
Exploring the coherent interaction in a hybrid system of hollow gold nanoprisms and cyanine dye J-aggregates: role of plasmon-hybridization mediated local electric-field enhancement†

Abstract
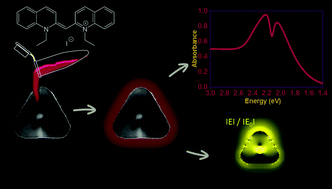
In this work, we probed the possibility of observing strong plasmon–exciton interactions in hollow gold nanoprism–J-aggregate nanocomposites. Several different hollow gold nanoprisms (HGNs) with different aspect ratios were synthesized. This allowed us to systematically tune the LSPR energies through the exciton energy of the PIC–J-aggregate, which in turn allowed us to have direct determination of the coupling strength of HGN–J-aggregate composites. Hybrid nanosystems were prepared by adsorbing and assembling 1,1′-diethyl-2,2′-cyanine (pseudoisocyanine or PIC) iodide onto the surface of hollow gold nanoprisms. Plasmon–exciton interactions were studied using extinction spectroscopy. The experimental results were analysed, and complemented by the results obtained from numerical simulations. Our results reveal that the HGN–PIC–J-aggregate hybrid nanosystem shows coherent coupling between the localized surface plasmons of the HGN and excitons of the PIC–J-aggregate, as obvious from the observation of a clear transparency dip and the formation of two new hybrid plexcitonic modes in the plexcitonic spectra. Anti-crossing behaviour of the plexcitonic modes, together with large Rabi splitting and coupling constant, asserts strong coupling between the plasmon and the exciton, overwhelming the decoherence effects, in our hybrid nanosystem. Analysis of the calculated near-field distribution establishes that the plasmon-hybridization mediated large electric-field enhancement holds the key to the strong coupling.
- This article is part of the themed collection: 2017 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

