
Kinetics of the simplest Criegee intermediate reaction with ozone studied using a mid-infrared quantum cascade laser spectrometer†
Yuan-Pin
Chang
 *ab,
Hsun-Hui
Chang
b and
Jim Jr-Min
Lin
bc
*ab,
Hsun-Hui
Chang
b and
Jim Jr-Min
Lin
bc
aDepartment of Chemistry, National Sun Yat-sen University, Kaohsiung 80424, Taiwan. E-mail: ypchang@mail.nsysu.edu.tw
bInstitute of Atomic and Molecular Sciences, Academia Sinica, Taipei 10617, Taiwan
cDepartment of Chemistry, National Taiwan University, Taipei 10617, Taiwan
First published on 17th November 2017
Abstract
The kinetics of the reaction of CH2OO with ozone has been studied by monitoring CH2OO using time-resolved infrared (IR) absorption spectroscopy, which utilized the fast chirped IR pulse train from a quantum cascade laser [J. Chem. Phys., 2017, 146, 244302]. CH2OO was prepared by photolyzing a gas mixture of CH2I2/O2/O3 at 352 nm; the photolysis wavelength was chosen to minimize the photodissociation of O3. The measured rate coefficient at 298 K and 30 Torr is (6.7 ± 0.5) × 10−14 cm3 s−1, independent of pressure from 30 to 100 Torr. The result indicates that previous ab initio calculations either underestimated or overestimated this reaction rate by one order of magnitude or more. The result also implies that, the reaction of the Criegee intermediate with ozone may play a role in laboratory studies of ozonolysis of alkenes. However, this reaction would not compete with other CH2OO sinks in the atmosphere.
Introduction
In the atmosphere, ozonolysis of alkenes produces highly reactive Criegee intermediates (CIs).1–4 The produced CIs may have excess internal energy and undergo unimolecular processes, such as isomerization or decomposition to form OH radicals; some of the CIs may be collisionally stabilized and then may react with other atmospheric species. In laboratory studies of ozonolysis, direct detection of CIs is difficult due to low steady-state concentrations. Direct spectroscopic and kinetic studies of CIs only became feasible after the work by Welz et al.,5 which demonstrated the efficient preparation of the simplest CI, CH2OO, via the reaction CH2I + O2 → CH2OO + I. Many recent studies on prototypical CIs have found that CIs play an important role in atmospheric chemistry, including the formation of OH radicals6–9 and oxidation of atmospheric species, like SO2, NO2, organic and inorganic acids, alkenes, and water vapor.5,10–31 The reactions of CIs may produce radicals or low-volatility organic species, which are key components in the formation of secondary organic aerosols.32The reactions of O3 with CIs are potentially important in both laboratory and atmospheric studies. For example, Novelli et al.7 have considered that the main loss paths of CIs in their ozonolysis experiments are unimolecular decomposition and reactions with ozone at early reaction times, whereas reactions with organic peroxy radicals, alcohols, aldehydes and organic peroxides become more important at later reaction times. However, to the best of our knowledge, there is no direct measurement for determining the rate of ozone reaction with CIs, while there have been a few theoretical works.33–36 For CH2OO, Kjaergaard et al.33 calculated its reaction rate with O3 by using the CCSD(T)//B3LYP level of theory and predicted the formation of a cycloaddition intermediate through a significant barrier, yielding small rate coefficients ranging from 4.0 × 10−16 to 1.2 × 10−18 cm3 s−1. Wei et al.34 also investigated the reaction mechanism by using the CCSD(T)//B3LYP level of theory, but they did not report any rate coefficient. Vereecken et al.35,36 calculated the rate of this reaction by using the CCSD(T)//M06-2X level of theory, and found that CH2OO and O3 will firstly form a prereactive complex without a barrier before passing through a submerged chain-addition transition state; they predicted a larger rate coefficient of 4 × 10−13 cm3 s−1. It appears that it is not easy to accurately estimate the rate coefficient with the present quantum chemistry approaches. Nonetheless, all the theoretical studies predicted CH2O + 2O2 as the final products.33–36
Recently, we have developed a high-resolution mid-infrared quantum cascade laser (QCL) spectrometer, and utilized it to study the spectrum of the ν4 band of CH2OO.37 In this work, we used the QCL spectrometer to study the kinetics of the reaction of CH2OO with O3. This method has the following advantages: (1) avoids byproduct interferences and baseline drifting by probing narrow spectral lines, (2) high sensitivity due to narrow laser linewidth and long optical path length, (3) long CH2OO lifetimes (up to 14 ms) due to low required concentration of CH2OO, and (4) efficient data acquisition using the fast chirped pulse train of the QCL.
Experimental methods
Most of the experimental procedures have been described in our previous work.37 Therefore, only a brief description is provided here. CH2OO was prepared in a flow cell following the well-established method of CH2I2/O2 photolysis:5 CH2I2 (1–7 mTorr) mixed with O2 (30 or 100 Torr) was photolyzed using an unfocused excimer laser beam at 352 nm (laser fluence: (1.3–4.4) × 1016 photon cm−2). We chose this photolysis wavelength (instead of 248 or 308 nm) to minimize the effect of O3 photolysis (a cross section of O3 ≈ 1.0 × 10−22 cm2 at 352 nm, and a photolysis probability of ≤4.4 × 10−6 at ≤4.4 × 1016 photon cm−2). The O3 gas was synthesized using a commercial ozone generator, collected by adsorption on silica gel at dry-ice temperature, and further purified by condensation in a stainless steel cylinder at liquid-nitrogen temperature, following the procedures described in our previous work.38 The O3 concentration was measured via its UV absorption by using an absorption cell with a path length of 5 cm, a deuterium lamp (Hamamatsu, L10671D), and a spectrometer (Ocean Optics, USB2000+UV-VIS-ES), right before the O3 gas entered the reactor cell. The flow rates of the used gases were controlled using mass flow controllers (Brooks, 5850E).We used a distributed-feedback quantum cascade laser (Alpes Lasers, CW-DFB-QCL) as a coherent IR source to probe CH2OO. The laser was driven by an intermittent CW driver (Alpes Lasers), powered by a DC-power supply (HAMEG Instruments, HMP2020). Its frequency coverage was 1279–1290 cm−1 and the practical spectral linewidth was about 0.002 cm−1 with a peak power over 10 mW. The temperature of the laser was controlled by a TEC cooling element inside the laser housing and a temperature controller. The laser was operated in the pulse-mode with a period of 18 μs or 120 μs and 40% duty cycle (a pulse duration of 7 μs or 48 μs). In each laser pulse, the laser frequency was down-chirped (1286.1–1285.5 cm−1 in 48 μs). During the experiment, the IR chirped pulses were repeatedly scanned through the Q branch of the ν4 fundamental band as shown in Fig. 1. The lifetime of CH2OO in our experiments was on the millisecond time scale, much longer than the IR pulse period. Thus, for a single photolysis event, we could obtain a full time profile of CH2OO from probing its transitions with the IR pulse train.
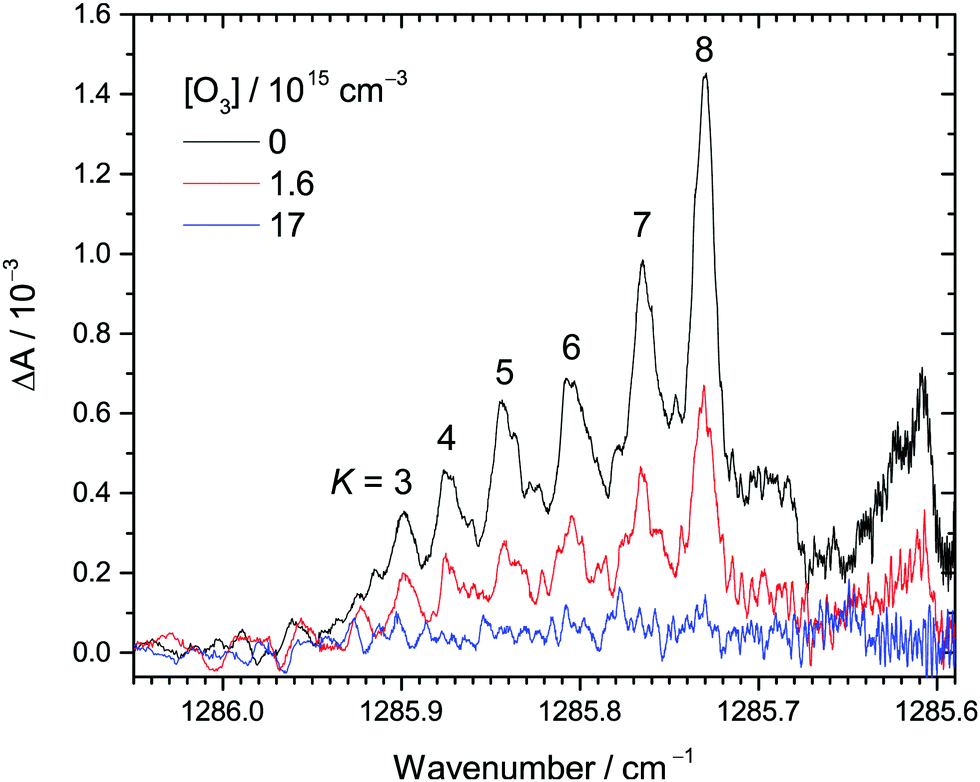 | ||
| Fig. 1 Examples of transient IR absorption spectra of CH2OO at different O3 concentrations at 30 Torr total pressure. The peaks correspond to the Q branch of the ν4 fundamental band of CH2OO. The K number is assigned to each (partially) resolved sub-band. The photolysis-probe delay time is 2.4 ms in this example. See Table S1 (ESI†) (expt. 1) for details of the experimental conditions. | ||
Our reactor cell was a glass tube (inner diameter 19 mm) with BaF2 windows at both ends, which were purged with N2. The photolysis excimer laser beam (352 nm, repetition rate 1 Hz) was combined with the IR probe beam and introduced into the flow cell by a high reflective BaF2 mirror at 352 nm (Eksma Optics, custom item). A BaF2 right-angle prism and a concave aluminium mirror (Edmund Optics, part # 43549) were placed before and after the flow cell, such that the probe IR beam was reflected back-and-forth between the two optics, creating a long overlap with the UV photolysis volume up to 3.9 meters (6 passes though the cell with an effective sample length of 65 cm). After leaving the multi-pass cell, the IR probe beam was guided to a HgCdTe (MCT) detector (Kolmar Technologies, KMPV11-1-J2), and the UV photolysis laser beam was reflected away by another BaF2 high reflective mirror. Each photolysis pulse was synchronized with the rising edge of one of the IR probe pulses by using a delay generator (SRS, DG535). The calibration of the IR laser wavelength was carried out by measuring a reference gas spectrum (3 Torr N2O) and an etalon signal (Ge etalon 3′′ in length, free spectral range = 0.0163 cm−1). The IR pulse train signals from the DC outputs of all MCT detectors were acquired using an oscilloscope (LeCroy, HDO4034, 12-bit vertical resolution) at a sampling rate of 1.25 GS s−1. For each [CH2OO] time profile measurement, we averaged the data for 100 UV laser shots to improve the signal-to-noise ratio.
Finally, we also utilized a UV transient absorption spectrometer to roughly estimate the absolute values of [CH2OO] under the experimental conditions of this work. A LED (Hamamatsu, LC-L2) with an emission profile centered at 365 nm was used as the probe light source. The probe light was projected into the flow cell by a convex lens, and then it was reflected once by the concave mirror to achieve two passes through the cell. After leaving the flow cell, the remaining probe light was guided to a balanced photodiode detector (Thorlab, PDB450A). The rest of the experimental configurations were the same as those described above. Since the system was not optimized for UV detection, the uncertainty in the absolute values of [CH2OO] would be a bit larger than those of our previous works.12
Results and discussion
Fig. 1 shows the selected transient IR absorption spectra of CH2OO at representative O3 concentrations. Transient absorption means the change in the IR absorption intensity with respect to that before the UV photolysis. In this example, the results at a photolysis-probe delay time of 2.4 ms are shown. As mentioned in the Experimental methods, the spectral range corresponds to the Q branch of the ν4 fundamental band of CH2OO.37 Note that each of the peaks between 1285.9 and 1285.7 cm−1 corresponds to congested transitions from the same K levels (from K = 3 to 8), while the broad feature peaked at 1285.61 cm−1 is the perturbed transitions of the higher K levels.37 See ref. 37 for detailed spectroscopic assignments and perturbation analysis. Upon increasing the O3 concentration, the CH2OO signal decreases accordingly, suggesting that CH2OO is consumed by its reaction with O3. The decay trends of these peaks are similar, indicating no rotational dependence.
Fig. 2 shows a few time profiles of [CH2OO] at various [O3]. Again, the increasing decay rates at higher [O3] indicate that CH2OO reacts with O3. The following reactions are involved in the formation and decay of CH2OO in our CH2I2/O2/O3 photolysis system:12
| CH2I2 + hν (352 nm) → CH2I + I |
| CH2I + O2 → CH2OO + I |
| CH2OO → products kfirst |
| CH2OO + O3 → products kO3 |
| CH2OO + I → products kI |
| CH2OO + CH2OO → products kself |
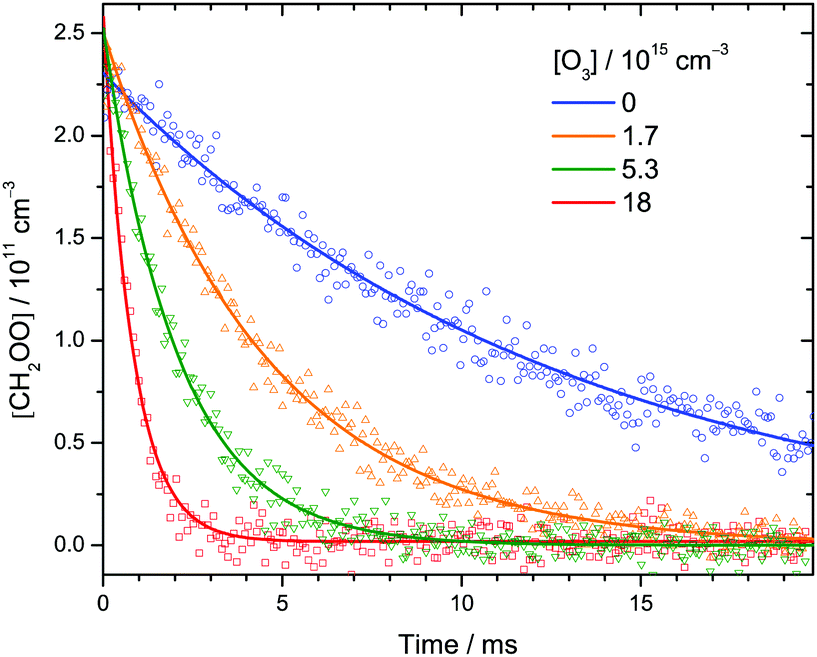 | ||
| Fig. 2 Representative time profiles of [CH2OO] at various [O3] at 30 Torr and 298 K. Each data point (symbol) is the integrated intensity of the highest peak (1285.71–1285.74 cm−1) of the Q branch of the CH2OO ν4 band (see Fig. 1) probed by each IR pulse. The lines are single-exponential fit to the data. See Table S1 (ESI†) (expt. 11) for details of the experimental conditions. | ||
In our first attempt of data analysis, we described the measured time profile of CH2OO as:
[CH2OO](t) = [CH2OO]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−kefft) exp(−kefft) | (1) |
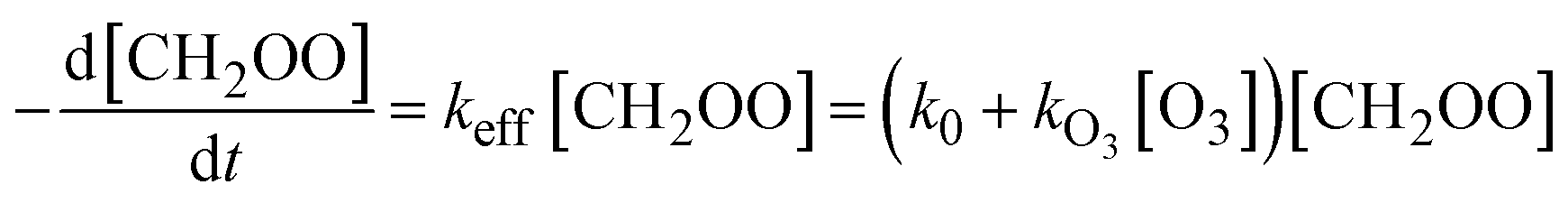 | (2) |
| k0 ≅ kfirst + kI[I] + 2kself[CH2OO] | (3) |
Fig. 3 plots the value of keff as a function of [O3] at 30 Torr and 298 K, and the solid lines are linear fits to the data points except those at [O3] = 0. At low [CH2OO]0 (≈2.4 × 1011 cm−3, Fig. 3(a)), keff shows a linear relationship with [O3], as expected from eqn (2). The slope (6.4 × 10−14 cm3 s−1) should correspond to a measured value of kO3. To our surprise, at higher [CH2OO]0 (for example, Fig. 3(b)), while keff is still a linear function of [O3] for [O3 ] > 0, this linear line does not go through the data points at [O3] = 0. Nonetheless, the keff values as a function of [O3] all exhibit a consistent slope for [O3] > 0. The detailed values are listed in Table S1 (ESI†) in the column of kO3. The averaged value (with one-sigma error bar) of the slope is (6.72 ± 0.46) × 10−14 cm3 s−1.
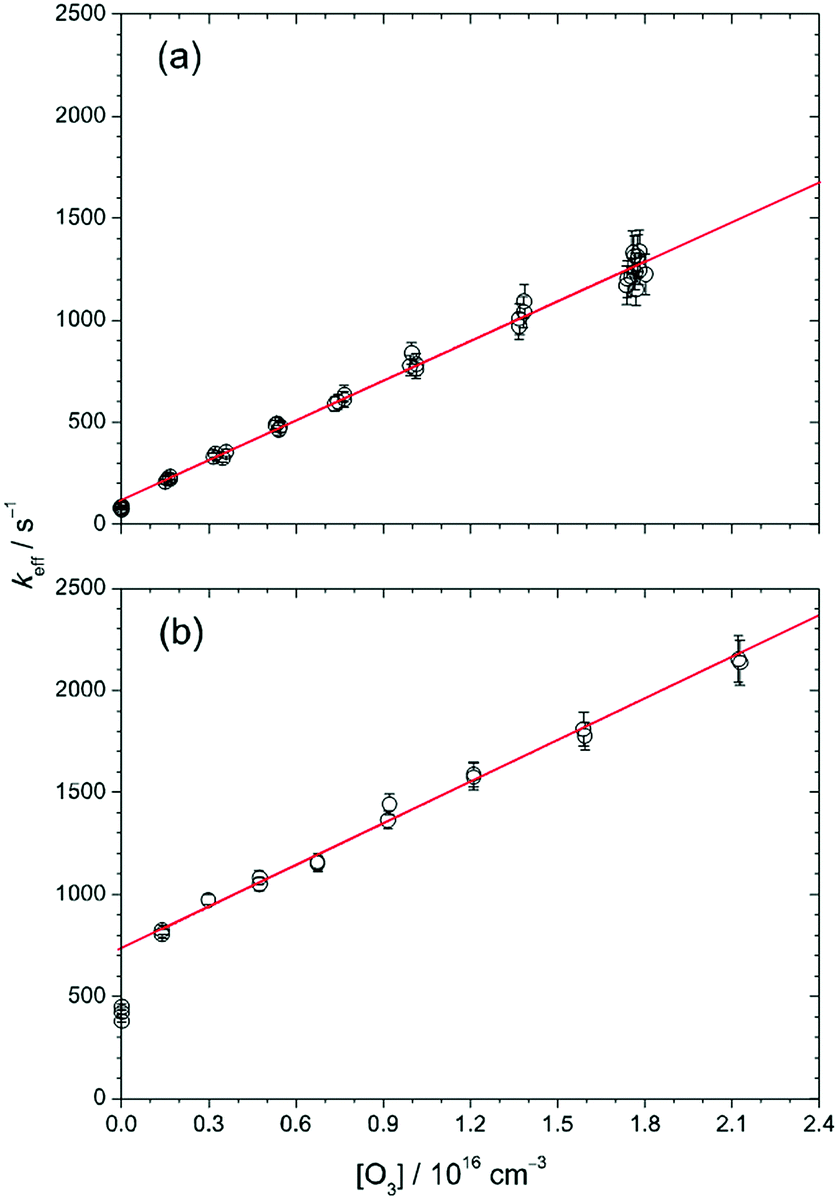 | ||
| Fig. 3 Effective first-order rate coefficient of CH2OO decay, keff, as a function of [O3] for [CH2OO]0 = (a) 2.4 × 1011 cm−3 (expt. 11) and (b) 2.1 × 1012 cm−3 (expt. 9). The error bars are 1σ uncertainties from the fitting of the [CH2OO] time profiles. The red line is a linear fit to keff excluding the data at [O3] = 0 (see text for details). The slope of the red line corresponds to the second-order rate coefficient of the CH2OO + O3 reaction. | ||
To investigate the possible source that causes the difference between the intercept value (kincpt) of keff (obtained from the linear fit of keff as a function of [O3] for [O3] > 0) and the measured k0 (keff at [O3] = 0), we performed several checking experiments as described below. First, one may wonder whether the O atoms from O3 photodissociation affect the measurements or not. To check this, we measured keff at different 352 nm laser fluences (by a factor of 2.8, which varied [O] by the same factor) and obtained very similar results (see Fig. S1, ESI†), indicating that no O3 photochemistry was involved. As mentioned in the Experimental methods, the probability of O3 photolysis is less than 4.4 × 10−6, thus the upper limit of [O] can be estimated to be (4.4 × 10−6)(2 × 1016 cm−3) = 9 × 1010 cm−3. The rate of the reaction of CH2OO with O atom is not known yet. If we assume that every collision between CH2OO and O atom leads to a reaction (the collision-limit), the rate coefficient would be about 3 × 10−10 cm3 s−1,40 and the effect of the O atoms on the CH2OO decay is still small (<27 s−1 in keff).
Because we synthesized and purified the O3 gas by ourselves, one may worry about its purity. The typical lifetime of our O3 gas (stored in a stainless steel cylinder at dry-ice temperature) is more than 20 hours. To check the effect of impurities, we deliberately warmed up the O3 gas to room temperature and waited until all the O3 molecules had decomposed (∼70 hours, verified using UV absorption). We called this gas “decomposed O3” which would contain a similar or higher level of impurity, compared to that of our fresh O3 gas. The results are shown in Fig. S3 (ESI†). The “decomposed O3” gas does not change the keff at all, indicating that the impurity in our O3 gas has a negligible effect.
As shown in Fig. 3, the difference between kincpt and k0 is more significant at higher [CH2OO]0. We found that this difference, kincpt − k0, is proportional to [CH2OO]0 as shown in Fig. S4 (ESI†). This observation gave us an idea that the chemistry may involve I atoms, of which the concentration is proportional to [CH2OO]0 in our preparation method. Thus, we propose that the following reactions should also be involved when O3 is present:
| O3 + I → IO + O2 kI+O3 |
| CH2OO + IO → products (possibly CH2O + OIO) kIO |
| kincpt ≅ kfirst + kI[I] + 2kself[CH2OO] + kIO[IO] | (4) |
| [I]0 = [I] + [IO] ≅ 2[CH2OO]0 | (5) |
We have also determined kincpt and k0 at various [CH2OO]0 (see Table S1, ESI†). Note that when O3 is present, the maximum concentration of IO is also controlled by [CH2OO]0, [IO]max ≅ [I]0 ≅ 2[CH2OO]0. Therefore the difference between kincpt and k0 would become:
| kincpt − k0 ≅ (kIO − kI) [I]0 ≅ 2(kIO − kI) [CH2OO]0. | (6) |
In brief summary, we have tested a few crucial experimental conditions. The results enable us to exclude the possibility of interferences which originate from the photolysis of O3 and the impurity in our O3 gas. In our system, I atoms would be quickly converted into IO when O3 is present; the side-reaction of CH2OO + I would be shifted to the reaction of CH2OO + IO and thus, changes the value of kincpt. For [O3] > 1.6 × 1015 cm−3, all the observed data of keff are linear with [O3] with a slope of (6.72 ± 0.46) × 10−14 cm3 s−1, which corresponds to the bimolecular rate coefficient of the reaction of CH2OO with O3. Finally, we found that the kinetics at a total pressure of 100 Torr is very similar to that at 30 Torr (Fig. S2, ESI†), indicating a weak pressure dependence in this pressure range.
As described in the Introduction, the mechanism of this reaction and the rate coefficient have been predicted by Kjaergaard et al.33 and Vereecken et al.35,36 Our measured rate coefficient is more close to that of Vereecken et al., 4 × 10−13 cm3 s−1, which has an uncertainty of at least 1 order of magnitude.35,36 Although O3 is isoelectronic with CH2OO, the self-reaction rate of CH2OO (∼8 × 10−11 cm3 s−1)12 is at least 10 orders of magnitude larger than that of O3. The reaction rate of CH2OO and O3 is somehow in between these two limiting cases. While the zwitterionic character of CH2OO leads to a pure attractive cycloaddition in its self-reaction,13,35 the interaction between CH2OO and O3 would lead to the formation of a pre-reactive complex instead, which has a barrier for either subsequent chain addition or cycloaddition, according to the predictions by Vereecken et al.35,36 A further clarification of the reaction mechanism via either theory or experiment is still required.
In our future work we would like to clarify the reaction mechanism via examining the reaction products. Theoretical works have predicted that CH2O and O2 are the final products of this reaction. We plan to utilize our QCL spectrometer to probe CH2O produced from the reactions, while a new QCL with a spectral range dedicated to detecting CH2O will be required. Vereecken et al.35,36 further predicted that the oxygen atom in the product CH2O is purely from O3, while the two oxygen atoms from CH2OO are released as O2. However, the cycloaddition mechanism predicted by Kjaergaard et al.33 would lead to a 1:1 ratio of oxygen atoms of CH2O originating from CH2OO or O3.36 To verify these predictions, our future works will also include isotopic labelling experiments and the IR identification of isotope-substituted CH2O.
Conclusions
In summary, we investigated the kinetics of CH2OO reaction with O3 at 298 K and 30 Torr using a high resolution mid-infrared quantum cascade laser spectrometer. No pressure dependence was observed from 30 to 100 Torr. Different from other reactant molecules like H2O, SO2, NO2, and organic/inorganic acids, ozone would react with I atoms to form IO, an unavoidable reaction in the current kinetic system. Based on our experimental observations, we suggest that CH2OO should react quickly with IO. Fortunately, the amount of IO is controlled by the initial amount of I atoms under our experimental conditions, where [O3] ≫ [I]. Thus, the kinetics of the CH2OO decay is still pseudo-1st-order and the observed keff is linear to [O3] for [O3] > 0.The measured rate coefficient of the reaction of CH2OO with O3 is (6.72 ± 0.46) × 10−14 cm3 s−1. This value differs from the predicted values in the literature by one order of magnitude or more, suggesting the need of multireference treatment for the quantum chemistry calculations. While the theory has made good predictions or estimations for a number of reaction rates of Criegee intermediates with water, H2S and some other molecules,24 it still seems tricky to calculate the rate of the reaction of CH2OO with O3. We hope the measured results of the present work could benefit future theoretical calculations of this reaction.
This measurement also indicates that the reaction of CH2OO with O3 should play a role in laboratory studies of ozonolysis, where the early-time decay of CH2OO may be controlled by its reactions with O3 and with the used alkene molecules. The rate coefficients of the reactions of CH2OO with simple alkenes have been reported by Buras et al.14 to be 2 × 10−15 to 11 × 10−15 cm3 s−1, which are smaller than that for the O3 reaction. As a result, if one wishes to produce a higher steady-state concentration of CH2OO in ozonolysis experiments, an ozone concentration lower than that of alkene may be desirable to slow down the decay of CH2OO due to its reaction with O3. For an ozonolysis study with 1 ppm of O3, the effective decay rate of CH2OO by O3 is about 2 s−1, which is slightly larger than the thermal decomposition rate of CH2OO (0.2 s−1).42 On the other hand, the reaction of CH2OO with atmospheric ozone is relatively slower (∼0.17 s−1 for 100 ppbv O3) than those with other CH2OO sinks, such as the reaction with water dimers (>1000 s−1),17 in the atmosphere.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Ministry of Science and Technology, Taiwan (MOST103-2113-M-001-019-MY3) and Academia Sinica. We thank Mr Wen Chao for his help with the measurements. We also thank Dr Mica Smith and Dr Kaito Takahashi for discussions about the reaction of CH2OO with IO.Notes and references
- R. Criegee, Angew. Chem., Int. Ed. Engl., 1975, 14, 745 CrossRef.
- C. A. Taatjes, D. E. Shallcross and C. J. Percival, Phys. Chem. Chem. Phys., 2014, 16, 1704 RSC.
- D. L. Osborn and C. A. Taatjes, Int. Rev. Phys. Chem., 2015, 34, 309 CrossRef CAS.
- Y.-P. Lee, J. Chem. Phys., 2015, 143, 020901 CrossRef PubMed.
- O. Welz, J. D. Savee, D. L. Osborn, S. S. Vasu, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Science, 2012, 335, 204 CrossRef CAS PubMed.
- N. M. Donahue, G. T. Drozd, S. A. Epstein, A. A. Presto and J. H. Kroll, Phys. Chem. Chem. Phys., 2011, 13, 10848 RSC.
- A. Novelli, L. Vereecken, J. Lelieveld and H. Harder, Phys. Chem. Chem. Phys., 2014, 16, 19941 RSC.
- F. Liu, J. M. Beames, A. S. Petit, A. B. McCoy and M. I. Lester, Science, 2014, 345, 1596 CrossRef CAS PubMed.
- N. M. Kidwell, H. Li, X. Wang, J. M. Bowman and M. I. Lester, Nat. Chem., 2016, 8, 509 CrossRef CAS PubMed.
- C. A. Taatjes, O. Welz, A. J. Eskola, J. D. Savee, A. M. Scheer, D. E. Shallcross, B. Rotavera, E. P. F. Lee, J. M. Dyke, D. K. W. Mok, D. L. Osborn and C. J. Percival, Science, 2013, 340, 177 CrossRef CAS PubMed.
- L. Sheps, A. M. Scully and K. Au, Phys. Chem. Chem. Phys., 2014, 16, 26701 RSC.
- W.-L. Ting, C.-H. Chang, Y.-F. Lee, H. Matsui, Y.-P. Lee and J. J.-M. Lin, J. Chem. Phys., 2014, 141, 104308 CrossRef PubMed.
- Y.-T. Su, H.-Y. Lin, R. Putikam, H. Matsui, M. C. Lin and Y.-P. Lee, Nat. Chem., 2014, 6, 477 CrossRef CAS PubMed.
- Z. J. Buras, R. M. I. Elsamra, A. Jalan, J. E. Middaugh and W. H. Green, J. Phys. Chem. A, 2014, 118, 1997 CrossRef CAS PubMed.
- D. Stone, M. Blitz, L. Daubney, N. U. M. Howes and P. Seakins, Phys. Chem. Chem. Phys., 2014, 16, 1139 RSC.
- O. Welz, A. J. Eskola, L. Sheps, B. Rotavera, J. D. Savee, A. M. Scheer, D. L. Osborn, D. Lowe, A. Murray Booth, P. Xiao, M. Anwar, H. Khan, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Angew. Chem., Int. Ed., 2014, 53, 4547 CrossRef CAS PubMed.
- W. Chao, J.-T. Hsieh, C.-H. Chang and J. J.-M. Lin, Science, 2015, 347, 751 CrossRef CAS PubMed.
- H.-L. Huang, W. Chao and J. J.-M. Lin, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10857 CrossRef CAS PubMed.
- M. C. Smith, C.-H. Chang, W. Chao, L.-C. Lin, K. Takahashi, K. A. Boering and J. J.-M. Lin, J. Phys. Chem. Lett., 2015, 6, 2708 CrossRef CAS PubMed.
- R. Chhantyal-Pun, A. Davey, D. E. Shallcross, C. J. Percival and A. J. Orr-Ewing, Phys. Chem. Chem. Phys., 2015, 17, 3617 RSC.
- E. S. Foreman, K. M. Kapnas and C. Murray, Angew. Chem., 2016, 128, 10575 CrossRef.
- L.-C. Lin, H.-T. Chang, C.-H. Chang, W. Chao, M. C. Smith, C.-H. Chang, J. Jr-Min Lin and K. Takahashi, Phys. Chem. Chem. Phys., 2016, 18, 4557 RSC.
- M. C. Smith, W. Chao, M. Kumar, J. S. Francisco, K. Takahashi and J. J.-M. Lin, J. Phys. Chem. A, 2017, 121, 938 CrossRef CAS PubMed.
- J. Jr-Min Lin and W. Chao, Chem. Soc. Rev., 2017 10.1039/c7cs00336f.
- L. Sheps, B. Rotavera, A. J. Eskola, D. L. Osborn, C. A. Taatjes, K. Au, D. E. Shallcross, M. A. H. Khan and C. J. Percival, Phys. Chem. Chem. Phys., 2017, 19, 21970 RSC.
- Z. C. J. Decker, K. Au, L. Vereecken and L. Sheps, Phys. Chem. Chem. Phys., 2017, 19, 8541 RSC.
- Y. Liu, F. Liu, S. Liu, D. Dai, W. Dong and X. Yang, Phys. Chem. Chem. Phys., 2017, 19, 20786 RSC.
- C. A. Taatjes, Annu. Rev. Phys. Chem., 2017, 68, 183 CrossRef CAS PubMed.
- R. L. Caravan, M. A. H. Khan, B. Rotavera, E. Papajak, I. O. Antonov, M.-W. Chen, K. Au, W. Chao, D. L. Osborn, J. J.-M. Lin, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Faraday Discuss., 2017, 200, 313 RSC.
- R. Chhantyal-Pun, M. R. McGillen, J. M. Beames, M. A. H. Khan, C. J. Percival, D. E. Shallcross and A. J. Orr-Ewing, Angew. Chem., Int. Ed., 2017, 56, 9044 CrossRef CAS PubMed.
- R. Chhantyal-Pun, O. Welz, J. D. Savee, A. J. Eskola, E. P. F. Lee, L. Blacker, H. R. Hill, M. Ashcroft, M. A. H. Khan, G. C. Lloyd-Jones, L. Evans, B. Rotavera, H. Huang, D. L. Osborn, D. K. W. Mok, J. M. Dyke, D. E. Shallcross, C. J. Percival, A. J. Orr-Ewing and C. A. Taatjes, J. Phys. Chem. A, 2017, 121, 4 CrossRef CAS PubMed.
- J. H. Kroll and J. H. Seinfeld, Atmos. Environ., 2008, 42, 3593 CrossRef CAS.
- H. G. Kjaergaard, T. Kurtén, L. B. Nielsen, S. Jørgensen and P. O. Wennberg, J. Phys. Chem. Lett., 2013, 4, 2525 CrossRef CAS.
- W. Wei, R. Zheng, Y. Pan, Y. Wu, F. Yang and S. Hong, J. Phys. Chem. A, 2014, 118, 1644 CrossRef CAS PubMed.
- L. Vereecken, H. Harder and A. Novelli, Phys. Chem. Chem. Phys., 2014, 16, 4039 RSC.
- L. Vereecken, A. R. Rickard, M. J. Newland and W. J. Bloss, Phys. Chem. Chem. Phys., 2015, 17, 23847 RSC.
- Y.-P. Chang, A. J. Merer, H.-H. Chang, L.-J. Jhang, W. Chao and J. J.-M. Lin, J. Chem. Phys., 2017, 146, 244302 CrossRef PubMed.
- B. Jin, M.-N. Su and J. J.-M. Lin, J. Phys. Chem. A, 2012, 116, 12082 CrossRef CAS PubMed.
- W.-L. Ting, Y.-H. Chen, W. Chao, M. C. Smith and J. J.-M. Lin, Phys. Chem. Chem. Phys., 2014, 16, 10438 RSC.
- J. H. Kroll, J. S. Clarke, N. M. Donahue and J. G. Anderson, J. Phys. Chem. A, 2001, 105, 1554 CrossRef CAS.
- M. E. Tucceri, T. J. Dillon and J. N. Crowley, Phys. Chem. Chem. Phys., 2005, 7, 1657 RSC.
- T. Berndt, R. Kaethner, J. Voigtländer, F. Stratmann, M. Pfeifle, P. Reichle, M. Sipilä, M. Kulmala and M. Olzmann, Phys. Chem. Chem. Phys., 2015, 17, 19862–19873 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp06653h |
| This journal is © the Owner Societies 2018 |