
Fragmentation of pure and hydrated clusters of 5Br-uracil by low energy carbon ions: observation of hydrated fragments†
M. C.
Castrovilli,  a
P.
Bolognesi,
a
P.
Rousseau,
b
S.
Maclot,
b
A.
Domaracka,
b
J.
Kočišek,
bd
L.
Avaldi
*a
a
P.
Bolognesi,
a
P.
Rousseau,
b
S.
Maclot,
b
A.
Domaracka,
b
J.
Kočišek,
bd
L.
Avaldi
*a
a
P.
Bolognesi,
a
P.
Rousseau,
b
S.
Maclot,
b
A.
Domaracka,
b
J.
Kočišek,
bd
L.
Avaldi
*a
Abstract
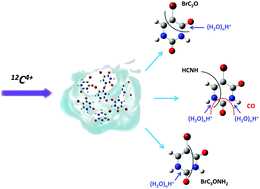
The fragmentation of the isolated 5-bromouracil (5BrU) molecule and pure and nano-hydrated 5BrU clusters induced by low energy 12C4+ ions has been studied. A comparison indicates that the environment, on the one hand, protects the system against the complete break-up into small fragments, but, on the other hand, triggers ‘new’ pathways for fragmentation, for example the loss of the OH group. The most striking result is the observation of several series of hydrated fragments in the hydrated cluster case, with water molecules bound to hydrophilic sites of 5BrU. This highlights the strong interaction between 5BrU and water molecules and the blocking of specific fragmentation pathways, such as the loss of the BrC2H group for example.
- This article is part of the themed collections: 2017 PCCP HOT Articles and XUV/X-ray light and fast ions for ultrafast chemistry
 Please wait while we load your content...
Please wait while we load your content...

