
Site-specific time-resolved FRET reveals local variations in the unfolding mechanism in an apparently two-state protein unfolding transition†

Abstract
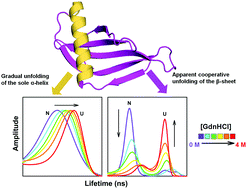
Protein folding/unfolding transitions between the native (N) and unfolded (U) states are usually describable as two-state, only because of the dominant presence of the N and/or U states, because of which high energy intermediates remain undetected. Delineation of the cooperativity underlying these transitions, and characterization of high energy intermediates that are populated sparsely, have been difficult challenges, especially under equilibrium conditions, and require the use of a sensitive probe that reports on both the structures and population distributions of the partially unfolded intermediates. In this study, the use of multisite time-resolved FRET to monitor structural change in five specific segments of the small protein monellin, has brought out local deviations from two-state behavior during unfolding. It is shown that in some segments of the protein structure, denaturant-induced unfolding proceeds first by gradual expansion of the N state, then by an all-or-none transition from the N state ensemble to the U state ensemble, followed finally by expansion of the U state. Segments encompassing the sole helix appear, however, to unfold completely through a gradual transition from the N to U states. Finally, it is shown that equilibrium unfolding of monellin is not only heterogeneous, but that the degree of non-cooperativity differs between the sole α-helix and different parts of the β-sheet.
- This article is part of the themed collection: Complex molecular systems: supramolecules, biomolecules and interfaces
 Please wait while we load your content...
Please wait while we load your content...

