
Interaction with prefibrillar species and amyloid-like fibrils changes the stiffness of lipid bilayers†

Abstract
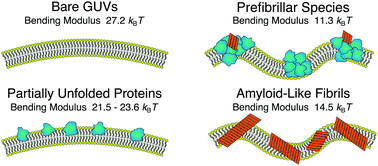
Evaluating the toxicity of self-assembled protein states is a key step towards developing effective strategies against amyloidogenic pathologies such as Alzheimer's and Parkinson's diseases. Such analysis is directly connected to quantitatively probing the stability of the cellular membrane upon interaction with different protein states. Using a combination of spectroscopic techniques, morphological observations, and spectral analysis of membrane fluctuations, we identify different destabilisation routes for giant unilamellar vesicles interacting with native-like states, prefibrillar species and amyloid-like fibrils of α-lactalbumin. These effects range from substantially lowering the bending rigidity of the membranes to irreversible structural changes and complete disruption of the lipid bilayers. Our findings clearly indicate how the wide heterogeneity in structures occurring during protein aggregation can result in different destabilisation pathways, acting on different length scales and not limited to enhanced membrane permeability.
- This article is part of the themed collection: 2017 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

