
Molecular details of the unique mechanism of chloride transport by a cyanobacterial rhodopsin†
 a
Mattia
Saita,
b
Alexandra
Hughes-Visentin,
a
Franziska
Pranga-Sellnau,
b
Ana-Nicoleta
Bondar,
c
Joachim
Heberle
*b
and
Leonid S.
Brown
*a
a
Mattia
Saita,
b
Alexandra
Hughes-Visentin,
a
Franziska
Pranga-Sellnau,
b
Ana-Nicoleta
Bondar,
c
Joachim
Heberle
*b
and
Leonid S.
Brown
*a
Abstract
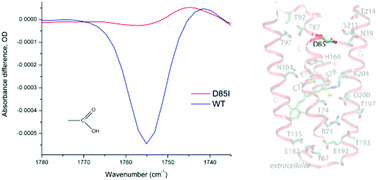
Microbial rhodopsins are well known as versatile and ubiquitous light-driven ion transporters and photosensors. While the proton transport mechanism has been studied in great detail, much less is known about various modes of anion transport. Until recently, only two main groups of light-driven anion pumps were known, archaeal halorhodopsins (HRs) and bacterial chloride pumps (known as ClRs or NTQs). Last year, another group of cyanobacterial anion pumps with a very distinct primary structure was reported. Here, we studied the chloride-transporting photocycle of a representative of this new group, Mastigocladopsis repens rhodopsin (MastR), using time-resolved spectroscopy in the infrared and visible ranges and site-directed mutagenesis. We found that, in accordance with its unique amino acid sequence containing many polar residues in the transmembrane region of the protein, its photocycle features a number of unusual molecular events not known for other anion-pumping rhodopsins. It appears that light-driven chloride ion transfers by MastR are coupled with translocation of protons and water molecules as well as perturbation of several polar sidechains. Of particular interest is transient deprotonation of Asp-85, homologous to the cytoplasmic proton donor of light-driven proton pumps (such as Asp-96 of bacteriorhodopsin), which may serve as a regulatory mechanism.
- This article is part of the themed collection: Complex molecular systems: supramolecules, biomolecules and interfaces
 Please wait while we load your content...
Please wait while we load your content...

