
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Protonation of N2O and NO2 in a solid phase†
Evgenii S.
Stoyanov
 *ab and
Irina V.
Stoyanova
a
*ab and
Irina V.
Stoyanova
a
aVorozhtsov Institute of Organic Chemistry, Siberian Branch of Russian Academy of Sciences, Novosibirsk 630090, Russia. E-mail: evgenii@nioch.nsc.ru
bDepartment of Natural Sciences, National Research University – Novosibirsk State University, Novosibirsk 630090, Russia
First published on 23rd November 2017
Abstract
Adsorption of gaseous N2O on the acidic surface Brønsted centers of the strongest known solid acid, H(CHB11F11), results in formation of the N≡N–OH+ cation. Its positive charge is localized mainly to the H-atom, which is H-bonded to the CHB11F11− anion forming an asymmetric proton disolvate of the L1–H+⋯L2 type, where L1 = N2O and L2 = CHB11F11−. NO2 protonation under the same conditions leads to the formation of the highly reactive cation radical NO2H˙+, which reacts rapidly with an NO2 molecule according to the equation N2OH+ + NO2 → [N2O4H+] → N2OH+ + O2 resulting in the formation of two types of N2OH+ cations: (i) a typical Brønsted superacid, N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N–OH+, with a strongly acidic OH group involved in a rather strong H-bond with the anion, and (ii) a typical strong Lewis acid, NN+–OH, with a positive charge localized to the central N atom and ionic interactions with the surrounding anions via the charged central N atom.
N–OH+, with a strongly acidic OH group involved in a rather strong H-bond with the anion, and (ii) a typical strong Lewis acid, NN+–OH, with a positive charge localized to the central N atom and ionic interactions with the surrounding anions via the charged central N atom.
Introduction
Weakly basic small molecules H2, N2, O2, CO2, CO, N2O, NO2 are interesting targets for protonation. No evidence has been obtained for the protonation of these simple gaseous molecules under ambient conditions in a “magic” superacid system, HSO3F–SbF5–SO3,1 one of the strongest known mixed Brønsted/Lewis acids. Nonetheless, a somewhat stronger HF/SbF5 mixed acid system can protonate CO, when it is dissolved, but under conditions of high pressure (up to 85 atm).2,3 The corresponding salt is not isolable. Using a newly synthesized strongest solid superacid, H(CHB11F11),4 we have been able to protonate CO under ambient conditions both through the C atom and via the O atom and to obtain in preparative quantities bulk salts of the H+CO and COH+ cations.3 Therefore, the H(CHB11F11) acid manifests itself as stronger than the “magic” acids, and is expected to protonate (under ambient conditions) other, less basic than CO, molecules such as N2O and NO2, which still cannot be protonated in a condensed phase. In a gas phase, the protonation of small molecules was proved by mass spectroscopy5–10 and IR spectroscopy.11–13 For example, N2O is protonated via the O atom, and the frequency of the O–H+ stretch was found to be 3331 cm−1 for the gas phase,12,13 and 43.3 cm−1 lower for the Ne matrix.14 The experimental difficulties with the protonation of the simple molecules are compensated so far by the research in this field on the basis of quantum-chemical calculations,15–24 that confirmed that the O-protonated isomer (NNOH+) is energetically more preferable (by 4.02 kcal mol−1)24 than the N-protonated isomer.Recently, it was reported25 that CO2 is protonated by H(CHB11F11) with the formation of a stable (at room temperature) salt of the symmetric disolvate OCO–H+–OCO. This seems unexpected because CO, being more basic than CO2, forms under the same conditions only salts of the L1–H+⋯L2 type cations with an asymmetric bridged proton, where L1 = CO and L2 = CHB11F11− anion.3 The basicity of CO is not sufficient to substitute L2 for the formation of symmetric OC–H+–CO. It is interesting to test whether other weakly basic molecules, N2O or NO2, can form the symmetric disolvates L–H+–L. Moreover, the carborane salts of the protonated nitrogen oxides must have superacidic properties, as salts of the COH+ cation,3 and can serve as the reagents for obtaining new types of functionalized carbocations.
In the present work, we studied the protonation of N2O and NO2 by the strongest known solid carborane superacid, H(CHB11F11) (Fig. 1), using the methods of infrared (IR) spectroscopy and quantum chemistry.
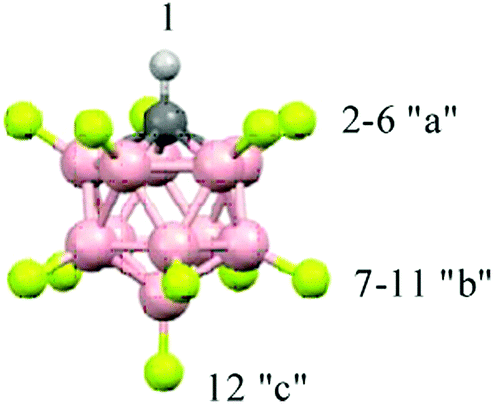 | ||
| Fig. 1 Icosahedral carborane anions, CHB11F11−, with the numbering of three types of F atoms differing in basicity. | ||
Experimental
Carborane acid H(CHB11F11) hereafter abbreviated as H{F11} was prepared as previously described.4 IR spectroscopic analysis of the interaction of N2O or NO2 with H{F11} was performed as follows. The solid acid was sublimed at 150–160 °C under a pressure of 10−5 Torr on cold Si windows in a specially designed IR cell reactor, forming a very thin translucent film of the amorphous acid.3 Dry gaseous N2O or NO2 (obtained from Sigma Aldrich, 99% purity), was injected anaerobically into the IR cell inside a dry box and reacted with the acid at room temperature. IR spectra were recorded at certain time intervals. Weighable amounts of the N2OH+ {F11−} salt were obtained by aging of a portion of H{F11} for 1 day in a Schleng tube filled with N2O.All procedures were performed in a Spectro-systems glovebox under an atmosphere of Ar (H2O < 1 ppm). The IR spectra were recorded on an Bruker Vector 22 spectrometer inside a dry box in either transmission or attenuated total reflectance (ATR) mode (525–4000 cm−1). The IR data were processed with the GRAMMS/A1 (7.00) software from Thermo Fisher Scientific.
Computational details
The geometric parameters of the species under study were optimized at the B3LYP-D3/def2-TZVPD level of theory26–29 with an ultrafine grid. The equilibrium structures of the specific compounds were also calculated in a dichloroethane (DCE) solution using the SMD solvation model.30 All stationary points were characterized as minima by a vibrational analysis (the number of imaginary frequencies was equal to zero). Zero-point energies were computed from the corresponding vibrational frequencies without scaling factors. (SMD-)B3LYP-D3/def2-TZVPD optimized structures were used in all subsequent computations.To compare the calculated and experimental vibrational frequencies, the (SMD-)B3LYP-D3/def2-TZVPD harmonic frequencies were scaled by the factor of 0.9674 as recommended by Kesharwani et al.31 Although application of the scaling factor to the highly anharmonic NH and OH stretch vibrations requires caution: a high-accuracy ab initio anharmonic force field study of N2OH+ showed23 that for the purposes of this work, the above mentioned scaling of the harmonic frequencies is reasonable. To obtain more accurate relative energies of some isomers, single-point high-level CCSD(T)/def2-TZVPD coupled-cluster computations32 within a frozen core approximation were additionally conducted.
The natural population analysis partial charges33,34 were calculated at the (SMD-)B3LYP-D3/def2-TZVPD theoretical level for the species of interest as implemented in Gaussian09,35 whereas natural resonance theory36–38 analysis was carried out at the B3LYP/TZ2P level of theory using scalar relativistic (SR) zero-order regular approximation Hamiltonian (core potentials were not used, and the quality of the Becke numerical integration grid was set to the keyword good)39 in the ADF2016 software suite.40–42
(SMD-)B3LYP-D3/def2-TZVPD and CCSD(T)/def2-TZVPD computations were performed using the Gaussian09 software.35 The def2-TZVPD basis sets were retrieved from the EMSL database.43,44
All the compounds were assumed to be in their ground state. The spin-unrestricted formalism was used for both density functional theory (DFT) and CCSD(T) calculations when computing radicals.
Results of calculations
The gas phase B3LYP-D3/def2-TZVPD calculations of {F11}-containing compounds do not fully describe the ionic interactions taking place in the solid state, leading to a bad agreement between some calculated and experimental vibrational frequency values of the cations (Fig. S1–S3, ESI†). For this reason, we performed calculations for the N2OH+⋯L and NO2H+⋯L model systems, where L = Ar, Kr, Xe, CO, or SO2. A wide range of L basicities, which includes the basicity of the {F11−} anion, allows us to interpret the experimental IR spectra more correctly (Fig. S4, ESI†). To model the effect of the environment playing an important role in crystals, we also conducted SMD-B3LYP-D3/def2-TZVPD calculations in a DCE solution for the compounds of interest (Fig. S5, ESI†).(N2O)H+ cation
Protonation of N2O is possible via terminal N and O atoms (Fig. S1, ESI†). The O-protonated structure is more stable (the energy difference between the O–H+ and N–H+ isomers is 3.9 kcal mol−1 at the CCSD[T]/def2-TZVPD//B3LYP-D3/def2-TZVPD level of theory; Fig. S1, ESI†), which is in line with other experimental12 and theoretical studies.17–19,22–24N2O described by the N-oxide valence formula, NN+–O−(Fig. S6, ESI†), has two valence frequencies (Table S1, ESI†), νasN2O at 2268 cm−1 and νsN2O at 1285 cm−1, which can be represented as the characteristic vibrations of the NN and NO stretches, respectively. The bent vibration is at 598 cm−1. Protonation of N2O leads to formation of the NN–OH+ cation (Fig. S7a, ESI†), with a significant decrease in the NO stretch (∼250 cm−1) and an increase in the NN stretch (by ∼90 cm−1; Table S2, ESI†) because the N–O and N ≡ N bonds approach the common single and triple bond respectively.
According to vibrational analysis, the solvation of N2OH+ by Ar, Kr, or Xe weakened the OH bond and strengthened the NO bond (Table S2, ESI†). The dependence of the νNO frequency (reflecting the N–O strength) on the proton affinity (PA) of the noble gases (L) is linear (Fig. 2) confirming that the N–O stretch is highly characteristic because the (N2O)H+–L bond is ionic. Nevertheless, the analogous dependences for νOH and νNN frequencies deviate from the linear function to a greater extent with the greater basicity of L because a decrease in the frequency of OH+ stretch and an increase in the frequency of the NN stretch result in their convergence with an enhancement of their interaction. This mixing becomes more notable when N2OH+ is solvated by the stronger bases, CO, SO2, and {F11−}, which formed a partially covalent bond with a cation. This situation leads to the formation of a rather asymmetric proton disolvate of the L1–H+⋯L2 type (L1 = N2O; L2 = SO2, {F11−}) with a further increase in the frequency of the N–O stretch (Table S2, ESI†).
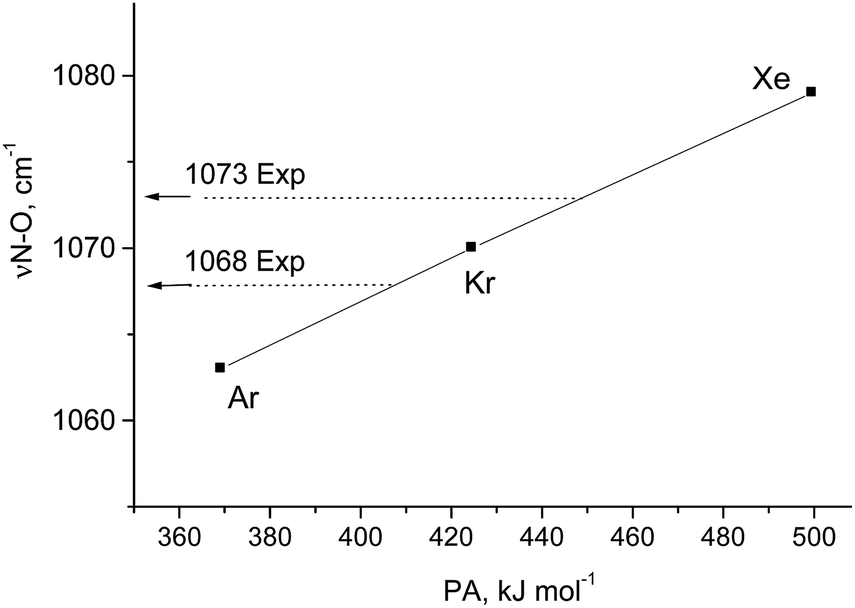 | ||
| Fig. 2 Dependence of the N–O stretch of N2O–H⋯L on the PA of L atoms. Experimental frequencies are given for comparison. | ||
(NO2)H+ cation
The NO2 molecule has two stretch vibrations, νasNO2 and νsNO2, with a frequency difference Δ = 295 cm−1 (Table S1, ESI†). After protonation, the radical cation NO2H+ is formed (I.B.1 structure; Fig. S2, ESI†) having NO and NO(H) stretches with increased and decreased frequencies, respectively, as compared to NO2 (Table S3, ESI†). Their difference Δ increased to 727 cm−1 indicating that the stretching vibrations acquire some characteristic nature. The solvation of (NO2)H+ with Ar decreases both NO stretches keeping their difference Δ actually unchanged. When the cation is solvated with stronger bases, Kr and Xe, the O–H+ bond continues to weaken (νOH+ decreases), thus strengthening the N–O(H+) bond and its frequency (Table S3, ESI†). With a further increase in the basicity of L (CO, SO2), H+ of (NO2)–H+⋯L became a typical bridged proton with stretch frequencies of 1500–1100 cm−1.(N2O4)H+ cation
Gaseous NO2 is always in equilibrium with N2O4. Accordingly, the protonation of NO2 may be accompanied by the protonation of N2O4. The latter is unstable, which is related to the large N–N bond length of 1.78 Å. After protonation, the optimized structure of N2O4H+ (Ic10 in Fig. S4, ESI†) showed a significant increase in the N–N distance (2.247 Å), which precluded its formation. Solvation of the N2O4H+ cation with such bases as Ar, CO, SO2, or {F11−}, reduced the N-N distance down to 2.179, 2.079, 2.023, and 1.909 Å respectively, but it was still big enough for the cation to exist. Calculations predicted that unstable N2O4H+⋯L can decompose in the simplest way into a (HNO3·NO+)⋯L compound (Fig. S4, S5 and Tables S4, S5, ESI†). In any case, it is expected that the protonation of N2O4 will lead to subsequent secondary reactions.Experimental results
N2O interaction with the H{F11} acid
After injection of N2O into the IR cell-reactor with the sublimed H{F11} acid, we started to register the IR spectra immediately. Subtraction from these spectra of the spectrum of gaseous N2O revealed a weak band at 2231 cm−1, which is very close to the νNN band at 2224 cm−1 of gaseous N2O, but without the fine vibrational structure (Fig. 3). Obviously, this pattern denotes N2O molecules absorbed by the acidic surface Brønsted centers of solid H{F11}. After vacuum removal of the gaseous N2O, the band at 2231 cm−1 persisted, but after fast heating up to 100 °C in a sealed vacuumed cell, this band disappeared and a weak spectrum of gaseous N2O appeared. Therefore, N2O molecules are indeed adsorbed to the surface Brønsted centers of the H{F11} acid, and this sorption is relatively strong. | ||
| Fig. 3 IR spectra of the compounds formed when H{F11} is aged under an atmosphere of N2O for 2 min (black solid and dashed curves), 1 h (blue curve) and 10 h (red curve). The spectra are shown before (dashed curve) and after subtraction (black solid curve) of the spectrum of gaseous N2O. The red spectrum was registered after vacuum removal of the gaseous phase. | ||
With time, in the IR spectra, two narrow bands of NN stretches of N2OH+ cations appeared and grew in intensity: at 2363 cm−1 and at 2320 cm−1 (Fig. 3). They belong to cations that bind to the most basic “b” and “c” sites of the {F11−} anion (hereinafter referred to as N2OH+b and N2OH+c), as is the case for H+CO binding to {F11−}.3 Simultaneously, the absorption corresponding to the free (unreacted) H{F11} acid decreases and after 8–10 h disappears (judging by the indicative band at 1616 cm−1 of the stretch vibration of the bridged proton in polymeric acid [H{F11}]n, Fig. 3). In the low-frequency region of the N–O(H+) stretches, weak complex bands appeared at 1068 and 1073 cm−1, which correspond to N2OH+c and N2OH+b, respectively (Fig. 5).
Fig. 4 shows the intensity dependences of the bands of the NN stretches from N2OH+b and N2OH+c (ANN at 2321 or 2364 cm−1 respectively) on the absorption intensity of the acid being used (an intensity decrease at 1616 cm−1, A1616). The figure shows that the formation of the N2OH+b and N2OH+c cations initially increased proportionally. Then, the filling of the most basic “c” site of {F11−} reached saturation, while the filling of the “b” site continued.
 | ||
| Fig. 4 Dependences of the intensity of the bands of νNN from cations N2OHb+ (at 2321 cm−1) and N2OHc+ (at 2364 cm−1) on the absorption of the acid being used (is equal to the decrease in intensity of the free acid at 1616 cm−1). | ||
The absorption corresponding to the O–H+ stretch is expected to be very broad and cannot be detected with certainty. To detect it, we used the following method. After completion of the reaction, gaseous N2O was removed by evacuation. The cell was sealed and heated to 100 °C for 5 min. The IR spectrum shows the emergence of a weak spectrum of gaseous N2O from the desorbed N2O. It should be noted that the broad band of the bridged proton of the free H{F11} acid at 1616 cm−1 appeared as well. That is, reaction (1) of N2OH+ decomposition takes place.
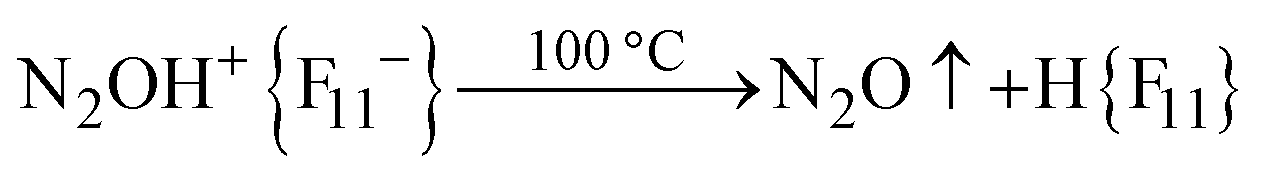 | (1) |
Partial decomposition of N2OH+ should decrease the absorption corresponding to the O–H+ stretch. This allows us, by calculating the difference in the spectra before and after heating, to detect the absorption corresponding to the O–H+ stretch with positive intensity, whereas the absorption corresponding to the H-vibrations of H{F11} will have negative intensity. As shown in Fig. 5 and Fig. S10 (in ESI†), ν(O–H+) emerges as a broad band at ca. 2000 cm−1.
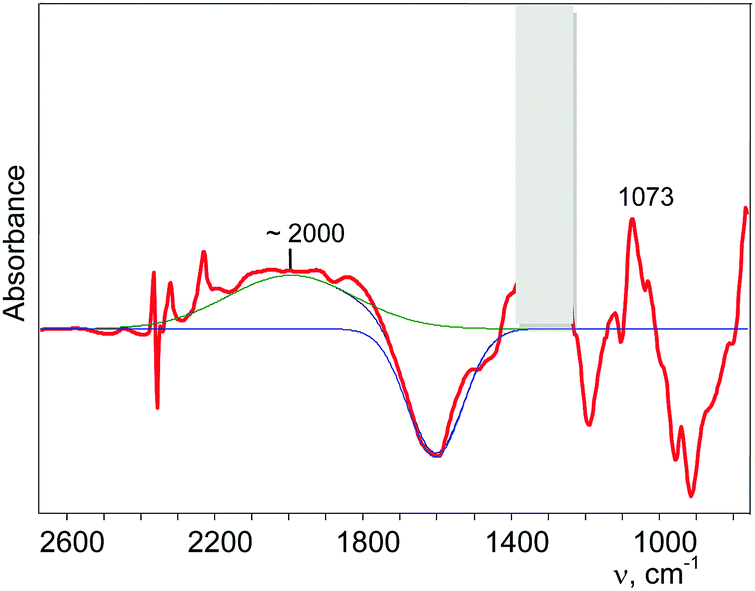 | ||
| Fig. 5 Determination of the band of the O–H+ stretch of the N2OH+ cation (for details see Fig. S10 in ESI†). | ||
Interaction of NO2 with the H{F11} acid
Interaction of gaseous NO2 with a thin film of the amorphous acid on the Si windows of the IR cell reactor results in a decreasing absorption band of the bridged proton of the H{F11} acid at 1616 cm−1 and the appearance of new bands of the formed compounds (Fig. 6). The reaction finished after ca. 3 h. | ||
| Fig. 6 IR spectra of the reaction products of a gas mixture NO2 + N2O4 with the H{F11} acid. Reaction times are 4 min (green, black curves), 3 h (purple curve) and 24 h (red curve). Spectra are shown without (green curve) and with digital subtraction (full for N2O and partial for N2O4) of the spectrum of the gaseous mixture (dark purple curve). The red spectrum was registered after vacuum removal of the gaseous mixture. νasNO2 of NO2 is marked with *, and ν9 of N2O4 is marked with **.45 The spectrum of H3O+{F11−} in the frequency region of OH stretches is given for comparison (dotted brown curve). | ||
IR spectra of the resulting products do not contain bands of the NO2H+⋯L type compounds predicted by calculations (Table S3, ESI†) but show bands of the stretch vibrations in the frequency region of 2300–2400 cm−1 belonging to the other compounds. One of these bands at 2364 cm−1 coincides exactly with that of N2OH+c. Moreover, as the reaction of NO2 with H{F11} proceeded, the band of the NN stretch at 2223 cm−1 of gaseous N2O emerged and increased in intensity (Fig. 6, inset). The dependence of the intensity of the band at 2364 cm−1 on that of 2223 cm−1 is strictly proportional (Fig. 7); this result confirmed the joint formation of N2OH+c and N2O in the course of the secondary reactions that take place between the initially formed NO2Hc+ and gaseous NO2.
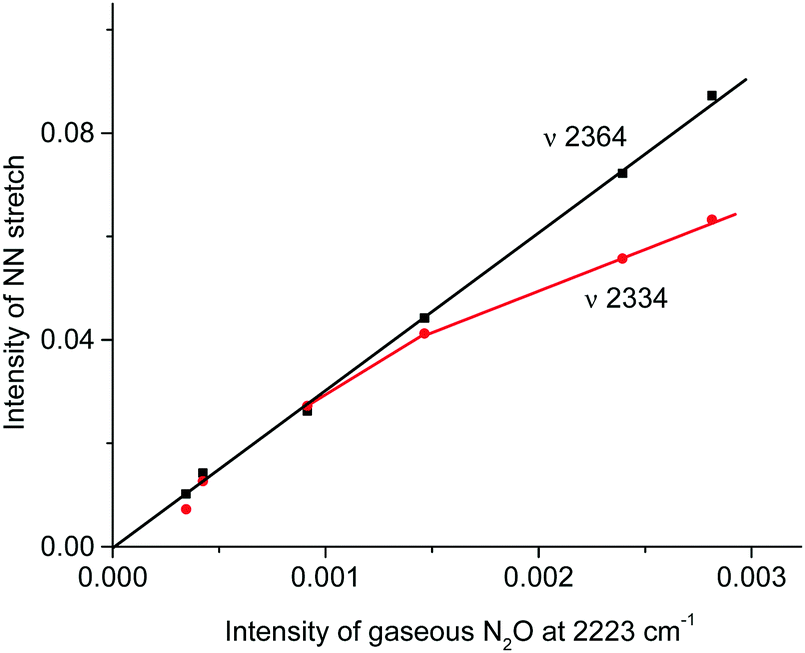 | ||
| Fig. 7 Dependences of the intensity of the NN stretch of the N2OH+ cation on the intensity of the band at 2223 cm−1 of the formed gaseous N2O. | ||
A distinctive band of the second compound at 2334 cm−1 is typical in terms of frequency for the NN stretch of the N2OH+ cation but did not coincide with that of N2OH+b discussed above. The character of changes in the intensity of this band is manifested in a certain relation with another band in the spectra at 3560 cm−1 (Fig. 6) which, without a doubt, belongs to OH stretch vibrations (IR spectra did not show the bands from the OH stretches of the H3O+ cation4). The dependence of the intensity of the band at 2334 cm−1 on that of νOH at 3560 cm−1 is directly proportional (Fig. 8), which means that they belong to one compound. A sample of this compound was obtained when a powder of the H{F11} acid (precipitated in liquid HCl during its synthesis) was aged in an atmosphere of NO2. At a low partial pressure of NO2 (ca. 0.2 atm) presumably N2OH+c is formed, whereas at a higher partial pressure (ca. 0.8 atm) the second compound mainly is formed (Fig. 9a). The only new band detected in the spectrum of the second compound is at 1045 cm−1, which is common for the N–O(H) stretch frequency (Fig. 9b and Table S2, ESI†).
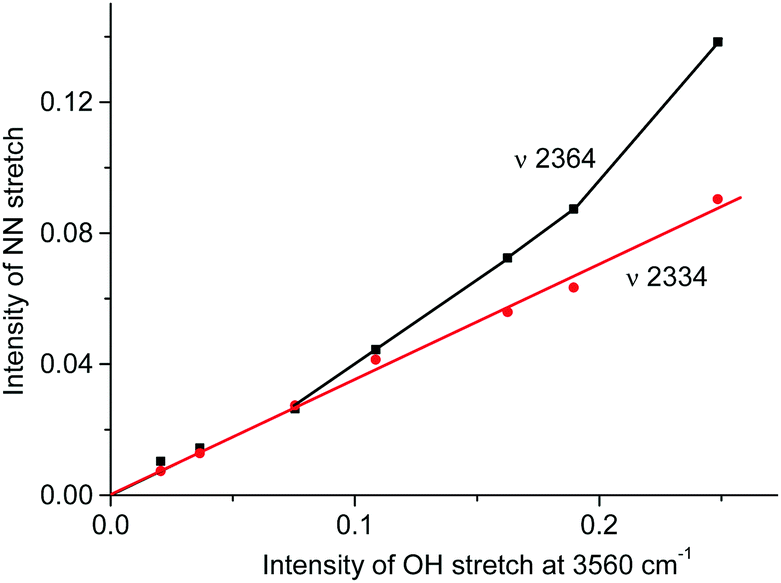 | ||
| Fig. 8 Dependences of intensity of the NN stretch of the N2OH+ cation on the intensity of the band of the OH stretch at 3560 cm−1. | ||
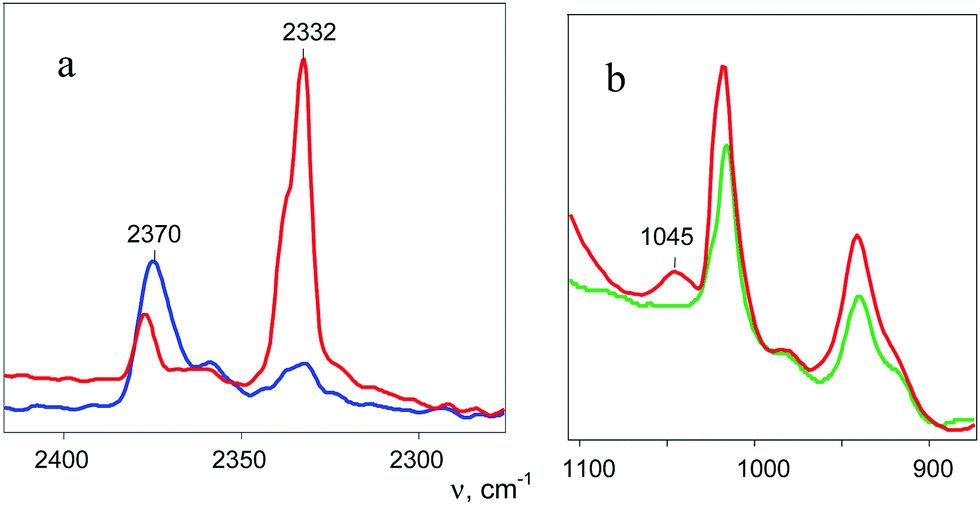 | ||
| Fig. 9 ATR IR spectra of N2OH+c{F11−} (blue curve) and N2OHfree+{F11−} (red curve) in comparison with the spectrum of the salt Cs{F11} (green curve), which allowed identification of the N–O(H+) stretch at 1045 cm−1 for N2OHfree+. | ||
This sample was placed on the bottom of the IR cell reactor, and after addition of a drop of water, the cell was sealed. The IR spectra registered the appearance of the absorption pattern of gaseous N2O. Thus, the second compound is the salt of the N2OH+ cation with a free OH bond (further denoted as N2OHfree+), which is decomposed by water with N2O elimination.
Finally, the third band in the frequency range of the NN stretch appears at 2307 cm−1 after long aging of the sample under an atmosphere of NO2 (more than 24 h, Fig. 6). We propose that it emerges due to water vapor penetration. We verified this idea by introducing water vapor into the IR cell along with the N2OH+{F11−} salt and observed rapid disappearance of the bands of the NN stretches of the N2OH+c and N2OHfree+ cations, but the band at 2307 cm−1 persisted and even increased in intensity (Fig. S11 in ESI†). This finding indirectly confirms the affiliation of this band with the hydrated species.
Discussion
Comparison of the empirical spectra of N2OH+b and N2OH+c cations (Table 1) with the calculated spectra (Table S2, ESI†) shows that these cations belong to the L1–H+⋯L2 type, where L1 = N2O and L2 = {F11−} anion with “b” and “c” basic sites. These empirical spectra show the greatest congruence with those calculated for the N2OH+⋯Kr and N2OH+⋯Xe solvates (Table S2, ESI†); in particular, the νN–O frequency almost coincides with that of N2OH+⋯Kr (Fig. 2). This result implies that an “effective” PA of {F11−} in the solid N2OH+{F11−} salt is close to that of the Kr atom. Moreover, the positive charge and electron density redistribution over the NN–O group of the N2OH+⋯Kr cation, as well as the geometric parameters of these groups, determined by means of calculations, should be very close to those of N2OH+b and N2OH+c. Thus, these cations can be described as having the NNO angle close to 180° with the triple NN (ca. 1.001 Å) and single N–O(H+) bonds (ca. 1.252 Å) in accordance with the N oxide valence formula NN+–OH. On the other hand, the O–H stretch at ca. 2000 cm−1 indicates strong H-bonding with the {F11−} anion having the positive charge mainly on the H atom (Scheme 1).
| Cation | νOH | νNN | νNO | δNOH |
|---|---|---|---|---|
| a Not identified. | ||||
| N2OHb+ | ∼2000 | 2321 | 1073 | |
| N2OHc+ | ∼2000 | 2364 | 1060 | |
| N2OHfree+ | 3560 | 2334 | 1045 | 1698 |
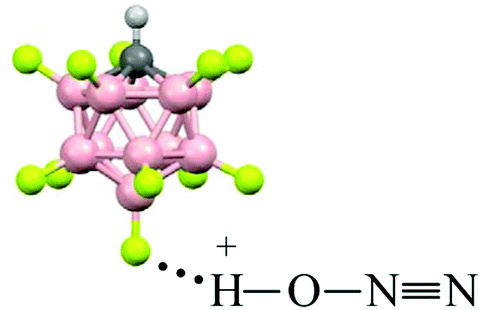 | ||
| Scheme 1 Representative structure of the N2OH+c cation in its salt with the {F11−} anion. | ||
Attempts to protonate NO2 led to an unexpected result: the spectrum of the cation radical NO2H˙+ predicted by calculations (Table S3, ESI†) is not registered, but the spectrum of N2OH+ cations appeared. Obviously, there is a rapid transition from NO2H+ to the N2OH+ cation, which can only take place viareaction (2)
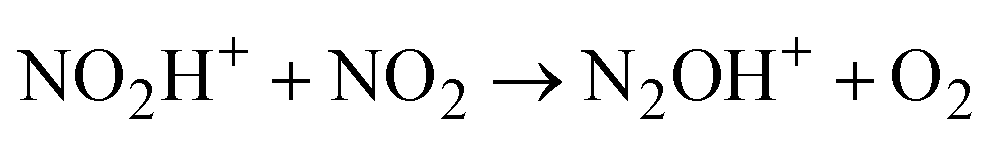 | (2) |
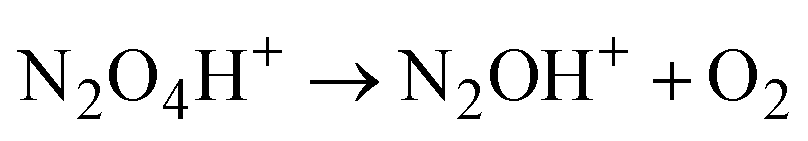 | (3) |
It was a surprise that one of the two N2OH+ cations formed in reactions (2) or (3) is N2OHfree+ with a free OH group, which is not H-bonded to the {F11−} anion. This means that N2OHfree+ exactly matches the N oxide valence formula NN+–OH with the positive charge located on the central N atom. The stretch O–H frequency of N2OHfree+ is higher (3560 cm−1) than that of the free cation in vacuum, both calculated (3332 cm−1) and empirically determined (3331 cm−1).12,13 This means that the interaction of N2OHfree+ with the neighboring {F11−} anions is purely ionic and proceeds via the N atom (Scheme 2) leading to polarization of the OH group and an increase in its stretch frequency so much that it even exceeds the value corresponding to naked N2OH+ in vacuum. Thus, N2OHfree+ is an unusual representative of a pure Lewis acid with a covalent OH group.
 | ||
| Scheme 2 Schematic presentation of N2OHfree+ in its salt with the {F11−} anion. | ||
Cation radical NO2H˙+, formed in the first step of NO2 protonation with H{F11}, according to calculations (Table S5, ESI†), is stable. Nevertheless, as experiments showed, it has high reactivity and quickly reacts with the next NO2 molecule (eqn (2)) forming an unstable intermediate, N2O4H+. According to calculations, the instability of N2O4H+ is caused by extension of the N–N bond up to 2.247 Å. The simplest route of its decomposition is formation of the HNO3·NO+ solvate (Fig. S4, S5 and Table S5 in ESI†). In contrast, the experiment shows that decomposition of N2O4H proceeds viaeqn (3) to N2OH+ + O2.
The fact that there is a proportionality between the amount of the formed N2OH+c and gaseous N2O (Fig. 2) indicates the existence of an equilibrium 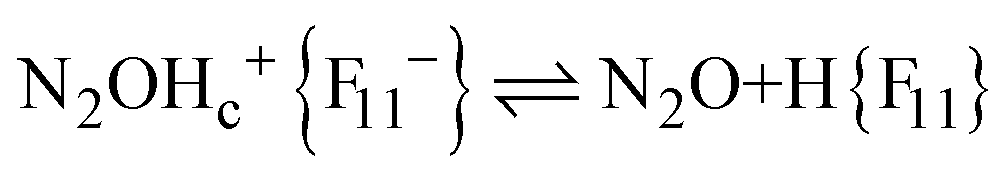 , which, however, is absent between N2OHfree+ and gaseous N2O. That is, N2OHfree+ is formed only through N2O4H+ decomposition.
, which, however, is absent between N2OHfree+ and gaseous N2O. That is, N2OHfree+ is formed only through N2O4H+ decomposition.
Conclusion
Gaseous N2O and NO2 are protonated under ambient conditions with the strongest known solid superacid, H{F11}. N2O is attached to the H atom of the polymeric (H{F11})n acid at the first stage via physical adsorption without breaking the bridge H-bond and proton transfer to the N2O molecule (Scheme 3, left). The adsorbed N2O molecules are not desorbed in vacuum at room temperature, but are easily detached at elevated temperatures. | ||
| Scheme 3 Illustration of N2O physically adsorbed by the surface Brønsted centers of the H{F11} acid (left), followed by the proton transfer to N2O. | ||
The second stage is the breakage of the –{F11}–H–{F11}– hydrogen bridge and proton transfer to the O atom of N2O (Scheme 3). The formed N2OH+ cation retains a fairly strong H bond with the {F11−} anion, attaching to its “b” or “c” site (Scheme 1). This compound can be regarded as an asymmetric proton disolvate, L1–H+⋯L2, with L1 = N2O and L2 = counterion.
Adsorption of NO2 on the surface of the H{F11} acid did not reveal the IR absorption pattern of physically adsorbed NO2 or protonated NO2 because the cation radical NO2H˙+, which obviously must be formed, has high reactivity. It quickly interacts with NO2 forming an unstable intermediate, N2O4H+, which decomposes (eqn (3)) forming two types of N2OH+ cations. The first one is N2OHc+ with a common H-bonding to the {F11−} ion. The second one, N2+OHfree, is unusual in that it has a free non-acid OH group with a positive charge localized to the central N atom, which enters into an ionic interaction with anions in the environment (Scheme 2). Thus, if the first N2OHc+ cation is a typical Brønsted superacid, then the second cation, N2+OHfree, is a strong Lewis acid that is formed only as a result of a chemical reaction, but not as a result of the sorption or desorption interaction.
The present work shows that N2O, just as CO studied earlier, during protonation by the H{F11} acid, cannot form a symmetric proton disolvate of the L–H+–L type in the solid phase. For this reason, the results of another article [ref. 25]—claiming that CO2, less basic than either N2O or CO, forms the stable salt of the proton disolvate under ambient conditions—are questionable, especially because the supporting experimental evidence is not convincing.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant # 16-13-10151 from the Russian Science Foundation. The authors thank Dr Anton S. Nizovtsev for providing the quantum chemical calculations. The synthesis of H(CHB11F11) acid was supported by the Russian Foundation for Basic Research (grant # 16-03-00357).References
- R. J. Gillespie and G. P. Pez, Inorg. Chem., 1969, 8, 1233 CrossRef CAS.
- P. J. F. Rege, J. A. Gladysz and I. T. Horvath, Science, 1997, 276, 776 CrossRef PubMed.
- E. S. Stoyanov and S. A. Malykhin, Phys. Chem. Chem. Phys., 2016, 18, 4871 RSC.
- M. Nava, I. V. Stoyanova, S. Cummings, E. S. Stoyanov and C. A. Reed, Angew. Chem., Int. Ed., 2014, 53, 1131 CrossRef CAS PubMed.
- F. H. Field and J. L. Franklin, J. Am. Chem. Soc., 1961, 83, 4509 CrossRef CAS.
- J. A. Burt, J. L. Dunn, M. J. McEwan, M. M. Sutton, A. E. Roche and H. I. Schiff, J. Chem. Phys., 1970, 52, 6062 CrossRef CAS.
- S. S. Jr, J. K. Kim, L. P. Theard and W. T. Huntress Jr., Chem. Phys. Lett., 1975, 32, 610 CrossRef.
- F. C. Fehsenfeld, W. Lindinger, H. I. Schiff, R. S. Hemsworth and D. K. Bohme, J. Chem. Phys., 1976, 64, 4887 CrossRef CAS.
- K. Hiraoka, T. Shoda, K. Morise, S. Yamabe, E. Kawai and K. Hirao, J. Chem. Phys., 1986, 84, 2091 CrossRef CAS.
- M. Polášek, M. Kaczorovwska and J. Hrušák, Chem. Phys. Lett., 2005, 402, 138 CrossRef.
- C. F. Neese, P. S. Kreynin and T. Oka, J. Phys. Chem. A, 2013, 117, 9899 CrossRef CAS PubMed and references therein.
- T. Amano, Chem. Phys. Lett., 1986, 130, 154 CrossRef CAS.
- T. Amano, Chem. Phys. Lett., 1986, 127, 101 CrossRef CAS.
- M. E. Jacox and W. E. Thompson, J. Chem. Phys., 2005, 123, 064501 CrossRef PubMed.
- U. Seeger, R. Seeger, J. A. Pople and P. v. R. Schleyer, Chem. Phys. Lett., 1978, 55, 399 CrossRef CAS.
- D. Sengupta, R. Sumathi and S. D. Peyerimhoff, Chem. Phys., 1999, 248, 147 CrossRef CAS.
- J. E. Del Bene, E. A. Stahlberg and I. Shavitt, Int. J. Quantum Chem., Quantum Chem. Symp., 1990, 24, 455 CrossRef CAS.
- K. Yamashita and K. Morokuma, Chem. Phys. Lett., 1986, 131, 237 CrossRef CAS.
- J. E. Del Bene and M. J. Frisch, Int. J. Quantum Chem., Quantum Chem. Symp., 1989, 23, 371 CAS.
- G. M. Chaban, N. M. Klimenko and O. P. Charkin, Izv. Akad. Nauk SSSR, Ser. Khim., 1992, 1, 126 Search PubMed.
- J. Grunenberg, R. Streubel, G. Frantzius and W. Marten, J. Phys. Chem., 2003, 119, 165 CrossRef CAS.
- J. L. M. Martin and T. J. Lee, J. Chem. Phys., 1993, 98, 7951 CrossRef CAS.
- X. Huang, R. C. Fortenberry and T. J. Lee, J. Chem. Phys., 2013, 139, 084313 CrossRef PubMed.
- M. C. McCarthy, O. Martinez, K. N. Crabtree, V. Lattanzi, S. E. Novick and S. Thorwirth, J. Phys. Chem. A, 2013, 117, 9968 CrossRef CAS PubMed.
- S. Cummings, H. P. Hratchian and C. A. Reed, Angew. Chem., Int. Ed., 2016, 55, 1382 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- B. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- M. K. Kesharwani, B. Brauer and J. M. L. Martin, J. Phys. Chem. A, 2015, 119, 1701 CrossRef CAS PubMed.
- G. D. Purvis III and R. J. Bartlett, J. Chem. Phys., 1982, 76, 1910 CrossRef.
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Peterssonet al., Gaussian 09, Revision D.01, Gaussian Inc., Wallingford, CT, 2013 Search PubMed.
- E. D. Glendening and F. Weinhold, J. Comput. Chem., 1998, 19, 593 CrossRef CAS.
- E. D. Glendening and F. Weinhold, J. Comput. Chem., 1998, 19, 610 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop and F. Weinhold, J. Comput. Chem., 1998, 19, 628 CrossRef CAS.
- E. van Lenthe, E.-J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597 CrossRef CAS.
- ADF2016, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com.
- C. F. Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391 CAS.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931 CrossRef CAS.
- D. Feller, J. Comput. Chem., 1996, 17, 1571 CrossRef CAS.
- K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045 CrossRef CAS PubMed.
- R. V. St. Louis and B. Crawford Jr., J. Chem. Phys., 1965, 42, 857 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp04474g |
| This journal is © the Owner Societies 2017 |