
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe dimer-approach to characterize opto-electronic properties of and exciton trapping and diffusion in organic semiconductor aggregates and crystals
Bernd
Engels
 * and
Volker
Engel
*
* and
Volker
Engel
*
Universität Würzburg, Institut für Physikalische und Theoretische Chemie, Am Hubland, Campus Nord, Emil-Fischer-Str. 42, 97074 Würzburg, Germany. E-mail: bernd.engels@uni-wuerzburg.de; volker.engel@uni-wuerzburg.de; Fax: +49 9313185331; Tel: +49 931 3185394 Tel: +49 931 3186376
First published on 4th May 2017
Abstract
A fundamental understanding of photo-induced processes in opto-electronic thin film devices is a prerequisite for the rational design of improved organic semiconductor materials. Absorption and emission spectra provide important insights into the complicated electronic structure of and relaxation processes in organic semiconductor aggregates and crystals. They are of interest because they often limit the efficiencies of the devices. For an assignment of the spectra a close interplay between experiment and theory is essential because simulations are often necessary to entangle the various effects which determine the features of the spectra. In the present perspective we describe the so called dimer-approach and provide a few examples in which this approach could successfully deliver an atomistic picture of photo-induced relaxation effects in perylene-based materials and characterize their optical spectra. The model Hamiltonians of standard monomer-based approaches are also briefly discussed to reveal the differences between both methods and to shed some light on their strengths and shortcomings.
1 Introduction
Functionalized polycyclic aromatic molecules are promising materials for opto-electronic thin film devices as light emitting diodes,1,2 thin film transistors3,4 and photovoltaics5,6 but, depending on the field, further efficiency enhancements are helpful or desperately needed for commercial applications.7,8 Efficient design and optimization strategies for new materials require an understanding of the underlying basic physical and chemical processes.9 Theoretical methods that reliably predict all relevant properties of yet unknown compounds would be extremely helpful because the time-consuming realization of the materials (synthesis, device fabrication and testing) could be focused.10 At present, such predictions are wishful thinking for obvious reasons. The advantage of a given material does not derive from a single but from the interplay between various processes.11–16 For a solar cell, the ideal donor material should be well suited for exciton transfer, allow an efficient charge separation at the interface, represent a good hole conductor, and enable an efficient crossing of the charges into the metal electrodes. Hence, reliable predictions need to comprise information about all or at least the most important processes. Furthermore, to realistically model devices, the simulations have to be performed for amorphous systems15,17–20 which are in particular troublesome because no accurate geometrical information exists. Due to the size of the systems, such studies are very expensive so that severe approximation have to be introduce which lowers their predictive power.21–25 Nevertheless, some applications that comprises various processes within model devices were recently performed,11,12,25 but most studies focus on single processes as exciton diffusion or hole transport alone.Another reason why reliable predictions are quite elaborate is the complicated electronic structure of the aggregates of organic semiconductors.23,26–29 Despite the complexity of their electronic structure, a quantitative picture can already be obtained in regarding the smallest aggregate, the dimer MM, consisting of two identical monomers M. Within a zeroth-order picture, there are two degenerated excited states which are characterized by an excitation of one or the other monomer, leading to configurations M*M and MM*, respectively. Here, the excitation energy is localized representing an ‘exciton’ residing on a single monomer (‘localized Frenkel-state’26). This situation is sketched in Fig. 1(left). Of course, both states interact via a coupling J which leads to a splitting of the degenerate levels where the energy difference is just 2|J|. The respective eigenstates are positive and negative linear combinations of the localized Frenkel states and thus are ‘delocalized Frenkel states’. The electronic coupling element J is often approximated by the simple Förster formula26 which is derived for point-dipoles interacting with each other at a separation R, and gives the typical 1/R3 distance dependence.
 | ||
| Fig. 1 Dimer excited state electronic level scheme. Starting from the localized configurations M*M, MM*, electronic coupling results in delocalized Frenkel states which are linear combinations of the former. In the same manner, the coupling of localized configurations M+M−, M−M+ yields delocalized charge transfer (CT) states. Additional couplings between the delocalized (diabatic) Frenkel- and CT states result in adiabatic electronic states with quantum numbers n = 1–4. | ||
Aggregates are specified according to their absorption spectra as J-aggregates and H-aggregates, respectively. In the first case, the name derives from the pioneering work of Jelly who discovered the characteristic red-shift of an aggregate spectrum if compared to that of the monomer.30,31 On the other hand, H-aggregates exhibit a blue-shift (Hypsochromic shift). These characteristic shifts are readily understood within a simple electronic dimer model as just described (Fig. 1, left). Depending on the dimer geometry, that is, the relative orientation of the monomer transition dipole vectors and the center of mass distance vector, photon absorption may lead to transitions exclusively to the lower (J-aggregate) or to the higher state (H-aggregate). In general, however, both states can be accessed leading to a double peak structure of the absorption lineshape.32
The picture involving only the Frenkel-states is not complete because, in general, two additional charge-transfer (CT) configurations have to be considered (Fig. 1, right hand side). Despite the high symmetry of the homo-dimer, Frenkel and CT states can mix28,33,34 which gives rise to the four adiabatic states which are sketched in the middle of the figure. The amount of mixing depends on the energy difference of Frenkel- and CT states which is very small for many dyes. In particular, for perylene-based aggregates, they are so small that the mixing and consequently the electronic structures of the states, vary strongly already as a function of the relative orientation of the monomers.29,33,35 Increasing the size of the aggregate to a trimer (tetramer) the number of states increase to 3 (4) Frenkel and 6 (12) CT states, and the energy separation between the upper and lower Frenkel state becomes larger. The limit for a large number of states (or large aggregates) is 4|J|.36
Measurements of absorption and emission spectra yield insight into the electronic structure and also allow the prediction of, e.g., exciton diffusion and thus are of importance for the characterization of aggregate properties.37–44 The simulation of such spectra has to take into account that light absorption induces intra-monomer vibrations.20,45–47 For many dyes, the monomer spectra exhibit a progression with a vibrational spacing around 1400 cm−1, as is shown in Fig. 2 (upper left panel) for the monomer absorption spectrum of 3,4,9,10-perylene tetracarboxylic acid bisimide48,49 (PBI, see Fig. 3). This vibrational motion has to be taken into account for the simulation of the aggregate absorption spectra because the vibrational spacing is in the order of the coupling strength |J|, i.e. the line shape is influenced by effective electron–phonon coupling.
For the simulation of the emission spectrum, relaxation effects have to be considered if light absorption mainly populates the upper Frenkel state, as it is the case for H-aggregates. Within a simple Frenkel dimer model, a red shift of the emission is expected to be about 2|J| because, according to Kasha's rule, the emission should originate from the lowest state, e.g. the lower Frenkel state. For PBI-aggregates, this simple model does not seem to work because the spectrum predicted by this simple approach (bottom right panel in Fig. 2, solid black line) differs considerably from its experimental counterpart (solid red line).48,49 The intense emission band at higher energies which is missing in the simulated aggregate emission spectra, resembles the monomer emission so that it can be assigned to the presence of monomers in the sample. Notably, the broad red-shifted band, which represents the most prominent feature of the aggregate emission spectrum, is also not seen in the simulated spectrum. This indicates that the simple Frenkel model does not incorporate all main effects.50 This failure persuaded us that, for the description of the emission spectroscopy of many organic aggregates, a more comprehensive approach has to be developed.29,33,34,51–55 This approach, which we coined as ‘dimer approach’, is the main topic of this perspective. However, to clarify the differences to standard approaches that are based on monomers as central units, we briefly summarize their model Hamiltonians in Section 2. The monomer-based (or site-based) methods have a long tradition47,56–58 and have successfully been used to describe various absorption experiments on, e.g., PBI-aggregates.48,59–62
 | ||
| Fig. 2 Absorption (upper panels) and emission spectra (lower panels) of PBI monomers (left) and aggregates (right). Shown are measured and calculated spectra, as indicated. The dashed blue line in the lower right panel results from simulations including inter-monomer torsional motion, see Section 3.3. | ||
 | ||
| Fig. 3 Structures of the monomers PBI (top), α-PTCDA (middle) and DIP (bottom). | ||
In Section 3 we discuss the main differences between the more traditional ansatz and the dimer approach and provide some exemplary studies. They are concerned with properties of aggregates consisting of PBI, α-PTCDA (tetracarboxylic dianhydride) and DIP (diindeno perylene) monomers whose structures are shown in Fig. 3. We also provide a path leading from the limited dimer ansatz to a more sophisticated aggregate approach.
2 Model hamiltonians
We start with a brief summary of the model Hamiltonians used in monomer-based approaches which have successfully been used to describe experiments. For more details we refer to the excellent recent reviews of Spano58 and Schröter et al.47 In the case of electronic levels and taking only Frenkel states into account, the monomer (m) is described by its electronic ground state |g,m〉 and an excited state |e,m〉 with energies Eg,m and Ee,m, respectively. The Frenkel–Hamiltonian for an aggregate with N identical monomers is | (1) |
|g〉 = |g,1〉![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |g,2〉…|g,m〉…|g,N〉, |g,2〉…|g,m〉…|g,N〉, | (2) |
| |em〉 = |g,1〉 |g,2〉…|e,m〉…|g,N〉, | (3) |
 | (4) |
To illustrate the structure of the excited-state Frenkel Hamiltonian, let us regard the homo-dimer. There, we have the two localized states |e1〉 and |e2〉 with equal energy ε1 = ε2. The ‘next-neighbor’ coupling between these states is just Jnm = J so that, the basis representation of this Hamiltonian is a 2 × 2 matrix of simple Hückel type.36
In a straight forward manner, one may now add charge-transfer states by introducing cationic and anionic monomer states |+,m〉 and |−,m〉. The excited states of energy Enm resulting from a hole residing at monomer (n) and an extra electron on monomer (m) then are:
| |n+;m−〉 = |g,1〉…|+,n〉…|−,m〉…|g,N〉. | (5) |
 | (6) |
To include the CT states, their relative energetic positions with respect to the Frenkel states has to be computed because experimental data are rare. Within the description of absorption and emission spectra of PTCDA given in Section 3.5 we will briefly discuss approximations to characterize the charge transfer properties which were used in treating perylene-based aggregates.
Finally, one adds additional couplings between the Frenkel- and CT states as:
 | (7) |
The crucial role of vibrational degrees of freedom was already discussed in the introduction. The model Hamiltonians described above can be generalized to include intra-monomer vibrations. Regarding the PBI as an example, the simple progressions seen in the spectra in Fig. 2, left panels, indicates that, in a first approach, it is sufficient to include a single monomer vibration within the harmonic approximation. The frequencies for the ground (ωg) and the excited state (ωe) can be taken from the measured spectra. Adjusting the shift xe in the equilibrium positions in going from the ground to the excited state, makes it possible to obtain the calculated absorption and emission spectra as included in Fig. 2.48 Formally, the ground and excited state monomer energies are replaced by the operators
 | (8) |
 | (9) |
 | (10) |
 | (11) |
Absorption spectra of many aggregates were successfully simulated using such monomer-based approaches.47,58 Regarding the PBI-systems, it is sufficient to use the Frenkel–Hamiltonian ĤF to simulate the temperature dependence of absorption spectra59 and also circular dichroism spectra measured under different experimental conditions.64 It is possible to generalize the vibronic Hamiltonians to include several intra-monomer vibrations.65 This was found to be necessary, for example, in the case of macro-cyclic squaraine dyes.66 Also, harmonic approximations are not essential and may be relaxed. As we will see, a more serious problem is that no attention has been payed yet to the inter-monomer degrees of freedom.
3 The dimer- and aggregate approach
3.1 General outline
Initiated by the failure of the monomer approach in the simulation of the emission spectrum of PBI-aggregates, we started to develop the so-called dimer-approach which represents the first step towards a more general aggregate-approach.29,50–54 In contrast to the monomer-based methods described in Section 2, the dimer-approach uses a dimer as central unit. This has the advantage that the interactions between two monomers are considered on a full quantum mechanical level. In particular, sufficiently accurate quantum chemical methods can predict the relative energy positions of Frenkel and CT states with high accuracy.21,67–69 Additionally, these methods also provide a reliable description about the mixing of both types of states. Finally, the dimer approach not only yields accurate vertical excitation energies for the Franck–Condon region, but is as well able to predict highly reliable potential energy surfaces (PES) for inter-monomer motions. This ability turned out to be most important for the description of emission spectra (see Section 3.3) and exciton diffusion (Section 3.4) because excited state relaxation processes seem to occur along such inter-monomer coordinates. Such relaxation pathways involving inter-monomer motions are related to exciton–phonon couplings that are expected to influence absorption and emission spectra of aggregates and crystals as well as charge carrier and exciton transport properties.23,26,70–72Monomer-based approaches include intra-monomer nuclear vibrations but usually neglect inter-monomer motions although they can be included, see, e.g.ref. 73 and 74. Another example was provided by Seibt and Eisfeld75 who used one-dimensional model potentials (based on quantum chemical calculation using the dimer-approach50) to investigate if two-dimensional optical spectroscopy can distinguish between relaxation processes caused by inter-monomer motions or by interactions with the surroundings.
In general, heuristic estimates of the surfaces for a given system, which are not based on QM-calculations, are troublesome. The shapes of the PESs are usually rather flat and include local extrema which prevents the usage of simple harmonic approximations. Furthermore, the small energy differences between the involved states (e.g. Frenkel and CT states) lead to various conical intersections. Fits to measured data may lead to good agreement with experiment but, due to the complicated nature of the PESs, it remains unclear if the agreement is obtained for the right reasons. Even qualitative assignments of absorption bands to CT and Frenkel states are often uncertain because both types of states mix considerably and can possess similar properties. For example, in a dimer both CT states and a dark Frenkel state feature vanishing transition dipole moments so that the nature of a low-lying dark state cannot be assigned beyond doubt if reliable computations are missing.
Within an aggregate approach which treats the complete aggregate quantum chemically, the shapes of the PESs of the involved states which determine the above mentioned exciton–phonon couplings and the relative positions of the states are in principle accessible, but severe approximations are necessary due to the arising high computational efforts. The strongest shortcoming of the dimer approach is the reduction of the aggregate within a polarizable environment to two monomers in vacuum. It thus gives only a rough description of the exciton–phonon couplings and underestimates the relative energy differences of the involved electronic states and also the density of states. Additionally, it might describe the wrong order of states because a polarizable environment may stabilize Frenkel- and CT states differently. Nevertheless, the limited dimer description seems to include all effects which determine the excited state relaxation within PBI-aggregates as we will shown in Section 3.3. However, concerning different systems, extensions of this simplest model were necessary. One example is described in Section 3.4 which is concerned with the variation in the exciton diffusion lengths found between crystalline α-PTCDA and DIP. Also, more extended calculations are performed for the assignment of absorption- and emission spectra of α-PTCDA crystals which are discussed in Section 3.5.
Overall, the employed dimer-approach was found to be in excellent agreement with all available experimental data. To ensure that this agreement does not arise due to error compensation we analyzed the impact of the various simplifications on the computed data. Details are provided in Section 3.6. After briefly discussing the technical details of the computations we will review the outcome of the mentioned studies that allow to assess possibilities and limits of our dimer approach.
3.2 Technical details
In a first step of the dimer approach, we determine potential energy surfaces of the inter-monomer motions, e.g. for the relative monomer center-of-mass shifts and the torsional motion. To keep the computations manageable, the intra-monomer degrees of freedom were not varied but frozen to the values obtained from the monomer ground-state optimization.28,29,33–35,51,52The ground state PESs were computed by Møller–Plesset perturbation theory to second-order in combination with spin-component-scaling (SCS-MP2) using the resolution of identity approximation.76–78 For all dimer computations, we employed the SVP (split valence plus polarization) basis sets.79–81 To compute the PESs of the excited states we use the high-level wave function based methods SCS-CC267,68 or SCS-ADC(2).69,82 The characters of the excited states were analyzed by the method proposed by Liu et al.83 All quantum chemical ab initio computations were performed using the Turbomole 6.1 program package.84
In previous studies we always used wave-function based approaches like SCS-CC2 because time-dependent density functional theory predicted a different order of the bright Frenkel (S2 state) and lower CT states (S3).33,34,85 Using SCS-CC2/TZVP as benchmark, pure GGA (BLYP, PBE) and the hybrid functionals as B3LYP predicted that the CT states should lie considerably below the corresponding Frenkel states, which was in contrast to the predictions of the more accurate SCS-CC2 method. For BHLYP/TZVP the energy difference between both states were smaller than 0.1 eV but the CT state lie still below the upper Frenkel state. To check if range-separated functionals are sufficiently accurate we also used CAM-B3LYP/TZVP86–88 but the results were virtually identical to the BHLYP results. This study also gave some very puzzling results. They are indicated in Fig. 4 which gives the potential energy curves as a function of γ describing the torsional motion of both monomers with respect to each other. The results obtained with SCS-CC2/TZV(P) are given on the left hand side while those computed with BHLYP/TZV(P) are given on the right hand side. Besides the potential energy curves Fig. 4 also gives the oscillator strengths for a characterization of the states (lower panels).
 | ||
| Fig. 4 Comparison of SCS-CC2 (left hand side) and BHLYP (right hand side) results on a PBI dimer. Upper part: Potential energy curves of the ground (black) and excited Frenkel (color, solid) and CT (color, dot-dashed) states as a function of the torsional angle γ. Lower part: Squared transition dipole moment of the neutral excited (solid) and charge-transfer (dot-dashed) states. The dashed lines are the sum of the squared transition dipole moments of the 1B1 (brown) and the 1B2 (blue) states. Taken from ref. 33. | ||
To discuss the resulting effects in some more detail we concentrate on the states possessing B2 symmetry (upper Frenkel and the lower CT states). SCS-CC2 predicts that the 11B2 state possesses a high transition dipole moment i.e. has predominantly Frenkel character. Its potential energy curve as a function of the torsional angle γ possesses a maximum at γ = 0° and a minimum around γ = 30°. For larger torsional angles, the energy slightly increases but no further maximum is found for γ < 80°. The slightly higher lying 21B2 state possesses a vanishing transition dipole moment i.e. is predicted to be a CT state. Its potential has a more complicated shape. For 0° < γ < 30° the potential of the 11B2 and 21B2 run parallel with an energy separation of less than 0.1 eV, i.e. also the 21B2 state shows a maximum at γ = 0° and a minimum at γ = 30°. For larger torsional angles the slope of the 21B2 is steeper than that of the 11B2 and the potential reach another maximum at around 60°. At a first glance the curves obtained with BHLYP are virtually identical to those computed with SCS-CC2. But in contrast to SCS-CC2, BHLYP predicts that the 21B2 state exhibits a high transition dipole moment while that of the 11B2 state vanishes i.e. BHLYP predicts the CT state to lie below the Frenkel state. The failure of BHLYP is understandable because the energy differences between both states are so small that an underestimation of the energy of the CT states of about 0.1 eV is already sufficient for a wrong energy order of both states. However, it is very puzzling that the electronic characters of both states switch while the shapes of the potential curves do not switch. The shape of the potential energy curve of the 11B2 state of BHLYP strongly resembled the shape of the 11B2 state of SCS-CC2. The same holds for the shapes of the 21B2 states. In other words, the CT state of BHLYP possessed the shape of the potential energy curve of the Frenkel state of SCS-CC2 while the potential energy curve of the Frenkel state of BHLYP is nearly identical to the potential energy curve of the CT state of SCS-CC2.
For a rationalization of these puzzling results it is important to note that the underlying diabatic state can interact with each other because they possess the same symmetry. Taken this into account it becomes clear that the shape of the adiabatic potential energy curves is dominated by the adiabatic interactions between the underlying diabatic states. Due to adiabatic interactions, the lower diabatic state is down shifted while the upper is up shifted. If in comparison to SCS-CC2 BHLYP erroneously places the diabatic CT state below the diabatic Frenkel state the wrong states are down and up-shifted. The shapes of the lower and upper adiabatic states are not changed because both approaches obviously predict similar adiabatic interactions. This picture could be supported by a detailed analyses of the electronic characters of both states. It revealed that both represent mixtures of CT and Frenkel characters, which would be expected if both diabatic states interact strongly. The strong influence of adiabatic interaction also explains why the potential energy surfaces of the intermolecular movements within a dimer, i.e. the torsional motion and the transversal and longitudinal shifts are not completely flat but possess various minima and maxima. This could not be explained by a simple dispersion interaction between both monomers.29
We also tested the ωB97XD functional but found the same error as found for CAM-B3LYP or BHLYP. However, our findings stood in contrast to a later study of Casanova.89 He computed potential energy curves for the longitudinal shift of two PBI with ωB97XD, which agreed nicely with the SCS-CC2 results of Settels and Liu.33 To investigate the difference we recomputed the curves and found that ωB97XD gives the right energy order if the geometry of the monomers were optimized with ωB97XD. However, if they are optimized with BLYP the resulting slight differences in the geometries of the monomers are already sufficient so that ωB97XD changes the energy order of the two 1B2 states as discussed above. This is not the case for SCS-CC2, which seems to be less sensitive. However, it might be of interest that in some cases the SCS approximation is necessary to obtain the right order of states.90 Because ωB97XD is considerably less demanding than SCS-CC2 or SCS-ADC(2) we were able to compute larger clusters e.g. trimers and tetramers of PTCDA.55 In this study, we also could show that even ZINDO/S gives very reasonable excitation energies for perylene-based compounds. However, it has to be coupled with an appropriate density functional or wave function based approach because it is not able to provide reliable information about the ground state energy surface. It is interesting to note that more sophisticated semi-empirical methods as the OMx-approaches of Thiel and coworkers91,92 seem to be less suitable.85 More information about these studies can be found in Section 3.6.
Having the PESs and possible non-adiabatic couplings at hand, the next step in the dimer approach is to perform quantum dynamical calculations, i.e. solve the time-independent or time-dependent Schrödinger equation (TDSE) for the nuclear motion. To illustrate the approach regard, as an example, the spectroscopy of the PBI (Section 3.3) leading to the spectra collected in Fig. 2. Although, using a dimer Hamiltonian of Frenkel type (eqn (1)) the calculated absorption spectrum is in rather good agreement with experiment, the emission spectrum is not.48 Therefore, it was proven to be necessary to extend the vibronic Frenkel–Hamiltonian and include inter-monomer motions into the theoretical model. Using the quantum chemically determined potentials Vn(γ) for the torsional motion (which are displayed in Fig. 5) we set up the ground and excited state Hamiltonians as:
| Hg(x1,x2,γ) = Hg,1(x1) + Hg,2(x2) + Tγ + V0(γ), | (12) |
| He1(x1,x2,γ) = He,1(x1) + Hg,2(x2) + Tγ + Vd(γ), | (13) |
| He2(x1,x2,γ) = Hg,1(x1) + He,2(x2) + Tγ + Vd(γ), | (14) |
 | (15) |
 | (16) |
 | ||
| Fig. 5 Potential energy curves of the PBI-dimer. Shown are curves for the three lowest electronic states (n = 0–2) as a function of the torsional angle γ. Also shown is the diabatic potential curve Vd(γ). Photoabsorption to the excited states starting from the Franck–Condon geometry at about 30° leads to the two peaks in the spectrum displayed in Fig. 2. Emission starts from the lower excited state at the parallel geometry where the de-stabilization in the ground state leads to a substantial red shift of the emission spectrum. | ||
Having defined the diabatic dimer Hamiltonian, it is then straight forward to integrate the time-dependent Schrödinger equation using grid-propagation schemes. In our examples we employed the Split-operator method93 which uses the coordinate and momentum representations to evaluate the action of operators depending on coordinate and momenta, respectively, on a wave functions.
3.3 PBI-aggregates: emission spectra and exciton trapping
As mentioned in the introduction, a Frenkel–Hamiltonian including a single intra-monomer vibration were sufficiently accurate to calculate absorption spectra of PBI-aggregates, whereas it failed to describe the emission. From electronic structure calculations on the dimer it emerged that the torsional motion of the monomers is an essential degree of freedom. Potential energy curves for the ground and the two first excited states are displayed in Fig. 5. According to our calculations,50 in the dimer ground state, the monomers are arranged at an equilibrium angle of about γ = 30° which agrees with single crystal orientations of the molecules.94 The arrows in the figure indicate one-photon transitions to the first two excited states. Concerning the level scheme shown in Fig. 1, we note that these states are already of mixed Frenkel- and CT-character so that the splitting obtained from the pure Frenkel states is not correct and one needs to regard the adiabatic energies with quantum numbers n = 1 and n = 2. The lowest excited state (n = 1), which serves as initial state for emission, has its minimum at the parallel configuration with γ = 0°. The potential curve is very flat as a function of the torsional angle around its minimum, whereas the ground state is destabilized by a large amount. This leads to the essential red shift of the emission spectrum and also causes the broad spectral lineshape.In the theoretical description, the vibronic Frenkel–Hamiltonian was extended to include a part Htor(γ) which describes the torsional dynamics and includes the ab initio determined potential curves (Sec. 3.2). The spectra were calculated by solving the time-dependent Schrödinger equation95,96 on a grid.93 These calculations predicted an emission spectrum which is also shown in Fig. 2, and an excellent agreement between theory and experiment is found, for details see ref. 50
While the mentioned simulations could nicely describe the absorption- and emission spectra, they seemed to be inappropriate to explain transient absorption measurements.51 In these experiments, femtosecond excitation at a photon energy which matches the energetically highest absorption peak (see Fig. 2) takes place. This means that the second excited state (n = 2) is optically accessed. From the recorded time-traces it was concluded that this state is de-populated on a time-scale of about 200 fs. This ultrafast depletion could not be explained in regarding the PESs of the torsional motion because the avoided crossing of both Frenkel states is too far away from the Franck–Condon region (for the case of a very strong interaction with a surrounding where relaxation takes place instantaneously, see the discussion in ref. 75). To analyze this effect we focused on the role of the CT states and their mixing with the Frenkel states because intra-molecular relaxation of the monomers alone could not explain the effect. Such mixing can occur if deformations of the dimer structure take place and, because complete geometry optimization for the excited states of the dimer are too expensive, we advised the following approach. Starting from the ground state dimer geometry, the first four excited state energies (Van(q = 0), n = 1–4) are calculated. Here, q denotes a reaction coordinate which, for the Franck–Condon geometry assumes a value of q = 0. Next, the cationic (M+) and anionic (M−) monomers are geometry-optimized separately and brought into the ground-state inter-monomer geometry of the dimer. This defines the reaction coordinate at a value of q = 1 and the calculated excited state energies are Van(q = 1). To obtain potentials as a continuous function of q, all atomic positions in the dimer are varied linearly from their initial values at the geometry q = 0 to their final values at q = 1. The resulting adiabatic potentials are given in Fig. 6, upper panel, which also contains calculated CT-characters (lower panel). Despite the rather small changes in the atomic positions in going from q = 0 to q = 1, which are in the order of 0.01 Å,53 the effect on the energetic order of the states is pronounced. The photoexcited state with quantum number n = 2 is of Frenkel-type and the energetically closest state (n = 3) is of CT-type. Upon increasing the value of q an avoided crossing between the two states occurs. In this region, the characters of the two adiabatic states interchange so that the second excited state assumes a large CT-character whereas the other one becomes of Frenkel-type. For still larger values of the reaction coordinate the CT-contribution to the n = 2 state decreases whereas in the lowest Frenkel state (n = 1), this character increases. The behavior documented in Fig. 6 shows that, even for smaller geometry deformations, there is a large coupling between states of merely Frenkel- and CT-character. This means that non-adiabatic transitions become important in the excited state dynamics of the PBI-dimer. Employing the ab initio adiabatic potentials Van(q) and CT-characters (see Fig. 6), we have performed a diabatization to arrive at a Hamiltonian including four excited dimer states which are coupled via potential coupling elements. The diabatic potentials (which are not shown in Fig. 6) smoothly connect between the adiabatic ones.51 Transformation matrices A(q) were determined such that a diagonalization of the diabatic potential matrix reproduced the ab initio potentials and simultaneously yielded nearly constant diabatic CT-characters. Taking the interaction with a surrounding into account we solved a stochastic Schrödinger equation,97 and the thus obtained population dynamics are in very good agreement with experiment.53
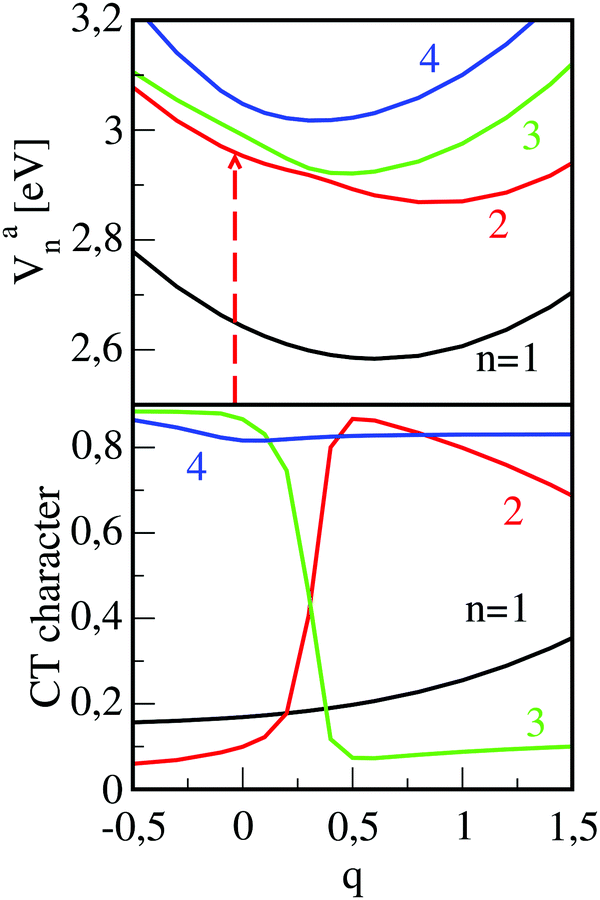 | ||
| Fig. 6 Upper panel: Excited state adiabatic potential curves Van(q) (n = 1–4) as a function of the reaction coordinate which relates the (M–M)* (q = 0) to the (M+–M−) (q = 1) configuration. The deformation results in a mixing of Frenkel- and CT states which can be taken from the CT-characters displayed in the lower panel. The arrow indicates the photo-excitation to the optically bright second state with quantum number n = 2. | ||
The overall picture of the dynamics which takes place in the dimer after femtosecond excitation of the higher lying Frenkel state (n = 2) is illustrated in Fig. 7, upper panel, and it reads as follows. Photo-absorption populates the bright Frenkel state (denoted as SD2 in the figure) and is followed by geometry distortion of the dimer. This leads to a strong coupling to a CT state and, while energy is dissipated into the surrounding, to an ultrafast non-adiabatic transition to the lower state with quantum number n = 1 (denoted as SD1). This process takes place on the femtosecond time-scale so that the dimer remains in its torsional geometry at γ = 30°. Afterwards, the torsional motion towards the parallel dimer configuration takes place on the picosecond time-scale. Note that in the latter geometry, emission to the ground state is forbidden which is the reason for the quite long fluorescence lifetime which was measured to be 32.7 ns (in contrast to that of the monomer which is 3.2 ns).49 This radiative decay defines the third time scale for the dynamical processes taking place after photo-excitation.
 | ||
| Fig. 7 Upper part: Illustration of relaxation mechanism taking place in PBI-aggregates as described in Section 3.3. The relaxation scheme evolving the analysis of the dynamics in PTCDA (Section 3.4) is sketched in the lower part of the figure. | ||
3.4 Exciton diffusion lengths: α-PTCDA versus DIP
As a second example, we discuss our investigations52 on the exciton diffusion lengths (LD) in α-PTCDA and in DIP which are about 22 nm98 and 60 nm,99 respectively. Employing a monomer hopping model in combination with the Marcus approach, predicts an LD value of 160 nm for α-PTCDA, which is almost an order of magnitude too large, while a reasonable value of 100 nm was computed for DIP.100,101 This indicates that the monomer-based model misses some important effects for α-PTCDA which seem to be less important or not active in DIP crystals. Simple disorder effects, which are expected to limit the LD for amorphous systems, should be less significant because both measurements were performed on single crystals.To investigate the underlying mechanisms leading to the strong differences between α-PTCDA and DIP crystals, we extended our dimer approach so that environmental effects are included in the model. We used a mechanical embedding hybrid approach which combines quantum mechanics (QM) to compute the excited states of a dimer, with molecular mechanics (MM) which takes account of the steric effects of the crystal environment.102 The latter is mimicked by a large cluster. It consists of two layers around the inner dimer which can adapt to its motions and a third layer which is fixed to the crystal geometry. To simulate the α-PTCDA environment, we employ the AMOEBA polarizable force field103 while a new force field had to be developed for DIP.104–106 For the computation of the inner dimer we used the SCS-CC2 approach,107 because multi-reference configuration interaction calculations108,109 are way too expensive for the given systems. As for our PBI studies described above, we used optimized monomer geometries and computed the PESs of the electronically excited states along the relevant inter-monomer relaxation coordinates. For α-PTCDA, they involve the shift coordinates along the long (longitudinal shift RL) and small (transversal shift RT) molecular axis. For DIP, we used a linear path between the Franck–Condon region and the conical intersection (CI) of the PESs of both Frenkel states.
Many investigations indicate that excitons are delocalized across more than two monomers, in particular in well-ordered systems as single crystals.110,111 Such a delocalization leads to an increase in the number of Frenkel- and CT states to be taken into account and increases the energy difference between the highest and the lowest Frenkel state.26,55 The increase in the number of states can only be accounted for by oligomer computations which failed due to hard- and software limitations. Hence, we only included the increase in the energy difference between upper and lower Frenkel state via the Apéry's constant.29,112 Consequently, the computed two Frenkel states of these corrected dimer calculations represent a sort of envelope states for the manifold of states which arise due to the delocalization of the exciton over several monomers.
Computed relevant potential energy surfaces for α-PTCDA are given as a function of the shifts RL and RT, in Fig. 8. To document the strong influence of the crystal environment in terms of delocalization and steric hindrances, the pure dimer (upper panel) as well as the full QM/MM computations (lower panel) are shown. The given excited states possess predominantly Frenkel character. The corresponding CT states are slightly higher in energy and hence were neglected in our description. The excitation energies in the Franck–Condon region of both Frenkel states are 2.4 and 3.1 eV, respectively. As expected for an H-aggregate, the upper state has a large transition dipole moment (1.15 a.u.) while, due to the high symmetry of the dimer, the transitions to the lower state is forbidden. Due to the shape of the PES of the upper Frenkel state, the system can barrier-less relax to a CI with the lower Frenkel state.
In order to estimate the time needed for a wave packet prepared in the Franck–Condon region by laser excitation from the ground state, to reach the conical intersection, we performed a quantum dynamical calculation. The propagation needs the construction of a kinetic energy operator in the reduced space spanned by the shift-coordinates and uses the adiabatic potential given of the upper Frenkel state displayed in Fig. 8 (lower panel), for details see ref. 52 It is found that a wave packet initially positioned in the Franck–Condon region, needs about 400–500 fs to reach the CI. The relaxation can take place despite the steric restrictions within the crystal because the relative motion of both monomers is quite small. After the transition to the lower Frenkel state, the system will further relax to the local minimum of the respective PES. The minimum (RL = 0.64 Å; RT = −0.49 Å) lies only about 1.5 eV above the ground state. This low excitation energy stems from a destabilization of the ground state relative to its global minimum (0.5 eV) going in hand with a stabilization of the lower Frenkel state by about 0.4 eV in comparison to the CI. For explanations, why the CI of both states lies below the Franck–Condon region although strong steric restrictions exist in a crystal we refer to our original paper.52Fig. 7, lower panel, summarizes the relaxation pathway which evolves from the analysis given above.
 | ||
| Fig. 8 Potential energy surfaces (PES) of α-PTCDA. Upper panel: Pure dimer computations; lower panel: dimer calculations including Apéry-factor and steric interactions within the crystal. The Franck–Condon region is indicated by a wave-packet and the arrows sketch the average direction of its motion. | ||
The corresponding potentials computed for DIP crystals differ considerably (not shown).52 As expected from the similar electronic structures of the PTCDA and the DIP dimers,34 also for DIP both Frenkel states cross. However, due to the steric restriction in the crystal the CI is higher in energy and the computations predict large barriers between the Franck–Condon region and the CI so that the latter is inaccessible. The difference in the PES readily explains the variations in the LD values of DIP vs. α-PTCDA and elucidates why a hopping model in combination with the Marcus approach successfully predicts the LD value for DIP but considerably overestimates the LD value for α-PTCDA. In the latter case, the exciton can readily relax to the lower Frenkel state along an inter-monomer mode included in the dimer model. Because the relaxation takes place on a time-scale of about 500 fs, the exciton can hop over several units, before it relaxes to the lower Frenkel state. Due to the energy loss accompanied by the relaxation, the exciton becomes trapped. For DIP, the corresponding CI is inaccessible so no trapping connected with an inter-monomer relaxation pathway takes place. Consequently, a monomer based hopping model which neglects such trapping effects but carefully considers relevant intra-monomer motions can provide reasonable LD values.100,101
Our examples given for the three perylene based aggregates PBI, PTCDA and DIP show that the dimer approach is necessary if fast trapping effects are important which involve relaxation pathways along inter-monomer coordinates leading to a population transfer to a lower lying state. Efficient trapping does not occur if the relaxation takes place in a single state because the corresponding PESs are very flat. Only for relaxation to lower lying states the accompanied energy loss of the exciton is considerably larger than stabilizations which results due to intra-monomer relaxation processes. Transitions to lower lying electronic states seem to be very common for such systems because of the small energy splitting between the four low-lying electronically excited states of the dimer. The strong interactions between the underlying diabatic states lead to various CIs between these four states and thus open up pathways for non-adiabatic transitions. Nevertheless, the example for DIP shows that steric effects within the solid state can lead to barriers which efficiently block the relaxation despite available CIs. This explains the experimentally found strong influence of the environment or of bulky substituents on the efficiency of exciton diffusion.113,114 The dimer approach seems to be necessary to describe the photo-physical processes in cases in which the upper Frenkel state is initially populated by light absorption (H-aggregates), because the shapes of the PESs and possible CIs cannot be extracted from experimental measurements or pure monomer computations. In other cases, e.g. J-aggregates, monomer approaches seems to be sufficient because inter-monomer relaxations are less important than their intra-monomer counterparts.
3.5 Optical properties of α-PTCDA films
We will use our investigations about the absorption and emission spectra of α-PTCDA thin films to discuss further differences between the monomer- and dimer-based approaches. The photo-physical behavior of α-PTCDA thin films is an ideal example because both models predict a completely different microscopic picture. The available experimental data37,40,41,115,116 which are collected in Fig. 9, can be summarized as follows: The absorption spectra exhibits a narrow (0.2–0.3 eV) band with a maximum at about 2.2 eV. A broader and more intense peak with a maximum at about 2.6 eV is found between 2.4 and 2.9 eV. Reasons for the shift in the peak maxima seen in the two measured spectra remain unclear because both experiments were performed at room temperature.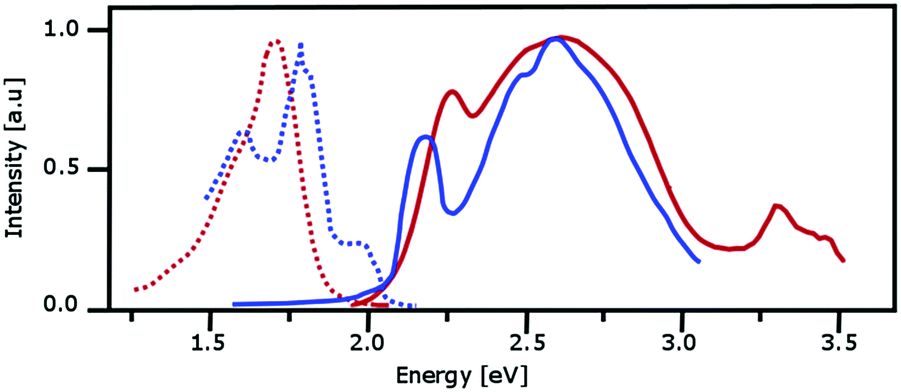 | ||
| Fig. 9 Measured absorption (solid lines) and emission spectra (dotted lines) for PTCDA. The red and blue curves are taken from the measurements reported in ref. 37, 115 andref. 40, 41 respectively. | ||
The emission spectra provided by Forrest and co-workers37,115 and Engel et al.40,41 resemble each other although the first group used pump-pulses at 2.72 eV and 2.05 eV while the second one employed pulses at 2.22 eV. The emission spectra provided by Forrest and co-worker possess one structureless band which starts somewhat below 1.5 eV and has its peak maximum at about 1.7 eV. The most intense peak maximum of the emission spectrum given by Engel et al. is at about 1.8 eV while two less intense peaks are found at about 1.6 and 1.9 eV. The differences between both spectra may result because the latter experiments were performed at a temperature of 10 K while the former ones were conducted at room temperature.
Monomer-based models37,115,117–121 assigned the lower band of the absorption spectrum to transitions into CT states which were computed to lie below the Frenkel state. Using ionization potentials and electron affinities and adding the electro-static attraction between anion and cation for monomer distances in the corresponding crystals, the relative energetic positions of CT states of the dimer were estimated. In this approach, stabilization effects resulting from the polarizable environment as well as effects resulting from charge delocalization and polarization are approximately taken into account.122–124 The model predicts that diabatic Frenkel- and CT states mix to some extent. However, the computations indicate that the lower states have predominantly CT character. A severe drawback of this approach is the assumption that the CT states are indeed real charge-separated states so that the cation and anion are localized. However, this is not the case because, due to the high symmetry, each CT state includes both local CT configurations. Consequently, the charges are completely delocalized and the dipole moments of both CT states vanish. This lowers the stabilizing effects from the Coulomb attraction of the anion and the cation and also from interactions with a polarizable environment which mainly result from the static dipole moments. Hence, stabilization of the CT states might be overestimated. For PBI thin films the same approach predicted the reverse order (Frenkel- below CT states), i.e. according to these computations PTCDA and PBI aggregates possess quite different electronic structures.119,122–124
Using the computed PES (Fig. 8) describing the inter-monomer motions along RL and RT, it is possible to explain the features of both, the absorption and the emission spectrum. However, our assignment differs from the ones discussed above. The bands seen in the absorption and the emission spectrum involve exclusively transitions to both Frenkel states because the CT states are predicted to lie above their Frenkel counterparts. Considering that the computed states represent envelope states for the manifold of all Frenkel states resulting from a delocalization of the exciton over various monomers, the computed vertical excitation energies of about 2.2. eV and 3.1 eV agree nicely with the positions and of the two absorption bands. The weak band at low energy and the broad intense band appearing at higher energies are assigned to transitions into the lower dark and higher bright Frenkel state, respectively. The PESs also explain why the emission spectra resemble each other for the different pump-pulses used in the experiments. Due to the shape of the PESs of both states and the CI between them, the upper state, which is initially populated from a pump pulse at 2.72 eV, decays to the global minimum of the lower Frenkel state which is computed to lie about 1.5 eV above the ground state. Pump-pulses at 2.05 or 2.2 eV which mainly populate the lower Frenkel state in the Franck–Condon region, lead to similar emission spectra because the shape of the PES of the lower Frenkel state also induces a relaxation to the global minimum of this state.
The differences in the assignment of the various transitions result because the monomer- and dimer-based theoretical approaches differ in the relative energetic order of Frenkel- and CT states. Additionally, while the dimer computations predict large similarities between PTCDA and PBI aggregates, the monomer-based models predict strong differences in their electronic structures. The question which model is correct is not that simple to answer. Both are able to describe features of the absorption- and emission spectra because the lower Frenkel and the CT states possess similar transition moments from/to ground states, i.e. both represent a lower lying ‘dark’ state which is necessary to explain the intensity pattern of the spectrum. Additionally, both approaches rest on various approximations which will be discuss in the next section.
3.6 Towards more realistic aggregate models
Monomer- and dimer-approaches both possess advantages and disadvantages. The main uncertainty of the monomer approach lies in the estimate of the relative position of CT- and Frenkel states. Inaccuracies in the dimer approach result from the reduction of the aggregate to a dimer and the neglect of a polarizable environment. Investigations about the influence of these approximations on the computed data were recently performed for PBI54 and PTCDA.55 Here, we concentrate on our studies on PTCDA thin films, because they go beyond the work on PBI.A major shortcoming of the dimer approach is the neglect of a polarizable environment, because the related stabilization was expected to shift the CT states below the Frenkel state. In contrast to this expectation, computations in which the environment was approximated by a polarizable continuum, stabilized the upper Frenkel state more than the CT states. This unexpected behavior results from the fact that both types of states possess vanishing static dipole moments due to the high symmetry of the system. For vanishing static dipoles the stabilization due to a polarizable environment correlates with the transition moment of the given state26,125 which is large for the upper Frenkel- but vanishes for both CT states. Non-symmetric environments which were mimicked by a QM/MM approach126,127 lead to a stronger stabilization of the CT states because they allow for a localization of the charges. This is not the case within a continuum description which preserves the symmetry of the system. Nevertheless, the CT state remains above the upper Frenkel state, i.e. the same order of states as for vacuum computations is predicted. A stronger stabilization of the CT states is only possible if the system adopts geometries optimized for the CT state. In this case, the lower CT state drops below the upper Frenkel state, but it remains above the lower Frenkel state.
The situation found for PTCDA resembles the situation discussed for PBI aggregates. Recent investigations showed that the ωB97XD functional128 in combination with a TZVP basis79,81 provides similar accurate structures, reorganization and excitation energies than more demanding wave-function based approaches as SCS-CC2 or SCS-ADC(2).21,85,129,130 Our computations for PTCDA dimer, trimers and tetramers showed that even ZINDO/S131,132,133 provides excellent vertical excitation energies.55 This enables us to extend the computations to larger out of plane stacks. An example of such a stack is sketched in Fig. 10, lower panel. Calculations for larger stacks are of interest because it is assumed that the exciton is delocalized over larger aggregates than dimers. In the upper part of Fig. 10 we present data including excitation energies, oscillator strengths and CT-characters for aggregates of selected size. The calculations indicate that also for larger aggregates the lower lying electronically excited states still possess Frenkel character. Additionally, an increase of the band width is seen. The upper Frenkel state which possesses the strongest transition probability also for larger stacks, is shifted into the manifold of the CT states. Due to the resulting mixing, its absorption intensity is distributed over various states. This indicates that the strong band in the PTCDA absorption spectrum results from transitions into several electronic states. Additional investigations revealed that interactions between two out of plane stacks do not change the order of the electronic states because the spectrum is dominated by the much stronger interaction within an out of plane stack. Assuming an average error of 0.3 eV, the predicted excitation energies agree excellently with experiment. This accuracy indicates another advantage of our ab initio based approach: it is able to predict the photo-physical properties of yet unknown materials because no experimental input is needed.
 | ||
| Fig. 10 Upper panel: Vertical excitation energies in eV and characters of the states (Frenkel (F), charge transfer (CT)) as predicted by ZINDO/S. The magnitude of the oscillator strength f is also indicated. The orientations of the monomers within an out of plane stack are shown in panel (lower panel). | ||
At this point the question arises, if dimer calculations are also needed for amorphous systems. Here, monomer approaches might be sufficient because the exciton is more localized than in an ordered structure. In contrast, trapping processes which are suppressed in well-ordered systems due to steric restrictions, might take place. Amorphous structures are less dense due to defects and grain-boundaries, so that the, in the average, reduced spatial confinement may allow for a relaxational motion. This assumption is underlined by computations in which the steric restrain in a DIP crystal was lifted. Hence, we predicted that an increased flexibility is one of the reasons for the strong differences in the exciton diffusion lengths of single crystals and of amorphous systems.52 An investigation about the interface processes in organic solar cell which combines fullerenes as p-semiconductor with various organic n-semiconductor revealed another reason.11,12 In these computations, we generated an amorphous film of DIP molecules and simulated the exciton diffusion within this film. Assuming that the excitons are localized on monomers, variations in their energies only depend on a varying environment. As a consequence, efficient exciton diffusion is predicted. However, if the exciton is delocalized over dimers, the energy disorder increases considerably because in dimers of an amorphous film the monomers adopt various orientations with respect to each other which results in considerably different excitation energies. Consequently, in amorphous materials trapping processes already occur if an exciton hops to a dimer which adopts an orientation with low excitation energies. Such trapping processes do not even depend on the possibility of relaxation motions of both monomers with respect to each other which might be prohibited by the dense packing of organic semiconductor thin films.
3.7 Summary and conclusions
Careful assignments of absorption and emission spectra of organic semiconductor aggregates allow a comprehensive understanding of their complicated electronic structure, which is a prerequisite for the design of improved materials. The necessary disentanglement of the various effects deserve a close collaboration between experiment and theory. The today's standard approach to model the spectra is monomer-based, i.e. it starts from the electronic structures of monomers and incorporates the interactions between the monomers of the aggregates via coupling matrix elements, which are partly computed and partly fitted to experimental data. In the present feature article, we compare this approach with the so-called dimer approach, which uses dimers as the smallest unit. The discussion focuses on strengths and weaknesses of both methods and gives some examples to show the applicability of the dimer approach. The advantages of the monomer-based approach can be summarized as follows: It contains all relevant interactions determining the energetic behavior of the Frenkel states and includes exciton–phonon couplings, which arise due to intra-monomer vibrations. On the other hand, inter-monomer motions are usually not considered. Further possible disadvantages result from the relative energies of Frenkel- and CT states. They have to be computed and uncertainties may arise because delocalization effects within the CT states are difficult to estimate.The dimer approach starts from a pair of monomers and uses sophisticated quantum chemical approaches to compute the potential energy surfaces (PESs) of the relevant inter-monomer motions. These PESs are subsequently used to model the influence of excited state relaxation processes on absorption and emission spectra and on exciton diffusion. This approach has the first advantage that the interaction between both monomers are treated quantum mechanically. A second advantage is that the employed quantum chemical methods treat Frenkel- and CT states on equal footing so that their energetic order, the energy differences and the mixing of the states are reliably predicted. Our given examples show, however, that the main advantage of the dimer approach results from an accurate treatment of the inter-monomer motions. They turned out to be of major importance for the development of atomistic pictures of exciton relaxation processes which are relevant to understand the photo-physics of many organic semiconductor aggregates.
We illustrate the power of the dimer approach regarding three examples which are the emission spectroscopy of PBI aggregates, the differences in the exciton diffusion within DIP and PTCDA single crystals and the assignment of optical spectra of PTCDA. In all three examples inter-monomer motions turned out to be central for excited state relaxation effects. The strongly red-shifted broad band of the emission spectra of PBI aggregates and transient absorption measurements could be fully explained by a two-step mechanism resulting from the interplay of intra- and inter-monomer motion (Fig. 7, upper part). In the first step a photo-induced intra-monomer motion, which distorts the monomers towards the cationic and anionic geometries leads to a conical intersection between the initially populated upper Frenkel- and the lower CT state. This induces a fast depopulation of the upper Frenkel state while the CT state acts only as a doorway state. Its population is quickly transferred to the lower Frenkel state because both states strongly couple along the deformation reaction coordinate. This population transfer is predicted to happen on the fs time scale, which is in nearly quantitative agreement with the experiment. In a second process which evolves on a much longer time scale, both monomers reorient with respect to each other. Because the lower Frenkel state is slightly stabilized but the ground state is strongly destabilized along the coordinate a strong red shift is seen in emission. The broad emission band results because the PES of the lower Frenkel state is rather flat whereas the ground state PES is steep.
Differences between the exciton diffusion length (LD) in single crystals of DIP (60–80 nm) and PTCDA (22 nm) can also be explained by relaxation processes along inter-monomer coordinates. This difference in the LD values is surprising because the orientation of the PTCDA molecules within the crystal environment is more appropriate for exciton hopping processes than the one of the DIP molecules. For this study, we had to improve the dimer approach used for the PBI aggregates. Steric restrictions arising from the crystal environment were included via QM/MM. Additionally we estimated effects, which arise from the delocalization of the exciton over larger aggregates by the Apéry factor. The two-dimensional quantum dynamical treatment based on the computed surfaces predicted that for PTCDA a photo-induced motion to a conical intersection between both Frenkel states leads to a population transfer from the initially populated upper to the lower Frenkel state. Despite the strong steric restrictions arising from the crystal environment, this process is predicted to happen on a similar time-scale than the exciton hopping. Consequently, the exciton can only hop a few times before it is quenched to the lower Frenkel state leading to the short diffusion length of only 22 nm. The electronic structures of DIP and PTCDA dimers are very similar, i.e. the conical intersection described above is also present in the PESs of the DIP dimer. However, the steric restrictions which arise within the DIP single crystal, lead to high barriers on the corresponding PES so that the conical intersection is not accessible. This shows that the differences in the LD values mainly result from the respective crystal environment. It also indicates that variations which decrease the mobility of the monomers within the aggregate network, should improve the exciton diffusion properties.
The computed PESs also provided an assignment of the absorption- and emission spectra of PTCDA, which differ from the interpretation of monomer-based approaches. The main difference results from the energy order of CT and Frenkel states. The monomer-based approaches predicted that the lowest state of PTCDA aggregates has CT character. Consequently, this state is responsible for the lower band of the absorption spectra and represents the initial state for emission. In contrast, our quantum chemical calculations predict that the CT states lie above the upper Frenkel state, i.e. they can influence the spectra only indirectly. Hence, our dimer-based computations assigned the lower band of the absorption spectra and the emission spectra to the lower Frenkel state.
The excellent agreements between computed and measured data support the applicability of the dimer approach and indicates that it considers the most important effects. Nevertheless, the method obviously possesses various shortcomings. The computational efforts needed to compute the PESs are its main disadvantage. It is much higher than that of standard monomer based approaches and constrains the introduction of various simplifications. The strongest approximation is the reduction of an aggregate within a given polarizable environment to a dimer in the vacuum. Both restrictions were already partly lifted in the DIP and PTCDA investigations but the interplay between the various effects is so complicated that it could not be excluded that our approach obtains the right answer for the wrong reason. Hence, for PTCDA and PBI we investigated how the various approximations influence the predictions. We incorporated environmental effects, increased the size of the aggregates and modeled the influence of intra- and inter-molecular vibrational motions. All these simulations show that only quantitative but no qualitative changes occur if the various approximations of the dimer approach are relaxed.
The latter investigations indicate a route to generalize the dimer model to a more appropriate aggregate model. This route is in principle feasible but the computational effort increases dramatically and the strongly increasing number of inter-monomer degrees of freedom leads to further problems. Hence, the question arises if a model is possible which combines the advantages of the monomer- and dimer-based approaches. One might think of, e.g., combining both approaches and start with a dimer which interacts with other dimers (or monomers) through the equations given in Section 2. The necessary coupling parameters could be taken from trimer (tetramer) computations. Work along these lines is under way.
Acknowledgements
Financial support by the DFG within the GRK 1221 and the FOR 1809 is gratefully acknowledged. Many people have been involved in the presented research and we thank all of them. In particular we mention the contributions of R. F. Fink, A. Schubert, V. Settels, W. Liu, and D. Bellinger.References
- B. Geffroy, P. Le Roy and C. Prat, Polym. Int., 2006, 55, 572–582 CrossRef CAS.
- H. Katz, A. Lovinger, J. Johnson, C. Kloc, T. Siegrist, W. Li, Y. Lin and A. Dodabalapur, Nature, 2000, 404, 478–481 CrossRef CAS PubMed.
- S. Shaheen, G. Jabbour, B. Kippelen, N. Peyghambarian, J. Anderson, S. Marder, N. Armstrong, E. Bellmann and R. Grubbs, Appl. Phys. Lett., 1999, 74, 3212–3214 CrossRef CAS.
- G. Horowitz, Adv. Mater., 1998, 10, 365–377 CrossRef CAS.
- C. Tang, Appl. Phys. Lett., 1986, 48, 183–185 CrossRef CAS.
- X. Liu, H. Chen and S. Tan, Renewable Sustainable Energy Rev., 2015, 52, 1527–1538 CrossRef CAS.
- M. A. Green and S. P. Bremner, Nat. Mater., 2017, 16, 23–34 CrossRef PubMed.
- S. Kim, B. Kim, J. Lee, H. Shin, Y.-I. Park and J. Park, Mater. Sci. Eng., R, 2016, 99, 1–22 CrossRef.
- A. A. Bakulin, A. Rao, V. G. Pavelyev, P. H. M. van Loosdrecht, M. S. Pshenichnikov, D. Niedzialek, J. Cornil, D. Beljonne and R. H. Friend, Science, 2012, 335, 1340–1344 CrossRef CAS PubMed.
- C. Groves, Rep. Prog. Phys., 2017, 80, 026502 CrossRef CAS PubMed.
- C. Brückner, F. Würthner, K. Meerholz and B. Engels, J. Phys. Chem. C, 2017, 121, 4–25 Search PubMed.
- C. Brückner, F. Würthner, K. Meerholz and B. Engels, J. Phys. Chem. C, 2017, 121, 26–51 Search PubMed.
- D. Beljonne, J. Cornil, L. Muccioli, C. Zannoni, J. L. Bredas and F. Castet, Chem. Mater., 2011, 23, 591–609 CrossRef CAS.
- M. Schulze, M. Hansel and P. Tegeder, J. Phys. Chem. C, 2014, 118, 28527–28534 CAS.
- S. R. Yost, E. Hontz, D. P. McMahon and T. Van Voorhis, in Multiscale Modelling of Organic and Hybrid Photovoltaics, ed. D. Beljonne and J. Cornil, Top. Curr. Chem., 2014, vol. 352, pp. 103–150 Search PubMed.
- D. Veldman, S. C. J. Meskers and R. A. J. Janssen, Adv. Funct. Mater., 2009, 19, 1939–1948 CrossRef CAS.
- Y. T. Fu, C. Risko and J. L. Bredas, Adv. Mater., 2013, 25, 878–882 CrossRef CAS PubMed.
- B. Baumeier, F. May, C. Lennartz and D. Andrienko, J. Mater. Chem., 2012, 22, 10971–10976 RSC.
- V. Ruhle, A. Lukyanov, F. May, M. Schrader, T. Vehoff, J. Kirkpatrick, B. Baumeier and D. Andrienko, J. Chem. Theory Comput., 2011, 7, 3335–3345 CrossRef PubMed.
- F. C. Spano, J. Chem. Phys., 2005, 122, 234701 CrossRef PubMed.
- C. Brückner and B. Engels, J. Phys. Chem. A, 2015, 119, 12876–12891 CrossRef PubMed.
- C. Brückner and B. Engels, J. Comput. Chem., 2016, 37, 1335–1344 CrossRef PubMed.
- J. L. Bredas, J. E. Norton, J. Cornil and V. Coropceanu, Acc. Chem. Res., 2009, 42, 1691–1699 CrossRef CAS PubMed.
- H. Yamagata, J. Norton, E. Hontz, Y. Olivier, D. Beljonne, J. L. Bredas, R. J. Silbey and F. C. Spano, J. Chem. Phys., 2011, 134, 204703 CrossRef CAS PubMed.
- T. Bauer, C. M. Jager, M. J. T. Jordan and T. Clark, J. Chem. Phys., 2015, 143, 044114 Search PubMed.
- V. May and O. Kühn, Charge and Energy Transfer Dynamics in Molecular Systems, Wiley-VCH, Weinheim, 3rd edn, 2011 Search PubMed.
- E. Collini and G. D. Scholes, Science, 2009, 323, 369–373 CrossRef CAS PubMed.
- R. F. Fink, J. Pfister, H. Zhao and B. Engels, Chem. Phys., 2008, 346, 275–285 CrossRef CAS.
- H. M. Zhao, J. Pfister, V. Settels, M. Renz, M. Kaupp, V. C. Dehm, F. Würthner, R. F. Fink and B. Engels, J. Am. Chem. Soc., 2009, 131, 15660–15668 CrossRef CAS PubMed.
- E. E. Jelly, Nature, 1936, 138, 1009 CrossRef.
- G. Scheibe, Angew. Chem., 1937, 50, 212 CrossRef CAS.
- N. J. Hestand and F. C. Spano, J. Am. Chem. Soc., 2017, 50, 341–350 CAS.
- W. Liu, V. Settels, P. H. P. Harbach, A. Dreuw, R. F. Fink and B. Engels, J. Comput. Chem., 2011, 32, 1971–1981 CrossRef CAS PubMed.
- V. Settels, W. Liu, J. Pflaum, R. F. Fink and B. Engels, J. Comput. Chem., 2012, 33, 1544–1553 CrossRef CAS PubMed.
- R. F. Fink, J. Pfister, A. Schneider, H. Zhao and B. Engels, Chem. Phys., 2008, 343, 353–361 CrossRef CAS.
- W. Kutzelnigg, Einführung in die Theoretische Chemie, Band 2, VCH, Weinheim, 1994 Search PubMed.
- V. Bulovic, P. E. Burrows, S. R. Forrest, J. A. Cronin and M. E. Thompson, Chem. Phys., 1996, 210, 1–12 CrossRef CAS.
- P. Peumans, A. Yakimov and S. R. Forrest, Chem. Phys., 2004, 95, 2938 CAS.
- M. Hoffmann, K. Schmidt, T. Fritz, T. Hasche, V. M. Agranovich and K. Leo, Chem. Phys., 2000, 258, 73–96 CrossRef CAS.
- E. Engel, M. Koschorreck, K. Leo and M. Hoffmann, J. Lumin., 2005, 112, 299–302 CrossRef CAS.
- E. Engel, M. Koschorreck, K. Leo and M. Hoffmann, Phys. Rev. Lett., 2005, 95, 157403 CrossRef CAS PubMed.
- H. Marciniak, M. Teicher, U. Scherf, S. Trost, T. Riedl, M. Lehnhardt, T. Rabe, W. Kowalsky and S. Lochbrunner, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 214204 CrossRef.
- M. Schlosser and S. Lochbrunner, J. Phys. Chem. B, 2006, 110, 6001–6009 CrossRef CAS PubMed.
- J. W. Cho, H. Yoo, J. E. Lee, Q. F. Yan, D. H. Zhao and D. Kim, J. Phys. Chem. Lett., 2014, 5, 3895–3901 CrossRef CAS PubMed.
- F. C. Spano, Annu. Rev. Phys. Chem., 2006, 57, 217–243 CrossRef CAS PubMed.
- C. Didraga, J. A. Klugkist and J. Knoester, J. Phys. Chem. B, 2002, 106, 11474–11486 CrossRef CAS.
- M. Schröter, S. Ivanov, J. Schulze, S. Polyutov, Y. Yani, T. Pullerits and O. Kühn, Phys. Rep., 2015, 567, 1–78 CrossRef.
- J. Seibt, P. Marquetand, V. Engel, Z. Chen, V. Dehm and F. Würthner, Chem. Phys., 2006, 328, 354–362 CrossRef CAS.
- Z. Chen, V. Stepanenko, V. Dehm, P. Prins, L. Siebbeles, J. Seibt, P. Marquetand, V. Engel and F. Würthner, Chem. – Eur. J., 2007, 13, 436–449 CrossRef CAS PubMed.
- R. F. Fink, J. Seibt, V. Engel, M. Renz, M. Kaupp, S. Lochbrunner, H.-M. Zhao, J. Pfister, F. Würthner and B. Engels, J. Am. Chem. Soc., 2008, 130, 12858–12859 CrossRef CAS PubMed.
- A. Schubert, V. Settels, W. Liu, F. Würthner, C. Meier, R. Fink, S. Schindlbeck, S. Lochbrunner, B. Engels and V. Engel, J. Phys. Chem. Lett., 2013, 4, 792–796 CrossRef CAS PubMed.
- V. Settels, A. Schubert, M. Tafipolski, W. Liu, V. Stehr, A. K. Topczak, J. Pflaum, C. Deibel, R. F. Fink, V. Engel and B. Engels, J. Am. Chem. Soc., 2014, 136, 9327–9337 CrossRef CAS PubMed.
- A. Schubert, M. Falge, M. Kess, V. Settels, S. Lochbrunner, W. T. Strunz, F. Würthner, B. Engels and V. Engel, J. Phys. Chem. A, 2014, 118, 1403–1412 CrossRef CAS PubMed.
- D. Bellinger, V. Settels, W. Liu, R. F. Fink and B. Engels, J. Comput. Chem., 2016, 37, 1601–1610 CrossRef CAS PubMed.
- D. Bellinger, J. Pflaum, C. Brüning, V. Engel and B. Engels, Phys. Chem. Chem. Phys., 2017, 19, 2434–2448 RSC.
- T. Förster, Ann. Phys., 1948, 437, 55–75 CrossRef.
- R. L. Fulton and M. Gouterman, J. Chem. Phys., 1964, 41, 2280–2286 CrossRef CAS.
- F. C. Spano, Acc. Chem. Res., 2010, 43, 429–439 CrossRef CAS PubMed.
- J. Seibt, T. Winkler, K. Renziehausen, V. Dehm, F. Würthner and V. Engel, J. Phys. Chem. A, 2009, 113, 13475–13482 CrossRef CAS PubMed.
- C. M. Pochas, K. A. Kistler, H. Yamagata, S. Matsika and F. C. Spano, J. Am. Chem. Soc., 2013, 135, 3056–3066 CrossRef CAS PubMed.
- P.-A. Plotz, S. P. Polyutov, S. D. Ivanov, F. Fennel, S. Wolter, T. Niehaus, Z. Xie, S. Lochbrunner, F. Würthner and O. Kühn, Phys. Chem. Chem. Phys., 2016, 18, 25110–25119 RSC.
- D. Bialas, C. Brüning, F. Schlosser, B. Fimmel, J. Thien, V. Engel and F. Würthner, Chem. – Eur. J., 2016, 22, 15011 CrossRef CAS PubMed.
- H. Yamagata, D. S. Maxwell, J. Fan, K. R. Kittilstved, A. L. Briseno, M. D. Barnes and F. C. Spano, J. Phys. Chem. C, 2014, 118, 28842–28854 CAS.
- J. Seibt, V. Dehm, F. Würthner and V. Engel, J. Chem. Phys., 2008, 128, 204303 CrossRef PubMed.
- J. Roden, A. Eisfeld, M. Dvorak, O. Bünemann and F. Stienkemeier, J. Chem. Phys., 2011, 134, 054907 CrossRef PubMed.
- C. Brüning, E. Welz, A. Heilos, V. Stehr, C. Walter, B. Engels, S. F. Völker, C. Lambert and V. Engel, J. Phys. Chem. A, 2015, 191, 6174–6180 Search PubMed.
- A. Hellweg, S. A. Grün and C. Hättig, Phys. Chem. Chem. Phys., 2008, 10, 4119–4127 RSC.
- N. O. C. Winter, N. K. Graf, S. Leutwyler and C. Hättig, Phys. Chem. Chem. Phys., 2013, 15, 6623–6630 RSC.
- A. Dreuw and M. Wormit, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 82–95 CrossRef CAS.
- G. D. Scholes and G. Rumbles, Nat. Mater., 2006, 5, 683–696 CrossRef CAS PubMed.
- F. C. Spano, S. C. J. Meskers, E. Hennebicq and D. Beljonne, J. Am. Chem. Soc., 2007, 129, 7044–7054 CrossRef CAS PubMed.
- M. Schwoerer and H. C. Wolf, Organic Molecular Solids, Wiley-VCH, Weinheim, 2007 Search PubMed.
- K. Hannewald, V. M. Stojanovic, J. M. T. Schellekens, P. A. Bobbert, G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 075211 CrossRef.
- B. Nebgen, F. L. Emmert and L. V. Slipchenko, J. Chem. Phys., 2012, 137, 084112 CrossRef PubMed.
- J. Seibt and A. Eisfeld, J. Chem. Phys., 2012, 136, 024109 CrossRef PubMed.
- S. Grimme, L. Goerigk and R. F. Fink, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 886–906 CrossRef CAS.
- F. Weigend and M. Häser, Theor. Chem. Acc., 1997, 97, 331–340 CrossRef CAS.
- F. Weigend, M. Häser, H. Patzelt and R. Ahlrichs, Chem. Phys. Lett., 1998, 294, 143–152 CrossRef CAS.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- K. Eichkorn, O. Treutler, H. Ohm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283–289 CrossRef CAS.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 CrossRef CAS.
- A. B. Trofimov and J. Schirmer, J. Phys., B, 1995, 28, 2299–2324 CrossRef CAS.
- W. L. Liu, B. Lunkenheimer, V. Settels, B. Engels, R. F. Fink and A. Köhn, J. Chem. Phys., 2015, 143, 084106 CrossRef PubMed.
- TURBOMOLE V6.5 2013, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available fromhttp://www.turbomole.com.
- C. Walter, V. Krämer and B. Engels, Int. J. Quantum Chem., 2017, 117, e25337 CrossRef.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- M. J. G. Peach, T. Helgaker, P. Salek, T. W. Keal, O. B. Lutnaes, D. J. Tozer and N. C. Handy, Phys. Chem. Chem. Phys., 2006, 8, 558–562 RSC.
- M. J. G. Peach, D. J. Tozer and N. C. Handy, Int. J. Quantum Chem., 2011, 111, 563–569 CrossRef CAS.
- D. Casanova, Int. J. Quantum Chem., 2015, 115, 442–452 CrossRef CAS.
- O. Oeltermann, C. Brand, B. Engels, J. Tatchen and M. Schmitt, Phys. Chem. Chem. Phys., 2012, 14, 10266–10270 RSC.
- M. Kolb and W. Thiel, J. Comput. Chem., 1993, 14, 775–789 CrossRef CAS.
- D. Tuna, Y. Lu, A. Koslowski and W. Thiel, J. Chem. Theory Comput., 2016, 12, 4400–4422 CrossRef CAS PubMed.
- M. D. Feit, J. A. Fleck and A. Steiger, J. Comput. Phys., 1982, 47, 412–433 CrossRef CAS.
- G. Klebe, F. Graser, E. Hadicke and J. Berndt, Acta Crystallogr., Sect. B: Struct. Sci., 1989, 45, 69–77 CrossRef.
- R. Schinke, Photodissociation Dynamics, Cambridge University Press, Cambridge, 1993 Search PubMed.
- D. J. Tannor, Introduction to Quantum Mechanics: A Time-dependent Perspective, University Science Books, Sausalito, 2007 Search PubMed.
- N. Gisin and I. C. Percival, J. Phys., A, 1992, 25, 5677–5691 CrossRef.
- R. R. Lunt, J. B. Benziger and S. R. Forrest, Adv. Mater., 2010, 22, 1233–1236 CrossRef CAS PubMed.
- D. Kurrle and J. Pflaum, Appl. Phys. Lett., 2008, 92, 133306 CrossRef.
- V. Stehr, R. F. Fink, M. Tafipolski, C. Deibel and B. Engels, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 6, 694–720 CrossRef CAS.
- V. Stehr, R. F. Fink, B. Engels, J. Pflaum and C. Deibel, J. Chem. Theory Comput., 2014, 10, 1242–1255 CrossRef CAS PubMed.
- H. M. Senn and W. Thiel, Angew. Chem., Int. Ed., 2009, 10, 1242–1255 Search PubMed.
- J. W. Ponder, C. Wu, P. Ren, V. S. Pande, J. D. Chodera, M. J. Schnieders, I. Haque, D. L. Mobley, D. S. Lambrecht, R. A. DiStasio Jr., M. Head-Gordon, G. N. I. Clark, M. E. Johnson and T. Head-Gordon, J. Phys. Chem. B, 2010, 114, 2549–2564 CrossRef CAS PubMed.
- M. Tafipolsky and B. Engels, J. Chem. Theory Comput., 2011, 7, 1791–1803 CrossRef CAS PubMed.
- K. Ansorg, M. Tafipolsky and B. Engels, J. Phys. Chem. B, 2013, 117, 10093–10102 CrossRef CAS PubMed.
- C. Grebner, J. Becker, D. Weber, D. Bellinger, M. Tafipolski, C. Brückner and B. Engels, J. Comput. Chem., 2014, 35, 1801–1807 CrossRef CAS PubMed.
- C. Hättig and F. Weigend, J. Chem. Phys., 2000, 113, 5154–5161 CrossRef.
- P. Bundgen, B. Engels and S. D. Peyerimhoff, Chem. Phys. Lett., 1991, 176, 407–412 CrossRef.
- M. Peric, B. Engels and S. D. Peyerimhoff, J. Mol. Spectrosc., 1991, 150, 70–85 CrossRef CAS.
- F. C. Spano and H. Yamagata, J. Phys. Chem. B, 2011, 115, 5133–5143 CrossRef CAS PubMed.
- F. F. So and S. R. Forrest, Phys. Rev. Lett., 1991, 66, 2649–2652 CrossRef CAS PubMed.
- J. D. F. Havil, Gamma: Exploring Euler's Constant, Princeton University Press, Princeton, 2003 Search PubMed.
- V. Kamm, G. Battagliarin, I. A. Howard, W. Pisula, C. Mavrinskiy, A. Li, K. Mullen and F. Laquai, Adv. Energy Mater., 2011, 1, 297–302 CrossRef CAS.
- P. Erwin and M. E. Thompson, Appl. Phys. Lett., 2011, 98, 223305 CrossRef.
- S. R. Forrest, Chem. Rev., 1997, 97, 1793–1896 CrossRef CAS PubMed.
- M. I. Alonso, M. Garriga, N. Karl, J. O. Osso and F. Schreiber, Org. Electron., 2002, 3, 23–31 CrossRef CAS.
- V. R. Gangilenka, L. V. Titova, L. M. Smith, H. P. Wagner, L. A. A. DeSilva, L. Gisslen and R. Scholz, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 155208 CrossRef.
- L. Gisslen and R. Scholz, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 115309 CrossRef.
- R. Scholz, A. Y. Kobitski, I. Vragovic, H. P. Wagner and D. R. T. Zahn, Org. Electron., 2004, 5, 99–105 CrossRef CAS.
- R. Scholz, A. Y. Kobitski, D. R. T. Zahn and M. Schreiber, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 245208 CrossRef.
- P. Petelenz and G. Mazur, Synth. Met., 2000, 109, 73–77 CrossRef CAS.
- Z. L. Shen and S. R. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 10578–10592 CrossRef CAS.
- Z. G. Soos, M. H. Hennessy and G. Wen, Chem. Phys., 1998, 227, 19–32 CrossRef CAS.
- M. H. Hennessy, Z. G. Soos, R. A. Pascal and A. Girlando, Chem. Phys., 1999, 245, 199–212 CrossRef CAS.
- N. S. Bayliss, J. Chem. Phys., 1950, 18, 292–296 CrossRef CAS.
- M. Mladenovic, R. F. Fink, W. Thiel, T. Schirmeister and B. Engels, J. Am. Chem. Soc., 2008, 130, 8696–8705 CrossRef CAS PubMed.
- M. Mladenovic, K. Junold, R. F. Fink, W. Thiel, T. Schirmeister and B. Engels, J. Phys. Chem. B, 2008, 112, 5458–5469 CrossRef CAS PubMed.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- C. Brückner, C. Walter, M. Stolte, B. Braida, K. Meerholz, F. Würthner and B. Engels, J. Phys. Chem. C, 2015, 119, 17602–17611 Search PubMed.
- C. Brückner, C. Walter and B. Engels, Int. J. Quantum Chem., 2016, 116, 1138–1152 CrossRef.
- G. Karlsson and M. C. Zerner, Int. J. Quantum Chem., 1973, 7, 35–49 CrossRef CAS.
- J. D. D. Neto and M. C. Zerner, Int. J. Quantum Chem., 2011, 81, 187–201 CrossRef.
- M. C. Zerner, G. H. Loew, R. F. Kirchner and U. T. Mueller-Westerhoff, J. Am. Chem. Soc., 1980, 102, 589–599 CrossRef CAS.
| This journal is © the Owner Societies 2017 |