
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Indirect NMR detection of transient guanosyl radical protonation in neutral aqueous solution†
O. B.
Morozova
ab,
N. N.
Fishman
ab and
A. V.
Yurkovskaya
 *ab
*ab
aInternational Tomography Center, Institutskaya 3a, 630090 Novosibirsk, Russia. E-mail: yurk@tomo.nsc.ru
bNovosibirsk State University, Pirogova 2, 630090 Novosibirsk, Russia
First published on 20th July 2017
Abstract
By using the time-resolved chemically induced dynamic nuclear polarization technique, we show that the neutral guanosyl radical, G(-H)˙, formed in the reaction of guanosine-5′-monophosphate with a triplet-excited 3,3′,4,4′-tetracarboxy benzophenone in neutral aqueous solution, protonates readily at the N7 position with the formation of a new guanosyl cation radical (G˙+)′.
The first studies on DNA photoreactions were started more than 50 years ago. Up to now a large amount of information about basic DNA photochemistry, in model systems as well as in isolated and cellular DNA, has been accumulated. Now it is well known that UV light can interact with DNA either by direct absorption or via photosensitization by endogenous or exogenous chromophores present in drugs, cosmetic agents, metabolites, etc.1,2 Photosensitizers cause certain types of DNA damage according to the properties of their excited states and their location in the cell, including reactions with photosensitizers under UV light, leading to the formation of short-lived radicals of easily oxidized nucleotides.1 Guanine is the main target of one-electron oxidation reactions, as it has the lowest oxidation potential among all DNA components.3 Upon one-electron oxidation, guanine is converted into a cation radical (G˙+). The guanine cation radical can deprotonate to form the neutral guanine radical G(-H)˙,4 and these two radicals are involved in the subsequent processes of pathological DNA damage.5 Therefore, there have been numerous publications on the formation and behavior of guanine radicals both as a nucleotide and as part of single strand and duplex DNA.6,7 However, despite continuous studies on guanine radical intermediates, there is still no common opinion on the structure and reactivity of these particles.
Recently Choi et al. using time-resolved resonance Raman spectroscopy combined with pulse radiolysis have proposed a new guanine cation radical species (G˙+)′ that results from protonation at the N7 position of the neutral guanine radical G(-H)˙.8 The authors reported that this reprotonation reaction rapidly occurs in neutral aqueous solution at a rate constant of 8.1 × 106 s−1. The common guanine cation radical G˙+ protonated at the N1 position has a pKa value of 3.9,9 and is not expected to be formed in neutral aqueous solution. The work by Choi et al. initiated a lively discussion about the possibility of formation of a new guanine cation radical (G˙+)′ and its structure.10 This hypothesis was surprisingly suitable for an explanation of the unusual behavior of the kinetics of CIDNP (chemically induced dynamic nuclear polarization) detected by us during the photoreaction of guanosine-5′-monophosphate (GMP) with the photosensitizer, 3,3′,4,4′-tetracarboxy benzophenone (TCBP). The unusual behavior was a change in sign of CIDNP for both reaction participants on the microsecond timescale, which was an indication of structural changes of at least one of the short-lived radicals formed in the photochemical reaction. In the present study, we approached the question of this CIDNP sign change from the perspective of the protonation reaction suggested by Choi et al. The term CIDNP means non-equilibrium nuclear spin-state populations produced in chemical reactions that involve radical pair intermediates. These are detected as enhanced absorptive or emissive signals in the NMR spectra of diamagnetic products of radical reactions. The amplitude and sign of the CIDNP signal depend on the magnetic parameters of the radicals (g-factors and HFC constants) and therefore allow determination of the radical structure.11 CIDNP is a time-dependent effect. CIDNP is formed in short-lived radical pairs and is maintained for the time of diamagnetic relaxation in the reaction products, the signals of which are detected by NMR. The time-resolved version of the CIDNP method (TR-CIDNP) allows us to easily separate the contributions from geminate and bulk processes and to determine the rate constants of the radical reactions.11 By using TR-CIDNP it is possible to follow the kinetics of radical transformations: when the magnetic resonance parameters of the secondary radical do not coincide with those of the primary one, the detected CIDNP kinetics differs from that obtained under conditions when no radical transformation takes place. This method is very helpful when radical reactions are not accompanied by changes in the optical absorption spectra, or when short-lived radicals have low absorption coefficients, or when radical absorption spectra are overlapped, or when EPR techniques are not applicable due to the low concentrations and short lifetimes of the radicals under study in aqueous solution at room temperature.
In the present work, CIDNP effects were detected in neutral aqueous solution during the cyclic photoreaction, including the photochemical formation of guanosyl radicals in the quenching reaction of the triplet-excited TCBP by GMP (see structures in Chart 1) and the subsequent radical termination which restores the initial compounds. We used GMP instead of dGMP that was used by Choi et al. However, since the structural changes studied here are associated with the guanyl base, we believe that the choice of the RNA nucleotide instead of the DNA one did not affect the obtained results. The choice of the dye is based on the fact that benzophenone (BP) is the classical model chromophore for the study of different photosensitized damages in nucleosides, oligonucleotides, and DNA.12 BP shows a high oxidizing ability and is able to oxidize all nucleobases.13 However, an essential drawback of using non-substituted BP for studying photo-induced electron transfer reactions is the poor solubility of BP in water. Introduction of hydrophilic substituents (e.g., carboxylic groups) into BP increases its solubility in water, and hence allows one to study the photooxidation reaction of DNA and its components in aqueous solution. We checked four commercially available water-soluble benzophenone derivatives for CIDNP in photoreactions with GMP, which are 3- and 4-carboxy benzophenones, 4,4′-dicarboxy benzophenone, and TCBP; only in the case of TCBP, the reaction of GMP with the triplet-excited dye was highly reversible and did not lead to the formation of byproducts.
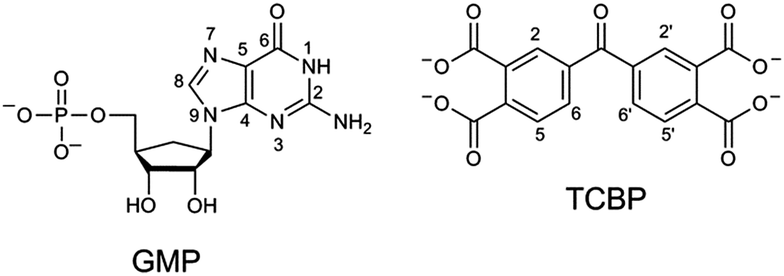 | ||
| Chart 1 Structures of the compounds studied. | ||
The photochemical reaction between the triplet excited TCBP and GMP in a wide pH range was investigated by us earlier.14 According to these findings, at neutral pH the reaction between the fully deprotonated 3TCBP and neutral guanosine proceeds via proton coupled electron transfer (PCET) and leads to the formation of a ketyl radical of TCBP (TCBPH˙) and a neutral guanosyl radical (G(-H)˙), which recombine giving rise to geminate CIDNP. The geminate reactions are as follows:
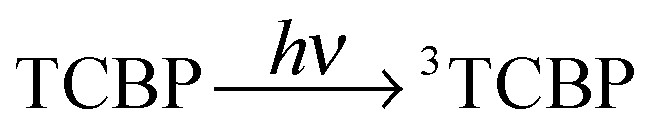 | (1) |
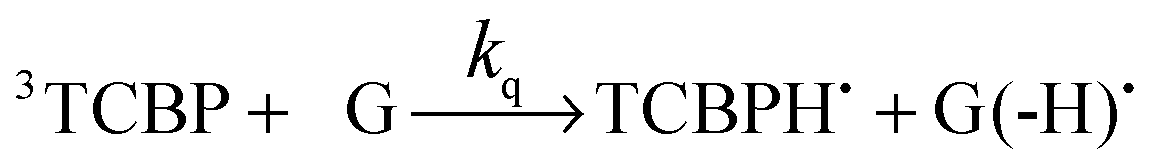 | (2) |
| TCBPH˙ + G(-H)˙ → TCBP + G | (3) |
In eqn (1)–(3), G stands for the guanyl base of GMP. Fig. 1 shows 1H CIDNP spectra at 400 MHz obtained in the photoreaction of TCBP and GMP in 50 mM sodium phosphate buffer at pH 6.9 at variable time delays after the laser pulse increases from 0 to 100 μs. The CIDNP sign, Γ, is determined in terms of simple CIDNP rules,15i.e., Γ = sgn(Δg) × sgn(A), for the geminate recombination product and for the triplet precursor of the radical pair. In this case, Δg = g1 − g2 is the difference in the g-factor of the radicals, where index 1 refers to the radical where the observed nucleus with the hyperfine coupling constant (HFCC) A is located, while index 2 refers to the partner radical. By this definition, the sign of Δg is opposite for the polarization of nuclei in the partner radical. NMR signals in the upper spectrum (detected without delay after the laser pulse) correspond to polarization formed in the geminate recombination processes, whereas the lower spectra (detected at 1–100 μs after the laser pulse) show the CIDNP formed in the termination of freely diffusing radical pairs (F-pairs) in bulk. For F-pairs, the CIDNP sign stays the same as for G-pairs with a triplet precursor.15 The lower spectrum enhanced absorption (A) is observed for H8 of GMP and for H5,5′ of TCBP (very small in the latter case) while emission (E) is observed for H2,2′ and H6,6′ of TCBP (Fig. 1a). In this case, the polarization signs correspond to signals originating from the pair consisting of a TCBP radical (g = 2.0035,16AH2,2′ < 0, AH6,6′ < 0, AH5,5′ > 017), and a neutral guanosyl radical (g = 2.0034, AH8 < 07). In the spectra that correspond to a delay of 3 μs and more after the laser pulse, the CIDNP signs for both GMP and TCBP are inverted: emission (E) for H8 of GMP and for H5,5′ of TCBP while enhanced absorption (A) is observed for H2,2′ and H6,6′ of TCBP. The reason for this CIDNP sign change can be a change in the sign of either Δg or HFCC. The inversion of the CIDNP sign for both reactants unambiguously indicates that the sign of Δg for freely diffusing radical pairs (F-pairs) is opposite to that for the geminate pairs.
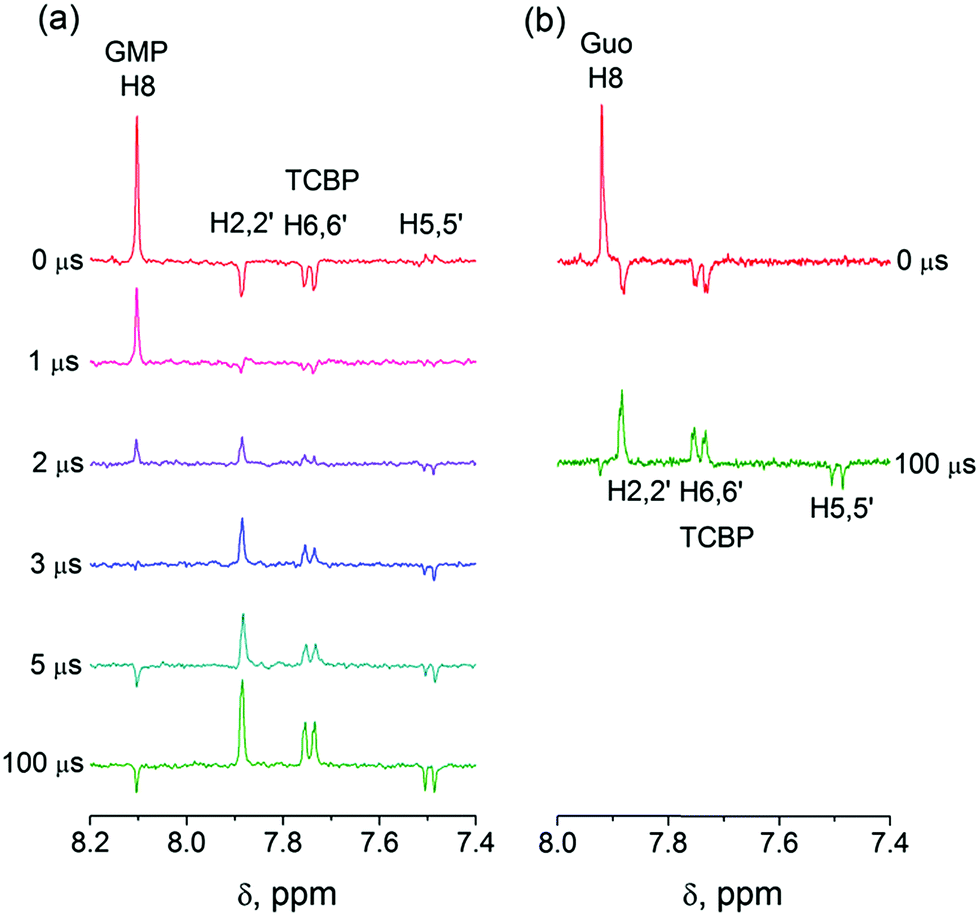 | ||
| Fig. 1 (a) 1H CIDNP spectra, detected in the photoreaction of 2 mM 3,3′,4,4′-tertracarboxy benzophenone and 20 mM guanosine-5′-monophosphate in 50 mM sodium phosphate buffer at pH 6.9 at variable time delays after the laser pulse. The delays are ascending from 0 (top) to 100 μs (bottom spectrum). (b) 1H CIDNP spectra detected in the photoreaction of 2 mM 3,3′,4,4′-tertracarboxy benzophenone and 2 mM guanosine in 50 mM sodium phosphate buffer at pH 6.9. The upper spectrum was taken immediately after the laser pulse, and the lower spectrum at 100 μs after the laser pulse. | ||
To make sure that the detected CIDNP sign inversion is associated with the structural change of the purine base but not of the phosphate group, we checked CIDNP effects in the photoreaction of TCBP and the nucleoside guanosine (Guo, Fig. 1b). The results are qualitatively the same: CIDNP spectra obtained in the photoreaction of Guo with the triplet-excited TCBP have opposite phases of the corresponding signals at time delays of 0 and 100 μs after the laser pulse. This result confirms that the CIDNP sign change is caused by a change in the radical structure of the purine base, but not by the protonation of the phosphate group.
Fig. 2 shows the CIDNP kinetics which is determined by structural changes resulting in Δg sign inversion. The larger amplitude of polarization at the opposite sign gained by TCBP in comparison with GMP is a consequence of the longer nuclear relaxation time of H6,6′ in the TCBPH˙ radical than that of H8 in the guanosyl radical (see ESI† for details). We propose the following explanation for the sign change in Δg. Since the reaction is fully reversible (no signals of products other than the starting compounds are observed in the CIDNP spectra), the only possible structural change responsible for the inversion of the Δg sign is a change in the protonation state of the radicals during protonation or deprotonation. The g-factor values are known to be noticeably different for guanosyl radicals in various protonation states: 2.0037 for the guanosyl cation radical protonated at the N1 position, 2.0034 for the neutral guanosyl radical, and 2.0036 for the guanosyl anion radical.7 The g-factor of the TCBP radical (gTCBP) is 2.0035.16 As observed in our previous investigation, the geminate CIDNP originating from pairs consisting of a TCBP radical and a guanosyl radical changed its sign twice upon pH variation from 1.3 to 13.2.14
The neutral guanosyl radical with pKa = 10.8 is stable on the timescale of 100 μs18 and deprotonates only in a highly basic solution in the reaction with OH−.14 Thus, we exclude deprotonation of the neutral guanosyl radical as the origin of the CIDNP sign change. The protonation at the N1 position with the formation of a guanosyl cation radical having pKa = 3.99 is thermodynamically unfavorable in neutral aqueous solution. The alternative is the protonation at the N7 position as suggested by Choi et al.8 It is reasonable to assume that the guanosyl radical protonated at the N7 position, as that protonated at the N1 position, has a g-factor, gC, higher than the g-factor of the neutral guanosyl radical, gN, and the g-factors increase in the sequence gN < gTCBP < gC, causing the CIDNP sign change in time.
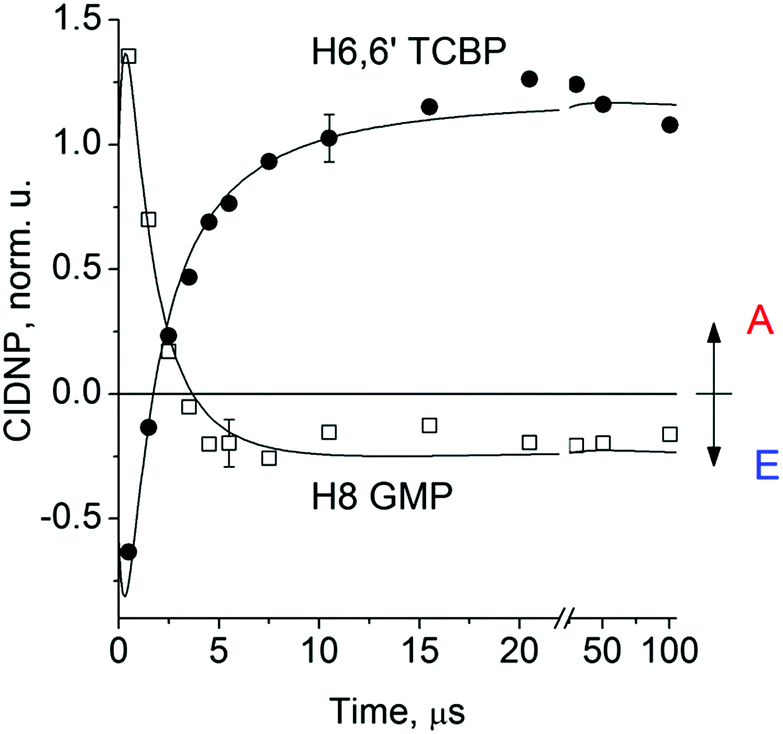 | ||
| Fig. 2 1H CIDNP kinetics, obtained by the photoreaction of 2 mM 3,3′,4,4′-tetracarboxy benzophenone and 20 mM guanosine-5′-monophosphate in 50 mM sodium phosphate buffer at pH 6.9: squares – for H8 of GMP and circles – for H6,6′ of TCBP. A and E denote enhanced absorption and emission of the NMR signals, respectively. | ||
Thus, the scheme of the bulk reactions is as follows:
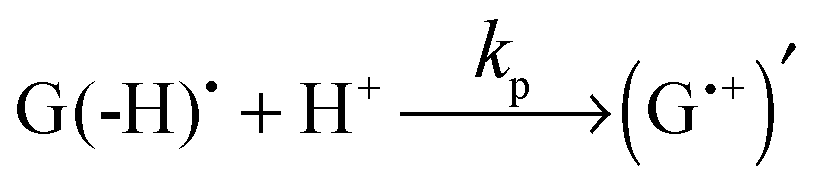 | (4) |
| TCBPH˙ + G(-H)˙ → TCBP + G | (5) |
| TCBPH˙ + (G˙+)′ → TCBP + G + H+ | (6) |
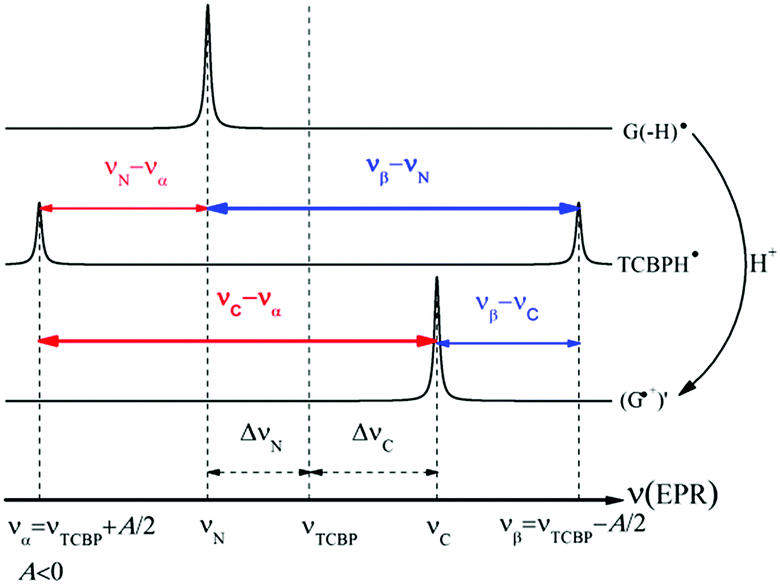 | ||
| Scheme 1 Schematic representation of triplet–singlet interconversion based on the EPR spectra of G(-H)˙ (top), TCBPH˙ (middle), and (G˙+)′ (bottom). Only a single spin-1/2 nucleus in TCBPH˙ with negative HFCC is considered. | ||
The CIDNP signs for all nuclei resulting from spin evolution of the two types of radical pairs are shown in Scheme 2. Thus, our observations are in agreement with the findings of Choi et al. who demonstrated that the neutral guanosyl radical G(-H)˙ is converted to the protonated guanosyl radical (G˙+)′ at a rate constant of 8.1 × 106 s−1 in 100 mM sodium phosphate buffer at pH 7.4.8 Based on the simulation of the CIDNP kinetics (see ESI†), the observed rate constant of protonation of G(-H)˙ was determined to be kp = (1.8 ± 0.4) × 106 s−1. This rate constant is of the same order of magnitude as reported by Choi,8 but is too low for quantitative agreement even if we take into account a two-fold decrease in rate constant due to the deuterium isotope effect.19 However, the experimental conditions of the present study and those from ref. 8 do not completely match, making a quantitative comparison impossible here.
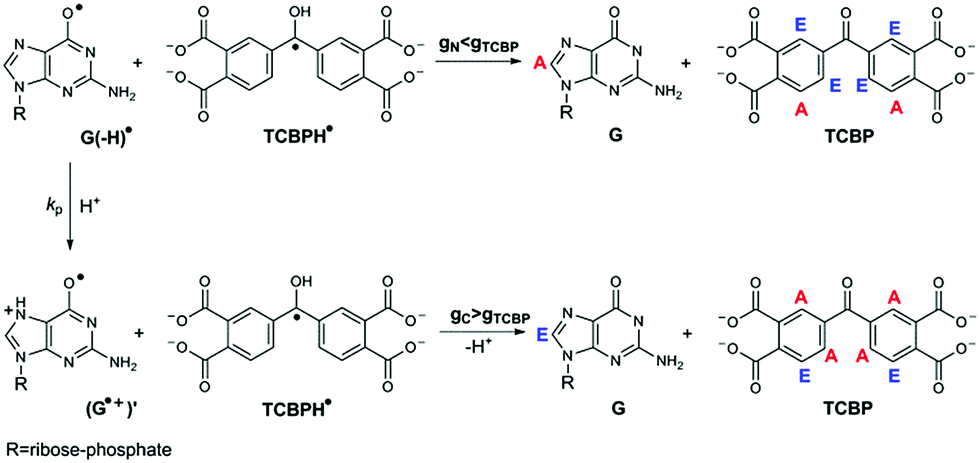 | ||
| Scheme 2 Protonation of the G(-H)˙ radical as a source of CIDNP sign change in products of reactions of guanosyl and TCBP radicals. The g-factors of the radicals G(-H)˙, TCBPH˙, and (G˙+)′ are denoted as gN, gTCBP, and gC, respectively. A and E denote enhanced absorption and emission of NMR signals for the corresponding protons, respectively. | ||
Conclusions
In conclusion, by using the TR CIDNP technique we have obtained irrefutable evidence for the protonation of the neutral guanosyl radical G(-H)˙ in neutral aqueous solution. The G(-H)˙ radical conversion and the formation of the secondary guanosyl cation radical (G˙+)′ are manifested in the inversion of the CIDNP sign for the TCBP and GMP protons in the course of the reaction indicating a change in the magnetic resonance parameters of the radical pair due to structural changes in the guanosyl radical. It is important to note that using only TCBP as a photosensitizer we could follow the protonation process by detecting the CIDNP sign change: the g-factor of the TCBP radical favorably lies in between the g-factors of the two types of guanosyl radicals, thus providing an indispensable piece of CIDNP evidence for the reaction under study. It should be noted that the recent work by Wasielewski et al. in which authors have observed the formation of the G cation radical in DNA hairpins linked with diphenylacetylenedicarboxamide20 corroborates the results presented here. Our work not only strongly supports Choi's findings but also allows us to give an estimate of the g-factor of the guanosyl radical protonated at N7 as being larger than the g-factor of the partner TCBP radical (2.0035) and being nearly equal to that of the guanosyl radical protonated at N1 (gC = 2.0037). At present, studies under systematic variation of experimental conditions aimed at determination of the pKa value of the elusive guanosyl radical protonated at N7 are in progress in our laboratory.Acknowledgements
This work was supported by the Russian Science Foundation (project number 15-13-20035). The authors acknowledge FASO Russia (project 0333-2016-0001) for the financing of the NMR facility.Notes and references
- J. Cadet, S. Mouret, J. L. Ravanat and T. Douki, Photochem. Photobiol., 2012, 88, 1048–1065 CrossRef CAS PubMed; J. Cadet and J. R. Wagner, Cold Spring Harbor Perspect. Biol., 2013, 5, 1–18 CrossRef PubMed.
- J. Cadet, T. Douki and J.-L. Ravanat, Photochem. Photobiol., 2015, 91, 140–155 CrossRef CAS PubMed.
- S. Steenken and S. V. Jovanovic, J. Am. Chem. Soc., 1997, 119, 617–618 CrossRef CAS.
- S. Steenken, J. P. Telo, H. M. Novais and L. P. Candeias, J. Am. Chem. Soc., 1992, 114, 4701–4709 CrossRef CAS.
- G. Pratviel and B. Meunier, Chem. – Eur. J., 2006, 12, 6018–6030 CrossRef CAS PubMed; W. L. Neeley and J. M. Essigmann, Chem. Res. Toxicol., 2006, 19, 491–505 CrossRef PubMed; J. R. Milligan, J. A. Aguilera, O. Hoang, A. Ly, N. Q. Tran and J. F. Ward, J. Am. Chem. Soc., 2004, 126, 1682–1687 CrossRef PubMed.
- D. Khanduri, A. Adhikary and M. D. Sevilla, J. Am. Chem. Soc., 2011, 133, 4527–4537 CrossRef CAS PubMed; A. W. Parker, C. Y. Lin, M. W. George, M. Towrie and M. K. Kuimova, J. Phys. Chem. B, 2010, 114, 3660–3667 CrossRef PubMed.
- A. Adhikary, A. Kumar, D. Becker and M. D. Sevilla, J. Phys. Chem. B, 2006, 110, 24171–24180 CrossRef CAS PubMed.
- J. Choi, C. Yang, M. Fujitsuka, S. Tojo, H. Ihee and T. Majima, J. Phys. Chem. Lett., 2015, 6, 5045–5050 CrossRef CAS PubMed.
- S. Steenken, Chem. Rev., 1989, 89, 503–520 CrossRef CAS.
- J. Choi, C. Yang, M. Fujitsuka, S. Tojo, H. Ihee and T. Majima, J. Phys. Chem. B, 2016, 120, 2987–2989 CrossRef CAS; M. D. Sevilla, A. Kumar and A. Adhikary, J. Phys. Chem. B, 2016, 120, 2984–2986 CrossRef PubMed.
- A. Yurkovskaya, O. Morozova and G. Gescheidt, Structures and Reactivity of Radicals Followed by Magnetic Resonance, Encyclopedia of Radicals in Chemistry, Biology and Materials, 2012 Search PubMed.
- M. C. Cuquerella, V. Lhiaubet-Vallet, J. Cadet and M. A. Miranda, Acc. Chem. Res., 2012, 45, 1558–1570 CrossRef CAS PubMed.
- T. Douki and J. Cadet, Int. J. Radiat. Biol., 1999, 75, 571–581 CrossRef CAS PubMed.
- N. N. Saprygina, O. B. Morozova, T. V. Abramova, G. Grampp and A. V. Yurkovskaya, J. Phys. Chem. A, 2014, 118, 4966–4974 CrossRef CAS PubMed.
- R. Kaptein, J. Chem. Soc., Chem. Commun., 1971, 14, 732–735 RSC.
- J. Säuberlich, O. Brede and D. Beckert, J. Phys. Chem., 1996, 100, 18101–18107 CrossRef.
- O. B. Morozova, K. L. Ivanov, A. S. Kiryutin, R. Z. Sagdeev, T. Köchling, H.-M. Vieth and A. V. Yurkovskaya, Phys. Chem. Chem. Phys., 2011, 13, 6619–6627 RSC.
- A. V. Yurkovskaya, O. A. Snytnikova, O. B. Morozova, Y. P. Tsentalovich and R. Z. Sagdeev, Phys. Chem. Chem. Phys., 2003, 5, 3653–3659 RSC.
- O. B. Morozova, A. V. Yurkovskaya, Y. P. Tsentalovich, M. D. E. Forbes and R. Z. Sagdeev, J. Phys. Chem. B, 2002, 106, 1455–1460 CrossRef CAS.
- M. A. Harris, A. K. Mishra, R. M. Young, K. E. Brown, M. R. Wasielewski and F. D. Lewis, J. Am. Chem. Soc., 2016, 138, 5491–5494 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods and sample preparation; simulation of CIDNP kinetics (PDF). See DOI: 10.1039/c7cp03797j |
| This journal is © the Owner Societies 2017 |