
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Imaging on-surface hierarchical assembly of chiral supramolecular networks†
Laerte L.
Patera
 *abc,
Zhiyu
Zou
ad,
Carlo
Dri
ace,
Cristina
Africh
a,
Jascha
Repp
b and
Giovanni
Comelli
ac
*abc,
Zhiyu
Zou
ad,
Carlo
Dri
ace,
Cristina
Africh
a,
Jascha
Repp
b and
Giovanni
Comelli
ac
aIOM-CNR Laboratorio TASC, Area Science Park, 34149 Trieste, Italy
bInstitute of Experimental and Applied Physics, University of Regensburg, 93053 Regensburg, Germany. E-mail: laerte.patera@ur.de
cDepartment of Physics, University of Trieste, 34127 Trieste, Italy
dAbdus Salam International Centre for Theoretical Physics, 34151 Trieste, Italy
eElettra – Sincrotrone Trieste S.C.p.A., 34149 Trieste, Italy
First published on 30th August 2017
Abstract
The bottom-up assembly of chiral structures usually relies on a cascade of molecular recognition interactions. A thorough description of these complex stereochemical mechanisms requires the capability of imaging multilevel coordination in real-time. Here we report the first direct observation of hierarchical expression of supramolecular chirality at work, for 10,10′-dibromo-9,9′-bianthryl (DBBA) on Cu(111). Molecular recognition first steers the growth of chiral organometallic chains and then leads to the formation of enantiopure islands. The structure of the networks was determined by noncontact atomic force microscopy (nc-AFM), while high-speed scanning tunnelling microscopy (STM) revealed details of the assembly mechanisms at the ms time-scale. The direct observation of the chirality transfer pathways allowed us to evaluate the enantioselectivity of the interchain coupling.
Introduction
Chirality recognition is essential in many biological systems, determining optical and pharmaceutical properties of enantiopure compounds.1,2 Furthermore, the realization of chiral catalysts, which allows the production of non-racemic chemicals with high enantioselectivity, has become of paramount importance in modern synthetic chemistry.3,4 However, the mechanisms underlying the selectivity of such processes remain poorly understood,5,6 limiting industrial applications.7,8In the field of supramolecular chemistry, it was shown that chirality can be expressed from single molecules to mesoscopic systems, by means of non-covalent assemblies.9–12 Direct observations, provided by in situ STM, highlighted the importance of dynamic mutual conformational adjustments for the chiral recognition to occur.13 In similar ways, exploiting recognition processes, enantiopure phases can be successfully synthesized on metal surfaces.14–16 Moreover, since the control of the supramolecular assembly critically depends on the interplay between molecule–molecule and molecule–substrate interactions,17–21 different strategies were developed for the realization of extended chiral networks, exploiting different molecular structures22,23 and bonding motifs.13,24–27
Recently, special attention has been devoted to systems presenting a hierarchical chirality transfer, which reveals the presence of sequentially-activated enantioselective mechanisms.24,28–35 Blüm et al.28 reported the fascinating case of rubrene (C42H28) assembling into pentagonal structures, which act as building blocks for a second generation decagons. Interestingly, both the resulting supramolecular structures are enantiopure, indicating the conservation of chirality during the coordination. Hierarchical organization can also be steered through metal–ligand interactions, where the presence of metal adatoms imposes distinctive multilevel binding processes.24,33 Furthermore, sequential self-assembly of nanoporous homochiral networks has been recently observed using porphyrin derivatives,29 representing an appealing platform for the realization of flexible functional materials. However, the atomic-scale mechanisms driving molecular recognition in hierarchical assemblies are yet to be clarified and the factors governing the enantioselectivity of these processes are largely unknown, limiting the possibility to design a priori tailored molecular units with programmed functionalities.
In this work, we report the study of chirality expression during 10,10′-dibromo-9,9′-bianthryl (DBBA) self-assembly on Cu(111), combining state of the art scanning probe microscopy (SPM) techniques. In fact, despite DBBA has been extensively studied on the Cu(111) surface as precursor for the on-surface synthesis of chiral graphene nanoribbons,36–38 little attention has been devoted to the molecular structure of Ullmann intermediates. In particular, the chiral properties of such compounds have not been properly addressed so far.
Upon deposition at 300 K, the molecules undergo debromination and subsequent assembly into chiral organometallic chains, which further interact forming 2D enantiopure islands. The atomic configurations of the resulting compounds are determined by means of high-resolution noncontact atomic force microscopy (nc-AFM), while high-speed scanning tunnelling microscopy (STM) measurements shed light on the assembly mechanisms, revealing the presence of co-existing coordination pathways. Finally, exploiting time-resolved STM sequences, the enantioselectivity of the interchain coupling has been evaluated.
Experimental
Experiments were carried out in two UHV systems, equipped with standard sample preparation facilities. Cu(111) single-crystal samples were cleaned by several sputtering (Ar+/Ne+, 1 keV) and annealing (773 K) cycles.Variable-temperature experiments were carried out with an Omicron VT-STM. High-speed STM measurements were performed using FAST,39 an add-on module for increasing the imaging frequency of commercial scanning probe microscopes up to video-rate.40 In the experiments here discussed, image time-series were acquired in quasi-constant height mode with frame rates between 10 and 36.5 Hz. The high-speed images are presented with horizontal fast scanning direction. Bias voltages are given as sample bias with respect to the tip. 10,10′-dibromo-9,9′-bianthryl molecules (AOKBIO, 98+% purity) were sublimed onto the sample held at ≅300 K.
Low-temperature experiments were carried out with a home-built combined scanning tunnelling/atomic force microscope, at a temperature of 6.8 K. The atomic force microscope's qPlus tuning fork41 was operated in the frequency modulation mode with sub-angstrom oscillation amplitudes.42 Bias voltages are given as sample bias with respect to the tip. A small amount of CO was dosed onto the surface for tip functionalization.43 NaCl islands were grown to facilitate the pick-up of CO molecules to the tip apex from these islands. AFM data was obtained with a CO-terminated tip apex. 10,10′-dibromo-9,9′-bianthryl molecules (AOKBIO, 98+% purity) were sublimed onto the sample kept at ≅8 K inside the microscope and subsequently annealed at ≅300 K in the preparation chamber.
Results and discussion
In gas-phase, DBBA is not planar, due to steric hindrance, presenting a large angle between the anthracene moieties (see Fig. 1), which confers axial chirality to the molecule. Upon surface deposition, the molecules remain non-planar, resulting in racemic ad-layers of two enantiomers, named α and β in Fig. 1. Moreover, on Cu(111), at surface temperatures above 240 K, debromination spontaneously occurs,36 generating biradical active species. While on Au(111) covalent coupling is readily triggered after the radical generation,44,45 for the case of similar molecules on Cu(111), metal adatoms have been shown to bind to the active C atoms, giving rise to C–Cu–C coordination (see Fig. 1),46–48 recently identified as possible intermediates of the Ullmann coupling reaction.46,49 | ||
| Fig. 1 Reaction scheme for the formation of chiral organometallic chains from DBBA enantiomers on Cu(111). Top (bottom) panel corresponds to α (β) enantiomer. | ||
Fig. 2a shows a STM image of the Cu(111) surface after DBBA deposition at 300 K. Molecular self-assembly gives rise to ordered zigzag-like chain structures, which can further coalesce into 2D islands. The absence of moiré patterns in uniform structures, exceeding 10 nm in length, suggests that the molecular orientation with respect to the substrate provides commensurability to the chains.
 | ||
| Fig. 2 DBBA self-assembly on Cu(111). (a) STM image of molecular chains and islands [−1 V, 0.5 nA, 295 K, 60 s per frame]. Circles highlight domains with opposite chirality. Fast and slow scanning directions are specified. (b) nc-AFM image of a dimer obtained with a CO tip [Δz = 2.9 Å, 6.8 K, STM setpoint: 1 V, 1.4 pA]. Arrows indicate the main intra-molecular features. (c) 3D model of the molecular structure imaged in (b). The arrows specified in (b) are superimposed. (d and e) Zoom-in on two regions of different chirality, extracted from the STM image in (a), overlaid with the proposed structures. A dashed line between the panels indicates the mirror symmetry between α and β domains (images were rotated to display the mirror plane along the vertical direction). | ||
Around room-temperature (RT), the molecular assembly presents equilibrium fluctuations, due to the balance between energy minimization from chain coordination and loss of conformational and translational entropy,50,51 resulting in the continuous diffusion of single molecules and chains (indicated by the fuzzy regions in Fig. 2a). As previously reported by Wintterlin et al.,50 this fact, along with the absence of qualitative structural evolution over long periods, reveals that the system has reached thermodynamic equilibrium.
In order to clarify the molecular organization within the chains, such structures have been characterized by means of nc-AFM with a CO-functionalized tip.43,52 Since the molecules are not planar, it is not possible to fully resolve their structure by constant-height AFM imaging.53 However, the image in Fig. 2b reveals intra-molecular features of a dimer (indicated by arrows on a molecule in Fig. 2b), which allow resolving the geometric arrangement of the chemical bonds located at the two protruding lobes of the molecule. In Fig. 2b, due to the non-planar geometry of the bi-anthracene, the protruding lobes (indicated by arrows in Fig. 2b and c) exhibit repulsive (less negative Δf) contrast in the AFM images, while the parts closer to the substrate are not visible. By superimposing a model on the image and rotating in-plane the anthracene units until the model fits the bright features in the AFM image, the orientation of the molecule can be determined (see Fig. S1 in the ESI†). In this way, we extracted an angle of about 82° between the azimuthal orientation of the anthracene long axis and the chain direction (see Fig. S1 and S2c in the ESI†). This observation indicates that the molecular coordination involves aryl group interaction, in agreement with previously proposed models based on STM images.36 However, the distance between the molecules (1.2 nm) is larger than the one found in anthracene chains on Au (0.85 nm),44 suggesting the presence of bridging Cu adatoms,36,38 usually not visible in STM images.54–56 Since the adatoms are expected to sit closer to the substrate than the molecular protruding lobes, they cannot be detected by constant-height AFM. Moreover, the observed distorted shape of diffusing chains (see below) suggests an important intermolecular interaction, supporting metal–organic coordination. Fig. 2c shows the resulting structural model of the organometallic chain, in line with recent results presented by Schulz et al.38 In Fig. 2b, arrows indicate the protruding C–H bonds within the molecule. The bright features located closer to the centre of the molecule (see blue arrows in Fig. 2b and c) correspond to bonds pointing out-of-plane due to steric hindrance, more than the other visible C–H bonds (indicated by red arrows in Fig. 2b and c). Instead, the ones adjacent to the aryl groups cannot be distinguished, indicating a different height with respect to the substrate, as result of a lateral bending of the anthracene towards the Cu adatoms. A slight difference between the protruding lobes of the single molecule can be observed in the AFM image (see Fig. 2b), revealing an asymmetry in the adsorption configuration, reflected also in some STM images (see below). This effect may be attributed to the possible presence of Br atoms underneath the tilted anthracene units, as reported by Bronner et al.57
Concerning the expression of supra-molecular chirality, a closer inspection reveals that the chains exhibit a high enantiopurity, indicating the formation of 1D chiral organometallic structures. Interestingly, also the islands are usually composed exclusively of either α or β enantiomers, as shown in Fig. 2. In fact, upon deposition at 300 K, the concentration of chiral defects is less than 10%, revealing that chirality is preserved in the 2D molecular assembly. In fact, the chains in the molecular islands are found to be aligned only along two groups of three-fold symmetric directions (see Fig. S3 in the ESI†),37 which indicates the presence of two distinguished inequivalent phases (i.e. α and β). This differs from the self-assembly behaviour of methylthiolate complexes on Au(111),58 where, in order to reduce the steric repulsion, chains of alternating chirality couple, leading to the formation of racemic islands. In contrast, in our case, molecular recognition mechanisms steer a hierarchical transfer of chirality from molecules to chains and from chains to islands, suggesting the presence of enantioselective assembly processes acting at different scales.
To study the self-assembly mechanisms, we investigated the time-evolution of the molecular network at temperatures between 290 and 370 K. Under these conditions, the assembly processes are too fast to be properly imaged through conventional STM. For this reason, we exploited the high-speed imaging capability provided by the FAST module, which allows acquiring STM time-series up to video-rate.39 In this way, it is possible to resolve in real-time chemical reactions steps with atomic resolution.59,60
Fig. 3 shows the three assembly mechanisms captured by high-speed STM imaging. The process in Fig. 3a and b advances by attachment of a single enantiomer to an organometallic chain. The centre of Fig. 3a presents a chain composed of four molecules. After 108 ms (Fig. 3b), an additional enantiomer of the same chirality is found to be attached at the termination on the bottom. This process involves the formation of organometallic bonds and leads to the growth of enantiopure 1D structures. A second mechanism is also characterized by organometallic coordination, where an individual molecule attaches to a chain located at the edge of a 2D island (Fig. 3c and d). Fig. 3(e and f) shows yet another process, involving coupling between preformed organometallic chains. In the STM sequence, the chain first approaches and then attaches to the island edge. Due to the lack of lateral functional groups, such interchain coupling is expected to be mediated by van der Waals forces.61
 | ||
| Fig. 3 Self-assembly mechanisms. (a and b) Enantiomer attachment to chain termination [−1 V, 1.4 nA, 294 K, 27 ms per frame]; (c and d) enantiomer attachment to the kink of island edge [−1.5 V, 0.3 nA, 343 K, 100 ms per frame]. In each frame, relevant molecules are highlighted with coloured dashes and red arrows highlight the position of the enantiomer located at the end of the chain. (e and f) Interchain coupling [−2 V, 0.5 nA, 295 K, 100 ms per frame]. Blue arrow indicates the chain attachment. The difference in the monomer appearance between a and b and c–f panels is likely due to the tip-dependent STM contrast. | ||
All the mechanisms are highly enantioselective and work identically for both molecular chiralities. Concerning the organometallic coordination, the clear correlation between molecular chirality and chain orientation with respect to the Cu(111) crystallographic directions (see Fig. S3 in the ESI†) indicates an important molecule–substrate interaction,36,37 to which we attribute the main reason for the high enantioselectivity of the organometallic coordination process. In fact, the rather strong coupling with the substrate does not allow heterochiral molecules to attach to the chains, while still maintaining a favourable substrate adsorption site. These arguments also explain why defect formation has been observed only for the radical attachment to single chain (see Fig. S2 in the ESI†), but not through its coordination at island edges. In fact, as compared to the extended islands, single chains can access a larger number of adsorption configurations, owing to their higher surface mobility, allowing for the attachment of radicals with opposite chirality.
Conversely, the formation of enantiopure islands is given by two distinct contributions: attractive van der Waals interactions between the chains and molecule–substrate adsorption energy.
While the former leads to the formation of 2D networks, steering the interchain coupling, the latter stabilizes the 1D organometallic structures along specific directions according to their chiralities,37 thereby providing enantioselectivity to the process.
Concerning the nature of the assembly processes, qualitative considerations can be done based on the observed event rates. Above RT, all the mechanisms shown in Fig. 3 are found to be reversible, i.e. it is possible to observe both formation and breaking of the molecular connections. However, the one shown in Fig. 3e and f exhibits a considerably higher disruption rate, compared to the first two processes, indicating that interchain coupling is less stable than organometallic bonding. This difference in the coordination stability is responsible for the general hierarchical behaviour of the self-assembly.
In order to shed light on the hierarchical chirality transfer, we investigated in detail the interchain coupling process, since it guides the coordination of 1D chiral structures, giving rise to second-generation enantiopure networks.
Fig. 4a shows three chiral domains, one composed of α enantiomers and two of β ones. In Fig. 4b, a β chain (four molecules long, highlighted in green) has attached to the β domain edge in the top-left corner. In Fig. 4c and d, this chain first moves to the β edge in the bottom-right (Fig. 4c), then goes back to the initial one (Fig. 4d). Interestingly, within the timescale of STM measurements (100 ms per frame), exchange events of the β chain appear to occur between β island edges only. In an extended STM time-series (≈15 s), we observed the chain attaching/detaching for 16 times at the β molecular island, while no detectable heterochiral coupling event can be clearly identified. This indicates a remarkable preference for homochiral coupling to occur, favouring the formation of enantiopure islands.
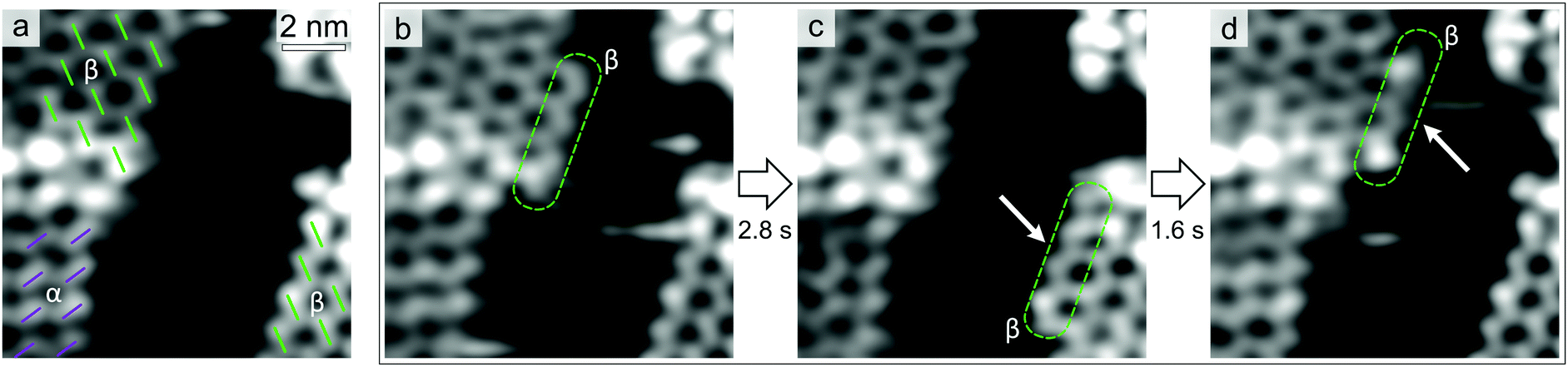 | ||
| Fig. 4 Enantioselectivity of interchain coupling. (a) STM image of three homochiral domains. The molecules are highlighted with coloured dashes according to their chirality. (b–d) Selected STM frames from a time-series. The exchanged chain is indicated in green. Arrows serve as a guide to the eye. [−1.6 V, 0.22 nA, 297 K, 100 ms per frame]. | ||
To better understand the molecular recognition interaction, we measured the residence times of different chains along the chiral edges. Fig. 5(a and c) shows two stable molecular islands (left: β island, right: α island), with two short oligomers of the opposite chirality moving in the separating region: a chain with three α enantiomers (marked with violet dashes in Fig. 5a) and one composed of three β ones (marked with green dashes in Fig. 5c). At RT, such short chains exhibit remarkable dynamics, resulting in a fast attachment/detachment process (see Movies in ESI†). For both the cases shown in Fig. 5(a and c), the chain position along the different molecular edges has been tracked by acquiring STM time-series with high temporal resolution (27 ms per frame). Despite the high mobility of the short chains, which prevents us from discriminating each exchange event, it is possible to quantify the probability to find the chain along the two edges, through the measurement of integrated residence time (IRT). Fig. 5(b and d) shows the average on 36 consecutive frames, after the subtraction of a background image (where the diffusing chain was not present), in which the preferential position of the moving chains turns to be enhanced compared with individual frames. In these images, the grey-scale contrast represents the IRT of the chain configurations with molecular resolution. Comparing the IRT intensities along the different island edges, it is possible to quantify the enantioselectivity of the interchain coupling process. For this purpose, we introduce the parameter S, which expresses the difference between IRT along the opposite homochiral edges:

 | ||
| Fig. 5 Enantioselectivity of interchain coupling. (a, c and e) STM frames extracted from extended movies acquired on the same region, for the case of: ααα, βββ and αββ chains, respectively. (b, d and f) IRT maps obtained from 36 consecutive frames (≈1 s) [−1 V, 1.4 nA, 294 K, 27 ms per frame]. | ||
For both the enantiopure chains, the main IRT features are localized along the edge of the same chirality, revealing a negligible adsorption along the heterochiral edge. S values of 0.99 and −0.96 indicate the high enantioselectivity of the interchain homocoupling process. Since the two configurations are symmetric due to geometrical reasons, the difference between the two reported absolute values of S is within the experimental error.
It is worth noting that, at the thermodynamic equilibrium, the residence-time distribution of different configurations reflects the relative binding energy difference. As mentioned above, for the system considered in this study, above RT, the large structural fluctuations combined with a lack of qualitative evolution over long times (up to few days) indicate that the equilibrium condition has been reached.50,51,62 This allows exploiting the IRT maps to estimate the binding energy difference between the homo- and heterochiral configurations. In fact, assuming the activation entropy to be the same for the two attachment processes, the ratio of the probability distribution can be expressed as:
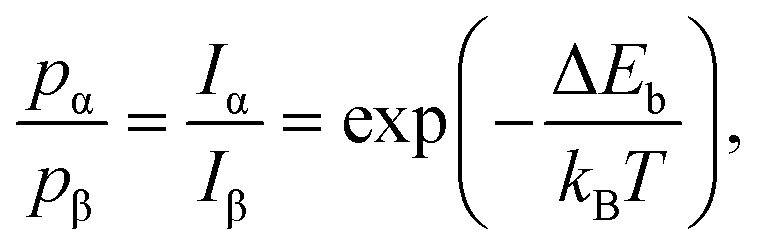
To further corroborate this argument, we examine the case of non-enantiopure oligomers. In Fig. 5e, the chain is composed of one α and two β molecules (details about the formation of defective organometallic chains are provided in the ESI†). In the corresponding IRT map (Fig. 5f), the absence of well-defined features along both island edges (S = −0.15) reveals a poor chain adsorption stability, highlighting how, for the case of short chains, the enantioselectivity of the process is importantly affected by the presence of chiral defects. This is in line with the thermodynamic control of the process selectivity. In fact, the (small) negative S value implies a higher adsorption binding energy along the β edge (in the order of 10 meV), which is expected given relative β excess in the chain composition (two β vs. one α). Despite the fact that the experimental uncertainty does not allow for a quantitative comparison between the homo- and heterocoupling energy gains, the negative S value indicates that, also for the case of non-enantiopure chains, the interaction is driven by thermodynamic arguments, steering the homochiral coupling.
Concerning the kinetic factors involved in the interchain coupling mechanism, in principle, relevant information can be obtained by measuring the attachment/detachment rates, since they are related to the difference in potential energy barriers between the homo- and heterocoupling configurations. However, the residence times along heterochiral edges are usually below our temporal resolution, preventing us from discriminating individual exchange events. Nevertheless, in the IRT maps of Fig. 5(b and d), the lack of a depletion region close to the heterochiral edge in the IRT maps suggests that the approaching barrier, if present, is at most comparable to the diffusion ones.
Conclusions
In summary, combining high-resolution nc-AFM and variable-temperature high-speed STM, we revealed the structures and self-assembly mechanisms of chiral organometallic networks on Cu(111). The molecule–substrate interaction is found to contribute importantly to the assembly enantioselectivity, dictating specific chirality-dependent orientations of the molecules. Through real-space imaging of molecular coordination at the ms time-scale, we observed hierarchical chirality transfer processes for the first time. By monitoring the molecular kinetics, we were able to evaluate the enantioselectivity of interchain coupling, providing information about energy gains and kinetic barriers involved in the chirality recognition process. These results represent a significant step forward in the understanding of enantioselective self-assembly, demonstrating the feasibility of monitoring complex chirality transfer mechanisms in real-time.Acknowledgements
We thank G. Troiano, S. Schillani, D. Giuressi, G. Cautero, P. Scheuerer, F. Albrecht and F. Berti for fruitful discussion and help. This project has received funding from the EU-H2020 Research and Innovation programme under grant agreement no. 654360 NFFA-Europe. Financial support from the Marie Curie Initial Training Network “MOLESCO” (no. 606728) is gratefully acknowledged. We thank Dr Friedrich Esch for his collaboration in the development and optimization of the Fast scanning module. Z. Z. acknowledges support by the “ICTP TRIL Programme, Trieste, Italy” in the framework of the agreements with the Elettra and CNR-IOM laboratories.References
- T. Verbiest, S. Van Elshocht, M. Kauranen, L. Hellemans, J. Snauwaert, C. Nuckolls, T. J. Katz and A. Persoons, Science, 1998, 282, 913–915 CrossRef CAS PubMed.
- J. McConathy and M. J. Owens, Prim. Care Companion J. Clin. Psychiatry, 2003, 5, 70–73 CrossRef PubMed.
- T. Mallat, E. Orglmeister and A. Baiker, Chem. Rev., 2007, 107, 4863–4890 CrossRef CAS PubMed.
- M. Heitbaum, F. Glorius and I. Escher, Angew. Chem., Int. Ed., 2006, 45, 4732–4762 CrossRef CAS PubMed.
- J. R. Cronin and S. Pizzarello, Science, 1997, 275, 951–955 CrossRef CAS PubMed.
- D. G. Blackmond, Cold Spring Harbor Perspect. Biol., 2010, 2, 1–17 Search PubMed.
- Z. Wang, K. Ding and Y. Uozumi, Handb. Asymmetric Heterog. Catal., 2008, 1–24 Search PubMed.
- H. U. Blaser, F. Spindler and M. Studer, Appl. Catal., A, 2001, 221, 119–143 CrossRef CAS.
- J. Weckesser, A. De Vita, J. V. Barth, C. Cai and K. Kern, Phys. Rev. Lett., 2001, 87, 96101 CrossRef CAS PubMed.
- S. Blankenburg and W. G. Schmidt, Phys. Rev. Lett., 2007, 99, 196107 CrossRef CAS PubMed.
- U. Schlickum, R. Decker, F. Klappenberger, G. Zoppellaro, S. Klyatskaya, W. Auwärter, S. Neppl, K. Kern, H. Brune, M. Ruben and J. V. Barth, J. Am. Chem. Soc., 2008, 130, 11778–11782 CrossRef CAS PubMed.
- R. Fasel, M. Parschau and K.-H. Ernst, Nature, 2006, 439, 449–452 CrossRef CAS PubMed.
- M. Lingenfelder, G. Tomba, G. Costantini, L. C. Ciacchi, A. De Vita and K. Kern, Angew. Chem., Int. Ed., 2007, 46, 4492–4495 CrossRef CAS PubMed.
- A. Schiffrin, A. Riemann, W. Auwärter, Y. Pennec, A. Weber-Bargioni, D. Cvetko, A. Cossaro, A. Morgante and J. V. Barth, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5279–5284 CrossRef CAS PubMed.
- A. Kühnle, T. R. Linderoth, B. Hammer and F. Besenbacher, Nature, 2002, 415, 891–893 CrossRef PubMed.
- R. Fasel, M. Parschau and K.-H. Ernst, Angew. Chem., Int. Ed., 2003, 42, 5178–5181 CrossRef CAS PubMed.
- J. V. Barth, J. Weckesser, G. Trimarchi, M. Vladimirova, A. De Vita, C. Cai, H. Brune, P. Günter and K. Kern, J. Am. Chem. Soc., 2002, 124, 7991–8000 CrossRef CAS PubMed.
- S. De Feyter, A. Gesquiere, F. De Schryver, C. Meiners, M. Sieffert and K. Mullen, Langmuir, 2000, 16, 9887–9894 CrossRef CAS.
- R. Otero, M. Schöck, L. M. Molina, E. Lægsgaard, I. Stensgaard, B. Hammer and F. Besenbacher, Angew. Chem., Int. Ed., 2005, 44, 2270–2275 CrossRef CAS PubMed.
- A. Kühnle, T. R. Linderoth and F. Besenbacher, J. Am. Chem. Soc., 2006, 128, 1076–1077 CrossRef PubMed.
- A. Kühnle, T. R. Linderoth and F. Besenbacher, J. Am. Chem. Soc., 2003, 125, 14680–14681 CrossRef PubMed.
- C. Bombis, S. Weigelt, M. M. Knudsen, M. Nørgaard, C. Busse, E. Lægsgaard, F. Besenbacher, K. V. Gothelf and T. R. Linderoth, ACS Nano, 2010, 4, 297–311 CrossRef CAS PubMed.
- S. Stepanow, N. Lin, F. Vidal, A. Landa, M. Ruben, J. V. Barth and K. Kern, Nano Lett., 2005, 5, 901–904 CrossRef CAS PubMed.
- H. Spillmann, A. Dmitriev, N. Lin, P. Messina, J. V. Barth and K. Kern, J. Am. Chem. Soc., 2003, 125, 10725–10728 CrossRef CAS PubMed.
- M. Böhringer, K. Morgenstern, W.-D. Schneider and R. Berndt, Angew. Chem., Int. Ed., 1999, 38, 821–823 CrossRef.
- M. Böhringer, W.-D. Schneider and R. Berndt, Angew. Chem., Int. Ed., 2000, 39, 792–795 CrossRef.
- V. Humblot, M. O. Lorenzo, C. J. Baddeley, S. Haq and R. Raval, J. Am. Chem. Soc., 2004, 126, 6460–6469 CrossRef CAS PubMed.
- M. C. Blüm, E. Ćavar, M. Pivetta, F. Patthey and W. D. Schneider, Angew. Chem., Int. Ed., 2005, 44, 5334–5337 CrossRef PubMed.
- D. Écija, K. Seufert, D. Heim, W. Auwärter, C. Aurisicchio, C. Fabbro, D. Bonifazi and J. V. Barth, ACS Nano, 2010, 4, 4936–4942 CrossRef PubMed.
- D. Bléger, D. Kreher, F. Mathevet, A. J. Attias, G. Schull, A. Huard, L. Douillard, C. Fiorini-Debuischert and F. Charra, Angew. Chem., Int. Ed., 2007, 119, 7548–7551 CrossRef.
- Y. Yang and C. Wang, Chem. Soc. Rev., 2009, 38, 2576–2589 RSC.
- W. Xu, R. E. A. Kelly, H. Gersen, E. Lægsgaard, I. Stensgaard, L. N. Kantorovich and F. Besenbacher, Small, 2009, 5, 1952–1956 CrossRef CAS PubMed.
- A. Dmitriev, H. Spillmann, M. Lingenfelder, N. Lin, J. V. Barth and K. Kern, Langmuir, 2004, 20, 4799–4801 CrossRef CAS PubMed.
- S. Katano, Y. Kim, H. Matsubara, T. Kitagawa and M. Kawai, J. Am. Chem. Soc., 2007, 129, 2511–2515 CrossRef CAS PubMed.
- S. M. Barlow, S. Louafi, D. Le Roux, J. Williams, C. Muryn, S. Haq and R. Raval, Langmuir, 2004, 20, 7171–7176 CrossRef CAS PubMed.
- K. A. Simonov, N. A. Vinogradov, A. S. Vinogradov, A. V. Generalov, E. M. Zagrebina, G. I. Svirskiy, A. A. Cafolla, T. Carpy, J. P. Cunniffe, T. Taketsugu, A. Lyalin, N. Mårtensson and A. B. Preobrajenski, ACS Nano, 2015, 9, 8997–9011 CrossRef CAS PubMed.
- P. Han, K. Akagi, F. F. Canova, H. Mutoh, S. Shiraki, K. Iwaya, P. S. Weiss, N. Asao and T. Hitosugi, ACS Nano, 2014, 8, 9181–9187 CrossRef CAS PubMed.
- F. Schulz, P. H. Jacobse, F. F. Canova, J. van der Lit, D. Z. Gao, A. van den Hoogenband, P. Han, R. J. M. Klein Gebbink, M.-E. Moret, P. M. Joensuu, I. Swart and P. Liljeroth, J. Phys. Chem. C, 2017, 121, 2896–2904 CAS.
- F. Esch, C. Dri, A. Spessot, C. Africh, G. Cautero, D. Giuressi, R. Sergo, R. Tommasini and G. Comelli, Rev. Sci. Instrum., 2011, 82, 53702 Search PubMed.
- For information on commercial availability of the FAST module contact http://ilo@elettra.eu.
- F. J. Giessibl, Appl. Phys. Lett., 2000, 76, 1470–1472 CrossRef CAS.
- T. R. Albrecht, P. Grütter, D. Horne and D. Rugar, J. Appl. Phys., 1991, 69, 668–673 CrossRef.
- L. Gross, F. Mohn, N. Moll, P. Liljeroth and G. Meyer, Science, 2009, 325, 1110–1114 CrossRef CAS PubMed.
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470–473 CrossRef CAS PubMed.
- A. Batra, D. Cvetko, G. Kladnik, O. Adak, C. Cardoso, A. Ferretti, D. Prezzi, E. Molinari, A. Morgante and L. Venkataraman, Chem. Sci., 2014, 5, 4419–4423 RSC.
- Q. Fan, C. Wang, Y. Han, J. Zhu, J. Kuttner, G. Hilt and J. M. Gottfried, ACS Nano, 2014, 8, 709–718 CrossRef CAS PubMed.
- W. Wang, X. Shi, S. Wang, M. A. Van Hove and N. Lin, J. Am. Chem. Soc., 2011, 133, 13264–13267 CrossRef CAS PubMed.
- M. Koch, M. Gille, A. Viertel, S. Hecht and L. Grill, Surf. Sci., 2014, 627, 70–74 CrossRef CAS.
- M. Di Giovannantonio, M. El Garah, J. Lipton-Duffin, V. Meunier, L. Cardenas, Y. Fagot Revurat, A. Cossaro, A. Verdini, D. F. Perepichka, F. Rosei and G. Contini, ACS Nano, 2013, 7, 8190–8198 CrossRef CAS PubMed.
- J. Wintterlin, J. Trost, S. Renisch, R. Schuster, T. Zambelli and G. Ertl, Surf. Sci., 1997, 394, 159–169 CrossRef CAS.
- S. Berner, M. Brunner, L. Ramoino, H. Suzuki, H. J. Güntherodt and T. A. Jung, Chem. Phys. Lett., 2001, 348, 175–181 CrossRef CAS.
- D. G. de Oteyza, P. Gorman, Y.-C. C. Chen, S. Wickenburg, A. Riss, D. J. Mowbray, G. Etkin, Z. Pedramrazi, H.-Z. Z. Tsai, A. Rubio, M. F. Crommie and F. R. Fischer, Science, 2013, 340, 1434–1437 CrossRef CAS PubMed.
- F. Albrecht, N. Pavliček, C. Herranz-Lancho, M. Ruben and J. Repp, J. Am. Chem. Soc., 2015, 137, 7424–7428 CrossRef CAS PubMed.
- U. Schlickum, R. Decker, F. Klappenberger, G. Zoppellaro, S. Klyatskaya, M. Ruben, I. Silanes, A. Arnau, K. Kern, H. Brune and J. V. Barth, Nano Lett., 2007, 7, 3813–3817 CrossRef CAS PubMed.
- N. Henningsen, R. Rurali, C. Limbach, R. Drost, J. I. Pascual and K. J. Franke, J. Phys. Chem. Lett., 2010, 2, 55–61 CrossRef PubMed.
- Z. Feng, S. Velari, A. Cossaro, C. Castellarin-Cudia, A. Verdini, E. Vesselli, C. Dri, M. Peressi, A. De Vita and G. Comelli, ACS Nano, 2015, 9, 8697–8709 CrossRef CAS PubMed.
- C. Bronner, J. Björk and P. Tegeder, J. Phys. Chem. C, 2015, 119, 486–493 CAS.
- O. Voznyy, J. J. Dubowski, J. T. Yates and P. Maksymovych, J. Am. Chem. Soc., 2009, 131, 12989–12993 CrossRef CAS PubMed.
- C. Africh, L. Köhler, F. Esch, M. Corso, C. Dri, T. Bucko, G. Kresse and G. Comelli, J. Am. Chem. Soc., 2009, 131, 3253–3259 CrossRef CAS PubMed.
- L. L. Patera, F. Bianchini, G. Troiano, C. Dri, C. Cepek, M. Peressi, C. Africh and G. Comelli, Nano Lett., 2015, 15, 56–62 CrossRef CAS PubMed.
- K. H. Ernst, Acc. Chem. Res., 2016, 49, 1182–1190 CrossRef CAS PubMed.
- C. Busse, S. Weigelt, L. Petersen, E. Lægsgaard, F. Besenbacher, T. R. Linderoth, A. H. Thomsen, M. Nielsen and K. V. Gothelf, J. Phys. Chem. B, 2007, 111, 5850–5860 CrossRef CAS PubMed.
- S. Kawai, A. S. Foster, T. Björkman, S. Nowakowska, J. Björk, F. Federici Canova, L. H. Gade, T. A. Jung and E. Meyer, Nat. Commun., 2016, 7, 11559 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp01341h |
| This journal is © the Owner Societies 2017 |