
Radical mechanism of a nucleophilic reaction depending on a two-dimensional structure†
Wenchuan
Lai
 ,
Yuehui
Yuan
,
Xu
Wang
,
Yang
Liu
,
Yulong
Li
and
Xiangyang
Liu
*
,
Yuehui
Yuan
,
Xu
Wang
,
Yang
Liu
,
Yulong
Li
and
Xiangyang
Liu
*
College of Polymer Science and Engineering, State Key Laboratory of Polymer Material and Engineering, Sichuan University, Chengdu 610065, People's Republic of China. E-mail: lxy6912@sina.com; Fax: +86 28 85405138; Tel: +86 28 85403948
First published on 24th November 2017
Abstract
The mechanism of nucleophilic substitution deserves more investigation to include more reaction systems such as two-dimensional (2D) materials. In this study, we used fluorinated graphene (FG) as a representative 2D material to reveal the in-depth mechanism of its defluorination and nucleophilic substitution reaction under attack of common nucleophiles to explore the chemistry of 2D materials and enrich the research on the nucleophilic substitution reaction. DFT calculations and electron paramagnetic resonance spectroscopy (EPR) demonstrated that defluorination of FG occurred via a radical mechanism after a single electron transfer (SET) reaction between the nucleophile and C–F bond, and a spin center was generated on the nanosheet and fluorine anion. Moreover, neither the SN1 nor SN2 mechanism was suggested to be appropriate for the substitution reaction of FG with a 2D structure due to the corresponding kinetics or thermodynamics disadvantage; hence, its nucleophilic substitution was proved to occur via a radical mechanism initiated by the defluorination step. The proposed substitution mechanism of FG demonstrates that nucleophilic substitution via a radical mechanism can also be applied to the attacking process of common nucleophiles without any particular conditions. Furthermore, it has been discovered that triethylamine without active hydrogen can be covalently attached to graphene nanosheets via a nucleophilic substitution reaction with FG; this further indicates a radical process for the nucleophilic substitution of FG rather than an SN1 or SN2 mechanism. The detailed process of the nucleophilic substitution reaction of FG was revealed to occur via a radical mechanism depending on the 2D structure of FG, which could also represent the typical characteristic of 2D chemistry.
1. Introduction
Nucleophilic reactions constitute a significant part of organic chemistry; among them, the nucleophilic substitution reaction has been paid significant attention.1–3 In 1933, Hughes and Ingold have postulated that two main mechanisms can be applied to nucleophilic substitution reactions in solution: the SN1 and SN2 processes.1,4,5 These two classical mechanisms have been continuously investigated and confirmed to be applicable to most nucleophilic substitution reaction systems including the similar unimolecular or bimolecular mechanism for the nucleophilic substitution reaction of aromatic molecules.1,6–8 However, it has been afterwards discovered that some nucleophilic substitution reaction systems cannot be reasonably explained using the SN1 or SN2 mechanism; this has brought about the emergence of nucleophilic substitution via a radical process instead of a polar process.9–11 Radical nucleophilic substitution (SRN1) indicates that some nucleophilic substitutions are analogous to the chain reactions including chain initiation, growth, and termination reactions.12,13 However, the present SRN1 mechanism requires rigorous reaction conditions including peculiar substrate molecules, attacking reagents or initiation conditions, which obviously limit its application range.10,12 The mechanism of nucleophilic substitution deserves more investigation to include more reaction systems such as two-dimensional (2D) materials, which have been a research hotspot in recent years.14–16Fluorinated graphene (FG), a representative 2D material and graphene derivative, has also received significant interest since its first report.17–19 It should be noted that its relatively simple chemical structure,17 high functionalization density,20 high-performance characteristics,21–23 and subsequent reactivity24,25 have made FG stand out from a variety of graphene derivatives. Moreover, these characteristics can enable FG to be an appropriate representative material for investigating the chemistry of 2D materials; accordingly, in recent years, some studies have been reported that shed light on the derivative chemistry, mainly including reductive defluorination26,27 and nucleophilic substitution, of FG.24,28–33 In these studies, the graphene functional material has been successfully prepared with desired structures based on the reactivity of FG;28,31–33 however, the detailed mechanism of the derivatization reactions is not revealed. For instance, the mechanism for defluorination of FG caused by reductive or alkaline reagents are still unknown, and the destination of fluorine atoms removed from FG is also unknown.24,31,34 Furthermore, during the displacement of the fluorine atoms, the nucleophilic substitution of the C–F bonds in FG has been designed to excessively depend on the substitution of the carbon–halogen bond of halohydrocarbon micromolecules,30,35 whereas the substitution process can be special for FG with 2D structures due to its special C–F bonds.36–38 A new thought should be introduced to the study of the derivatization reactions of FG, especially its nucleophilic substitution reactions, as well as to the research of the 2D chemistry.
Herein, we take FG as a representative 2D material to investigate the in-depth mechanism of its derivatization reactions, the chemistry of 2D materials, as well as enrich the research on nucleophilic substitution in the field of organic chemistry. We have studied the detailed mechanism of derivatization reactions of FG under attack of usual nucleophiles such as amines, phosphines, and potassium hydroxide. DFT calculations and electron paramagnetic resonance spectroscopy (EPR) demonstrated that the defluorination of FG occurred via a radical mechanism after a single-electron transfer (SET) reaction39–41 between the nucleophile and C–F bond; this resulted in a spin center on the nanosheet and fluorine anion. More importantly, it has been indicated that both the SN1 and SN2 mechanism are inappropriate for describing the nucleophilic substitution reaction of FG with a rigid 2D structure due to relevant thermodynamics or kinetics disadvantage. In contrast, the nucleophilic substitution of the C–F bonds in FG is also thought to occur via a radical mechanism initiated by the defluorination step, which is defined as a DR mechanism and is analogous to the SRN1 mechanism. The DR mechanism was then proven using some experimental results including the covalent attachment of a triethylamine fragment to graphene nanosheets by the nucleophilic substitution of FG. The mechanism of the nucleophilic substitution reaction of FG was first revealed and also used to explore the chemistry of 2D materials.
2. Experimental
2.1 Materials
Graphene was purchased from the Sixth Elementary (Changzhou) Materials Technology Co., Ltd. A F2/N2 (10 vol% for F2) mixture with a purity of 99.99% was obtained from Chengdu Kemeite Fluorine Industry Plastic Co., Ltd. N-tert-Butyl-alpha-phenylnitrone (PBN), 2-methyl-2-nitrosopropane (MNP), ethanediamine (EDA), hydrazine (N2H4), diphenylamine (DPA), diphenyl phosphine (DPP), potassium hydroxide (KOH), and triethylamine (TEA) were purchased from the Adamas Reagent, Ltd and were of analytical grade (the chemical structures of the abovementioned reagents are shown in Fig. S1, ESI†). All other chemical reagents were of commercially analytical grade and used without further purification.2.2 Methods for preparing fluorinated graphene and its subsequent derivatization reactions
The preparation of fluorinated graphene (FG) was conducted in accordance with our previous study.24,33,42 Herein, 0.5 g of graphene was placed in a closed stainless steel (SUS316) chamber (20 L) equipped with a vacuum line and fluorinated with an F2/N2 mixture at 80 kPa and 180 °C for 1 h.The detection of intermediate radicals during the nucleophilic reaction between FG and nucleophilic reagents, including EDA, N2H4, DPA, DPP, KOH, and TEA, was performed using PBN or MNP as a radical trapper. Herein, 0.1 mmol of radical trapper and 0.3 mmol of the nucleophilic reagent were first dissolved in 10 mL of ethanol and mixed adequately by 10 min of sonication. Then, 10 mg of FG was added to the resulting solution followed by 30 min of sonication to accomplish the reaction. The reaction mixture was then centrifuged for 20 min, and the liquid supernatant was used for EPR measurements to detect the radical intermediates.
The main derivatization reactions of FG with various nucleophilic reagents were performed using a solution method. Herein, 60 mg of FG was adequately dispersed in 50 mL of ethanol by 30 min of sonication in a round bottom flask. Then, 10 mmol of the nucleophilic reagent was gradually added. After 4 h of reaction, the mixture was centrifuged for 20 min, whereas the sediment was filtered. The filter cake was washed using a Soxhlet extractor for 24 h to remove any adsorbed nucleophilic reagent and then dried for 4 h at 60 °C under vacuum conditions. The derivatization reaction of FG with FM (formamide) in the blank group was performed using a similar method, whereas that in the control group was performed in the same way under ultra-violet irradiation for 30 min at the beginning of the reaction.
2.3 Characterization
X-ray photoelectron spectroscopy (XPS) was carried out using a Kratos ASAM 800 spectrometer (Kratos Analytical Ltd, UK) at a base vacuum higher than 10−6 Pa under non-monochromatized Al Kα (1486.6 eV) X-ray source at a voltage of 15 kV and power of 250 W. The Fourier transform infrared (FTIR) spectrum was obtained using a Nicolet 560 Fourier transform spectrometer in the range 4000–400 cm−1. Electron paramagnetic resonance (EPR) measurements were carried out via Bruker EPR EMX Plus (Bruker Beijing Science and Technology Ltd, USA) of a frequency of 9.8 GHz approximately using a standard microwave power of 0.02 mW. Elemental analysis was performed using the ELEMENT vario EL cube (German). Measurement of the fluorine anions in the solution after performing the reaction was carried out using an ICS-90 Ion chromatograph. The microstructures of the fluorinated graphene sample were characterized using HR-TEM (Tecnai G2 F20 S-TWIN).2.4 Computational details
DFT calculations were performed using the DMol3 module43 implemented in Materials Studio 8.0 to calculate the reaction energy for the dissociation of the C–F bond via various pathways and nucleophilic substitution in the SN2 process of model molecules. Geometry optimization and a TS search of the reactions of the model molecules were performed using the generalized gradient approximation (GGA) of the Perdew–Burke–Ernzerhof (PBE) functional due to its wide application in the research of carbon nanomaterials,44 and the basis was set as DNP with the basis file 3.5.45 The self-consistent-field calculation had a convergence criteria of 10−5 Hartree.45 The vibration analysis was performed to confirm the transition state of the calculated reactions.3. Results and discussion
3.1 The defluorination mechanism
The derivatization reaction of FG means the defluorination reaction, which can occur under conditions such as thermal treatment,22,46,47 ultra-violet radiation,26 and electron donors.33,41 For chemical defluorination caused by electron donors including reductive reagents and nucleophiles,29,34,35 the defluorination mechanism of FG still remains unknown; herein, DFT calculations were employed to explore the energy difference of defluorination (namely the dissociation of the C–F bond) via several pathways using FG as a model molecule. The calculated defluorination reaction energy is exhibited in Fig. 1, including radical dissociation (R1), ionic dissociation (R2, namely dissociation in the SN1 process), and partial radical dissociation (R4) after a single electron transfer (SET) reaction between the C–F bond and electron donor (R3). It is shown that the dissociation R1 is thermodynamically favourable (103.19 kcal mol−1) as compared to the dissociation R2 in the SN1 pathway (271.05 kcal mol−1); this is consistent with the results previously reported.35 However, under the conditions of an electron donor such as a nucleophilic reagent, the SET reaction will occur, leading to the formation of the C–F− structure, which dissociates into a carbon radical and fluorine anion in the following R4 step. The dissociation in the R4 pathway is thermodynamically favourable with a defluorination reaction energy of only 10.69 kcal mol−1. In addition, it should be noted that the C–F− structure is even more energetically stable than the C–F structure under the conditions of the electron donors; this demonstrates the better chance of defluorination occurring via the R3 and R4 pathway. Moreover, the SET reaction of the C–F bond with particular electron donors, especially the nucleophiles discussed in this study, was conducted. The nucleophiles were set as ethanediamine (EDA), hydroxide ion (OH−), and triethylamine (TEA), which were involved with the SET reaction, as shown in step 1 of Scheme 1 (the molecule structures used for calculations are shown in Fig. S2, ESI†), and the calculated results are provided in Table 1. It can be found that the SET reaction of all the particular nucleophiles with the C–F bond in the FG model molecule is also thermodynamically favorable. The abovementioned results imply the most possible defluorination mechanism of FG in the presence of electron donor reagents such as nucleophiles.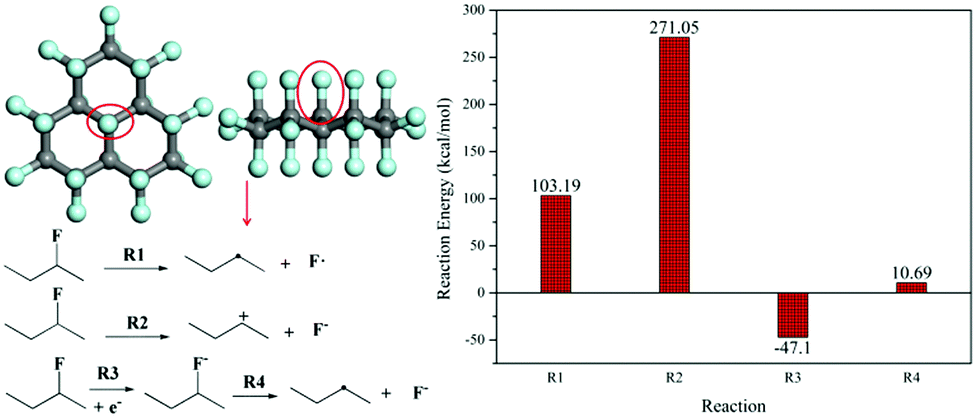 | ||
| Fig. 1 The reaction energy (dissociation of the C–F bond) for various FG defluorination pathways based on DFT calculations. | ||
 | ||
| Scheme 1 The proposed defluorination mechanism for FG under nucleophilic attack. GF means the chemical structure of FG. | ||
| SET reaction system | EDA (Nu–H) | OH− (Nu−) | TEA (N–R3) |
|---|---|---|---|
| Energy of reaction (kcal mol−1) | −2.10 | −5.21 | −8.78 |
On the other hand, the single electron transferred to the C–F bond during the SET process was donated by the electron donor. This SET process between fluorinated carbon materials (FCMs) and electron donors has been proposed in previous reports, whereas the related donors are mainly restricted in active metals41 and do not include usual nucleophilic reagents such as amines or anionic nucleophiles. However, as the following content suggests, the SET process between FCMs, such as FG and conventional nucleophiles, can also occur. The detailed R3 reaction in the presence of nucleophiles is shown as step 1 in Scheme 1, which would result in a radical intermediate related to the nucleophile. However, due to its very short life, it is usually difficult to demonstrate the existence of this radical intermediate to prove the rationality of the defluorination process step 1 in Scheme 1. A radical trapping technique was then introduced into the defluorination study of FG using N-tert-butyl-alpha-phenylnitrone (PBN) and 2-methyl-2-nitroso-propane (MNP) as radical trappers.48,49 The EPR spectra of the various radical intermediates captured during the defluorination step by nucleophiles, including amines, phosphines, and sodium hydroxide, are exhibited in Fig. 2. For the PBN-FG blank group without the nucleophilic reagent, no radical intermediate micromolecule was discovered as no evident EPR signal was observed. In contrast, the defluorination groups, including MNP-FG-EDA (ethanediamine), MNP-FG-N2H4 (hydrazine), PBN-FG-DPA (diphenylamine), PBN-FG-DPP (diphenylphosphine), and PBN-FG-KOH (potassium hydroxide), have evident EPR spin signals, which are likely attributed to the corresponding captured radical intermediates depicted in Fig. 2. The corresponding possible radical intermediates may be neutral in charge, as depicted, or in their electropositive form (perhaps [˙Nu–H]+) originating from their relevant nucleophilic reagent. It should be noted that even defluorination caused by potassium hydroxide may be involved with the hydroxyl radical resulting from the SET reaction between FG and the hydroxyl anion; this is quite interesting and beyond common thought. However, the successful detection of these radical intermediates has first proved the mechanism step 1 in Scheme 1 proposed for the defluorination of FG caused by conventional nucleophiles, including the defluorination reaction under nucleophilic attack from potassium hydroxide, which is usually thought to be non-reductive. Moreover, the deciduous fluorine atoms after defluorination of FG caused by nucleophiles, such as EDA, were confirmed to be in the form of fluorine anions, as suggested by the fluorine ion chromatography curve shown in Fig. S5 (ESI†); this further demonstrated the reasonability of step 2 in Scheme 1.
 | ||
| Fig. 2 The EPR spectra of the various radical intermediates captured by the radical trappers PBN and MNP during the derivatization reactions of FG. | ||
The EPR spectra of FG and FG treated with various nucleophilic reagents (T–FG) are shown in Fig. 3. Compared to FG, the T–FG samples have a relatively more intense EPR signal, implying that a number of new spin centers are generated on the graphene nanosheets during the defluorination step caused by the nucleophilic reagents; this is consistent with step 2 in Scheme 1. The existence of these spin centers on the nanosheets depends on the stabilizing effect of the π-conjugation system and rigid structure of the FG derivatives.1,50–53 After step 2, the coupled reaction of two closer spin centers on a nanosheet may occur and thus result in a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, as shown in step 3 of Scheme 1, whereas some of the spin centers can survive and remain in the final graphene products. In addition, it appeared that two spin centers on two different nanosheets can also couple; this leads to the formation of a C–C bond and cross-linking of two nanosheets (step 4 Scheme 1). Herein, it has been demonstrated that the defluorination of FG caused by nucleophilic reagents occurs via a radical mechanism generating spin centers on the nanosheets, and the subsequent coupled reaction of the spin centers will generate CC bonds; this means that the defluorination step has been accomplished. It is found that these spin centers play an important role in the defluorination process of FG. In addition to this, it must be emphasized that these spin centers will also affect the nucleophilic substitution of FG to a great extent, just as we has discussed in the following section.
C bond, as shown in step 3 of Scheme 1, whereas some of the spin centers can survive and remain in the final graphene products. In addition, it appeared that two spin centers on two different nanosheets can also couple; this leads to the formation of a C–C bond and cross-linking of two nanosheets (step 4 Scheme 1). Herein, it has been demonstrated that the defluorination of FG caused by nucleophilic reagents occurs via a radical mechanism generating spin centers on the nanosheets, and the subsequent coupled reaction of the spin centers will generate CC bonds; this means that the defluorination step has been accomplished. It is found that these spin centers play an important role in the defluorination process of FG. In addition to this, it must be emphasized that these spin centers will also affect the nucleophilic substitution of FG to a great extent, just as we has discussed in the following section.
 | ||
| Fig. 3 The EPR spectra of FG and FG treated with various nucleophilic reagents. | ||
3.2 Mechanism for the nucleophilic substitution of FG
To date, the nucleophilic substitution mechanism of the C–F bonds in FG is always regarded as an SN2 process as it is thermodynamically favourable,35 whereas its substitution via an SN1 process is undesirable as it is thermodynamically unfavourable, as suggested in Fig. 1. However, from this viewpoint, the SN2 or SN1 process has still been designed excessively depending on the usual substitution of the halohydrocarbon micromolecule, whereas the substitution reaction can be special for FG with a 2D structure due to its special C–F bonds.36Fig. 4 depicts a schematic of the nucleophilic attack process of the nucleophilic reagent to the C–F bond of the FCMs. It can be concluded that the SN2 process, where the nucleophile starts attacking from the opposite direction of the fluorine atoms, is impossible for fluorinated carbon nanotubes (F-CNTs) and unilaterally fluorinated graphene (U-FG) on the substrate. Even if the common fluorinated graphene is taken into consideration, nucleophilic attack from the back direction of the C–F bond will occur with an inversion of configuration.1,2 However, any single substitution accompanied by inversion of configuration (transformed from pyramid to planar and to pyramid structure) will be kinetically unfavourable for the C–F bonds located on a rigid 2D structure. Furthermore, the DFT calculations in Table 2 demonstrate the kinetics and thermodynamics disadvantage of the nucleophilic substitution reaction of the C–F bond located in the rigid structure of the FG model as compared to the case of C–F bond in the micromolecule fluoromethane, whose configuration can be easily changed (for the molecule structures used for the calculations, see Fig. S3, ESI†). As a result, the classical SN2 mechanism was also inapplicable for the nucleophilic substitution process of FG with a 2D structure.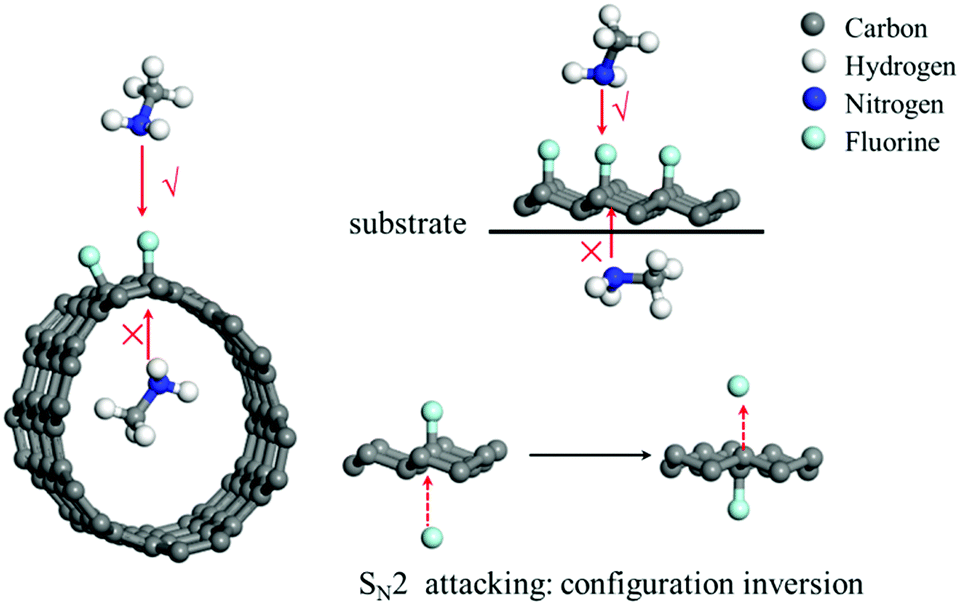 | ||
| Fig. 4 A schematic of the fluorinated carbon nanotubes (F-CNTs), unilaterally fluorinated graphene deposited on substrate (U-FG), and common FG, which have undergone nucleophilic attack by the nucleophilic reagent methylamine or fluorine anion from various directions. | ||
| Substrate molecule | Energy of reaction (kcal mol−1) | Energy of barrier (kcal mol−1) |
|---|---|---|
| Fluoromethane | −0.01 | 48.21 |
| FG model molecule | 17.36 | 104.72 |
As abovementioned, a classical nucleophilic substitution mechanism was not appropriate for the substitution process of C–F bonds in FG; therefore, a new thought should be introduced into this field. Since configuration transformation is difficult for the C–F bonds located on the rigid 2D structure, a substitution process without excessive configuration transformation will be kinetically favourable. Moreover, as Fig. 3 shows, the EPR spectra of the FG derivatives display much more intense spin signals than that of the original FG sample. This implied that a number of spin centers were formed after the reaction of FG with the nucleophiles, which would be involved in both the defluorination as well as the nucleophilic substitution mechanism. Moreover, it was demonstrated that almost all the nucleophilic substitution reactions of FG in previous studies were accompanied by its defluorination reaction; this suggested the extraordinary correlation between these two kinds of derivatization reactions of FG.
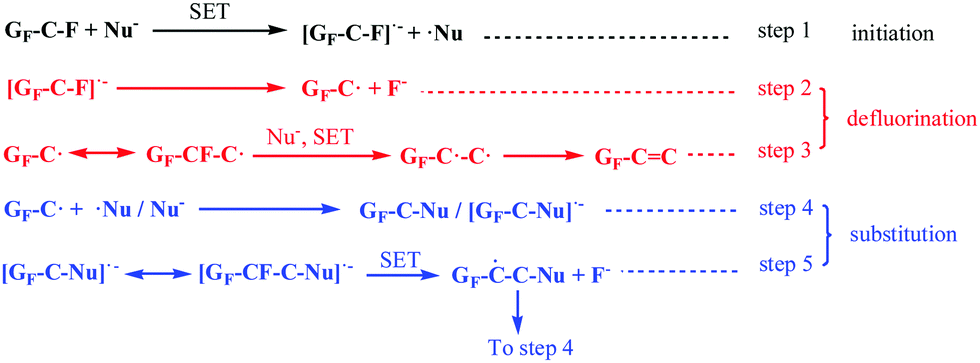
Based on these discussions, we first proposed the whole derivatization reaction process of FG via a radical mechanism including its defluorination and substitution reactions, which was named as the DR mechanism and depicted in Scheme 2. The proposed mechanism is analogous to the radical nucleophilic substitution (SRN1) mechanism of micromolecules, whose application is limited by the required rigorous reaction conditions including peculiar substrate molecules, attacking reagents or initiation conditions. However, it is demonstrated by the DR mechanism that nucleophilic substitution via a radical mechanism can also be applied to attacking process of common nucleophiles, such as amines or hydroxyl ions, without special conditions. The whole derivatisation reaction via a radical mechanism shown in Scheme 2 clearly reveals the reaction process of FG under attack by a neutral nucleophilic reagent (with active hydrogen). The DR mechanism contains three types of reaction: the initiation, defluorination, and substitution reactions. The initiation and defluorination process have been discussed above, including the SET process between the nucleophile and C–F bonds in FG (step 1) followed by dissociation of the C–F− structure that results in a spin center on the graphene nanosheet and fluorine anion (or HF, step 2), as well as the following formation of the CC bond (step 3).
 | ||
| Scheme 2 A schematic of the proposed radical derivatization mechanism of FG under attack of neutral nucleophilic reagents. | ||
The substitution reaction of FG also subsequently occurs via a radical mechanism. As step 4 shows, on the one hand, the coupled reaction between the spin center on the nanosheet and radical center of the micromole ˙Nu from the SET process in step 1 may happen, which brings about the formation of the C–Nu bond or the accomplishment of nucleophilic substitution of the C–F bond. On the other hand, the nucleophile can directly attack the spin center on the nanosheet, generating the [GF–C–Nu]˙− structure. The [GF–C–Nu]˙− structure can be regarded as an intermediate, allowing the existence of an additional unpaired electron, which is delocalized and distributed in the –C–Nu– structure regions on the graphene nanosheets. Furthermore, as step 5 suggests, the intramolecular SET reaction of the intermediate with a neighbouring C–F bond will result in another new spin center adjacent to the original spin as well as the deciduous fluorine anion, which is similar to the intramolecular transfer of spin centers. The new spin center formed in step 5 can be involved with another substitution reaction in step 4. It was discovered from the analysis of step 4 and step 5 that the radical chain reaction was started after the initiation and defluorination process. Moreover, as step 6 suggests, the interaction of GF–C˙ with Nu–H results in hydrogen abstraction to provide GF–C–H, and ˙Nu can also happen, which brings about the formation of another substitution product of FG.
The abovementioned explanations explicitly demonstrate the detailed process of the nucleophilic substitution reaction of FG via a radical mechanism instead of the classical SN2 mechanism, which is initiated by the SET reaction between the C–F bonds and nucleophilic reagent. It should be noted that due to the pyramidal configuration of the spin centers on the nanosheets, the nucleophilic substitution process in the proposed mechanism was not accompanied by the evident configuration transformation; this demonstrated the thermodynamic and kinetic advantages of this kind of substitution process in comparison to the case of other substitution reactions with an inversion of configuration. The 2D structure where the C–F bonds in FG are located leads to the unfavourable kinetics of nucleophilic substitution via an SN2 mechanism as well as the stable existence of spin centers on the nanosheets due to their rigid structure and conjugation effects;50–53 this brings about a greater possibility of nucleophilic substitution via a radical process. It can be deduced that the radical process can also represent the typical characteristics of the chemistry of 2D materials.
Similar to the reaction process shown in Scheme 2, the derivatization reaction of FG under attack of an electronegative nucleophile also occurs via a radical mechanism, as shown in Scheme 3. The difference between the two processes is that the SET reaction between a neutral nucleophile and the C–F bond will result in a cation radical [˙Nu–H]+, which will dissociate into a Nu˙ radical and a proton. It can be discovered from Schemes 1 and 2 that in the DR mechanism, the defluorination process of FG, generating spin centers on the graphene nanosheets, becomes the requirement of the subsequent nucleophilic substitution. This is consistent with the previous reports stating that the substitution of FG is always accompanied by a defluorination reaction. It was also demonstrated in a previous study that the grafting ratio of a nucleophilic amino group on the FG nanosheets was improved as the nucleophilicity or reducibility of amine increased; this was in agreement with the increasing degree of defluorination. The relationship between these two kinds of reactions is not a total competitive relation, which is another feature of the DR mechanism, distinguished from that of the typical SN2 process.
 | ||
| Scheme 3 A schematic of the proposed radical derivatization mechanism of FG under attack of an electronegative nucleophilic reagent. GF represents the chemical structure of FG. | ||
3.3 Further experiments supporting the substitution mechanism in FG
In most cases, a tertiary amine reagent without active hydrogen cannot participate in the nucleophilic substitution reaction via a conventional SN1 or SN2 mechanism. However, different observations were obtained in the abovementioned proposed DR mechanism. It has been experimentally found that some nitrogen-containing reagents without active hydrogen, such as trimethylamine (TEA), can defluorinate FG as well, whereas this kind of defluorination mechanism has not been revealed in Scheme 2. Despite this, the defluorination mechanism can be similar by deducing the DR mechanism. The proposed defluorination process is exhibited in Scheme 4. It is suggested that the SET reaction between TEA and the C–F bond of FG can also occur, resulting in the generation of the C–F− structure and subsequent defluorination. Moreover, a cationic radical [˙N–R3]+ is produced, whose affiliation is an interesting question. The radical trapping method was then introduced into the reaction between FG and TEA, and the EPR spectrum (Fig. S4, ESI†) demonstrated the existence of the radical intermediate in the form of the radical cation [˙N–R3]+. More importantly, it is proposed by step 4 and step 5 that the coupled reaction between the radical cation and spin center on the nanosheet can occur, thus generating the [GF–C–N–R3]+ structure, which is followed by another SET reaction leading to the covalent attachment of the nitrogen group to the nanosheet and the formation of the RF fragment. The existence of RF fragment indicates that some fluorine atoms are in the form of non-ionic fluorinated molecules. As a result, there are less deciduous fluorine anions in the FG-TEA reacting solution as compared to the case of other reaction systems such as the reacting solution of FG-EDA. The fluorine anion chromatography curves of the FG-EDA and FG-TEA solutions in Fig. S4 (ESI†) show that there is indeed less fluorine anions in the FG-TEA solution; this implies the additional destination of fluorine atoms during the defluorination by TEA. This further suggests the rationality of the proposed nucleophilic attack shown in Scheme 4. Moreover, it appears in Scheme 4 that the TEA fragment group (–NR2) can be grafted onto the graphene nanosheet in some way; this is impossible for nucleophilic substitution via an SN1 or SN2 mechanism. | ||
| Scheme 4 A schematic of the proposed nucleophilic attack of triethylamine (TEA) to FG. R represents the ethyl group. | ||
Furthermore, by comparing the XPS spectra obtained for the FG and FG-TEA samples in Fig. 5, the different signature of FG-TEA whereby a significantly less intense peak for the C–F group and an enhanced peak for the CC bond in the C 1s spectrum, as well as the new emerging peak in the N 1s spectrum were observed. Therefore, it can be tentatively concluded that the TEA fragment has been attached to the graphene nanosheets; this is also supported by the FTIR spectrum of FG-TEA shown in Fig. S6 (ESI†). The elemental analysis given in Table S1 (ESI†) further demonstrates that the new introduced species into the graphene nanosheet is a covalently attached group in the form of –N(CH2CH3)2 instead of a physically absorbed species N(CH2CH3)3 because of the increased molar ratio for H/N of around 10 rather than 15 in the FG-TEA sample. The negligible nitrogen element in the blank group G-TEA (graphene treated by TEA) further confirmed that the increased nitrogen-related group in FG-TEA was indeed a covalently attached group. However, for nucleophilic substitution via an SN2 mechanism, the TEA fragment cannot replace fluorine atom at all. These discussions effectively demonstrate the rationality of Scheme 4 as well as the proposed DR mechanism shown in Scheme 2, in which the nucleophilic substitution of FG occurs via a radical mechanism after the SET and defluorination reactions. In addition, it should be noted that the oxygen element content of the derivative sample FG-TEA is slightly increased as compared to that of the FG sample (O/C ratio 0.24 vs. 0.18); this has occurred in almost all the previously reported derivatization reactions of FG.29,33,34 Considering the spin centers on the graphene nanosheets, it is suggested by Scheme S1 (ESI†) that the increasing oxygen element originates from the coupled reaction between the spin centers and oxygen molecules.54,55 The reaction mechanism for the introduction of oxygen has again showed the significance of the spin centers in the derivatization reactions of FG.
 | ||
| Fig. 5 The XPS C 1s spectra of the FG and FG-TEA samples. The insets correspond to the F 1s spectrum of FG and N 1s spectrum of FG-TEA. | ||
As we have emphasized above, the DR mechanism shows a special characteristic different from the SN1 or SN2 mechanisms in which the defluorination process is the requirement for the subsequent nucleophilic substitution reaction of FG. In the case of the nucleophilic reaction of FG with a relatively inert nucleophile, the DR process cannot be adequately initiated; this results in a low degree of defluorination and low grafting ratio of nucleophile. In contrast, the introduction of a reductive reagent, such as pyridine, into the reaction system will promote the defluorination step as well as the degree of substitution; this may be the reason of the catalytic effect of pyridine during the derivatisation of FCMs.33,34,56 In addition, it has been suggested that FG can be defluorinated using ultra-violet radiation; this leads to the formation of new spin centers on the graphene nanosheets (Fig. S7, ESI†), which can be the attaching position of relatively inert nucleophiles on the nanosheets. A grafting experiment using formamide (FM) under ultra-violet irradiation was then performed, and the grafting amount is given in Table S2 (ESI†). It was demonstrated that ultra-violet irradiation indeed promoted the defluorination of FG under attack of FM as well as the substitution reaction; this resulted in a higher degree of defluorination and a higher grafting ratio of formamide on the graphene nanosheets. These results further indicate the rationality of our proposal that the nucleophilic substitution of FG occurs via a radical mechanism, which is dependent on the preceding defluorination process generating spin centers on the nanosheets. It will be easier to understand the derivative chemistry of FG with 2D structures based on the discussions proposed above.
4. Conclusions
In summary, we have studied fluorinated graphene as a representative 2D material to reveal the detailed mechanism of its defluorination and nucleophilic substitution reactions under attack with conventional nucleophiles and explored the chemistry of 2D materials. It has been demonstrated using DFT calculations that homolytic dissociation of C–F bond after the single electron transfer (SET) reaction between the nucleophile and C–F bond of FG is thermodynamically favourable. The relevant radical intermediates and changes in the spin centers of FG were confirmed via the EPR method; this indicated that defluorination of FG occurred via a radical mechanism. More importantly, it has been discovered that neither the SN1 nor SN2 mechanism is appropriate for the substitution process of C–F bonds located in the 2D structure of FG. Instead, the nucleophilic substitution of FG occurs via a radical mechanism initiated by the preceding defluorination step. Upon deducing the proposed substitution mechanism of FG, it has been demonstrated that tertiary amine nucleophiles, such as triethylamine, can also be covalently attached to graphene nanosheets via a nucleophilic reaction with FG; this has never been previously reported in the literature. The detailed process of derivative nucleophilic reaction of FG including defluorination and substitution has been first revealed to be via a radical mechanism depending on its 2D structure. This study also suggests the potential radical characteristics of the chemistry of 2D materials and enriches the previous research on the nucleophilic substitution in the field of organic chemistry. Moreover, the revelation of the nucleophilic reaction mechanism of FG can enlighten us on how to control the reaction of graphene derivatives to prepare accurate graphene products.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 51633004 and Grant No. 51573105) and State Key Laboratory of Polymer Materials Engineering (Grant No. sklpme2017-2-03). The authors acknowledge the Analytical & Testing Centre of Sichuan University, College of Polymer Science and Engineering of Sichuan University and the State Key Laboratory of Polymer Materials Engineering (Sichuan University) for characterization and computational simulation.Notes and references
- C. K. Ingold, Structure and mechanism in organic chemistry, Cornell University Press, Ithaca, New York, 1953 Search PubMed.
- L. P. Hammett, Physical organic chemistry, 1940 Search PubMed.
- M. B. Smith and J. March, March's advanced organic chemistry: reactions, mechanisms, and structure, John Wiley & Sons, 2007 Search PubMed.
- E. D. Hughes and C. K. Ingold, J. Chem. Soc., 1935, 244–255 RSC.
- E. Uggerud, J. Phys. Org. Chem., 2006, 19, 461–466 CrossRef CAS.
- J. Miller, 1968.
- J. F. Bunnett and R. E. Zahler, Chem. Rev., 1951, 49, 273–412 CrossRef CAS.
- W. R. Bowman and J. M. D. Storey, Chem. Soc. Rev., 2007, 36, 1803 RSC.
- R. A. Rossi, A. B. Pierini and A. B. Peñéñory, Chem. Rev., 2003, 103, 71–168 CrossRef CAS PubMed.
- J. F. Bunnett and J. K. Kim, J. Am. Chem. Soc., 1970, 92, 7463–7464 CrossRef CAS.
- D. Lexa, J. M. Saveant, S. K. Binh and D. L. Wang, J. Am. Chem. Soc., 1988, 110, 7617–7625 CrossRef CAS.
- G. A. Russell, J. Hershberger and K. Owens, J. Am. Chem. Soc., 1979, 101, 1312–1313 CrossRef CAS.
- B. Janhsen, C. G. Daniliuc and A. Studer, Chem. Sci., 2017, 8, 3547–3553 RSC.
- M. Xu, T. Liang, M. Shi and H. Chen, Chem. Rev., 2013, 113, 3766–3798 CrossRef CAS PubMed.
- J. Yu, J. Li, W. Zhang and H. Chang, Chem. Sci., 2015, 6, 6705–6716 RSC.
- P. Miro, M. Audiffred and T. Heine, Chem. Soc. Rev., 2014, 43, 6537–6554 RSC.
- W. Feng, P. Long, Y. Feng and Y. Li, Adv. Sci., 2016, 3 Search PubMed.
- M. Pumera and Z. Sofer, Chem. Soc. Rev., 2017, 46, 4450–4463 RSC.
- M. Inagaki and F. Kang, J. Mater. Chem. A, 2014, 2, 13193–13206 CAS.
- R. R. Nair, W. Ren, R. Jalil, I. Riaz, V. G. Kravets, L. Britnell, P. Blake, F. Schedin, A. S. Mayorov and S. Yuan, Small, 2010, 6, 2877–2884 CrossRef CAS PubMed.
- V. Mazánek, O. Jankovský, J. Luxa, D. Sedmidubský, Z. Janoušek, F. Šembera, M. Mikulics and Z. Sofer, Nanoscale, 2015, 7, 13646–13655 RSC.
- W. Lai, D. Xu, X. Wang, Z. Wang, Y. Liu, X. Zhang and X. Liu, Phys. Chem. Chem. Phys., 2017, 19, 19442–19451 RSC.
- X. Wang, Y. Dai, W. Wang, M. Ren, B. Li, C. Fan and X. Liu, ACS Appl. Mater. interfaces, 2014, 6, 16182–16188 CAS.
- B. Li, T. He, Z. Wang, Z. Cheng, Y. Liu, T. Chen, W. Lai, X. Wang and X. Liu, Phys. Chem. Chem. Phys., 2016, 18, 17495–17505 RSC.
- S. B. Bon, L. Valentini, R. Verdejo, J. L. Garcia Fierro, L. Peponi, M. A. Lopez-Manchado and J. M. Kenny, Chem. Mater., 2009, 21, 3433–3438 CrossRef.
- M. Ren, X. Wang, C. Dong, B. Li, Y. Liu, T. Chen, P. Wu, Z. Cheng and X. Liu, Phys. Chem. Chem. Phys., 2015, 17, 24056–24062 RSC.
- X. Wang, W. Wang, Y. Liu, M. Ren, H. Xiao and X. Liu, Phys. Chem. Chem. Phys., 2016, 18, 3285–3293 RSC.
- X. Ye, L. Ma, Z. Yang, J. Wang, H. Wang and S. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 7483–7488 CAS.
- P. Lazar, C. K. Chua, K. Hola, R. Zboril, M. Otyepka and M. Pumera, Small, 2015, 11, 3790–3796 CrossRef CAS PubMed.
- V. Mazanek, A. Libanska, J. Sturala, D. Bousa, D. Sedmidubsky, M. Pumera, Z. Janousek, J. Plutnar and Z. Sofer, Chem. – Eur. J., 2017, 23, 1956–1964 CrossRef CAS PubMed.
- D. D. Chronopoulos, A. Bakandritsos, P. Lazar, M. Pykal, K. Cepe, R. Zboril and M. Otyepka, Chem. Mater., 2017, 29, 926–930 CrossRef CAS PubMed.
- V. Urbanova, K. Hola, A. B. Bourlinos, K. Cepe, A. Ambrosi, A. H. Loo, M. Pumera, F. Karlicky, M. Otyepka and R. Zboril, Adv. Mater., 2015, 27, 2305–2310 CrossRef CAS PubMed.
- B. Li, K. Fan, X. Ma, Y. Liu, T. Chen, Z. Cheng, X. Wang, J. Jiang and X. Liu, J. Colloid Interface Sci., 2016, 478, 36–45 CrossRef CAS PubMed.
- R. Stine, J. W. Ciszek, D. E. Barlow, W. K. Lee, J. T. Robinson and P. E. Sheehan, Langmuir, 2012, 28, 7957–7961 CrossRef CAS PubMed.
- M. Dubecky, E. Otyepkova, P. Lazar, F. Karlicky, M. Petr, K. Cepe, P. Banas, R. Zboril and M. Otyepka, J. Phys. Chem. Lett., 2015, 6, 1430–1434 CrossRef CAS PubMed.
- W. Zhang, M. Dubois, K. Guerin, P. Bonnet, H. Kharbache, F. Masin, A. P. Kharitonov and A. Hamwi, Phys. Chem. Chem. Phys., 2010, 12, 1388–1398 RSC.
- H. F. Bettinger, K. N. Kudin and G. E. Scuseria, J. Am. Chem. Soc., 2001, 123, 12849–12856 CrossRef CAS PubMed.
- R. Taylor, Russ. Chem. Bull., 1998, 47, 823–832 CrossRef CAS.
- N. Kornblum, R. E. Michel and R. C. Kerber, J. Am. Chem. Soc., 1966, 88, 5660–5662 CrossRef CAS.
- E. C. Ashby, Acc. Chem. Res., 1988, 21, 414–421 CrossRef CAS.
- S. Chakraborty, W. Guo, R. H. Hauge and W. E. Billups, Chem. Mater., 2008, 20, 3134–3136 CrossRef CAS.
- X. Wang, W. Wang, Y. Liu, M. Ren, H. Xiao and X. Liu, Anal. Chem., 2016, 88, 3926–3934 CrossRef CAS PubMed.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- X. Chen, J. Chang, H. Yan and D. Xia, J. Phys. Chem. C, 2016, 120, 28912–28916 CAS.
- J. Plsek, K. A. Drogowska, V. Vales, J. Ek Weis and M. Kalbac, Chem. – Eur. J., 2016, 22, 8990–8997 CrossRef CAS PubMed.
- S. D. Costa, J. Ek Weis, O. Frank, Z. Bastl and M. Kalbac, Carbon, 2015, 84, 347–354 CrossRef CAS.
- E. G. Janzen, Acc. Chem. Res., 1971, 4, 31–40 CrossRef CAS.
- K. P. Madden and H. Taniguchi, J. Am. Chem. Soc., 1991, 113, 5541–5547 CrossRef CAS.
- J. Hioe and H. Zipse, Org. Biomol. Chem., 2010, 8, 3609–3617 CAS.
- H. Zipse, Radicals in Synthesis I, Springer, 2006, pp. 163–189 Search PubMed.
- J. Hioe and H. Zipse, Encyclopedia of Radicals in Chemistry, Biology and Materials, John Wiley & Sons, New York, 2012, pp. 449–476 Search PubMed.
- T. Shida, E. Haselbach and T. Bally, Acc. Chem. Res., 1984, 17, 180–186 CrossRef CAS.
- K. U. Ingold, Acc. Chem. Res., 1969, 2, 1–9 CrossRef CAS.
- S. I. Kuzina, A. P. Kharitonov, Y. L. Moskvin and A. I. Mikhailov, Russ. Chem. Bull., 1996, 45, 1623–1627 CrossRef.
- V. N. Khabashesku, W. E. Billups and J. L. Margrave, Acc. Chem. Res., 2002, 35, 1087–1095 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp06708a |
| This journal is © the Owner Societies 2018 |