
The mechanism of cesium ions immobilization in the nanometer channel of calcium silicate hydrate: a molecular dynamics study

Abstract
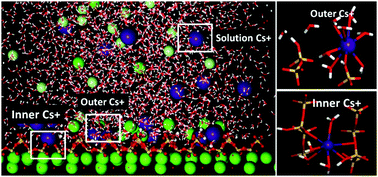
The cement-based matrices are preferred candidates in disposing nuclear waste due to the immobilization role of the calcium–silicate–hydrate (C–S–H) gel. To better understand the immobilization mechanism of cementitious materials, molecular dynamics was utilized to investigate the intensity distribution, local structure and dynamics properties of Cs+ ions in the vicinity of the calcium silicate surface. The strong inner-sphere adsorbed cesium ions were restricted by coordinated oxygen atoms in bridging and pair silicate tetrahedron and water molecules were fixed in the silicate channel by H-bonds network. On the other hand, the adsorption of chloride ion, repulsed by the negatively charged silicate surface, is mainly attributed to the formation of the cation–anion ionic pair near the interface. As compared with those of the solvated ions in the solution, the relaxation time of water in the hydration shell of adsorbed Cs+ is significantly increased and the diffusion coefficient of adsorbed Cs+ is dramatically reduced. Furthermore, based on the intensity profile and resident-time analysis, the adsorption capacities of monovalent cations on the C–S–H surface increase with decrease in the ionic radius, following the sequence of Na+ ≫ K+ > Cs+. This study provides a molecular-level understanding of the immobilization mechanism of different ions in the C–S–H gel pores.
- This article is part of the themed collection: 2017 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

