
Intriguing transport dynamics of ethylammonium nitrate–acetonitrile binary mixtures arising from nano-inhomogeneity†
Alessandro
Mariani,  ‡*a
Matteo
Bonomo,
a
Boning
Wu,
b
Danilo
Dini,
a
Edward W.
Castner, Jr.
b
and
Lorenzo
Gontrani
*a
‡*a
Matteo
Bonomo,
a
Boning
Wu,
b
Danilo
Dini,
a
Edward W.
Castner, Jr.
b
and
Lorenzo
Gontrani
*a
‡*a
Matteo
Bonomo,
a
Boning
Wu,
b
Danilo
Dini,
a
Edward W.
Castner, Jr.
b
and
Lorenzo
Gontrani
*a
Abstract
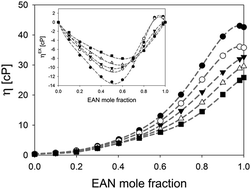
Binary mixtures of ethylammonium nitrate and acetonitrile show interesting properties that originate from the structural and dynamical nano-heterogeneity present in ionic liquids. These effects are most pronounced when the ionic liquid is the minority compound. In this study the transport properties of such mixtures are studied, including viscosity, self-diffusion and conductivity. The results strongly support the presence of structural inhomogeneity and show an interesting composition-dependent behaviour in the mixtures.
- This article is part of the themed collection: 2017 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

