased sharply to only 12 μmolMeOH gcat−1 h−1 over a period of 20 h due to the sintering of the Cu and ZnO NPs and the collapse of MOF-5. Although the microscopic details of the Cu/ZnO interface were not studied, the remarkably high initial activity obtained with a very low Cu loading (1.4 wt%) indicated superior interfacial contact between the Cu and ZnO nanophases. Using a similar loading method, the same group also prepared Au/MOx@MOF-5 (M = Zn, Ti; x = 1, 2) composites for liquid-phase benzyl alcohol oxidation.157 Very small Au/ZnO NPs were distributed homogeneously over the entire MOF-5 framework in Au/ZnO@MOF-5. In contrast, the TiO2 particles in the Au/TiO2@MOF-5 material were mainly located on the external surface, and larger Au NPs were distributed over the MOF-5 matrix. XPS analysis showed that the O 1s binding energies of Au/TiO2@MOF-5 and Au/ZnO@MOF-5 were higher than those of TiO2@MOF-5 and ZnO@MOF-5, respectively. These findings indicated that TiO2 (or ZnO) strongly interacted with the Au NPs loaded in MOF-5. Both Au/ZnO@MOF-5 and Au/TiO2@MOF-5 exhibited higher activity and selectivity for liquid-phase benzyl alcohol oxidation in the presence of K2CO3 than Au@MOF-5. In another study, Pd NPs and TiO2 were encapsulated in the mesopores of MIL-101.158 The Pd/TiO2@MIL-101 composite not only exhibited enhanced photocatalytic activity for RhB degradation but also exhibited excellent catalytic performance and stability in benzophenone hydrogenation and the dehydrogenation of aromatic alcohols by C–H activation.Highly dispersed platinum species together with phosphotungstic acid were encapsulated in the mesoporous MOF NH2-MIL-101(Al).159 Two different Pt species, Pt(0) and Pt(II), were formed when the precursor was treated at 473 K in the presence of hydrogen. Increasing the temperature to 573 K led to complete reduction of platinum to Pt(0) and partial reduction of tungsten to W(V), and intermetallic Pt–W species were subsequently formed. The reduced Pt/PTA-NH2-MIL-101(Al) catalyst exhibited higher activity for both CO oxidation and the preferential oxidation (PROX) of CO reaction than a commercial Pt/Al2O3 catalyst. In particular, enhanced selectivity was observed in the PROX reaction because the formation of intermetallic Pt–W species facilitated the adsorption of tilted CO molecules, which strongly weakened the C–O bond and hindered H2 chemisorption.
Multiple active sites supported on MOFs could catalyse tandem reactions. Ru NPs were prepared on a PTA/MIL-101 support (Ru–PTA/MIL-100) using the so-called ship-in-a-bottle approach, followed by reduction with H2.160 Cellulose and cellobiose were selectively converted to sorbitol over the Ru–PTA/MIL-100 catalyst under aqueous conditions in the presence of H2 with yields of 57.9% and 95.1%, respectively. In these reactions, cellulose and cellobiose were first hydrolysed by the Brønsted acid PTA to give glucose as the major product. The subsequent conversion of glucose to sorbitol was catalysed by the Ru sites in the presence of H2 with very high selectivity. The same group also used the similar Ru/NENU-3 catalyst (PTA encapsulated in HKUST-1) to synthesize ethylene glycol from cellulose.161 Whereas the Ru–PTA/MIL-100 catalyst gave sorbitol as the main product in cellulose hydrolysis,160 an ethylene glycol (EG) yield of 50.2% was obtained when Ru/NENU-3 was used in the presence of H2 at 245 °C. The PTA active species not only catalysed cellulose hydrolysis but also promoted the C–C bond cleavage of the cellulose-derived sugars to give glycolaldehyde. This intermediate was then hydrogenated to the ethylene glycol product at the Ru NP sites. However, the recovered Ru/NENU-3 catalyst was deactivated by HKUST-1 decomposition at 245 °C under high H2 pressure.
5. Multifunctional MOFs for synergistic photocatalysis and tandem reactions
Many MOFs can act as semiconductor materials because they exhibit broad UV-Vis absorption in the range of typical band gap values.23 The numerous tunable metal centres and organic ligands in MOFs combined with the unique porous structures make MOFs promising semiconductor materials for photocatalytic reactions, including water splitting, CO2 photoreduction, and organic molecule degradation and/or transformations.23,162–164 Thus, MOFs could be used to address energy and environmental problems. The tunable SBUs and/or functional organic ligands in MOFs can be employed as antennas to absorb light to produce photogenerated electron–hole pairs for photocatalysis. Furthermore, due to their large surface areas and tunable pores/cages, MOFs can provide a platform for anchoring or encapsulating photoactive species for solar energy conversion. Then, the metal ions or clusters and functional ligands can act cooperatively with the encapsulated active sites to synergistically enhance photocatalytic reactions. Here, the discussion of the progress in synergistic photocatalysis and tandem photocatalytic reactions promoted by MOF-based materials is organized into two sections: (i) systems based on the MOF SBUs and functional ligands and (ii) synergistic host–guest systems.5.1 SBUs and linkers used for synergistic photocatalysis and tandem reactions
The UV-Vis absorption bands of MOFs are generally ascribed to a localized metal-to-ligand charge transfer (MLCT), a ligand-to-metal charge transfer (LMCT) or a π–π* transition of the aromatic ligand.23 The energy transfer between the metal nodes and ligands in MOFs has been proven to synergistically enhance photocatalytic reactions. Motivated by the enhanced activity of Ti-containing zeolite photocatalysts,165 Li and coworkers developed an amino-functionalized Ti-based MOF photocatalyst (NH2-MIL-125(Ti)) that could reduce CO2 under visible light irradiation.166 NH2-MIL-125(Ti) not only had a higher CO2 adsorption capacity than MIL-125(Ti) (Ti8O8(OH)4(bdc)6) but also exhibited a visible light absorption band extending to approximately 550 nm. Under visible light (420–800 nm) irradiation, NH2-MIL-125(Ti) produced 8.4 μmol HCOO− in the presence of the triethanolamine (TEOA) sacrificial agent after 10 h. Upon light absorption in the LMCT band, the excited electrons from the amino-functionalized ligands were transferred to Ti(IV), reducing it to Ti(III) and thereby promoting HCOO− production from the adsorbed CO2 (Scheme 30). Although the photoactivity of NH2-MIL-125(Ti) for CO2 conversion was very low, the activity can be significantly improved by changing the MOF composition and structure. The same group further explored CO2 photoreduction over Fe-based MIL-101(Fe), MIL-53(Fe) and MIL-88B(Fe) MOF materials.167 Although the UV-Vis DRS spectra of the Fe-based MOFs exhibited similar broad, intense absorption bands in the 200–450 nm region, the mesoporous MIL-101(Fe) exhibited the highest activity for CO2 photoreduction, producing 59 μmol HCOO− in 8 h in the presence of TEOA. The amount of HCOO− produced over MIL-53(Fe) reached 29.7 μmol in 8 h, whereas only 9.0 μmol HCOO− was obtained over MIL-88B(Fe). These results were attributed to the fact that MIL-101(Fe) has the highest CO2 adsorption capacity (26.4 g cm−3) due to its very high surface area and coordinatively unsaturated Fe metal sites. Similarly to NH2-MIL-125(Ti), the amino-functionalized Fe-based MOF materials (NH2-MIL-101(Fe), NH2-MIL-53(Fe), and NH2-MIL-88B(Fe)) exhibited enhanced absorption in the visible light region extending to ca. 700 nm. Furthermore, compared with their unfunctionalized MOF counterparts, all the amino-functionalized Fe-based MOF materials exhibited not only higher CO2 adsorption capacities but also enhanced activities for photocatalytic CO2 reduction. In particular, 178 μmol HCOO− was produced over NH2-MIL-101(Fe) in 8 h, which was approximately three times that produced over the parent MIL-101(Fe) under similar conditions. The amount of HCOO− produced over NH2-MIL-53(Fe) and NH2-MIL-88B(Fe) reached 46.5 and 30.0 μmol, respectively, in 8 h. For the unmodified Fe-based MOF materials, electrons were transferred from O2− to Fe(III) in the Fe–O clusters under visible light irradiation, resulting in Fe(III) reduction to Fe(II). CO2 was subsequently reduced by Fe(II); TEOA acted as an electron donor (Fig. 12). In addition to the direct excitation of the Fe–O clusters, another excitation pathway existed in the amino-functionalized Fe-MOFs. In this pathway, the NH2 functionality was first excited, and an electron was then transferred from the excited organic linker to the metal centre to generate Fe(II). These findings indicated that the existence of dual excitation pathways and the synergistic effect between these two pathways promoted the photocatalytic CO2 reduction over amino-functionalized Fe-based MOFs (Fig. 12). | ||
| Scheme 30 Proposed mechanism for CO2 photoreduction over NH2-MIL-125(Ti) under visible light irradiation. Reproduced from ref. 166 with permission from Wiley-VCH. | ||
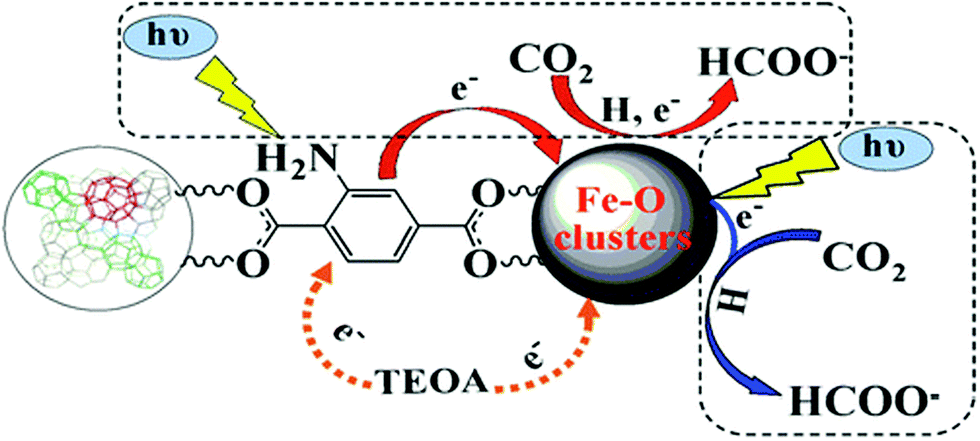 | ||
| Fig. 12 Dual excitation pathways over amino-functionalized Fe-based MOFs. Reproduced from ref. 167 with permission from American Chemical Society. | ||
Interestingly, the amino-functionalized NH2-MIL-101(Fe) and UiO-66-NH2 MOFs could catalyse the tandem reaction consisting of the photooxidation of aromatic alcohols168 and subsequent Knoevenagel condensation with active methylene compounds under visible light.169,170 In particular, NH2-MIL-101(Fe) and UiO-66-NH2 served as antennas to absorb visible light to oxidize aromatic alcohols to aldehydes, which subsequently reacted with methylene compounds via a Knoevenagel condensation catalysed by the basic amino groups of the MOFs.
It is well known that porphyrins and metalloporphyrins have chromophoric properties and photocatalytic activities due to their highly conjugated, aromatic electron system.171 Accordingly, Wu and coworkers prepared a 3D Zn-MOF based on a photoactive tin(IV)-porphyrin, which exhibited excellent activity for the photooxidation of 1,4-dihydroxybenzene and sulfides under Xe lamp irradiation.172 Because the sulfide compound sulfur mustard is one of the most toxic and dangerous chemical agents to humans and the environment, it should be degraded.173 Interestingly, sulfur mustard was photooxidized by a dual functional PCN-222/MOF-545 catalyst based on a photoactive porphyrin.174,175 Under visible light (blue LED) irradiation, triplet oxygen was converted to singlet oxygen by the photoactive PCN-222/MOF-545. Sulfur mustard (2-chloroethyl ethyl sulfide) was then selectively oxidized to the non-toxic 2-chloroethyl ethyl sulfoxide product by the singlet oxygen within several minutes. More importantly, the toxic sulfone product was not detected. In another study, a singlet oxygen-generating porous coordination network (SO-PCN) based on a photoactive porphyrin and the photoswitch ligand 1,2-bis(2-methyl-5-(pyridin-4-yl)thiophen-3-yl)cyclopent-1-ene was irradiated with visible light (λ > 450 nm) to generate singlet oxygen for 1,5-dihydroxynaphthalene oxidation to juglone.176
The use of highly active and enantioselective homochiral MOFs in asymmetric photocatalysis is attractive for obtaining chiral products. For example, the homochiral Zn–PYI1 and Zn–PYI2 MOFs were constructed from the chiral stereoselective L- and D-pyrrolidin-2-ylimidazole organocatalysts, respectively, and the triphenylamine photosensitizer. These MOFs catalysed the asymmetric α-alkylation of aliphatic aldehydes with diethyl 2-bromomalonate under the irradiation of a common fluorescent lamp (Fig. 13).177 A moderate yield of 74% and excellent enantioselectivity (92% ee) were achieved with the Zn–PYI1 catalyst. The red shift in the Zn–PYI1 absorption after aldehyde adsorption indicated that the aldehyde CO group interacted with Zn–PYI1, possibly forming a highly π-nucleophilic enamine. The significant decrease in the luminescence intensity of Zn–PYI1 upon diethyl 2-bromomalonate adsorption suggested that a photoelectron transfer from Zn–PYI1* to the diethyl 2-bromomalonate molecule occurred. Under photoirradiation, the excited photoelectrons initiated by the triphenylamine electron donor functionality in the MOF were transferred to the α-alkylation agent diethyl 2-bromomalonate to produce active radicals. These radicals cleaved the σ-bond to give active intermediates for aliphatic aldehyde α-alkylation. At the same time, the chiral PYI ligands acted as cooperative active sites, facilitating the combination of the highly π-nucleophilic enamine and the electron-deficient radical to drive the asymmetric photocatalysis.
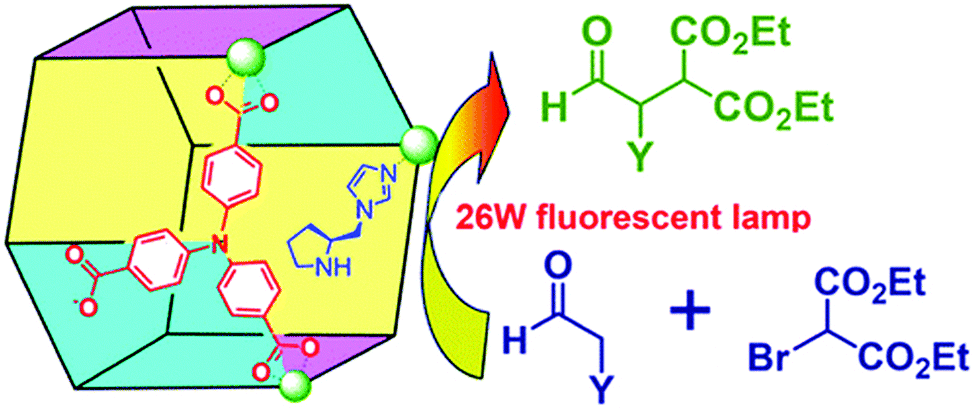 | ||
| Fig. 13 Asymmetric α-alkylation of aliphatic aldehydes with diethyl 2-bromomalonate catalysed by Zn–PYI1 or Zn–PYI2 under the irradiation of a fluorescent lamp. Reproduced from ref. 177 with permission from American Chemical Society. | ||
5.2 MOF composites for synergistic photocatalysis
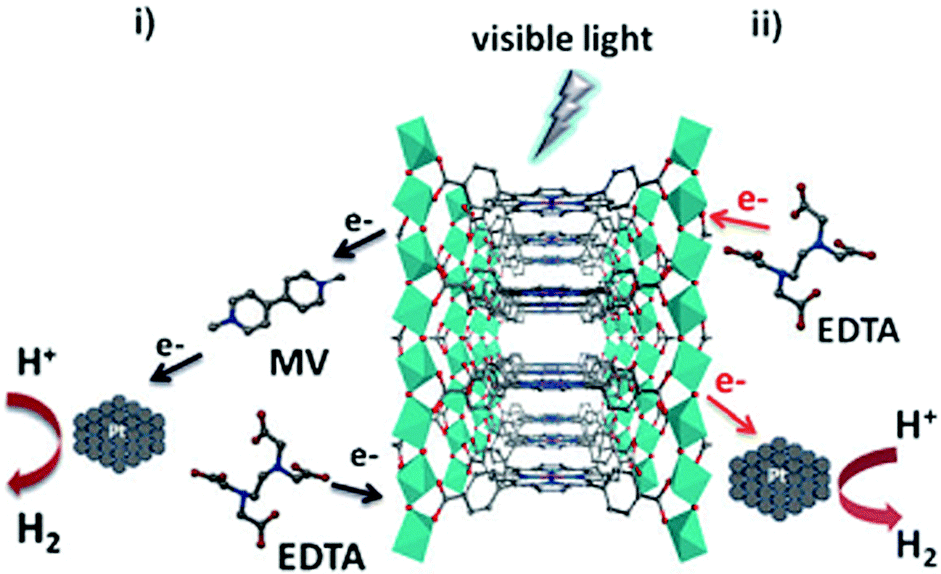 | ||
| Fig. 14 Synergistic photocatalytic hydrogen evolution by Al-PMOF-supported Pt NPs in the (I) presence and (II) absence of MV. Reproduced from ref. 178 with permission from Wiley-VCH. | ||
To improve the energy transfer between MNPs and MOF functionalities, Lin and coworkers inserted Pt NPs into the cavities of phosphorescent UiO-type MOFs, which were constructed from [Ir(ppy)2(bpy)]+-derived dicarboxylate ligands (ppy = 4-phenyl-2-pyridine, bpy = 5,5′-dicarboxylate-2,2′-bipyridine) and the Zr6(μ3-O)4(μ3-OH)4(carboxylate)12 SBU.179 Interestingly, K2PtCl4 was photoreduced in situ in the presence of TEA to produce Pt NPs with diameters of 2–3 nm and 5–6 nm encapsulated in the cages of the MOFs, respectively. Pt NPs larger than the MOF cages were obtained due to partial framework distortion/degradation during their formation. The [Ir(III)(ppy)2(bpy)]+ moiety was excited to a 1MLCT state under visible light irradiation, and this excited state efficiently underwent intersystem crossing to a 3MLCT state. The photoexcited [Ir(III)(ppy)2(bpy)]+ species was reductively quenched by triethylamine (TEA) to generate the [Ir(III)(ppy)2(bpy˙−)] reduced radical. These radicals then transferred electrons to the Pt NPs, which catalysed proton reduction to produce hydrogen (Fig. 15). Consequently, the Pt@MOF composite exhibited high photocatalytic activities for hydrogen evolution under visible light (>420 nm) irradiation. The TON of 7000 obtained with the MOF composite material was 4.7 times higher than that obtained with the homogeneous catalyst system ([Ir(ppy)2(bpy)]Cl/K2PtCl4). The enhanced activities of the Pt@MOFs were attributed to the highly efficient electron transfer from the unstable [Ir(III)(ppy)2(bpy˙−)] species to the Pt NPs, which slowed down the decomposition of the Ir complexes. Pt–Pd NCs were encapsulated in the pores of ZIF-8, which only absorbs UV light, to obtain a Pt–Pd@ZIF-8 composite material. This material exhibited high activity for the synergistic photocatalytic conversion of adsorbed ethylene into CO2 and H2O at room temperature.140 MNPs such as Au, Ag, and Cu NPs exhibit strong surface plasmon resonance (SPR) effects that could promote photocatalysis.183 Pd NPs not only exhibit excellent catalytic performance in hydrogenation reactions but also exhibit SPR absorption that can utilize solar energy to drive photoreactions.184 Recently, Jiang and coworkers introduced Pd nanocubes (17 ± 3 nm), which exhibited a weak broad SPR absorption band at 220–700 nm, into ZIF-8 crystals.185 Consequently, the Pd@ZIF-8 composite exhibited enhanced activity for photocatalytic olefin hydrogenation due to the synergistic effect of the Pd NPs and the SPR-derived photothermal effect at room temperature. In the dark, a 1-hexene hydrogenation yield of only 37% was achieved after 30 min at room temperature. Surprisingly, the yield of the desired product improved significantly to 66% under full-spectrum irradiation with a 100 mW cm−2 Xe lamp. More interestingly, the Pd@ZIF-8 catalyst afforded the hydrogenation product in 52% yield under visible light irradiation (100 mW cm−2) at room temperature, whereas a temperature of 50 °C was required to achieve the same yield in the dark. The superior catalytic performance was attributed to the synergistic benefits of the plasmonic photothermal effects of the Pd nanocubes and the large number of accessible Pd active sites protected by ZIF-8.
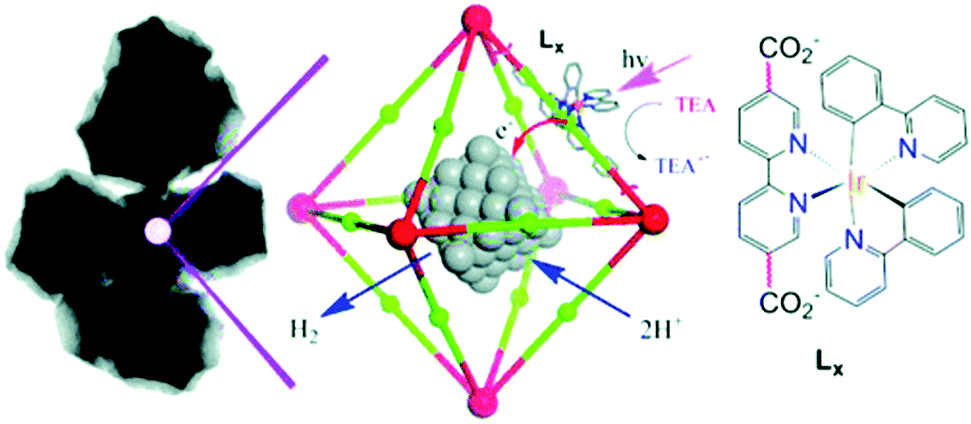 | ||
| Fig. 15 Synergistic photoreduction of water to hydrogen catalysed by Pt NPs encapsulated in UiO-type MOFs based on [Ir(III)(ppy)2(bpy)]+. Reproduced from ref. 179 with permission from American Chemical Society. | ||
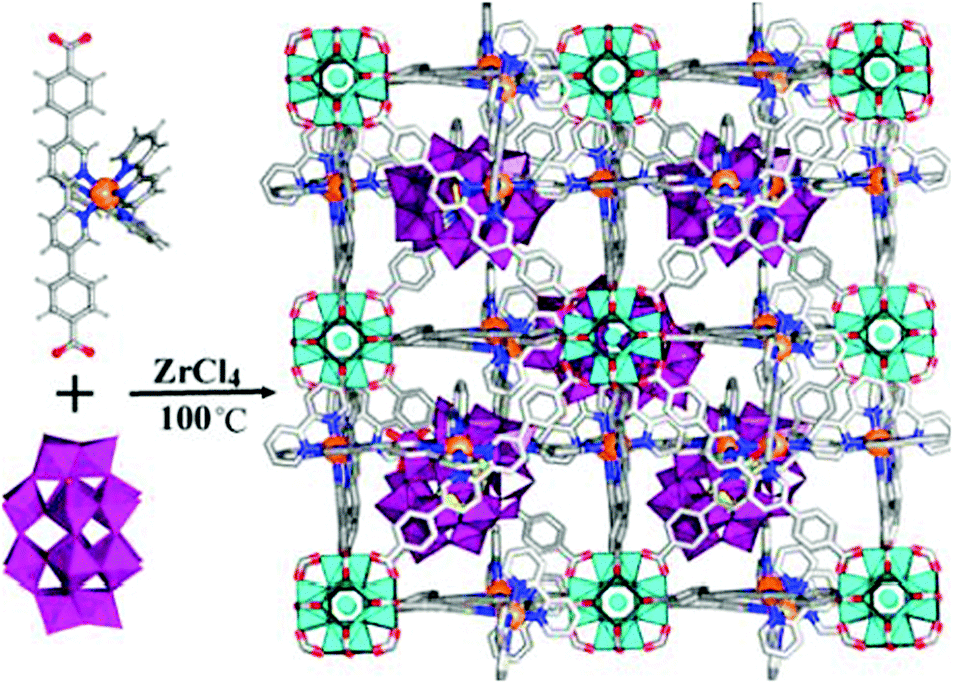 | ||
| Fig. 16 Synthesis of [P2W18O62]6− encapsulated in the pores of a UiO MOF based on [Ru(bpy)3]2+. Reproduced from ref. 186 with permission from American Chemical Society. | ||
Furthermore, photoactive sites and MOF metal nodes can act cooperatively to catalyse organic transformations under light irradiation. Duan and coworkers incorporated the ruthenium(III)-substituted [SiW11O39Ru(H2O)]5− into the pores of copper(II)-bipyridine MOFs for organic catalysis (Fig. 17).188 The resulting material, named CR-BPY1, exhibited an absorption band centred at 398 nm in the solid-state UV-Vis spectrum. The N-phenyl-tetrahydroisoquinoline substrate could be activated by the [SiW11O39Ru(H2O)]5− moiety to form an iminium intermediate under light irradiation. Meanwhile, the acetophenone substrate coordinated to the copper node to form an enol intermediate, which acted as a nucleophile in the oxidative coupling C–C bond formation. The enhanced activity and selectivity of the composite was attributed to the synergistic effects of the [SiW11O39Ru(H2O)]5− photocatalyst and the metal catalyst in the copper(II)-bipyridine MOF.
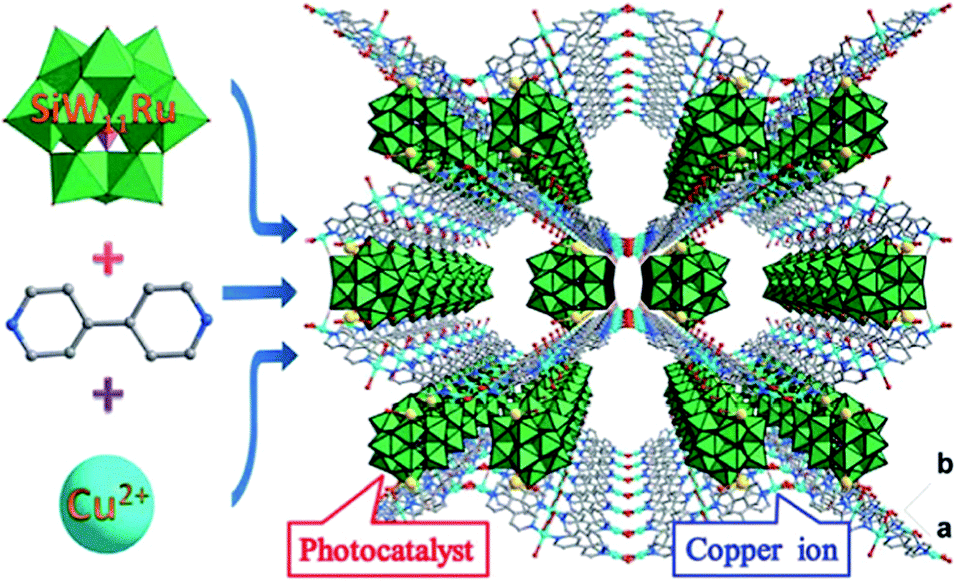 | ||
| Fig. 17 Synthesis route for the 3D CR-BPY1 MOF constructed from [SiW11O40Ru]7− anions and Cu–bpy sheets. Reproduced from ref. 188 with permission from Royal Society of Chemistry. | ||
POMs not only serve as photoactive catalysts but also reduce and stabilize MNPs. In one report, Pt NPs were reduced and stabilized by H3PW12O40 (PW12) and subsequently supported on the amino-functionalized NH2-MIL-53.189 The resulting composite exhibited synergistic activity for hydrogen evolution with a TON of ca. 66 in 6 h under visible light irradiation. The excited electrons of NH2-MIL-53 produced by visible light irradiation were transferred to PW12 and then to the Pt NPs, which catalysed H2 evolution.
The ternary CdS@Cd(II)-MOF@TiO2 composite material was prepared by a TiO2-induced gel-to-crystal approach.192 The ternary composite exhibited a high H2 evolution rate of up to 1604 μmol g−1 h−1 under visible light irradiation. The high activity was attributed to the synergistic effect of the three CdS/Cd(II)-MOF/TiO2 components. Mott–Schottky measurements demonstrated that under visible light irradiation, the excited photoelectrons were stabilized by the Cd-MOF, resulting in efficient charge separation. The excitons were then transferred to TiO2 at the external surface, which reduced the protons to H2.
Substrate adsorption and activation are key factors for improving quantum efficiencies. Xiong et al. prepared a Cu3(BTC)2@TiO2 (BTC = benzene-1,3,5-tricarboxylate) core–shell structure with a Cu3(BTC)2 core and a TiO2 inorganic semiconductor shell for enhanced CO2 reduction.193 Although the microporous Cu3(BTC)2 external surfaces were covered by the TiO2 shells, the hybrid material had a CO2 adsorption capacity of 49.17 cm3 g−1 at 298 K. Therefore, the activity of Cu3(BTC)2@TiO2 for CH4 production was higher than that of bare TiO2 (2.64 μmol gTiO2−1 h−1 CH4 produced vs. 0.52 μmol gTiO2−1 h−1 CH4 produced). More interestingly, the selectivity to CH4 achieved with the core–shell structure was considerably higher than that achieved with bare TiO2 due to improved electron–hole separation. Ultrafast transient absorption spectroscopy indicated that the formation of the interface states between TiO2 and Cu3 (BTC)2 most likely blocked the electron-transfer channels from the TiO2 conduction band (CB) to its surface states. The kinetic behaviour suggested that the photoexcited electrons localized in the Cu3(BTC)2@TiO2 interface states were sufficiently long-lived, which should be beneficial for activating CO2 adsorbed on Cu3(BTC)2. It can be concluded that TiO2 was photoexcited to generate electron–hole pairs and the electrons were subsequently transferred to Cu3(BTC)2. It was proposed that the CO2 photoreduction occurred at the Cu sites, whereas the oxidation occurred on TiO2. TiO2 and CPO-27-Mg (Mg2(dobdc)) also acted cooperatively in photocatalytic CO2 reduction to CO and CH4.194 The observed enhancement in the activity was attributed to the high CO2 adsorption capacity of the composite and the existence of open alkaline metal sites in CPO-27-Mg, which could inhibit the competitive reduction of H2O to H2.
Graphitic carbon nitride (g-C3N4) is a new metal-free polymeric semiconductor material that has been widely employed in visible light-driven photocatalytic reactions, such as CO2 activation and water splitting.195 Incorporating porous MOFs into g-C3N4 can enhance CO2 capture and promote charge separation.196 In one report, g-C3N4 was integrated with a cobalt-containing zeolitic imidazolate framework (Co-ZIF-9) for use in CO2 photoreduction to CO.197 In this reaction, g-C3N4 acted as a semiconductor photocatalyst, whereas Co-ZIF-9 acted as a co-catalyst. PL measurements showed that Co-ZIF-9 significantly inhibited the recombination of the photoinduced electron–hole pairs in the g-C3N4 semiconductor. Although the composite catalysts had a low TON of 35, they could be reused without loss in activity for seven runs due to the good stability of Co-ZIF-9 and g-C3N4. To improve the CO2 photoreduction activity, Ye and coworkers incorporated nanosized g-C3N4 nanosheets (CNNS) into UiO-66.198 Compared with the CNNS, the UiO-66/CNNS composite exhibited a higher CO yield of 59.4 μmol g CN−1 after 6 h under light illumination. The photoelectrons generated on the CNNS were transferred to UiO-66, which effectively suppressed electron–hole recombination. Therefore, long-lived electrons were available for the reduction of the CO2 molecules adsorbed in UiO-66.
The interactions between semiconductors, MNPs and MOFs could improve their photocatalytic performance. Au@CdS/MIL-101 heterostructures were fabricated via a two-step approach, in which CdS was selectively coated on Au NPs to form a core–shell structure.199 The three-component Au@CdS/MIL-101 composite had a H2 production rate of 250 μmol h−1 10 mg−1, which was 2.6 times higher than that of pure CdS. The high performance of the composite was attributed to the fact that the large MIL-101 surface area enabled efficient photoelectron transfer from MIL-101 to the Au NPs, which exhibited strong SPR absorption that accelerated the charge transfer to CdS.
Cohen and coworkers incorporated the [FeFe](dcbdt)(CO)6 (dcbdt = 1,4-dicarboxylbenzene-2,3-dithiolate) proton reduction catalyst into a highly robust UiO-type MOF by a ligand-exchange approach to form UiO-[FeFe](dcbdt)(CO)6.202 When [Ru(bpy)3]2+ and ascorbate were used as the photosensitizer and electron donor, respectively, UiO-[FeFe](dcbdt)(CO)6 exhibited higher activity for photocatalytic hydrogen evolution than UiO-66 and [FeFe](dcbdt)(CO)6. The enhanced catalytic performance was attributed to the structural stabilization of [FeFe](dcbdt)(CO)6 due to its confinement in the MOF pores and to the inhibition of the undesirable charge recombination with oxidized ascorbate (Fig. 18). Using a similar Ru-based photosensitizer [Ru(dmb)3]2+ (dmb = 4,4′-dimethyl-2,2′-bipyridine), the same group synthesized a UiO-67-type MOF based on Mn(bpda)(CO)3Br (bpda = 5,5′-dicarboxylate-2,2′-bipyridine) and used it to catalyse CO2 photoreduction to formate in DMF/triethanolamine under visible light irradiation (Fig. 19).203 The Mn-containing MOF exhibited a TON of 110 in 18 h, exceeding that of the corresponding homogeneous catalyst. The enhanced activity was attributed to the isolation of the Mn active sites in the framework, preventing their dimerization to form a singly reduced Mn complex.
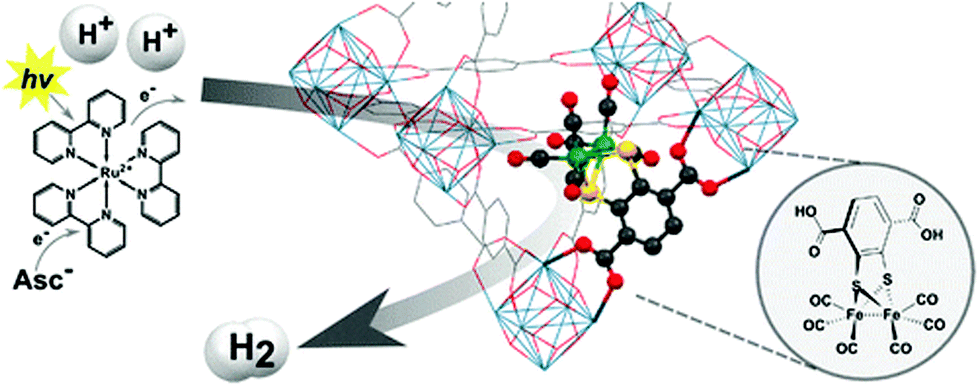 | ||
| Fig. 18 Synergistic photocatalytic hydrogen evolution by UiO-[FeFe](dcbdt)(CO)6 and [Ru(bpy)3]2+. Reproduced from ref. 202 with permission from American Chemical Society. | ||
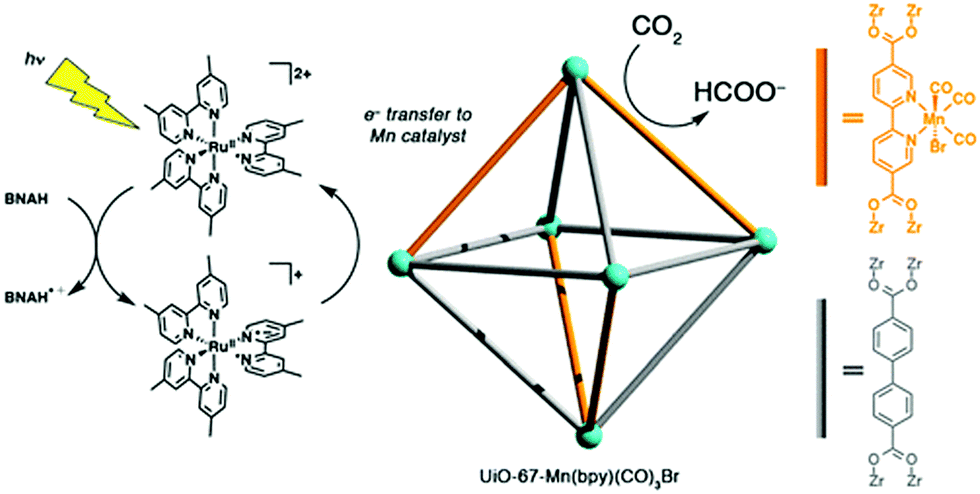 | ||
| Fig. 19 Synergistic photoreduction of CO2 by a UiO-67-type MOF based on Mn(bpda)(CO)3Br and [Ru(dmb)3]2+. Reproduced from ref. 203 with permission from American Chemical Society. | ||
A platinum complex was immobilized in microporous MOF-253, which has 2,2′-bipyridine linkers, to form MOF-253–Pt by PSM.205 This bifunctional material was used for enhanced photocatalytic hydrogen production under visible light irradiation. After the Pt complex was immobilized in the MOF, an extra absorption band centred at 410 nm was observed, and the absorption edge was extended to 650 nm. The low-energy absorption in Pt–MOF-253 was attributed to a MLCT transition (Pt(II) → bipyridine π*). The activity of MOF-253–Pt for photocatalytic hydrogen evolution using TEOA as a sacrificial electron donor under visible light irradiation was approximately five times higher than that of the homogeneous Pt(bpda)Cl2 catalyst. The enhanced water-splitting activity of MOF-253–Pt was attributed to the close interactions of the Pt⋯Pt pairs and to efficient electron transfer within the porous framework. However, after repeated runs, the MOF-253–Pt photocatalyst was obviously deactivated. Nevertheless, combining porous MOFs with photoactive species will open new avenues for artificial photosynthesis because their chemical and physical properties can be modified by designing their electronic and textural structures.
6. Conclusions and perspectives
As demonstrated in this review, MOFs have recently emerged as a powerful platform for engineering multifunctional heterogeneous catalysts for synergistic catalysis and tandem reactions. The advantages of MOFs include the wide variety of possible compositions, structures and functionalities; tunable pore sizes and volumes; and high porosity, which make them excellent candidates for heterogeneous catalysts. MOF materials that contain multiple catalytic centres can be easily designed and prepared by direct synthesis or PSM. The open metal nodes and functional linkers, as well as grafted or encapsulated guest species, such as MNPs, POMs and metal complexes, can serve as catalytic active sites.The production of defects or CUMs upon the removal of coordinated solvent molecules to create open metal centres enables MOFs to be used as Lewis acid catalysts and/or redox catalysts. Furthermore, the functional linkers can be designed with active sites such as amino groups that are suitable for organocatalysis. In addition to direct synthesis, other active groups can be incorporated into MOF pores by different PSM methods, including coordination to the CUMs, covalently bonding to the functional linkers and anion–cation exchange. Moreover, solid–solution MOFs can be obtained by using mixed metals and/or mixed ligands or by a metal- or ligand-exchange method. These MOFs could have a greater number of different active centres for synergistically enhanced catalysis. Therefore, the combination of open metal sites and/or functional groups, especially acid–base active sites, in MOFs is suitable for synergistic catalysis and tandem reactions and is difficult to achieve in homogeneous catalysis.
The unique pore structures of MOFs and the various functional groups provide many advantages for loading guest species, including MNPs, POMs, metal oxides and metal complexes, into the pores. In particular, the pore confinement effect could limit the growth of MNPs and metal oxides and facilitate the fabrication of ultrafine nano-objects. Moreover, the interactions between the organic functional groups and guest species could stabilize them to prevent them from leaching during catalysis. The cooperative action of guest species and active sites on the MOF framework, such as metal centres or organic functional groups, in synergistic catalysis and/or tandem reactions has been demonstrated. Notably, although great progress has been made in the preparation of representative MNP/MOF composites, no reports of metal atomic MNPs supported on MOFs have appeared in the literature. Accordingly, the fabrication of dispersed metal atomic MNPs embedded in MOFs would be of great interest because these atom-based catalysts could exhibit significantly enhanced catalytic activity. Furthermore, sharp high-resolution transmission electron microscopy (HRTEM) images of small MNPs are usually difficult to obtain due to the charges that tend to accumulate under the electron beam, which results in distorted images. Thus, the lattice planes of the MNPs, especially those encapsulated in MOF pores, are difficult to observe, and their relation to the catalytic mechanism cannot be ascertained. Therefore, new characterization techniques must be developed. Spherical aberration-corrected high-resolution electron microscopy and cryo-electron microscopy are powerful techniques that could circumvent the problems with other techniques.206 In addition, the spatial location of MNPs and their interactions with MOF supports play important roles in determining the catalytic performance of MOF composites.207 However, few reports address the control of the spatial location of MNPs, and the interactions between MNPs and MOF supports are rarely studied.13 Calorimetric measurements,207 X-ray synchrotron radiation experiments,98in situ characterization methods and theoretical calculations208 could provide useful information about the interfacial interactions between MNPs and MOF supports and help to explain synergistically enhanced catalytic reactions. It should be noted that compared with MNPs, the use of other guest species, such as POMs, semiconductors, metal complexes and enzymes, with MOFs to achieve synergistic catalysis and promote tandem reactions is rare and should be explored more in the future.
Remarkable advances in MOF-supported bimetallic alloy NPs for synergistically enhanced catalysis have been made. However, it is difficult to determine the NP structure by EDS line scanning or elemental mapping, and therefore, verifying the formation of bimetallic alloy NPs embedded in MOFs can be challenging, especially for small NPs prepared by precursor insertion into the pores, followed by reduction. Thus, new characterization technologies such as X-ray synchrotron radiation could be useful for determining the structures of bimetallic NPs. Moreover, the synergistic effects of bimetallic alloy NPs on their catalytic performance are not well understood. X-ray synchrotron radiation techniques and theoretical calculations can provide detailed structure information. In addition, although bimetallic core–shell NPs can exhibit enhanced catalytic activity due to their unique structure and interparticle interactions, only a few examples of bimetallic core–shell NPs in MOFs have been reported in the literature. More facile fabrication methods for core–shell MNPs encapsulated in MOFs are expected to be developed in the future.
The development of multifunctional MOFs for synergistic photocatalysis and tandem reactions, including water-splitting, CO2 photoreduction, and organic photosynthesis and/or photodegradation, is one of the fastest growing research fields today, and therefore, progress in this field was summarized based on the composition and functionality of the materials. Although some impressive results have been achieved in synergistic photocatalysis by MOFs, significant improvements must be made in the future. First, to satisfy practical requirements, the photocatalytic efficiencies need to be improved considerably by modifying the metal nodes, functional linkers, active species encapsulated in the pores and co-catalysts. Second, most photocatalytic MOF systems require sacrificial agents and/or noble metals such as Pt and Ru, making them expensive and environmentally unfriendly. Developing photosensitive materials such as C3N4, CuO2, CdS, and TiO2 to avoid the use of noble metals is one way to address this problem. Third, the advantages of sunlight and MOFs could be exploited simultaneously by developing MOF composites that promote tandem reactions involving photocatalytic reactions and reactions that can be catalysed by open metal centres or organic functionalities.
Compared with other porous materials such as zeolites and carbon materials, homochiral MOFs can be easily prepared by using enantiotropic ligands or by introducing chiral groups by PSM. Recently, the use of homochiral MOFs in heterogeneous asymmetric catalysis has been extensively studied.25,26 However, only a few examples of homochiral MOF catalysts for synergistic asymmetric catalysis and tandem reactions have been reported in the literature.69,105,106 Another interesting but challenging area of research is the fabrication of chiral inorganic NPs for asymmetric catalysis using homochiral MOFs. For example, Tang et al. used a homochiral MOF based on an N-(4-pyridylmethyl)-L-leucine template to arrange helical CdS nanotubes and Ag NPs.209,210 However, the chiral framework of the MOF was degraded, and the helically arranged CdS and Ag NPs were not utilized for catalysis. Nevertheless, this pioneering work will open a new avenue for developing multifunctional chiral materials. In particular, combining helical inorganic NPs and homochiral MOFs will provide new opportunities for constructing multifunctional materials for synergistic asymmetric organic catalysis and photocatalysis.
Stability is a very important issue in heterogeneous catalysis and is related to the catalyst lifetime and recyclability. Compared with inorganic porous materials, MOFs have relatively low thermal and chemical stability.211 However, this limitation could be overcome by developing more robust MOFs, such as Zr-based MOFs.212 The transformation of MOFs to porous stable carbons and/or other related nanostructured functional materials for catalysis is one possible strategy for circumventing this problem.213
In summary, although some challenges still exist and the development of MOF materials for synergistic catalysis and tandem reactions is a relatively new field, it will continue to grow considerably in the near future.
List of acronyms and abbreviations
| AB | Ammonia borane |
| AlPF-1 | Al(OH)(hfipbb) |
| BDC-NH2 | 2-Amino-benzene-1,4-dicarboxylate |
| BET | Brunauer–Emmett–Teller |
| BOC | t-Butyloxy carbonyl |
| BPDA | 2,2′-Bipyridine-5,5′-dicarboxylic acid |
| Bpy | 2,2′-Bipyridine |
| CB | Conduct bond |
| CMOF-1 | Chiral metal–organic framework-1 |
| CNNS | g-C3N4 nanosheets |
| COD | 1,5-Cyclooctadiene |
| COT | 1,3,5-Cyclooctatriene |
| BTC | Benzene-1,3,5-tricarboxylate |
| CUMs | Coordinatively unsaturated metal centers |
| CUS | Coordinatively unsaturated sites |
| KA-oil | Cyclohexanone and cyclohexanol |
| Dcbdt | 1,4-Dicarboxylbenzene-2,3-dithiolate |
| DFT | Density functional theory calculations |
| Dmb | 4,4′-Dimethyl-2,2′-bipyridine |
| Dobdc | 2,5-Dioxidoterephthalate |
| DRIFTS | Diffuse reflectance infrared Fourier transform spectroscopy |
| DRS | Diffuse reflectance spectra |
| DSM | Double solvent method |
| ED | Ethylenediamine |
| EDS | Energy disperse spectroscopy |
| EDTA | Ethylenediaminetetraacetic acid |
| ee | Enantiomeric excess |
| EXAFS | Extended X-ray absorption fine structure |
| FA | Formic acid |
| GaPF-1 | Ga(OH)(hfipbb) |
| g-C3N4 | Graphitic carbon nitride |
| H2hfipbb | 4,4′-(Hexafluoroisopropylidene)bis-(benzoicacid) |
| H3-TCA | Tricarboxytriphenylamine |
| HER | Hydrogen evolution reaction |
| Hf-2 | Fe-porphyrin struts of Hf-NU-1000 |
| HOMO | Highest occupied molecular orbital |
| HRTEM | High resolution transmission electron microscopy |
| InGaPF | InxGa1−x(O2C2H4)0.5(hfipbb) |
| IRMOF | Isoreticular metal–organic framework |
| LMCT | Ligand-to-metal charge transfer |
| LUMO | Lowest unoccupied molecular orbital |
| MIBK | Methyl isobutyl ketone |
| MIL | Matérial Institut Lavoisier |
| MIL-101–Cr | (Cr3(F)(H2O)2O[(O2C)C6H4(CO2)]3) |
| MIL-125(Ti) | Ti8O8(OH)4(bdc)6 |
| MIL-53 | Al(OH)[O2C–C6H4–CO2] |
| MLCT | Metal-to-ligand charge transfer transition |
| MM-MIL-53 | Hierarchically micro- and mesoporous MIL-53 |
| MNPs | Metal nanoparticles |
| MO | Mesityl oxide |
| MOCVD | Metal–organic chemical vapour deposition |
| MOFs | Metal–organic frameworks |
| MV2+ | Methyl viologen dication |
| NCs | Nanocrystals |
| (Ni4P2) | [Ni4(H2O)2(PW9O34)2]10 |
| PCN | Porous coordination network |
| PCPs | Porous coordination polymers |
| POMs | Polyoxometallates |
| Ppy | 4-Phenyl-2-pyridine |
| PROX | Preferential oxidation |
| PSE | Post-synthetic exchange |
| PSM | Post-synthetic modification |
| PTA | Phosphotungstic acid |
| PVP | Polyvinylpyrrolidone |
| PW12 | H3PW12O40 |
| PXRD | Powder X-ray diffraction |
| PYI | L- or D-Pyrrolidin-2-ylimidazole |
| RGO | Reduced-graphene-oxide |
| SHE | Standard hydrogen electrode |
| SO-PCN | Singlet oxygen-generating porous coordination network |
| SPR | Surface plasmon resonance |
| TBAB | n Bu4NBr |
| TEM | Transmission electron microscopy |
| TEOA | Triethanolamine |
| TMSCN | Trimethylsilyl cyanide |
| TOF | Turnover frequency |
| TONs | Turnover numbers |
| USTC | University of Science and Technology of China |
| XPS | X-ray photoelectron spectroscopy |
| ZIF | Zeolitic imidazolate frameworks |
Acknowledgements
The authors gratefully acknowledge the financial support of the 973 Program (2014CB845605 and 2013CB933200), NSFC (21671188, 21273238, 21521061, and 21331006), Strategic Priority Research Program of the Chinese Academy of Sciences (XDB20000000), Youth Innovation Promotion Association, CAS (2014265), and Chunmiao Project of the Haixi Institute of the Chinese Academy of Sciences (CMZX-2014-004).References
- F.-X. Felpin and E. Fouquet, ChemSusChem, 2008, 1, 718–724 CrossRef CAS PubMed.
- A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633–658 RSC.
- D. J. Martin, G. Liu, S. J. A. Moniz, Y. Bi, A. M. Beale, J. Ye and J. Tang, Chem. Soc. Rev., 2015, 44, 7808–7828 RSC.
- H. You, S. Yang, B. Ding and H. Yang, Chem. Soc. Rev., 2013, 42, 2880–2904 RSC.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- J.-F. Capon, F. Gloaguen, P. Schollhammer and J. Talarmin, Coord. Chem. Rev., 2005, 249, 1664–1676 CrossRef CAS.
- A. Dhakshinamoorthy and H. Garcia, ChemSusChem, 2014, 7, 2392–2410 CrossRef CAS PubMed.
- J.-C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001–1020 CrossRef CAS PubMed.
- O. M. Yaghi and H. Li, J. Am. Chem. Soc., 1995, 117, 10401–10402 CrossRef CAS.
- S. Subramanian and M. J. Zaworotko, Angew. Chem., Int. Ed. Engl., 1995, 34, 2127–2129 CrossRef CAS.
- C. J. Kepert and M. J. Rosseinsky, Chem. Commun., 1998, 31–32 RSC.
- C. Janiak and J. K. Vieth, New J. Chem., 2010, 34, 2366–2388 RSC.
- C. R. Kim, T. Uemura and S. Kitagawa, Chem. Soc. Rev., 2016, 45, 3828–3845 RSC.
- D. Zhao, D. J. Timmons, D. Yuan and H.-C. Zhou, Acc. Chem. Res., 2011, 44, 123–133 CrossRef CAS PubMed.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed.
- Z.-J. Lin, J. Lü, M. Hong and R. Cao, Chem. Soc. Rev., 2014, 43, 5867–5895 RSC.
- Y. He, B. Li, M. O'Keeffe and B. Chen, Chem. Soc. Rev., 2014, 43, 5618–5656 RSC.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, Chem. Soc. Rev., 2015, 44, 1922–1947 RSC.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkovc and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC.
- Y. Liu, W. Xuan and Y. Cui, Adv. Mater., 2010, 22, 4112–4135 CrossRef CAS PubMed.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed.
- K. Fujie and H. Kitagawa, Coord. Chem. Rev., 2016, 307, 382–390 CrossRef CAS.
- M. Giménez-Marqués, T. Hidalgo, C. Serre and P. Horcajada, Coord. Chem. Rev., 2016, 307, 342–360 CrossRef.
- L. M. Aguirre-Díaz, F. Gándara, M. Iglesias, N. Snejko, E. Gutiérrez-Puebla and M. Á. Monge, J. Am. Chem. Soc., 2015, 137, 6132–6135 CrossRef PubMed.
- L. Mitchell, P. Williamson, B. Ehrlichov, A. E. Anderson, V. R. Seymour, S. E. Ashbrook, N. Acerbi, L. M. Daniels, R. I. Walton, M. L. Clarke and P. A. Wright, Chem. – Eur. J., 2014, 20, 17185–17197 CrossRef CAS PubMed.
- A. Schejn, A. Aboulaich, L. Balan, V. Falk, J. Lalevée, G. Medjahdi, L. Aranda, K. Mozet and R. Schneider, Catal. Sci. Technol., 2015, 5, 1829–1839 CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- H. Yang, X.-W. He, F. Wang, Y. Kang and J. Zhang, J. Mater. Chem., 2012, 22, 21849–21851 RSC.
- Z. Sun, G. Li, Y. Zhang, H. Liu and X. Gao, Catal. Commun., 2015, 59, 92–96 CrossRef CAS.
- H. Deng, C. J. Doonan, H. R. Furukawa, B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Science, 2010, 327, 846–850 CrossRef CAS PubMed.
- Y.-R. Lee, Y.-M. Chung and W.-S. Ahn, RSC Adv., 2014, 4, 23064–23067 RSC.
- P. V. Dau and S. M. Cohen, Inorg. Chem., 2015, 54, 3134–3138 CrossRef CAS PubMed.
- H. Yamamoto and K. Futatsugi, Angew. Chem., Int. Ed., 2005, 44, 1924–1942 CrossRef CAS PubMed.
- Z.-R. Jiang, H. Wang, Y. Hu, J. Lu and H.-L. Jiang, ChemSusChem, 2015, 8, 878–885 CrossRef CAS PubMed.
- E. D. Bloch, D. Britt, C. Lee, C. J. Doonan, F. J. Uribe-Romo, H. Furukawa, J. R. Long and O. M. Yaghi, J. Am. Chem. Soc., 2010, 132, 14382–14384 CrossRef CAS PubMed.
- F. Vermoortele, B. Bueken, G. L. Bars, B. Van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. Van Speybroeck, C. Kirschhock and D. E. De Vos, J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- B. Li, K. Leng, Y. Zhang, J. J. Dynes, J. Wang, Y. Hu, D. Ma, Z. Shi, L. Zhu, D. Zhang, Y. Sun, M. Chrzanowski and S. Ma, J. Am. Chem. Soc., 2015, 137, 4243–4248 CrossRef CAS PubMed.
- F. Vermoortele, R. Ameloot, A. Vimont, C. Serre and D. D. Vos, Chem. Commun., 2011, 47, 1521–1523 RSC.
- Y. Yang, H.-F. Yao, F.-G. Xi and E.-Q. Gao, J. Mol. Catal. A: Chem., 2014, 390, 198–205 CrossRef CAS.
- A. M. Rasero-Almansa, A. Corma, M. Iglesias and F. Sánchez, ChemCatChem, 2014, 6, 1794–1800 CrossRef CAS.
- J. Gascon, U. Aktay, M. Hernandezalonso, G. Vanklink and F. Kapteijn, J. Catal., 2009, 261, 75–87 CrossRef CAS.
- F. X. Llabrés i Xamena, F. G. Cirujano and A. Corma, Microporous Mesoporous Mater., 2012, 157, 112–117 CrossRef.
- P. Wu, J. Wang, Y. Li, C. He, Z. Xie and C. Duan, Adv. Funct. Mater., 2011, 21, 2788–2794 CrossRef CAS.
- P. Valvekens, M. Vandichel, M. Waroquier, V. Van Speybroeck and D. De Vos, J. Catal., 2014, 317, 1–10 CrossRef CAS.
- J. Kim, S.-N. Kim, H.-G. Jang, G. Seo and W.-S. Ahn, Appl. Catal., A, 2013, 453, 175–180 CrossRef CAS.
- F. Zhang, Y. Wei, X. Wu, H. Jiang, W. Wang and H. Li, J. Am. Chem. Soc., 2014, 136, 13963–13966 CrossRef CAS PubMed.
- C. Chizallet, S. Lazare, D. Bazer-Bachi, F. Bonnier, V. Lecocq, E. Soyer, A.-A. Quoineaud and N. Bats, J. Am. Chem. Soc., 2010, 132, 12365–12377 CrossRef CAS PubMed.
- J. Park, J. R. Li, Y. P. Chen, J. Yu, A. A. Yakovenko, Z. U. Wang, L. B. Sun, P. B. Balbuena and H. C. Zhou, Chem. Commun., 2012, 48, 9995–9997 RSC.
- T. Toyao, M. Fujiwaki, Y. Horiuchi and M. Matsuoka, RSC Adv., 2013, 3, 21582–21587 RSC.
- R. Ameloot, F. Vermoortele, J. Hofkens, F. C. D. Schryver, D. E. D. Vos and M. B. J. Roeffraers, Angew. Chem., Int. Ed., 2013, 52, 401 CrossRef CAS PubMed.
- F. Douelle, A. S. Capes and M. F. Greaney, Org. Lett., 2007, 9, 931 CrossRef PubMed.
- S. Hasegawa, S. Horike, R. Matsuda, S. Furukawa, K. Mochizuki, Y. Kinoshita and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 2607 CrossRef CAS PubMed.
- R. Srirambalaji, S. Hong, R. Natarajan, M. Yoon, R. Hota, Y. Kim, Y. H. Ko and K. Kim, Chem. Commun., 2012, 48, 11650–11652 RSC.
- Y. Huang, T. Liu, J. Lin, J. Lü, Z. Lin and R. Cao, Inorg. Chem., 2011, 50, 2191–2198 CrossRef CAS PubMed.
- M. Pintado-Sierra, A. M. Rasero-Almansa, A. Corma and M. I. Félix Sánchez, J. Catal., 2013, 299, 137–145 CrossRef CAS.
- A. M. Rasero-Almansa, A. Corma, M. Iglesias and F. Sánchez, ChemCatChem, 2013, 5, 3092–3100 CrossRef CAS.
- M. H. Beyzavi, N. A. Vermeulen, A. J. Howarth, S. Tussupbayev, A. B. League, N. M. Schweitzer, J. R. Gallagher, A. E. Platero-Prats, N. Hafezi, A. A. Sarjeant, J. T. Miller, K. W. Chapman, J. F. Stoddart, C. J. Cramer, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 13624–13631 CrossRef CAS PubMed.
- D. Feng; Z.-Y. Gu, J.-R. Li, H.-L. Jiang, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef PubMed.
- W. Morris, B. Volosskiy, S. Demir, F. Gándara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Inorg. Chem., 2012, 51, 6443–6445 CrossRef CAS PubMed.
- M. H. Beyzavi, R. C. Klet, S. Tussupbayev, J. Borycz, N. A. Vermeulen, C. J. Cramer, J. F. Stoddart, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2014, 136, 15861–15864 CrossRef CAS PubMed.
- B. Li, Y. Zhang, D. Ma, L. Li, G. Li, G. Li, Z. Shi and S. Feng, Chem. Commun., 2012, 48, 6151–6153 RSC.
- A. Arnanz, M. Pintado-Sierra, A. Corma, M. Iglesias and F. Sánchez, Adv. Synth. Catal., 2012, 354, 1347–1355 CrossRef CAS.
- J. A. Gladysz, Chem. Rev., 2002, 102, 3215–3216 CrossRef CAS PubMed.
- F. Song, C. Wang, J. M. Falkowski, L. Ma and W. Lin, J. Am. Chem. Soc., 2010, 132, 15390–15398 CrossRef CAS PubMed.
- F. Song, C. Wang and W. Lin, Chem. Commun., 2011, 47, 8256–8258 RSC.
- P. Falcaro, R. Ricco, A. Yazdi, I. Imaz, S. Furukawa, D. Maspoch, R. Ameloot, J. D. Evans and C. J. Doonan, Coord. Chem. Rev., 2016, 307, 237–254 CrossRef CAS.
- H. R. Moon, D.-W. Limb and M. P. Suh, Chem. Soc. Rev., 2013, 42, 1807–1824 RSC.
- Q.-L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468–5512 RSC.
- A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2012, 41, 5262–5284 RSC.
- C. Rösler and R. A. Fischer, CrystEngComm, 2015, 17, 199–217 RSC.
- Y. Pan, B. Yuan, Y. Li and D. He, Chem. Commun., 2010, 46, 2280–2282 RSC.
- H. Liu, Y. Li, R. Luque and H. Jiang, Adv. Synth. Catal., 2011, 353, 3107–3113 CrossRef CAS.
- H. Liu, T. Jiang, B. Han, S. Liang and Y. Zhou, Science, 2009, 326, 1250 CrossRef CAS PubMed.
- I. E. Ertas, M. Gulcan, A. Bulut, M. Yurderi and M. Zahmakiran, J. Mol. Catal. A: Chem., 2015, 410, 209–220 CrossRef CAS.
- F. G. Cirujano, F. X. L. i Xamena and A. Corma, Dalton Trans., 2012, 41, 4249–4254 RSC.
- F. G. Cirujano, A. Leyva-Pérez, A. Corma and F. X. L. i Xamena, ChemCatChem, 2013, 5, 538–549 CrossRef CAS.
- Y.-Z. Chen, Y.-X. Zhou, H. Wang, J. Lu, T. Uchida, Q. Xu, S.-H. Yu and H.-L. Jiang, ACS Catal., 2015, 5, 2062–2069 CrossRef CAS.
- X. Huang, X. Wang, X. Wang, X. Wang, M. Tan, W. Ding and X. Lu, J. Catal., 2013, 301, 217–226 CrossRef CAS.
- H. Liu, Y. Li, H. Jiang, C. Vargas and R. Luque, Chem. Commun., 2012, 48, 8431–8433 RSC.
- L. Kesavan, R. Tiruvalam, M. H. Ab Rahim, M. I. Bin Saiman, D. I. Enache, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, S. H. Taylor, D. W. Knight, C. J. Kiely and G. J. Hutchings, Science, 2011, 331, 195 CrossRef CAS PubMed.
- X. Li, Z. Guo, C. Xiao, T. W. Goh, D. Tesfagaber and W. Huang, ACS Catal., 2014, 4, 3490–3497 CrossRef CAS.
- R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977–1986 RSC.
- Y.-B. Huang, M. Shen, X. Wang, P.-C. Shi, H. Li and R. Cao, J. Catal., 2015, 330, 452–457 CrossRef CAS.
- K. Shimizu, K. Kon, M. Seto, K. Shimura and S. S. M. A. Hakim, J. Catal., 2013, 300, 242 CrossRef CAS.
- Y. Huang, Z. Zheng, T. Liu, J. Lü, Z. Lin, H. Li and R. Cao, Catal. Commun., 2011, 14, 27–31 CrossRef CAS.
- Y. Huang, S. Gao, T. Liu, J. Lü, X. Lin, H. Li and R. Cao, ChemPlusChem, 2012, 77, 106–112 CrossRef CAS.
- Y. Huang, S. Liu, Z. Lin, W. Li, X. Li and R. Cao, J. Catal., 2012, 292, 111–117 CrossRef CAS.
- M. Martis, K. Mori, K. Fujiwara, W.-S. Ahn and H. Yamashita, J. Phys. Chem. C, 2013, 117, 22805–22810 CAS.
- B. Loges, A. Boddien, F. Gartner, H. Junge and M. Beller, Top. Catal., 2010, 53, 902–914 CrossRef CAS.
- S.-T. Gao, W. Liu, C. Feng, N.-Z. Shang and C. Wang, Catal. Sci. Technol., 2016, 6, 869 CAS.
- Y. Qi, Y. Luan, X. Peng, M. Yang, J. Hou and G. Wang, Eur. J. Inorg. Chem., 2015, 5099–5105 CrossRef CAS.
- M. Zhao, K. Deng, L. He, Y. Liu, G. Li, H. Zhao and Z. Tang, J. Am. Chem. Soc., 2014, 136, 1738–1741 CrossRef CAS PubMed.
- K. M. Choi, K. Na, G. A. Somorjai and O. M. Yaghi, J. Am. Chem. Soc., 2015, 137, 7810–7816 CrossRef CAS PubMed.
- X. Li, T. W. Goh, L. Li, C. Xiao, Z. Guo, X. C. Zeng and W. Huang, ACS Catal., 2016, 6, 3461–3468 CrossRef CAS.
- H. Pan, X. Li, D. Zhang, Y. Guan and P. Wu, J. Mol. Catal. A: Chem., 2013, 377, 108–114 CrossRef CAS.
- S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- X.-S. Wang, Y.-B. Huang, Z.-J. Lin and R. Cao, Dalton Trans., 2014, 43, 11950–11958 RSC.
- Q. Han, C. He, M. Zhao, B. Qi, J. Niu and C. Duan, J. Am. Chem. Soc., 2013, 135, 10186–10189 CrossRef CAS PubMed.
- Q. Han, B. Qi, W. Ren, C. He, J. Niu and C. Duan, Nat. Commun., 2015, 6, 10007 CrossRef PubMed.
- D. Wang and Y. Li, Adv. Mater., 2011, 23, 1044–1060 CrossRef CAS PubMed.
- A. K. Singh and Q. Xu, ChemCatChem, 2013, 5, 652–676 CrossRef CAS.
- F. Schröder, S. Henke, X. Zhang and R. A. Fischer, Eur. J. Inorg. Chem., 2009, 3131–3140 CrossRef.
- Z. Liu, E. T. Ada, M. Shamsuzzoha, G. B. Thompson and D. E. Nikles, Chem. Mater., 2006, 18, 4946–4951 CrossRef CAS.
- S. Hermes, M.-K. Schröter, R. Schmid, L. Khodeir, M. Muhler, A. Tissler, R. W. Fischer and R. A. Fischer, Angew. Chem., Int. Ed., 2005, 44, 6237–6241 CrossRef CAS PubMed.
- F. Schröder, D. Esken, M. Cokoja, M. W. E. van den Berg, O. I. Lebedev, B. Walaszek, G. Buntkowsky, H.-H. Limbach, B. Chaudret and R. A. Fischer, J. Am. Chem. Soc., 2008, 130, 6119–6130 CrossRef PubMed.
- S. Proch, J. Herrmannsdorfer, R. Kempe, C. Kern, A. Jess, L. Seyfarth and J. Senker, Chem. – Eur. J., 2008, 14, 8204–8212 CrossRef CAS PubMed.
- J. Hermannsdörfer, M. Friedrich, N. Miyajima, R. Q. Albuquerque, S. Kümmel and R. Kempe, Angew. Chem., Int. Ed., 2012, 51, 11473–11477 CrossRef PubMed.
- M. S. El-Shall, V. Abdelsayed, A. E. R. S. Khder, H. M. A. Hassan, H. M. El-Kaderi and T. E. Reich, J. Mater. Chem., 2009, 19, 7625–7631 RSC.
- X. Gu, Z.-H. Lu, H.-L. Jiang, T. Akita and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11822–11825 CrossRef CAS PubMed.
- Y. K. Hwang, D. Y. Hong, J. S. Chang, S. H. Jhung, Y. K. Seo, J. Kim, A. Vimont, M. Daturi, C. Serre and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 4144 CrossRef CAS PubMed.
- J.-M. Yan, Z.-L. Wang, L. Gu, S.-J. Li, H.-L. Wang, W.-T. Zheng and Q. Jiang, Adv. Energy Mater., 2015, 5, 1500107 CrossRef.
- H. Dai, N. Cao, L. Yang, J. Su, W. Luo and G. Cheng, J. Mater. Chem. A, 2014, 2, 11060–11064 CAS.
- H. Dai, B. Xia, L. Wen, C. Du, J. Su, W. Luo and G. Cheng, Appl. Catal., B, 2015, 165, 57–62 CrossRef CAS.
- K. Tedsree, C. W. A. Chan, S. Jones, Q. Cuan, W.-K. Li, X.-Q. Gong and S. C. E. Tsang, Science, 2011, 332, 224 CrossRef CAS PubMed.
- Y. Huang, Z. Lin and R. Cao, Chem. – Eur. J., 2011, 17, 12706–12712 CrossRef CAS PubMed.
- Y. Huang, T. Ma, P. Huang, D. Wu, Z. Lin and R. Cao, ChemCatChem, 2013, 5, 1877–1883 CrossRef CAS.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. D. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- A. K. Singh and Q. Xu, ChemCatChem, 2013, 5, 3000–3004 CrossRef CAS.
- N. Cao, L. Yang, H. Dai, T. Liu, J. Su, X. Wu, W. Luo and G. Cheng, Inorg. Chem., 2014, 53, 10122–10128 CrossRef CAS PubMed.
- L. Wen, X. Du, J. Su, W. Luo, P. Cai and G. Cheng, Dalton Trans., 2015, 44, 6212–6218 RSC.
- B. Xia, N. Cao, H. Dai, J. Su, X. Wu, W. Luo and G. Cheng, ChemCatChem, 2014, 6, 2549–2552 CrossRef CAS.
- H. Liu, G. Chen, H. Jiang, Y. Li and R. Luque, ChemSusChem, 2012, 5, 1892–1896 CrossRef CAS PubMed.
- J. Long, H. Liu, S. Wu, S. Liao and Y. Li, ACS Catal., 2013, 3, 647–654 CrossRef CAS.
- H. Liu, R. Fang, Z. Li and Y. Li, Chem. Eng. Sci., 2015, 122, 350–359 CrossRef CAS.
- M. Imperor-Clerc, D. Bazin, M.-D. Appay, P. Beaunier and A. Davidson, Chem. Mater., 2004, 16, 1813 CrossRef CAS.
- Y.-B. Huang, M. Shen, X. Wang, P. Huang, R. Chen, Z.-J. Lin and R. Cao, J. Catal., 2016, 333, 1–7 CrossRef CAS.
- A. Aijaz, A. Karkamkar, Y. J. Choi, N. Tsumori, E. Ronnebro, T. Autrey, H. Shioyama and Q. Xu, J. Am. Chem. Soc., 2012, 134, 13926 CrossRef CAS PubMed.
- Q.-L. Zhu, J. Li and Q. Xu, J. Am. Chem. Soc., 2013, 135, 10210–10213 CrossRef CAS PubMed.
- J. Li, Q.-L. Zhu and Q. Xu, Chem. Commun., 2014, 50, 5899–5901 RSC.
- J. Li, Q.-L. Zhu and Q. Xu, Catal. Sci. Technol., 2015, 5, 525–530 CAS.
- K. Yang, L. Zhou, X. Xiong, M. Ye, L. Li and Q. Xia, Microporous Mesoporous Mater., 2016, 225, 1–8 CrossRef CAS.
- G. Lu, S. Li, Z. Guo, O. K. Farha, B. G. Hauser, X. Qi, Y. Wang, X. Wang, S. Han, X. Liu, J. S. DuChene, H. Zhang, Q. Zhang, X. Chen, J. Ma, S. C. J. Loo, W. D. Wei, Y. Yang, J. T. Hupp and F. Huo, Nat. Chem., 2012, 4, 310–316 CrossRef CAS PubMed.
- Y. Huang, Y. Zhang, X. Chen, D. Wu, Z. Yi and R. Cao, Chem. Commun., 2014, 50, 10115–10117 RSC.
- C. Rösler, D. Esken, C. Wiktor, H. Kobayashi, T. Yamamoto, S. Matsumura, H. Kitagawa and R. A. Fischer, Eur. J. Inorg. Chem., 2014, 5514–5521 CrossRef.
- L. Chen, X. Chen, H. Liu and Y. Li, Small, 2015, 11, 2642–2648 CrossRef CAS PubMed.
- D.-W. Lim, J. W. Yoon, K. Y. Ryu and M. P. Suh, Angew. Chem., Int. Ed., 2012, 51, 9814 CrossRef CAS PubMed.
- F. Ke, L. Wang and J. Zhu, Nanoscale, 2015, 7, 8321–8325 RSC.
- Z. Li, R. Yu, J. Huang, Y. Shi, D. Zhang, X. Zhong, D. Wang, Y. Wu and Y. Li, Nat. Commun., 2015, 6, 8248 CrossRef CAS PubMed.
- Y. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorjai and A. P. Alivisatos, Science, 2004, 304, 711–714 CrossRef CAS PubMed.
- H. Zhang, T. Watanabe, M. Okumura, M. Haruta and N. Toshima, Nat. Mater., 2012, 11, 49–52 CrossRef PubMed.
- H.-L. Jiang, T. Akita, T. Ishida, M. Haruta and Q. Xu, J. Am. Chem. Soc., 2011, 133, 1304–1306 CrossRef CAS PubMed.
- Y. Sun and Y. Xia, Science, 2002, 298, 2176 CrossRef CAS PubMed.
- S. Alayoglu, A. U. Nilekar, M. Mavrikakis and B. Eichhorn, Nat. Mater., 2008, 7, 333 CrossRef CAS PubMed.
- Y. Wei, S. Han, D. A. Walker, P. E. Fuller and B. A. Grzybowski, Angew. Chem., Int. Ed., 2012, 51, 7435–7439 CrossRef CAS PubMed.
- A. Aijaz, T. Akita, N. Tsumori and Q. Xu, J. Am. Chem. Soc., 2013, 135, 16356–16359 CrossRef CAS PubMed.
- Y.-Z. Chen, Q. Xu, S.-H. Yu and H.-L. Jiang, Small, 2015, 11, 71–76 CrossRef CAS PubMed.
- L. Chen, B. Huang, X. Qiu, X. Wang, R. Luque and Y. Li, Chem. Sci., 2015, 7, 228–233 RSC.
- W. Salomon, C. Roch-Marchal, P. Mialane, P. Rouschmeyer, C. Serre, M. Haouas, F. Taulelle, S. Yang, L. Ruhlmann and A. Dolbecq, Chem. Commun., 2015, 51, 2972–2975 RSC.
- M. Müller, S. Hermes, K. Kähler, M. W. E. van den Berg, M. Muhler and R. A. Fischer, Chem. Mater., 2008, 20, 4576–4587 CrossRef.
- M. Müller, S. Turner, O. I. Lebedev, Y. Wang, G. van Tendeloo and R. A. Fischer, Eur. J. Inorg. Chem., 2011, 1876–1887 CrossRef.
- D. Tilgner, M. Friedrich, J. Hermannsdörfer and R. Kempe, ChemCatChem, 2015, 7, 3916–3922 CrossRef CAS.
- E. V. Ramos-Fernandez, C. Pieters, B. van der Linden, J. Juan-Alcañiz, P. Serra-Crespo, M. W. G. M. Verhoeven, H. Niemantsverdriet, J. Gascon and F. Kapteijn, J. Catal., 2012, 289, 42–52 CrossRef CAS.
- J. Chen, S. Wang, J. Huang, L. Chen, L. Ma and X. Huang, ChemSusChem, 2013, 6, 1545–1555 CrossRef CAS PubMed.
- S. Wang, J. Chen and L. Chen, Catal. Lett., 2014, 144, 1728–1734 CrossRef CAS.
- S. Wang and X. Wang, Small, 2015, 11, 3097–3112 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, A. M. Asiri and H. García, Angew. Chem., Int. Ed., 2016, 55, 5414–5445 CrossRef CAS PubMed.
- K. Meyer, M. Ranocchiari and J. A. van Bokhoven, Energy Environ. Sci., 2015, 8, 1923–1937 CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- D. Wang, R. Huang, W. Liu, D. Sun and Z. Li, ACS Catal., 2014, 4, 4254–4260 CrossRef CAS.
- M. A. Nasalevich, M. G. Goesten, T. J. Savenije, F. Kapteijn and J. Gascon, Chem. Commun., 2013, 49, 10575–10577 RSC.
- D. Wang and Z. Li, Catal. Sci. Technol., 2015, 5, 1623–1628 CAS.
- T. Toyao, M. Saito, Y. Horiuchi and M. Matsuoka, Catal. Sci. Technol., 2014, 4, 625–628 CAS.
- W.-Y. Gao, M. Chrzanowski and S. Ma, Chem. Soc. Rev., 2014, 43, 5841–5866 RSC.
- M.-H. Xie, X.-L. Yang, C. Zou and C.-D. Wu, Inorg. Chem., 2011, 50, 5318–5320 CrossRef CAS PubMed.
- N. B. Munro, S. S. Talmage, G. D. Griffin, L. C. Waters, A. P. Watson, J. F. KIng and V. Hauschild, Environ. Health Perspect., 1999, 107, 933–974 CrossRef CAS PubMed.
- Y. Liu, A. J. Howarth, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2015, 54, 9001–9005 CrossRef CAS PubMed.
- Y. Liu, S.-Y. Moon, J. T. Hupp and O. K. Farha, ACS Nano, 2015, 9, 12358–12364 CrossRef CAS PubMed.
- J. Park, D. Feng, S. Yuan and H.-C. Zhou, Angew. Chem., Int. Ed., 2015, 54, 430–435 CrossRef CAS PubMed.
- P. Wu, C. He, J. Wang, X. Peng, X. Li, Y. An and C. Duan, J. Am. Chem. Soc., 2012, 134, 14991–14999 CrossRef CAS PubMed.
- A. Fateeva, P. A. Chater, C. P. Ireland, A. A. Tahir, Y. Z. Khimyak, P. V. Wiper, J. R. Darwent and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2012, 51, 7440–7444 CrossRef CAS PubMed.
- C. Wang, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2012, 134, 7211–7214 CrossRef CAS PubMed.
- J. He, J. Wang, Y. Chen, J. Zhang, D. Duan, Y. Wang and Z. Yan, Chem. Commun., 2014, 50, 7063–7066 RSC.
- Y. Horiuchi, T. Toyao, M. Saito, K. Mochizuki, M. Iwata, H. Higashimura, M. Anpo and M. Matsuoka, J. Phys. Chem. C, 2012, 116, 20848 CAS.
- L. Shen, M. Luo, L. Huang, P. Feng and L. Wu, Inorg. Chem., 2015, 54, 1191–1193 CrossRef CAS PubMed.
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473–486 RSC.
- R. Long, K. Mao, X. Ye, W. Yan, Y. Huang, J. Wang, Y. Fu, X. Wang, X. Wu, Y. Xie and Y. Xiong, J. Am. Chem. Soc., 2013, 135, 3200–3207 CrossRef CAS PubMed.
- Q. Yang, Q. Xu, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 3685–3689 CrossRef CAS PubMed.
- Z.-M. Zhang, T. Zhang, C. Wang, Z. Lin, L.-S. Long and W. Lin, J. Am. Chem. Soc., 2015, 137, 3197–3200 CrossRef CAS PubMed.
- X.-J. Kong, Z. Lin, Z.-M. Zhang, T. Zhang and W. Lin, Angew. Chem., Int. Ed., 2016, 55, 6411–6416 CrossRef CAS PubMed.
- D. Shi, C. He, B. Qi, C. Chen, J. Niu and C. Duan, Chem. Sci., 2015, 6, 1035–1042 RSC.
- W. Guo, H. Lv, Z. Chen, K. P. Sullivan, S. M. Lauinger, Y. Chi, J. M. Sumliner, T. Lian and C. L. Hill, J. Mater. Chem. A, 2016, 4, 5952–5957 CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- L. Zhang, P. Cui, H. Yang, J. Chen, F. Xiao, Y. Guo, Y. Liu, W. Zhang, F. Huo and B. Liu, Adv. Sci., 2016, 3, 1500243 CrossRef PubMed.
- C.-W. Zhao, Y.-A. Li, X.-R. Wang, G.-J. Chen, Q.-K. Liu, J.-P. Ma and Y.-B. Dong, Chem. Commun., 2015, 51, 15906–15909 RSC.
- R. Li, J. Hu, M. Deng, H. Wang, X. Wang, Y. Hu, H. Jiang, J. Jiang, Q. Zhang, Y. Xie and Y. Xiong, Adv. Mater., 2014, 26, 4783–4788 CrossRef CAS PubMed.
- M. Wang, D. Wang and Z. Li, Appl. Catal., B, 2016, 183, 47–52 CrossRef CAS.
- J. Liu, H. Wang and M. Antoniettia, Chem. Soc. Rev., 2016, 45, 2308–2326 RSC.
- H. Wang, X. Yuan, Y. Wu, G. Zeng, X. Chen, L. Leng and H. Li, Appl. Catal., B, 2015, 174–175, 445–454 CrossRef CAS.
- S. Wang, J. Lin and X. Wang, Phys. Chem. Chem. Phys., 2014, 16, 14656–14660 RSC.
- L. Shi, T. Wang, H. Zhang, K. Chang and J. Ye, Adv. Funct. Mater., 2015, 25, 5360–5367 CrossRef CAS.
- Y. Wang, Y. Zhang, Z. Jiang, G. Jiang, Z. Zhao, Q. Wu, Y. Liu, Q. Xu, A. Duan and C. Xu, Appl. Catal., B, 2016, 185, 307–314 CrossRef CAS.
- S. Han, Y. Wei and B. A. Grzybowski, Chem. – Eur. J., 2013, 19, 11194–11198 CrossRef CAS PubMed.
- S. Wang, W. Yao, J. Lin, Z. Ding and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 1034–1038 CrossRef CAS PubMed.
- S. Pullen, H. Fei, A. Orthaber, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2013, 135, 16997–17003 CrossRef CAS PubMed.
- H. Fei, M. D. Sampson, Y. Lee, C. P. Kubiak and S. M. Cohen, Inorg. Chem., 2015, 54, 6821–6828 CrossRef CAS PubMed.
- M. A. Nasalevich, R. Becker, E. V. Ramos-Fernandez, S. Castellanos, S. L. Veber, M. V. Fedin, F. Kapteijn, J. N. H. Reek, J. I. van der Vlugt and J. Gascon, Energy Environ. Sci., 2015, 8, 364–375 CAS.
- T. Zhou, Y. Du, A. Borgna, J. Hong, Y. Wang, J. Han, W. Zhang and R. Xu, Energy Environ. Sci., 2013, 6, 3229–3234 CAS.
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo, D. Zang, B. Wu, G. Fu and N. Zheng, Science, 2016, 352, 797–801 CrossRef CAS PubMed.
- J. A. Farmer and C. T. Campbell, Science, 2010, 329, 933–936 CrossRef CAS PubMed.
- L. B. Vilhelmsen, K. S. Walton and D. S. Sholl, J. Am. Chem. Soc., 2012, 134, 12807–12816 CrossRef CAS PubMed.
- X. Kuang, Y. Ma, C. Zhang, H. Su, J. Zhang and B. Tang, Chem. Commun., 2015, 51, 5955–5958 RSC.
- X. Kuang, S. Ye, X. Li, Y. Ma, C. Zhang and B. Tang, Chem. Commun., 2016, 52, 5432–5435 RSC.
- J. Gascon, A. Corma, F. Kapteijn and F. X. L. i Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- J.-K. Sun and Q. Xu, Energy Environ. Sci., 2014, 7, 2071–2100 CAS.
| This journal is © The Royal Society of Chemistry 2017 |