
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From E to Z and back again: reversible photoisomerisation of an isolated charge-tagged azobenzene
James N.
Bull
 ,
Michael S.
Scholz
,
Eduardo
Carrascosa
and
Evan J.
Bieske
*
,
Michael S.
Scholz
,
Eduardo
Carrascosa
and
Evan J.
Bieske
*
School of Chemistry, University of Melbourne, Parkville, VIC 3010, Australia. E-mail: evanjb@unimelb.edu.au
First published on 29th November 2017
Abstract
Substituted azobenzenes serve as chromophores and actuators in a wide range of molecular photoswitches. Here, tandem ion mobility spectrometry coupled with laser excitation is used to investigate the photoisomerisation of selected E and Z isomers of the charge-tagged azobenzene, methyl orange. Both isomers display a weak S1(nπ*) photoisomerisation response in the blue part of the spectrum peaking at 440 nm and a more intense S2(ππ*) photoisomerisation response in the near-UV with maxima at 370 and 310 nm for the E and Z isomers, respectively. The 60 nm separation between the S2(ππ*) photo-response maxima for the two isomers allows them to be separately addressed in the gas phase and to be reversibly photoisomerised using different colours of light. This is an essential characteristic of an ideal photoswitch. The study demonstrates that a sequence of light pulses at different stages in an ion mobility spectrometer can be deployed to generate and probe isomers that cannot be electrosprayed directly from solution or produced through collisions in the ion source.
Introduction
Photoswitch molecules can be converted between two or more distinct geometric forms using light. Natural photoswitches include the retinal chromophore in opsin proteins that are involved in the visual transduction cycle and bacterial photosynthesis,1 and photoactive proteins, such as the photoactive yellow protein, found in certain bacteria.2 Over the last decade, photoswitches based on azobenzenes have been developed and deployed for a range of applications, including as actuators in molecular machines,3 to control solution pH or ionic liquid properties with light,4,5 in optogenetics,6 to control peptide and lipid structure,7,8 and for targeted activation or release of drugs in photopharmacology.9 The appeal of azobenzenes as core photoswitching units stems from their rapid photoisomerisation between E and Z isomers with a high quantum yield (0.2–0.3), and the possibility of reversible control using different colours of light.10,11 To realise the full potential of azobenzene molecules and substituted derivatives as chromophores and actuators in photoswitching applications, it is desirable to characterise the spectroscopic and photoisomerisation properties of the E and Z isomers for a series of benchmark molecules. For most azobenzenes and azoarenes, the Z isomer is less stable than the E isomer, meaning Z–E thermal reversion limits the ability to cleanly isolate and spectroscopically probe the Z isomer in solution. Most condensed phase spectroscopic and ultrafast studies rely on creating a photostationary state and subtracting the contribution of the E isomer to yield the contribution of the Z isomer.12 Ambiguities associated with this subtraction approach are exacerbated if the E and Z isomers have similar properties or if other species in the sample generate interfering signals.Recently, our group has combined tandem ion mobility spectrometry (IMS) and laser excitation to probe the photoisomerisation of protonated azobenzenes, azoheteroarenes and other cationic azoheteroarenes in the gas phase or in solution.13–16 For the gas-phase photoisomerisation studies, charged isomers drifting under the influence of an electric field through a buffer gas are separated according to their drift speeds, which depend on their collision cross-sections. Typically, the target isomer is selected in a primary IMS stage and then exposed to wavelength tunable light, with the photo-isomers separated in a secondary IMS stage. Using this approach, the E and Z isomers are spatially and temporally separated allowing one to record isomer-specific photoisomerisation action (PISA) spectra. In an ideal case, the photoisomerisation quantum yield is independent of wavelength, so the PISA spectrum corresponds to the absorption spectrum. The technique provides a direct means to characterise the photoresponse of selected isomer, avoiding complications associated with overlapping spectra of coexisting isomers that sometimes bedevil other spectroscopic techniques such as photodissociation spectroscopy. In previous studies of protonated azobenzene, we obtained PISA spectra for E → Z isomerisation but were thwarted in attempts to observe the reverse process because we were unable to obtain sufficient amounts of the Z isomer.16 Parallel investigations of protonated aminoazobenzene suffered the added complexity of multiple possible gas-phase protomers, which may have different absorption profiles and photochemical behaviour.17 Spectroscopic measurements on gas-phase molecules also allows straightforward connections with theory, providing confidence regarding predictions for photoisomerisation properties of similar molecules.
Here, we investigate photoisomerisation of the E and Z isomers of methyl orange anions (4-[4-(dimethylamino)phenyl]diazenylbenzene-1-sulfonate, see Fig. 1) using the PISA technique. Methyl orange is a charge-tagged azobenzene that has been incorporated into push–pull switching dendrimers that can respond to both light and solution pH.18,19 Femtosecond photoisomerisation measurements20 of methyl orange in aqueous solution show that E–Z photoisomerisation occurs on an ultrafast 0.6–0.9 ps timescale, similar to E-azobenzene in either the gas phase and in solution.11,21–23 The crucial issues we wish to address for methyl orange anions are whether we can produce both E and Z isomers in the gas phase, and whether they can be reversibly photoswitched using different colours of light.
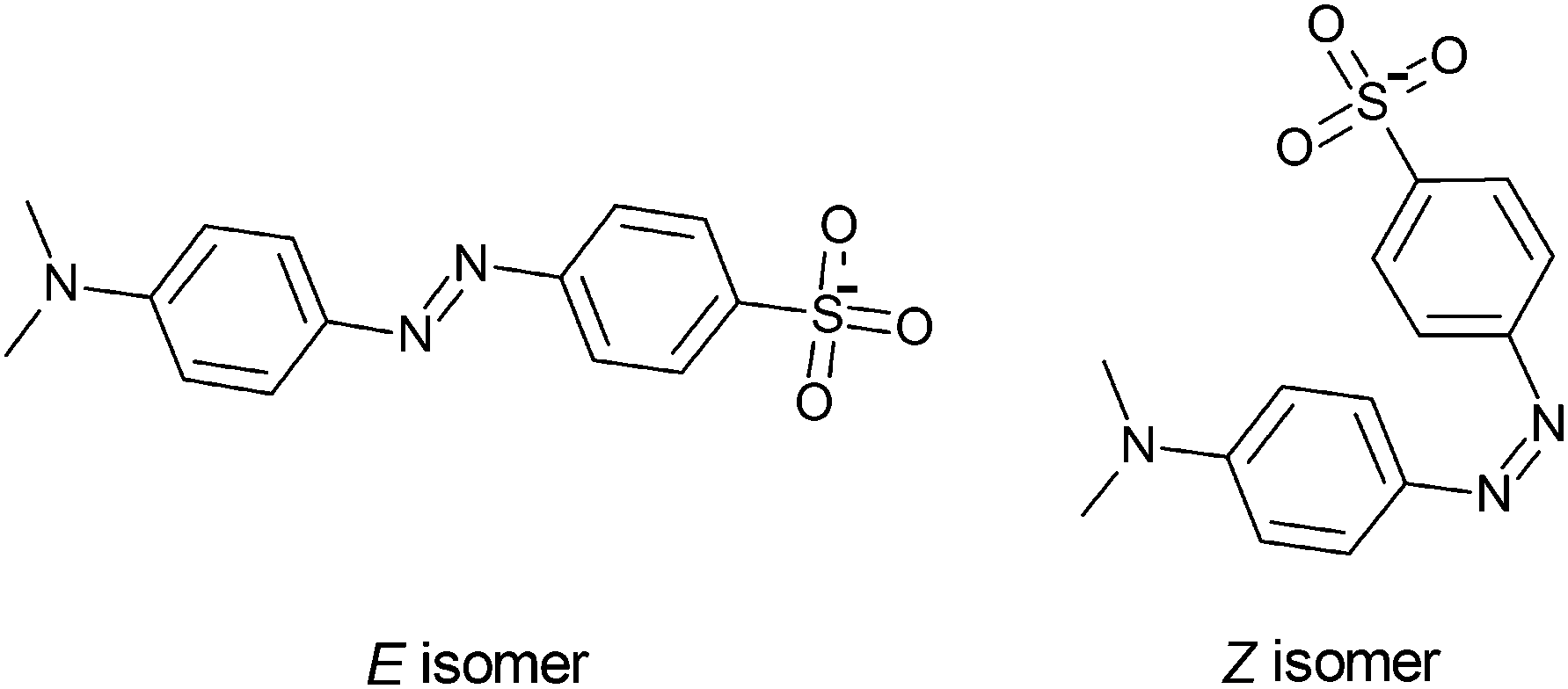 | ||
| Fig. 1 E and Z isomers of the methyl orange anion. | ||
Experimental methods
The photoisomerisation of methyl orange anions was investigated in a custom tandem ion mobility spectrometer (IMS-IMS) coupled with a quadrupole mass filter (QMF).24,25 The IMS-IMS-QMF apparatus is illustrated schematically in Fig. 2. Briefly, methyl orange anions were produced through electrospray ionisation of a ≈10 μmol L−1 solution of sodium methyl orange (Sigma Aldrich) dissolved in methanol (voltage −3 kV, flow rate ≈10 μL min−1). Electrosprayed ions were transferred via a heated capillary into a RF ion funnel (IF1), which radially gathered and confined the ions. An ion gate (IG1) at the end of IF1 injected ≈100 μs packets of ions at 40 Hz into the first IMS drift region (IMS1) where they were propelled by an electric field (44 V cm−1) through N2 buffer gas at a pressure of ≈6 Torr. The E and Z isomers were separated spatially and temporally because more extended ions (E isomers) have larger collision cross-sections with the buffer gas than more compact ions (Z isomers).26 After traversing the drift regions (IMS1 + IMS2), a second ion funnel (IF2) collected the ions and introduced them into a differentially pumped octupole ion guide and QMF that mass-selected the ions before they reached the ion detector. The detector was connected to a multichannel scaler that produced a histogram of ion counts against arrival time, t, corresponding to an arrival time distribution (ATD). In all presented ATDs, t = 0 corresponds to the opening of IG1. The mobility resolution, t/Δt, for a singly-charged anion is typically 80–90.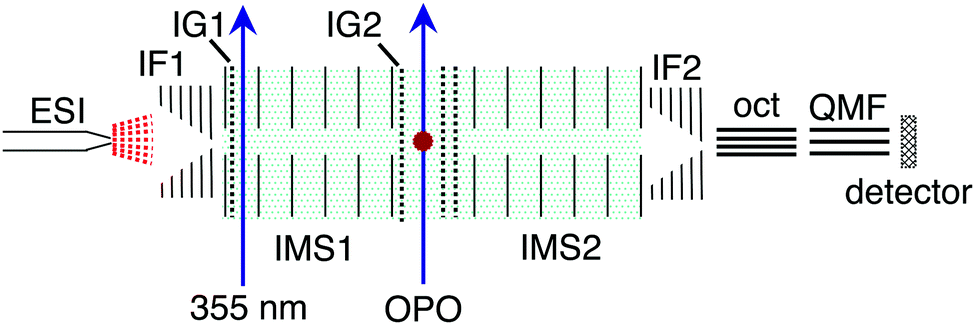 | ||
| Fig. 2 Schematic illustration of the IMS-IMS-QMF instrument. Key: ESI – electrospray ionisation; IF1 and IF2 – ion funnels; IG1 and IG2 – ion gates; IMS1 and IMS2 – ion mobility drift regions; OPO – light beam passing through the photoisomerisation zone; oct – octupole ion guide; QMF – quadrupole mass filter. Total drift region length (IMS1 + IMS2) is 0.9 m. Further details of the instrument are given in Adamson et al.24,25 This figure is adapted from ref. 14. | ||
For the PISA spectroscopy measurements, packets of ions with similar collision cross-sections were selected using a Bradbury–Nielsen ion gate after IMS1 (IG2, ≈100 μs opening time). Immediately after this, the mobility-selected ions were excited with a pulse of light from an optical parametric oscillator (OPO, EKSPLA NT342B). Any change in the ions' collision cross-section due to photoisomerisation was manifested as a shift in arrival time following passage through a second IMS drift region (IMS2). The OPO was operated at 20 Hz, half the rate of ion injection, allowing accumulation of light-on and light-off ATDs. The difference between the light-on and light-off ATDs reflected the photoresponse (photo-action ATD). PISA spectra were derived by integrating the photo-action ATD signal, and normalizing with respect to light pulse fluence and total laser-off signal at each wavelength. The PISA measurements were performed with a light pulse fluence of <1 mJ cm−2 to minimise multiphoton absorption and dissociation producing SO3−. The Z isomer was synthesised in the gas phase by irradiating electrosprayed ions with a pulse of 355 nm light (Quantel Big Sky Nd:YAG, 40 Hz) immediately after the ions were injected into the drift region of IMS1.
Theoretical methods
Supporting electronic structure calculations for the methyl orange isomers were performed using the Gaussian 16 and ORCA 4.0.0.2 software packages.27,28 Geometrical optimisations, vibrational frequencies and isomerisation transition state searches were performed at the ωB97X-D/aug-cc-pVDZ level of theory,29,30 followed by single-point energy calculations at the DLPNO-CCSD(T)/aug-cc-pVTZ level of theory.31 Vertical excited state energies were computed at the ωB97X-D/aug-cc-pVDZ level of theory within the TD-DFT framework. Collision cross-sections were calculated using MOBCAL with the trajectory method parametrised for N2 buffer gas.32,33 Input charge distributions were computed at the ωB97X-D/aug-cc-pVDZ level of theory with the Merz–Singh–Kollman scheme constrained to reproduce the electric dipole moment.34 Sufficient trajectories were computed to give standard deviations of ±1 Å2 for the calculated values.Results and discussion
The arrival time distribution (ATD) of electrosprayed methyl orange anions exhibits a single peak (Fig. 3a), that can be assigned to the E isomer based on calculations that suggest it is 21 kJ mol−1 more stable than the Z isomer. We attempted to generate the Z isomer by several means. First, exposing the methyl orange solution to 532, 425 or 365 nm light before electrospray ionisation produced no evidence for a second isomer in the ATD, indicating Z → E thermal reversion in methanol occurs in less than a few seconds, consistent with previous observations.20,35,36 Second, we tried synthesising the Z isomer by subjecting the E isomer ions to energetic collisions in IF1 prior to their injection into the drift region, again without success. Finally, we managed to generate Z isomer ions by irradiating the E isomer ions with a pulse of 355 nm light immediately after their injection into the drift region of the IMS. This resulted in an additional, faster ion packet (Fig. 3b), that can be assigned to the more compact Z isomer. The ATD peak assignments are consistent with calculated collision cross-sections of 196 and 174 Å2 for the E and Z isomers, respectively. Because the E and Z isomers are associated with clear, baseline-resolved ATD peaks and can be individually selected, the timescale for thermal reversion in the drift region must be long relative to the drift time (≈13 ms).37 This is consistent with the present calculated Z → E thermal reversion barrier of 179 kJ mol−1.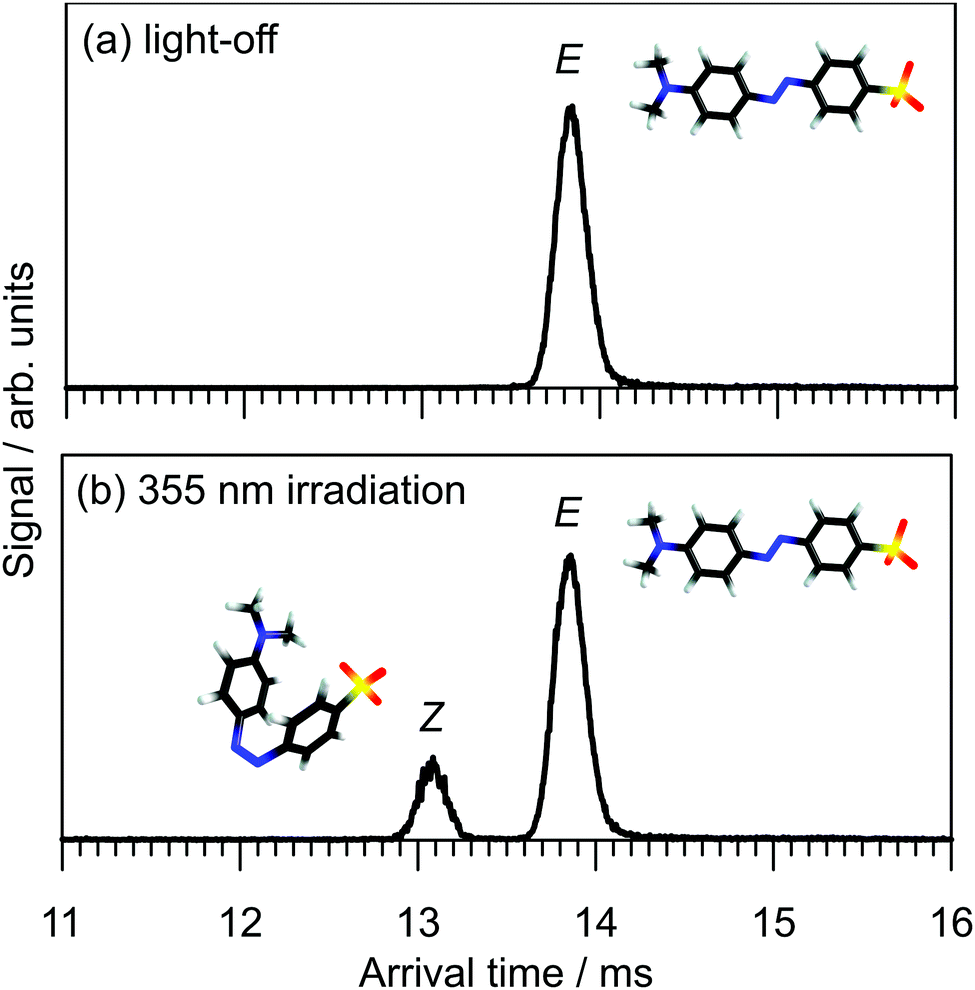 | ||
| Fig. 3 Arrival time distributions for methyl orange anions: (a) light-off and (b) 355 nm irradiation of the gas-phase ion packet at the start of the drift region. Peak resolutions (t/Δt ≈ 90) are consistent with singly-charged ions. | ||
The photoisomerisation of E and Z isomers of methyl orange anions in the gas phase was probed by selecting one of the isomers using an ion gate (IG2) following mobility-separation in the first drift region and exposing the ion packet to a pulse of tunable wavelength light (see Fig. 2). Fig. 4a shows light-off, light-on, and photo-action (light-on–light-off) ATDs obtained by exposing the E isomer to 370 nm light. The E–Z PISA spectrum, generated by plotting the Z photo-isomer signal against wavelength and normalizing with respect to light pulse intensity and total laser-off ion signal, is presented in Fig. 4b. The PISA spectrum exhibits a weak S1(nπ*) band over the 410–530 nm range, peaking at 440 nm, and an intense S2(ππ*) band in the 280–410 nm range, peaking at 370 nm. The Z–E PISA spectrum, shown in Fig. 4c, features a similar S1(nπ*) band in the 370–540 nm range, peaking at 440 nm. However, the Z–E S2(ππ*) PISA band peaks at 310 nm, blue-shifted by 60 nm from the E–Z PISA band. The clear separation of the S2(ππ*) PISA bands for the two isomers means that gas-phase methyl orange anions can be reversibly photo-switched with light of different colours, as required for an ideal photoswitch. Specifically, exposing methyl orange anions to 370 nm light (E–Z PISA maximum) drives E → Z photoisomerisation, whereas irradiation with 310 nm light (or 440 nm) drives net Z → E photoisomerisation.
 | ||
| Fig. 4 Photoisomerisation action spectra for E and Z isomers of the methyl orange anion: (a) example light-off, light-on and photo-action ATDs at 370 nm for the mobility-selected E isomer; (b) E–Z PISA spectrum and (c) Z–E PISA spectrum. An absorption spectrum of methyl orange in methanol is included in (b). Vertical bars in (b and c) represent calculated transition wavelengths and oscillator strengths (f). | ||
The transition wavelengths and relative intensities for the S1(ππ*) ← S0 and S2(ππ*) ← S0 transitions of the E and Z isomers agree reasonably well with the calculated vertical excitation energies and oscillator strengths (vertical bars in Fig. 4b and c). The absorption spectrum of methyl orange in methanol, included in Fig. 4b, differs markedly from the gas-phase E–Z PISA spectrum. Specifically, the main S2(ππ*) absorption band in solution is strongly red-shifted, peaking at 420 nm, with perhaps a small contribution from the S1(nπ*) band appearing as a shoulder at 460 nm.11 The solution absorption spectrum shows evidence for two further absorption bands at 315 and 280 nm, that are not evident in the E–Z PISA spectra, but which may correspond to the S3(ππ*) ← S0 and S4(ππ*) ← S0 transitions, which, in the gas phase, are calculated to lie at 309 and 284 nm with oscillator strengths of f > 0.01 and f > 0.02, respectively. The absence of these bands in the E–Z PISA spectrum may be due to low isomerisation yields for the S3(ππ*) and S4(ππ*) states (i.e. direct internal conversion to S0), as is the case for neutral azobenzene.21
Returning to a comparison of the spectra of methyl orange in solution and in the gas phase, we note that the S1(ππ*) ← S0 band of methyl orange in methanol is red shifted with respect to the corresponding band in the PISA spectrum (peak at 420 nm versus 370 nm). Such a solvent induced red shift is in line with observations for other azobenzenes. For example, the absorption bands of substituted azobenzenes such as butter yellow (methyl orange without the –SO3− charge tag) are red shifted in polar solvents compared to non-polar solvents.38 It is also possible that the PISA spectrum does not mirror the absorption spectrum due to a wavelength dependent photoisomerisation yield. Ultimately, the absorption spectra of the E and Z isomers of methyl orange in the gas phase should be revealed through spectroscopic measurements of mobility-selected ions contained in a cryogenic trap and tagged with helium atoms. Such action spectra should allow a rigorous assessment of the effect of wavelength dependent photoisomerisation quantum yield on the PISA spectra and the effect of solvent on the absorption bands.
Conclusions
In summary, we have investigated photoisomerisation of selected E and Z isomers of methyl orange anions in the gas phase. Both isomers have similar, weak S1(nπ*) PISA bands in the visible and more intense S2(ππ*) PISA bands in the near-UV. Notably, the S2(ππ*) PISA bands for the two isomers are separated by ≈60 nm meaning that methyl orange can be reversibly switched between E and Z forms using light of different colours, a requirement of an ideal photoswitch. The present study illustrates that ions which cannot normally be generated through electrospray ionisation due to short lifetimes in solution can be produced at the start of the drift region of an ion mobility spectrometer using an intense laser pulse and probed downstream with a second tunable laser pulse. Significantly, we have shown that it is possible to photoswitch a population of methyl orange anions in the gas phase from E to Z and back again, demonstrating unprecedented control over the isomeric state of an ion population.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded through the Australian Research Council Discovery Project scheme (DP150101427 and DP160100474). J. N. B. acknowledges resources from the Australian National Computational Infrastructure (NCI) through Early Career Allocation ya1 and a Microsoft Azure Research Award. M. S. S. thanks the Australian government for an Australian Postgraduate Award scholarship. E. C. acknowledges support by the Austrian Science Fund (FWF) through a Schrödinger Fellowship (No. J4013-N36).References
- O. P. Ernst, D. T. Lodowski, M. Elstner and P. Hegemann, Chem. Rev., 2013, 114, 126–163 CrossRef PubMed
.
- T. E. Meyer, G. Tollin, J. H. Hazzard and M. A. Cusanovich, Biophys. J., 1989, 56, 559–564 CrossRef CAS PubMed
- S. Silvi, M. Venturi and A. Credi, Chem. Commun., 2011, 47, 2483–2489 RSC
- C. E. Weston, R. D. Richardson and M. J. Fuchter, Chem. Commun., 2016, 52, 4521–4524 RSC
- L. C. Branco and F. Pina, Chem. Commun., 2009, 6204–6206 RSC
- T. Fehrentz, M. Schönberger and D. Trauner, Angew. Chem., Int. Ed., 2011, 50, 12156–12182 CrossRef CAS PubMed
- C. Renner, U. Kusebauch, M. Löweneck, A. G. Milbradt and L. Moroder, J. Pept. Res., 2005, 65, 4–14 CrossRef CAS PubMed
- Y. Einaga, T. Yamamoto, T. Sugai and O. Sato, Chem. Mater., 2002, 14, 4846–4850 CrossRef CAS
- M. Dong, A. Babalhavaeji, S. Samanta, A. A. Beharry and G. A. Woolley, Acc. Chem. Res., 2015, 48, 2662–2670 CrossRef CAS PubMed
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC
- T. Nägele, R. Hoche, W. Zinth and J. Wachveitl, Chem. Phys. Lett., 1997, 272, 489–495 CrossRef
- J. N. Bull, M. S. Scholz, N. J. A. Coughlan, A. Kawai and E. J. Bieske, Anal. Chem., 2016, 88, 11978–11981 CrossRef CAS PubMed
- J. N. Bull, M. S. Scholz, N. J. A. Coughlan and E. J. Bieske, Phys. Chem. Chem. Phys., 2017, 19, 12776–12783 RSC
- J. N. Bull, E. Carrascosa, M. S. Scholz, N. J. A. Coughlan and E. J. Bieske, Analyst, 2017, 142, 2100–2103 RSC
- M. S. Scholz, J. N. Bull, N. J. A. Coughlan, E. Carrascosa, B. D. Adamson and E. J. Bieske, J. Phys. Chem. A, 2017, 121, 6413–6419 CrossRef CAS PubMed
- J. N. Bull, N. J. A. Coughlan and E. J. Bieske, J. Phys. Chem. A, 2017, 121, 6021–6027 CrossRef CAS PubMed
- H. Stephan, H. Spies, B. Johannsen, C. Kauffmann and F. Vögtle, Org. Lett., 2000, 2, 2343–2346 CrossRef CAS PubMed
- A. Dirksen, E. Zuidema, R. M. Williams, L. De Cola, C. Kauffmann, F. Vögtle, A. Roque and F. Pina, Macromolecules, 2002, 35, 2743–2747 CrossRef CAS
- M. Takei, H. Yui, Y. Hirose and T. Sawada, J. Phys. Chem. A, 2001, 105, 11395–11399 CrossRef CAS
- T. Schultz, J. Quenneville, B. Levine, A. Toniolo, T. J. Martínez, S. Lochbrunner, M. Schmitt, J. P. Shaffer, M. Z. Zgierski and A. Stolow, J. Am. Chem. Soc., 2003, 125, 8098–8099 CrossRef CAS PubMed
- E. M. M. Tan, S. Amirjalayer, S. Smolarek, A. Vdovin, F. Zerbetto and W. J. Buma, Nat. Commun., 2015, 6, 5860 CrossRef PubMed
- T. Fujino, S. Y. Arzhantsev and T. Tahara, J. Phys. Chem. A, 2001, 105, 8123–8129 CrossRef CAS
- B. D. Adamson, N. J. A. Coughlan, R. E. Continetti and E. J. Bieske, Phys. Chem. Chem. Phys., 2013, 15, 9540–9548 RSC
- B. D. Adamson, N. J. A. Coughlan, P. B. Markworth, R. E. Continetti and E. J. Bieske, Rev. Sci. Instrum., 2014, 85, 123109 CrossRef CAS PubMed
-
G. A. Eiceman, Z. Karpas and H. H. Hill, Ion Mobility Spectrometry, CRC Press, 3rd edn, 2013 Search PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 16 Revision A.03, Gaussian Inc., Wallingford CT, 2016 Search PubMed
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC
- T. H. Dunning, Jr., J. Chem. Phys., 1989, 90, 1007 CrossRef
- C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, J. Chem. Phys., 2013, 139, 134101 CrossRef PubMed
- I. Campuzano, M. F. Bush, C. V. Robinson, C. Beaumont, K. Richardson, H. Kim and H. I. Kim, Anal. Chem., 2012, 84, 1026–1033 CrossRef CAS PubMed
- M. F. Mesleh, J. M. Hunter, A. A. Shvartsburg, G. C. Schatz and M. F. Jarrold, J. Phys. Chem., 1996, 100, 16082–16086 CrossRef CAS
- B. H. Besler, K. M. Merz, Jr. and P. A. Kollman, J. Comput. Chem., 1990, 11, 431–439 CrossRef CAS
- A. M. Sanchez, M. Barra and R. H. de Rossi, J. Org. Chem., 1999, 64, 1604–1609 CrossRef CAS PubMed
- A. Yano, Y. Konno, E. Kinoshita and R. Yano, J. Photochem. Photobiol., A, 2017, 346, 411–415 CrossRef CAS
- S. Poyer, C. Comby-Zerbino, C. M. Choi, L. MacAleese, C. Deo, N. Bogliotti, J. Xie, J.-Y. Salpin, P. Dugourd and F. Chirot, Anal. Chem., 2017, 89, 4230–4237 CrossRef CAS PubMed
- H. Bisle, M. Römer and H. Rau, Ber. Bunsen-Ges., 1976, 80, 301–305 CrossRef CAS
| This journal is © the Owner Societies 2018 |