
Stereochemical effects on dynamics in two-component systems of gelators with perfluoroalkyl and alkyl chains as revealed by vibrational circular dichroism†
 *a
Tomoko
Yajima
b
and
*a
Tomoko
Yajima
b
and
Abstract
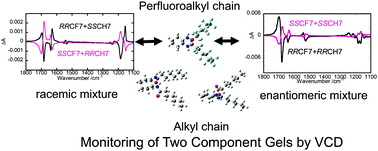
A mixture of chiral low-molecular weight gelators, (1R,2R)- or (1S,2S)-N,N′-diperfluoroheptanoyl-1,2-diaminocyclohexane (denoted as RR- or SS-CF7, respectively) and (1R,2R)- or (1S,2S)-N,N′-diheptanoyl-1,2-diaminocyclohexane (denoted as RR- or SS-CH7, respectively), was used as a two-component gelator in acetonitrile solvent. The process of gelation was investigated by time-step vibrational circular dichroism (VCD) spectroscopy. The method enabled us to study the dynamical behavior of CF7 and CH7 molecules independent of their characteristic vibrational bands. We focused on the effects of the chirality relation between the two gelators. In the case of the enantiomeric mixtures (RR-CF7/RR-CH7 or SS-CF7/SS-CH7), the two components exhibited different time-courses in their VCD spectra. As for CF7, the couplet peak intensities assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching increased with time, while those for CH7 remained low. In the case of the racemic mixture (RR-CF7/SS-CH7 or SS-CF7/RR-CH7), the intensities of the peaks assigned to the CO stretching vibration for both CF7 and CH7 molecules maintained a constant level with time. The VCD results were compared with the SEM images of the freeze-dried gel samples taken at various time intervals. Furthermore, the mechanisms for the gelation of two-component systems are discussed.
O stretching increased with time, while those for CH7 remained low. In the case of the racemic mixture (RR-CF7/SS-CH7 or SS-CF7/RR-CH7), the intensities of the peaks assigned to the CO stretching vibration for both CF7 and CH7 molecules maintained a constant level with time. The VCD results were compared with the SEM images of the freeze-dried gel samples taken at various time intervals. Furthermore, the mechanisms for the gelation of two-component systems are discussed.
- This article is part of the themed collection: Complex molecular systems: supramolecules, biomolecules and interfaces
 Please wait while we load your content...
Please wait while we load your content...

