
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHill kinetics as a noise filter: the role of transcription factor autoregulation in gene cascades†
Anna
Ochab-Marcinek
 *,
Jakub
Jędrak
and
Marcin
Tabaka‡
*,
Jakub
Jędrak
and
Marcin
Tabaka‡
Institute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: ochab@ichf.edu.pl
First published on 25th July 2017
Abstract
An intuition based on deterministic models of chemical kinetics is that population heterogeneity of transcription factor levels in cells is transmitted unchanged downstream to the target genes. We use a stochastic model of a two-gene cascade with a self-regulating upstream gene to show that, counter to the intuition, there is no simple mapping (bimodal to bimodal, unimodal to unimodal) between the shapes of the distributions of transcription factor numbers and target protein numbers in cells. Due to the presence of the two regulations, the system contains two nonlinear transfer functions, defined by the Hill kinetics of transcription factor binding. The transfer function of the regulator can “interfere” with the transfer function of the target, converting the bimodal input into a unimodal output or vice versa. We show that this effect can be predicted by a geometric construction. As an example application of the method, we present a case study of a system of several downstream genes of different sensitivities, controlled by a common transcription factor which also regulates its own transcription. We show that a single regulator can induce qualitatively different patterns (binary or graded) of responses to a signal in different downstream genes, depending on whether the sensitivity regions of the transfer functions of the upstream and downstream genes overlap or not. Alternatively, the same model can be interpreted as describing a single downstream gene that has different sensitivities in different cell lines due to mutations. Our model shows, therefore, a possible kinetic mechanism by which different genes can interpret the same biological signal in a different manner.
1 Background
Understanding the contributions of the basic building blocks of gene regulatory circuits is an important step towards the knowledge of the design principles of larger gene networks. This step cannot be missed out because even the most simple network motifs may produce counter-intuitive outputs due to the interplay of the stochasticity and nonlinearity of biochemical reactions.In our earlier work,1 we have shown the possibility of such effects in as simple a system as a two-gene regulatory cascade without feedback and with even only one transcription factor binding site. Due to the cell-to-cell variability of transcription factor (TF) levels, different cells have different propensities to transcribe2 from the target gene. Our model has shown that, counter to intuition, the unimodal distribution of TF numbers may give rise to a bimodal distribution of target protein numbers, resulting in a division of a genetically identical cell population into two phenotypically different subpopulations. This effect of noise filter-induced bimodality occurred due to the nonlinear kinetics of the binding of the TF to the operator of the target gene. It should be noted that only one transfer function, defined by Hill kinetics, was present in the above model because the TF was able to bind only to the target operator. In the present paper, the regulatory gene additionally regulates its own transcription, which introduces the second transfer function describing the binding of the TF to its own operator, which makes the behaviour of the system more complex.
It was shown in experimental studies that similar mechanisms of noise filtering by nonlinear kinetics, which distort the input distribution and produce a bimodal output, may be present in the MAPK/ERK signalling network in human embryonic kidney cells3,4 and in the hypoxia-inducible factor-mediated response network in HCT116 cells.5 However, the input–output pathways in these systems were composed of multiple intermediate steps of a rather complex5 or not fully known4 topology, which opens the question of how the particular building blocks of a pathway, being themselves nonlinear, contribute to the final shape of the distribution of protein levels.
Recently, Sherman et al.2 demonstrated that the cell-to-cell variability of transcription rates is the main source of extrinsic noise for the SSA1 promoter in Saccharomyces cerevisiae. This observation supports the importance of theoretical models that capture the distribution of TF levels in cells. A noteworthy point in their analysis is that it indicates fast switching between the active and inactive states of the promoter. Fast on/off switching has also been recently observed in the lysogeny maintenance promoter of phage lambda in Escherichia coli.6 These findings seem to support the use of Hill kinetics in the modelling of promoter activity, as we do in the present paper.
Noise propagation through gene cascades has been usually studied in terms of noise measures such as the Fano factor or coefficient of variation,7,8 but less attention has been devoted to the study of how the shapes of protein number distributions vary as the levels of an external signal, regulating an activity of TFs, are varied. Such variation may give rise to a binary response (the distribution of target protein numbers changing from unimodal through bimodal to unimodal) or a graded response (the distribution remaining unimodal with its peak shifting as the signal intensity changes). It has been observed that the same gene system can have binary or graded responses to the same stimulus under different environmental conditions (e.g., Gal1 regulated by Gal4 and Mig1, in response to glucose9) and that the binary or graded response of an upstream component of a network does not necessarily translate itself into the same shape of the response of downstream components (e.g., the behaviour of the MAPK cascade module is itself ultrasensitive but some MAPK pathways have a graded response to a stimulus, whereas the response of others is binary;10 on the other hand, some regulators of type III secretion in Salmonella exhibit a graded response but the response of their targets is binary;11 however, this was a dynamic response to a single level of the inducer). Causes of these phenomena are largely unknown.
In the present paper we study the onset of the binary and graded responses, induced by the Hill kinetics, in a simple model system of a two-gene cascade with an autoregulation of the upstream gene. Positive autoregulation of TFs is widespread; for example, it plays an important role in two-component systems (see ref. 12 for a recent extensive review) where the positive feedback allows a single regulator to control both the more and less sensitive promoters, with the more sensitive ones responding to the uninduced regulator (early response) and the less sensitive ones responding to the induced regulator (late response).
We ask whether the bimodal or unimodal expression of the self-regulating upstream gene is accurately transferred downstream or whether it is re-shaped by the nonlinear kinetics of the binding of the TF to the target. To answer this question, we will adapt the method of geometric construction, used previously to study cascades with an unregulated regulator1 and single autoregulated genes.13 As an example use of our approach, we will explore the case study of a system where a single, positively autoregulated upstream gene regulates multiple downstream genes. Using the method of geometric construction, we will test whether the downstream genes can have different types of responses, binary or graded, to varying levels of a signal that activates the TFs.
2 Theory & model
2.1 Distribution of transcription rates
The binding of a TF to an operator can be described by a Hill function, under the assumption of strong cooperativity and fast TF binding and unbinding: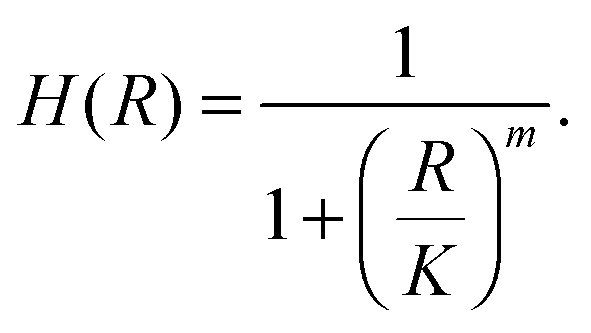 | (1) |
The transcription rate of the regulated gene is kmh(R), where km is the maximal gene transcription rate (at full activation/null repression) and h(R) is a relative transcription rate that depends on the number of TFs:13
| h(R) = H(R)(1 − ε) + ε. | (2) |
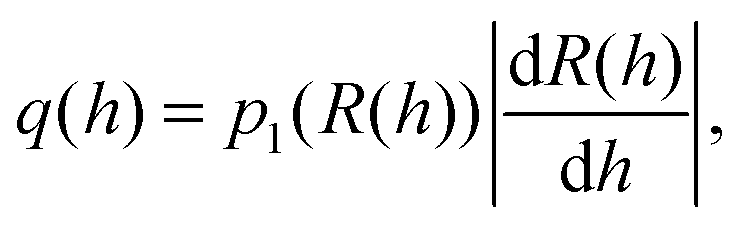 | (3) |
2.2 Model: cascade with an autoregulated upstream gene
Our model system consists of gene 1 (regulatory and autoregulated) and gene 2 (target), as shown in Fig. 1 and Table S2 (ESI†). The binding of the TF to the operators of the i-th gene is described by Hill kinetics with the transfer function hi(R) as in eqn (2), signal parameter Ki, leakage εi = kmli/kmi, and cooperativity n (upstream gene) or m (downstream gene), see the ESI,† Section S1. Gene 1 produces an input distribution of TF numbers, p1(R), which is transferred through the nonlinear filter h2(R) and as a result it produces an output distribution of transcription rates q(h2) of gene 2.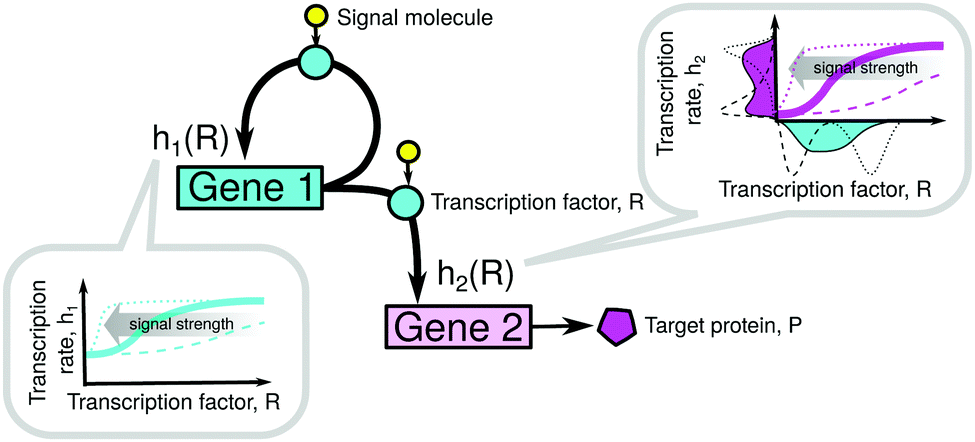 | ||
| Fig. 1 Model system of a two-gene cascade where the upstream gene is self-regulating. Binding of the transcription factor (TF) to both operators is governed by Hill kinetics described by the transfer functions h1(R) and h2(R) that depend on the number of TF molecules, R. The shape of the transfer function depends on the intensity of an external signal (e.g. concentration of effector molecules or phosphorylation) which activates a fraction of TFs. The distribution of TF numbers in a cell population is nonlinearly transformed by the transfer function h2 into a distribution of transcription rates of the downstream gene, which results in a distribution of the number of target proteins produced by that gene. | ||
The assumption of Hill kinetics is often made in gene expression modelling.14,15 Our method is suitable for the analysis of systems where Hill kinetics are conserved in both genes, and where the timescales are separated: TF activation/deactivation and the dominating rates of TF binding/unbinding are assumed to be greater than other reaction rates (steady state approximation). In the simulations shown in this paper, we have used approximately the lowest TF binding/unbinding rates at which the system still behaves as predicted by our model, see Fig. S1 (ESI†). Recent experimental data indicate that promoters in S. cerevisiae2 and E. coli6 can exhibit fast on/off switching (in particular, faster than mRNA degradation6), which seems to be consistent with our assumption of Hill kinetics. In principle, a reasonable assumption for our model of noise filter to work is that the downstream promoter should experience an approximately constant TF level compared to the timescale of its own transcription. Interestingly, in our simulations this assumption is somewhat relaxed – while the fluctuations of the total number of TFs are indeed slower than the fluctuations in the target mRNA number, the fluctuations of the number of active TFs, which can bind the operator, are much faster than the mRNA noise (see the ESI,† Section S7 and Fig. S2). In spite of that, the results show a very good agreement with the theoretical predictions, probably because the active–inactive TF cycling is so fast that the target promoter sees only its mean value (adiabatic regime).
Since both genes share a common TF, both Ki values are inversely proportional to the signal-dependent active fraction of TFs (eqn (S5), ESI†), under the assumptions of our model, i.e. strong cooperativity and negligible binding of inactive TFs. K1 is, therefore, proportional to K2 at any signal level, so the signal strength can be measured by one of these parameters.
2.3 Intrinsic noise representation
In the next sections, we will study the behaviour of the transcription rate distribution q(h2). Here, we give an argument why it is sufficient to analyse q(h2) instead of the full distribution of target protein numbers in systems where the intrinsic noise generated by the downstream gene is relatively low.1 The full distribution reflects the distribution of transcription rates q(h2) from the target gene, but the stochastic processes of mRNA and protein bursting add an overlay of intrinsic noise g(P;km2h2), which blurs the distribution's shape. The distribution of target protein numbers is given by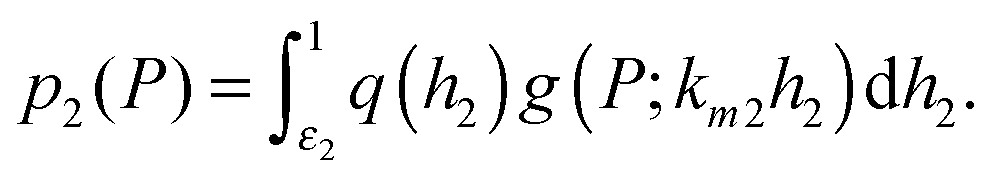 | (4) |
In this paper, we exploit the fact that when the intrinsic noise of the target gene is sufficiently small compared to the noise introduced by the non-homogeneous distribution of TF numbers among cells, then the transcription rate distribution itself gives significant information about the shape of the target gene expression. Under the assumption that the lifetime of target mRNA is shorter than the lifetime of the target protein,16g can be a negative binomial distribution, as used in ref. 1, or a gamma distribution as its continuous limit17,18 (see the ESI,† Section S2). Then, the intrinsic noise of the target gene decreases as the mean burst frequency a = km2/kdp2 increases (kdp2 being the target protein degradation rate). For large a, the functions g(P;km2h2) are sufficiently narrow, such that p2(P) can be approximated1 by the rescaled distribution of transcription rates, 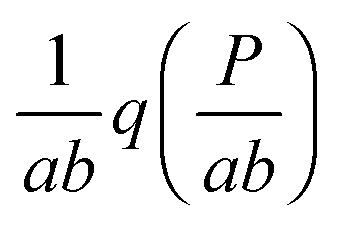 (b = kp2/kdm2 being the mean burst size, with the target protein production rate kp2 and target mRNA degradation kdm2; see the ESI,† Section S2). In the following sections, we will demonstrate this by comparison of the distributions of transcription rates q(h2) with simulated distributions of protein numbers, obtained from mesoscopic simulations using the Gillespie algorithm19 (see the ESI,† Section S6), at protein levels typical for E. coli20,21 (see the ESI,† Section S7, for details) and at rate constants that give rise to low intrinsic noise. The degradation rates of both TFs and target proteins are assumed to be equal, as in bacteria, where dilution and cell division are the dominating processes that decrease cellular protein concentrations.22
(b = kp2/kdm2 being the mean burst size, with the target protein production rate kp2 and target mRNA degradation kdm2; see the ESI,† Section S2). In the following sections, we will demonstrate this by comparison of the distributions of transcription rates q(h2) with simulated distributions of protein numbers, obtained from mesoscopic simulations using the Gillespie algorithm19 (see the ESI,† Section S6), at protein levels typical for E. coli20,21 (see the ESI,† Section S7, for details) and at rate constants that give rise to low intrinsic noise. The degradation rates of both TFs and target proteins are assumed to be equal, as in bacteria, where dilution and cell division are the dominating processes that decrease cellular protein concentrations.22
3 Results
3.1 Geometric construction predicts whether uni/bimodal expression of the upstream gene is transferred to the downstream gene
Under the biologically justified assumption of fast mRNA degradation and exponentially-distributed sizes of protein bursts, the distribution of the number of TFs produced by self-regulating gene 1 among different cells is given by13,18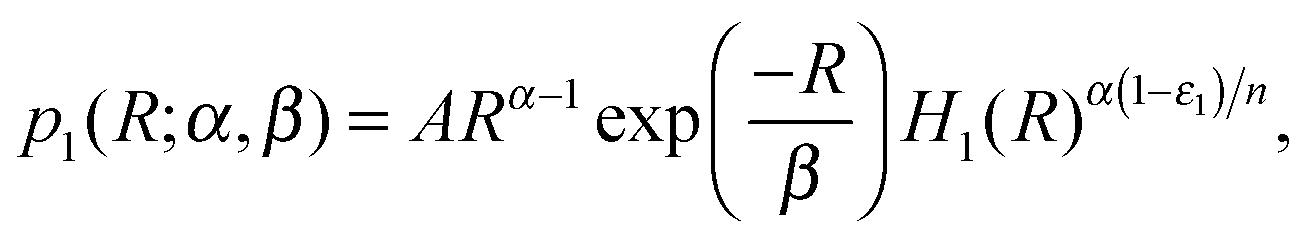 | (5) |
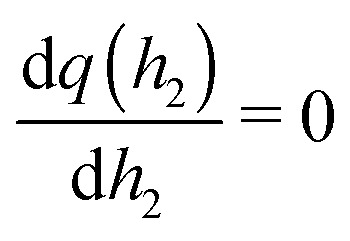 . As a result, we obtain the geometric construction (see the ESI,† Section S3.2, for derivation):
. As a result, we obtain the geometric construction (see the ESI,† Section S3.2, for derivation):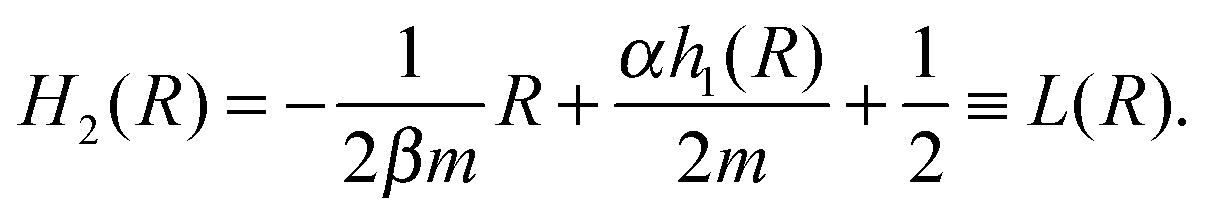 | (6) |
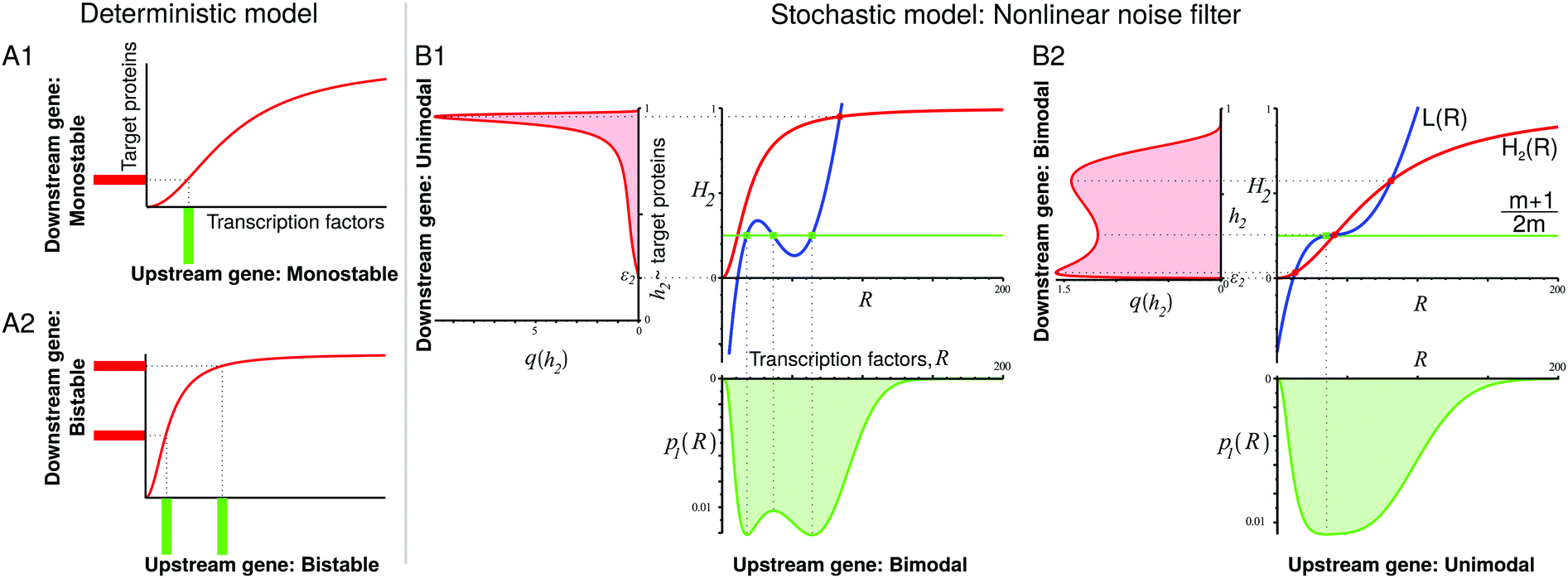 | ||
| Fig. 2 In deterministic models of gene cascades, a single steady state of TF levels is mapped onto a single steady-state response of the downstream gene (A1), and a bistable input leads to a bistable output (A2). Contrastingly, there is no such simple input-to-output mapping in our stochastic model (B). Here, the downstream gene's transfer function transforms the input distribution of TF levels in a nonlinear manner, possibly changing the number of its maxima: a bimodal input may lead to a unimodal output (B1), or a unimodal input may give rise to a bimodal output (B2). H2(R) is the rescaled transfer function of the downstream gene. L(R) contains the transfer function of the upstream gene. The intersections of H2(R) and L(R) indicate the minima and maxima of the transcription rate distribution of the downstream gene. The intersections of the green line (m + 1)/(2m) with L(R) indicate the minima and maxima of the distribution of TF numbers. Parameters: (B1) n = m = −2, α = 60, β = 2, ε1 = 0.1, ε2 = 0.2, K1 = 64, K2 = 19.2. (B2) n = m = −2, α = 25, β = 5, ε1 = 0.15, ε2 = 0.01, K1 = K2 = 70. | ||
After the change of the sign of the downstream gene regulation, m → −m, the geometric construction and the transcription rate distribution q(h2) become mirror images of those for m, such that h2 → −h2 + ε2 + 1. This means that the positive or negative regulation of the downstream gene does not change the bimodality or unimodality of the distribution.
We can also compare on one plot (Fig. 2B1 and B2) the properties of the distribution p1(R) of TF numbers and the distribution of transcription rates q(h2) of gene 2. The extrema of p1(R) are also given by a geometric construction, which is a transformed version of that shown in ref. 13 (see the ESI,† Section S4, for derivation):
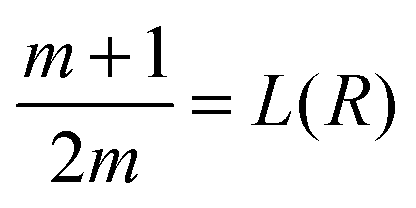 | (7) |
Within the deterministic description, there should be a simple mapping: one steady state of the transcription of the upstream gene produces one steady state of the transcription of the downstream gene, and bistability produces bistability (Fig. 2A). However, if the expression levels of the upstream gene are randomly distributed in the cell population, then the output of the downstream gene depends both on the shape of the input distribution of TF numbers and on the shape of the transfer function h2 which describes the binding of those TFs to the operator.
In Fig. 2B we show example constructions for cascades with positively autoregulated TFs. When the upstream gene has a bimodal expression but the downstream gene is more sensitive to TFs, then the bimodal distribution of TF numbers transforms itself into an almost maximal expression of the downstream gene. The nonlinearity of the transfer function h2 results in the amplification of one peak of the TF number distribution, whereas the other peak does not contribute much to the transcription rate distribution and thus it does not produce a second peak of q(h2) (Fig. 2B1). When the upstream gene has a unimodal expression but it has the same sensitivity to TFs as the downstream gene, then the overlap of the sensitivity regions of the transfer functions of both genes may cause the noise filter-induced bimodality of the expression of the downstream gene (Fig. 2B2). In the ESI,† we compare the above examples from Fig. 2B with their deterministic counterparts, and show that their behaviour is different (Section S9, ESI†). We also analyse the dependence of distribution shapes on the amount of leakage. Here, in most cases, we have assumed a significant basal transcription, as it is frequently observed in wild-type genes,23,24 see also ref. 13. Our geometric construction suggests that the significant amount of leakage in the regulatory gene may lead to a greater diversity of behaviours of the studied gene cascade (Section S10, ESI†).
In some exotic cases, at very high cooperativities, trimodal distributions are also possible (Fig. S15, ESI†); however, the multiple peaks are then very close to each other, such that they may be easily blurred by intrinsic noise.
3.2 Example application: multiple downstream genes regulated by a common positively self-regulated TF can have both binary and graded responses to the same signal
The analysis of the geometric construction at all possible combinations of the parameter values (cooperativities of both genes, their TF affinities, protein burst parameters α and β, different levels of leakiness of the promoters) is beyond the scope of this article. Therefore, we present in this section a selected case study of a system in which the regulatory gene responds in a graded manner to a signal but the interplay of the non-linearities of the upstream and downstream regulation can introduce both graded and binary responses in the downstream genes, an effect which would be impossible to obtain in the same system upon removal of the feedback.We consider an upstream gene producing TFs that regulate several downstream genes, whose promoters have different affinities for TFs (Fig. 3A). The diversity in promoter–TF affinities may be due to different promoter architectures and also due to the gene surroundings (DNA conformation or the presence of other transcription factors25,26). We model the different affinities by different K1/K2 ratios. We assume that the number of TFs is large enough so that the downstream promoters do not compete for TFs. We want to know how the genes respond to an external signal that effectively varies K1 and K2. Since K1 is proportional to K2 under our assumptions, we simply measure the signal strength using one of these parameters (e.g. K1 proportional to 1/(active TF fraction), i.e. 1/signal, see eqn (S5), ESI†), similarly to that in ref. 13. We ask whether the shape of the TF response to varying levels of the signal is reproduced downstream and, if not, how it depends on the differences between the TF affinities of the upstream and downstream genes. This problem can also be interpreted in terms of robustness to mutations: if the upstream gene regulates a single downstream gene (Fig. 3B), how will the response pattern change after a mutation in the downstream promoter that changes its affinity to TFs?
 | ||
| Fig. 3 Self-regulating upstream gene, regulating downstream genes that have different sensitivities to TFs: single regulator for several downstream genes (A). An alternative interpretation of the model: a single cascade, where the promoter–TF affinity of the downstream gene varies from cell to cell due to mutations (B). | ||
In Fig. 4, we show the geometric construction which predicts the positions of the minima and maxima of the distributions of protein levels for both TFs and target proteins. The construction demonstrates that the number of maxima of the protein number distributions depends on the overlap (or lack thereof) of the sensitivity regions of the transfer functions of both genes. The corresponding distributions are shown in Fig. 5 and their behaviour is in excellent agreement with the predictions from Fig. 4.
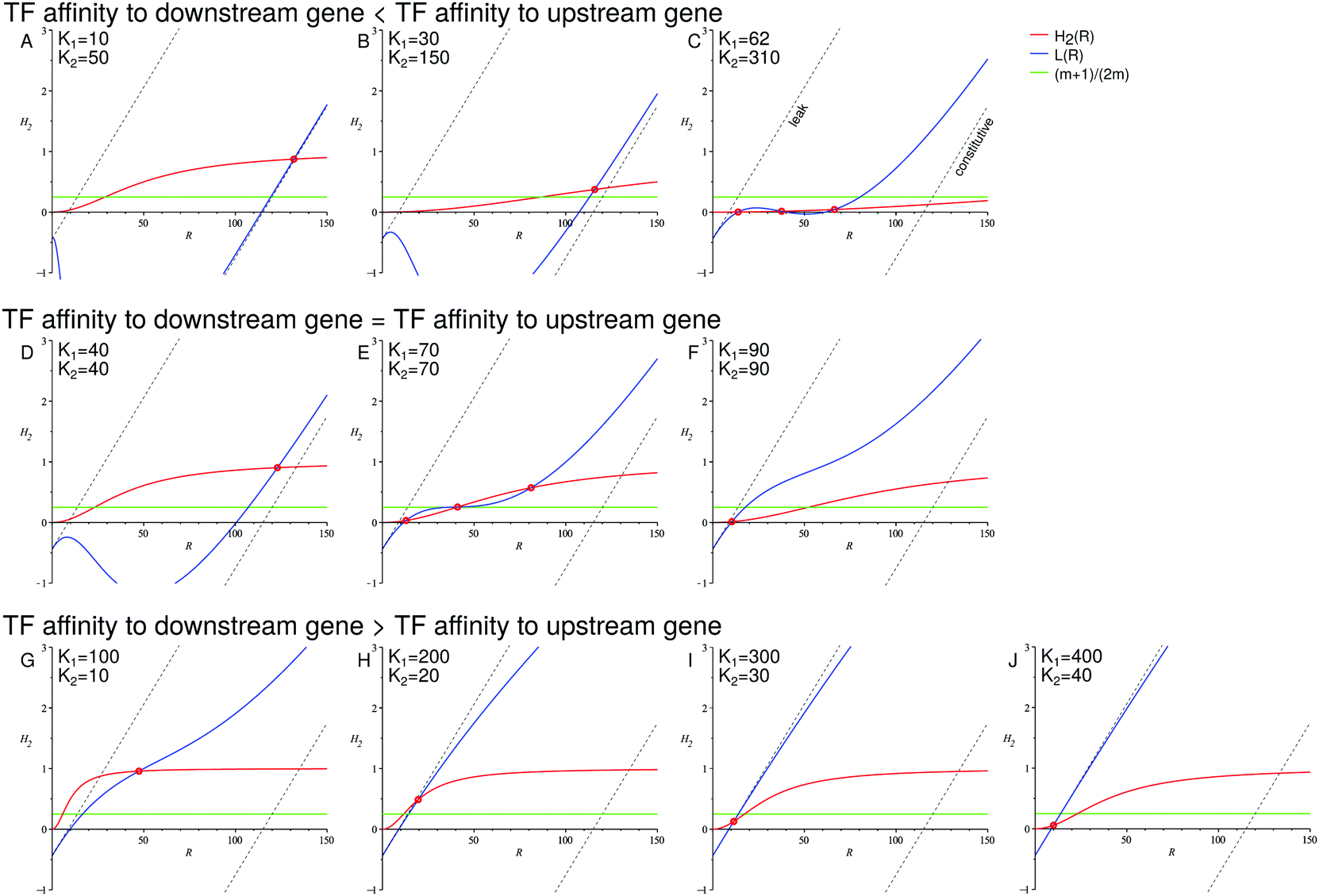 | ||
| Fig. 4 The overlap of the sensitivity regions of the transfer functions of both genes facilitates the onset of a binary response. The intersections of the red curve H2(R) and the blue curve L(R) indicate the minima and maxima of the downstream gene's transcription rate distribution. The intersections of the green line (m + 1)/(2m) and the blue curve L(R) indicate the minima and maxima of the TF number distribution. Dashed lines: limits of the leaky expression and constitutive expression. Intersections of the dashed straight lines and the red curve H2(R) correspond to the extrema of the transcription rate distributions of the downstream gene when the upstream gene has no feedback (left dashed line: minimal expression of the upstream gene; right dashed line: maximal expression of the upstream gene). Parameters: n = −2, m = −2, α = 25, β = 5, a = 250, b = 5, ε1 = 0.15, ε2 = 0.01. Constructions shown in A–C correspond to the distributions shown in Fig. 5A and D. Constructions D–F correspond to Fig. 5B and E. Constructions G–J correspond to Fig. 5C and F. | ||
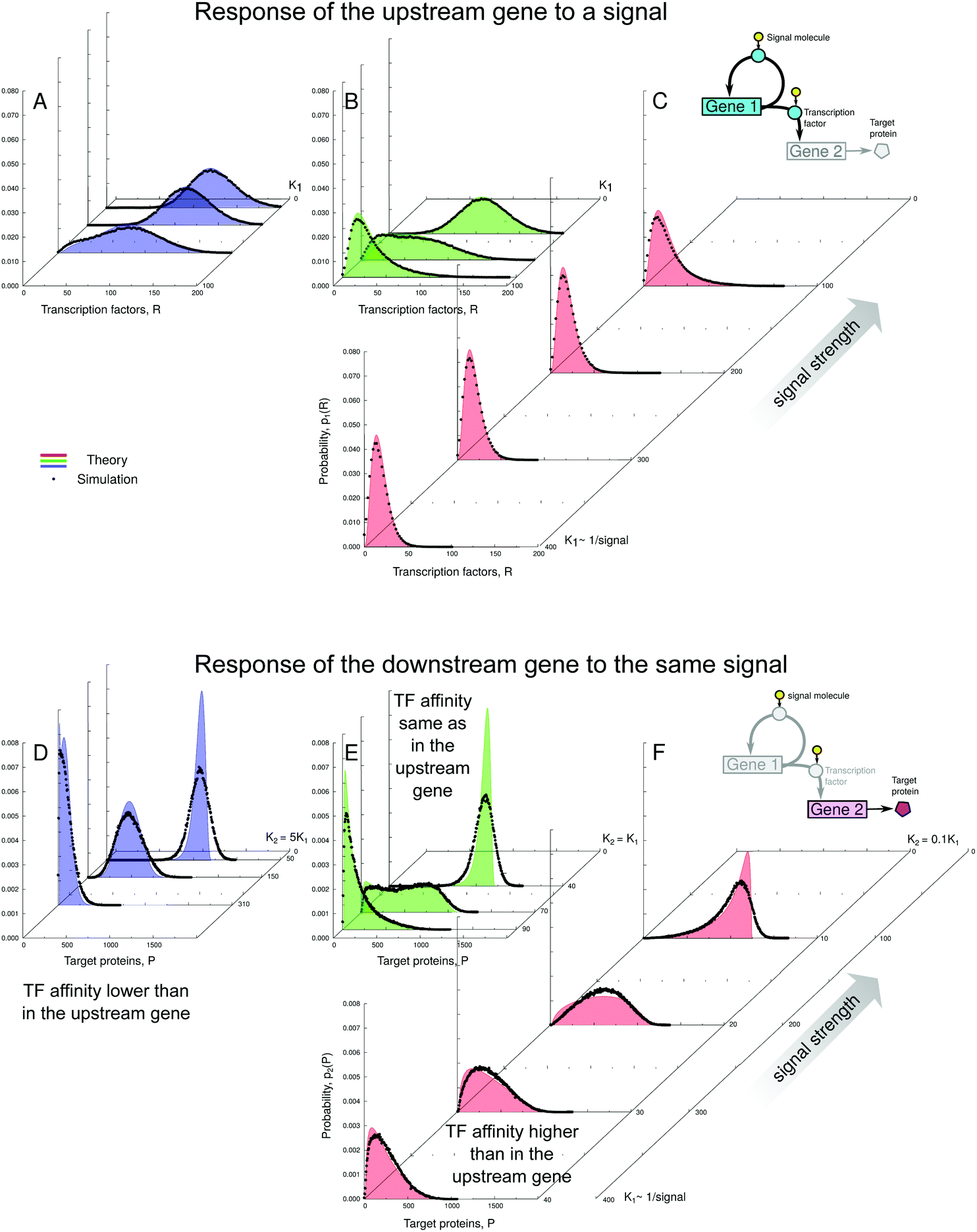 | ||
Fig. 5 Response of the self-regulating upstream gene to a varying signal (shown in A–C by the total number of TFs, both active and inactive) is not simply mirrored by the downstream gene (D–F). The response of the downstream gene can be binary or graded, which depends on whether the sensitive regions of the transfer functions of both genes overlap or not (see the geometric construction in Fig. 4 and explanations in the text). Distributions shown in A and D correspond to constructions shown in Fig. 4A–C. Distributions shown in B and E correspond to constructions shown in Fig. 4D–F. Distributions shown in C and F correspond to constructions shown in Fig. 4G–J. Black dots: simulated protein distributions p2(P). Filled curves: theoretical distributions, p1(R) in (A–C), and 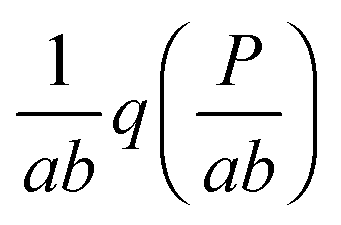 in (D–F). The theoretical curve for K2 = 310 in (D) has two maxima but the bimodality disappears in the simulation due to intrinsic noise (see the zoomed-in view in Fig. S16, ESI†). The parameters are the same as in Fig. 4. The values of simulation parameters are given in Table S3 (ESI†). in (D–F). The theoretical curve for K2 = 310 in (D) has two maxima but the bimodality disappears in the simulation due to intrinsic noise (see the zoomed-in view in Fig. S16, ESI†). The parameters are the same as in Fig. 4. The values of simulation parameters are given in Table S3 (ESI†). | ||
We have chosen a set of parameters such that the upstream gene shows a graded response to the signal when self-regulated, whereas the downstream gene shows a binary response in the presence of that autoregulation. However, upon removal of the feedback in the upstream gene, the downstream gene would exhibit a graded response (shown in detail in the ESI,† Section S8); for the minimal and constitutive expression of the open-loop regulatory gene, the extrema of the downstream gene's transcription rate distributions are given by the intersections of the dashed straight lines and the red curve H2(R) in Fig. 4.
The downstream genes have different promoter–TF affinities relative to the upstream gene: K2 = 0.1K1, K1, 5K1. The noise 1/a of the downstream gene is low. We measured the signal strength using the parameter K1. We assumed that the cooperativity of the TF binding is n = −2 for the upstream gene (positive regulation), and the downstream gene can be positively or negatively regulated with m = ±2 (note that homodimerisation is typical for regulators in two-component systems12).
In our example system, the TF response is graded, although at the intermediate signal strength (K1 = 70) it becomes wide, nearly bimodal, which is represented by the blue curve L(R) almost tangent to the green straight line (m + 1)/(2m) (Fig. 4E).
At the same time, the response pattern of the downstream genes depends on their promoter–TF affinity and can be binary or graded (Fig. 5D–F):
(1) When the downstream gene has the same affinity to the TF as the upstream gene (K2 = K1, Fig. 5E), its response becomes binary. The downstream transfer function transforms the wide unimodal input distribution into a clearly bimodal output distribution.
(2) However, if the downstream gene is much more sensitive to the TF than the upstream gene (K2 = 0.1K1, Fig. 5F), its response becomes graded. The geometric construction reveals that the upstream gene remains almost fully inactivated, with only a low expression level caused by the leakiness of its promoter. Depending on the signal strength, a certain fraction of this small number of TFs becomes activated and only the downstream gene with the strongest promoter responds sensitively to the variation of that fraction. The behaviour of the system is close to that of the cascade with the minimally active upstream gene without feedback (compare with Fig. S4, ESI†).
(3) An interesting case is the one of the downstream gene whose promoter is less sensitive to the TF than the regulatory promoter (K2 = 5K1, Fig. 5D). Here, the target response is, in principle, graded and sharper than that of the regulatory gene. The geometric construction shows that at a certain signal strength (K1 = 62) the transcription rate distribution should become slightly bimodal. However, this bimodality is “compressed” by the transfer function of the downstream gene, which is close to its lower limit. Moreover, the bimodality becomes completely blurred by intrinsic noise (see Fig. S16, ESI,† for a zoomed-in view).
The presence of TF autoregulation may allow different target genes to display diverse types of responses – not only early or late but also binary or graded – depending on their affinities to TFs, even though they are regulated by the same TF. In our example, this behaviour would be impossible if the upstream gene were unregulated.
On the other hand, it does not matter whether the regulation of the downstream gene is positive or negative: the sign of regulation changes the response direction but not its binary or graded shape.
3.3 Relative distribution width as a measure of imprecision of a gene's response to an external signal
Because we analyse the problem of a gene’s response to varying levels of a signal, the most natural way to view the distribution plots is to compare their width to the range of possible gene expression levels induced by the signal. The standard measures of noise, the coefficient of variation (standard deviation to mean, CV) and the Fano factor (variance to mean, FF), ignore the dependence on the signal range. We can see that the CV and FF are increased when the sensitivity regions of the transfer functions overlap (Fig. S17A and B, ESI†). But when the distributions are plotted in the scale which ranges from the minimal to maximal mean expression levels, the distributions do not appear to be the widest or maximally bimodal at their maximal CV or FF (ESI,† Section S13). For this reason, we propose the relative distribution width as the measure which can quantify the imprecision of the gene's response to a range of signal levels (greater width implies a less precise response):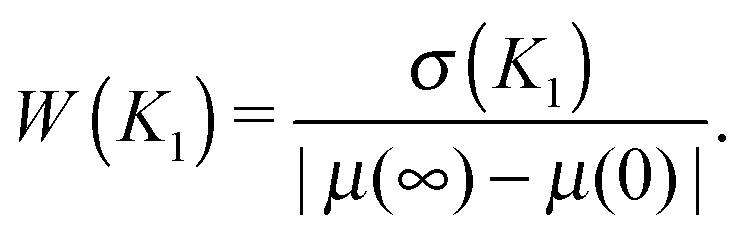 | (8) |
Fig. 6 shows that the precision of the downstream gene's response to the external signal is the greatest when the downstream gene is less sensitive to the TF than the self-regulating upstream gene (indeed, the distributions in Fig. 5 are the most narrow in case D). In general, the precision of the response depends on the contribution of the intrinsic noise of the downstream gene. If that noise is gamma-distributed, then the relative width of the distribution of target protein numbers differs from that of the distribution of transcription rates by a term dependent on the mean burst frequency a of the target gene (the derivation, using the intrinsic noise representation (eqn (4)), is presented in the ESI,† Section S14):
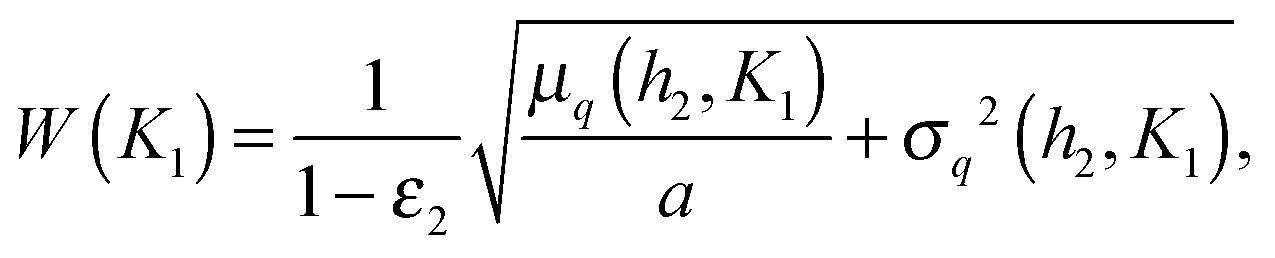 | (9) |
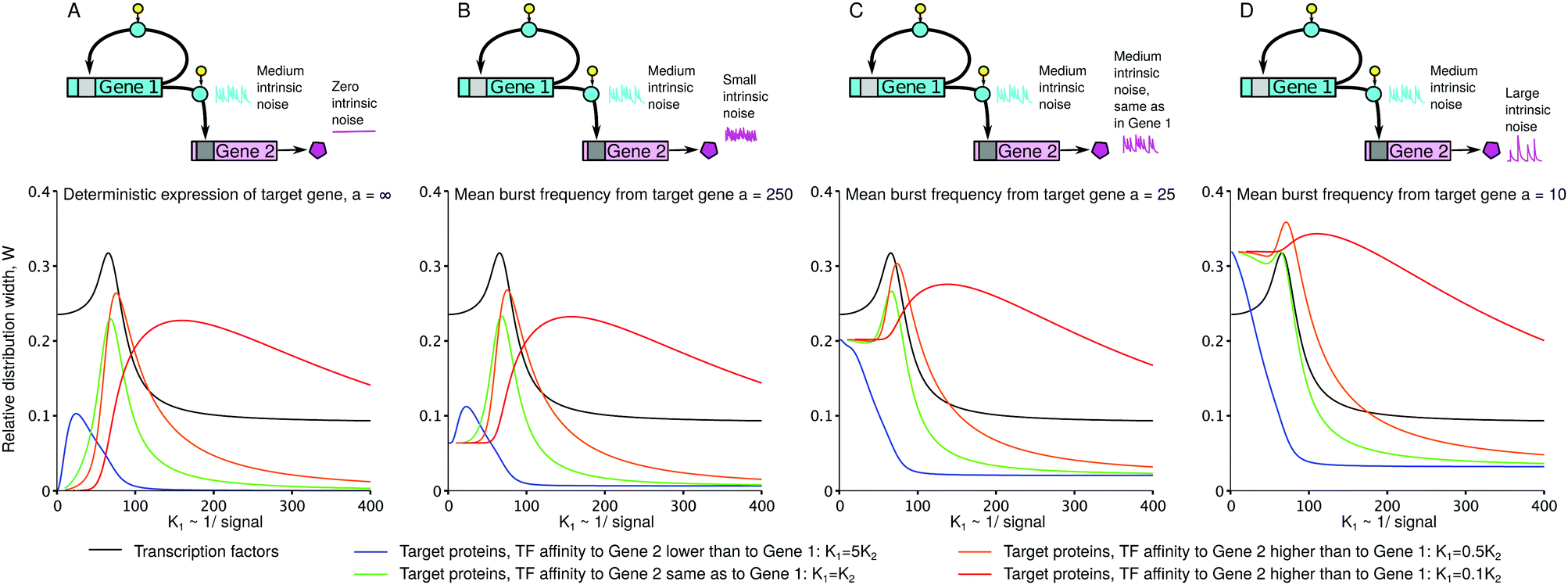 | ||
| Fig. 6 Imprecision W of the target's response may be smaller than that of its regulator (A–C). When the intrinsic noises of both the regulator and target are equal, the target's response may still be more precise (C) if the regulatory promoter is more leaky (which is the case for the parameter set chosen for this figure: ε1 = 0.15, ε2 = 0.01). Here, the target's response can be less precise than the regulator's response (D), when the target's intrinsic noise is greater than the regulator's noise. Intrinsic noises of the regulator and the target are defined by 1/α and 1/a, respectively. α = 25 and a = ∞, 250, 25, 10 are the maximal mean burst frequencies. The other parameters and the corresponding colours of lines are the same as in Fig. 4 and 5; we added the intermediate value of 0.5K1 (orange). W was numerically calculated using the analytical formulas for p1(R) (viaeqn (8), TFs) and q(h2) (viaeqn (9), target proteins). | ||
Interestingly, the response of the downstream gene to a signal may be more precise than the response of the upstream gene (Fig. 6). In particular, this is possible even when the intrinsic noise of the downstream gene is equal to the intrinsic noise of the upstream gene (Fig. 6C). In our example, the latter is due to the high leakage ε1 of the upstream gene, which confines the αβ(1 − ε1) range of its mean expression.
3.4 Within the tested parameter range, the model can describe cell systems where the TF and target protein lifetimes are equal
We tested different target protein lifetimes, corresponding to the protein degradation rates kdp2. Fig. 7 shows that there are two constraints on the target protein lifetime for our analytical method to be valid. (i) If the target protein lives much longer than the TF, then its levels average out even the slow fluctuations of the transcription rate (magenta histogram in Fig. 7). (ii) If the target protein degrades very quickly, then its fluctuations become correlated with the on/off switching of the promoter, which generates bimodality due to transcriptional pulsing:1 the left peak of the resulting distribution (red and green histograms in Fig. 7) is shifted to zero, which corresponds to the “off” state of the promoter. These results demonstrate that, at least for this choice of parameters, the model can be valid for cell systems where the TF and target protein lifetimes are the same, as in bacteria, where the main factors that decrease protein levels are dilution and cell division.22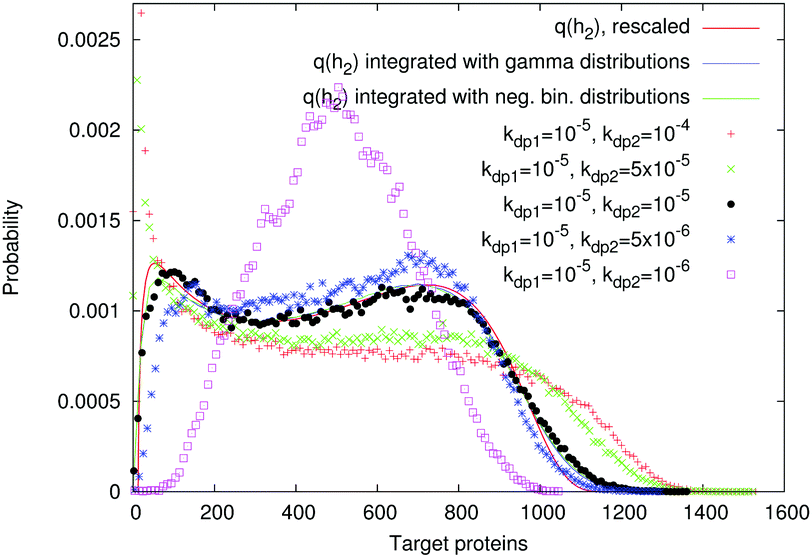 | ||
Fig. 7 Simulated protein number distributions for a cascade with a self-regulated TF quite faithfully reflect the rescaled transcription rate distribution 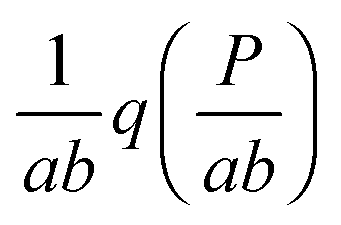 . Theoretical curves, which represent q(h2) integrated with the corresponding gamma (S9, ESI†) or negative binomial (S40, ESI†) distributions, show that the intrinsic noise is sufficiently low to compare the simulated distributions with the sole transcription rate distribution. The TF degradation rate is kdp1 = 10−5. We tested different degradation rates of the target protein, as shown in the legend. Parameters: n = −2, m = −2, α = 25, a = 250, β = b = 5, ε1 = 0.15, ε2 = 0.01, K1 = K2 = 70. The values of other parameters are shown in Table S7 (ESI†). . Theoretical curves, which represent q(h2) integrated with the corresponding gamma (S9, ESI†) or negative binomial (S40, ESI†) distributions, show that the intrinsic noise is sufficiently low to compare the simulated distributions with the sole transcription rate distribution. The TF degradation rate is kdp1 = 10−5. We tested different degradation rates of the target protein, as shown in the legend. Parameters: n = −2, m = −2, α = 25, a = 250, β = b = 5, ε1 = 0.15, ε2 = 0.01, K1 = K2 = 70. The values of other parameters are shown in Table S7 (ESI†). | ||
4 Discussion
In the first part of this section (Section 4.1), we provide a summary of our theoretical findings. In the second part (Section 4.2), we discuss their possible relevance to known biological circuits.4.1 Conclusions
We consider the behaviour of a gene cascade in which the upstream gene is self-regulating, and the random distribution of transcription factor (TF) numbers in cells is transformed by the nonlinear kinetics of the binding of the TF to the operator of the downstream gene, which results in a distribution of target protein numbers. Depending on the strength of the external signal that activates the TFs, the expression of both regulatory and target gene changes not only in terms of its mean level but also in terms of its qualitative shape (unimodal or bimodal).The nonlinearity and cooperativity of biochemical reactions are important features modulating gene regulation.27–30 Due to the presence of the two regulations, there are two nonlinear transfer functions in the system, potentially having different sensitivity ranges. Altszyler et al.15 studied, using deterministic models, how the overlap (or lack thereof) of the sensitivity ranges of the upstream and downstream modules influences the sensitivity of the system. However, deterministic modelling assumes that if the TF expression is monostable or bistable, then this property is transmitted downstream in the cascade. We point out, on the other hand, that if TFs are randomly distributed among cells, then their distribution may be strongly distorted during transmission downstream and attain or lose bimodality.
In order to gain insight into this effect, we refer to the concept of intrinsic noise representation. This approach, which we previously used to study gene cascades without self-regulation,1 is similar to the Poisson representation,31,32 where the protein number distribution was represented by a superposition of Poisson distributions, or to the method applied by Thomas et al.,33 who used a mixture of Gaussian distributions. However, the distribution mixing in these references was a formal procedure facilitating calculations, without the clearly defined biological meaning of the distributions and the mixing functions. The advantage of our approach is that the representation of the protein number distribution in the form of a distribution mixture has a clear biological interpretation: differences in the number of TFs in the cell population lead to a different propensity to transcribe2 in each cell, which constitutes extrinsic noise. The distribution of target protein numbers can be obtained by integration of the distribution of transcription rates in the population (the mixing function) with the distributions of intrinsic noise (e.g., negative binomial or gamma) corresponding to each transcription rate.
From this conceptual approach, it follows that for those downstream genes which have low intrinsic noise we do not need to calculate the full distribution of target protein numbers. Instead, it suffices to analyse the behaviour of the mixing function, i.e., the transcription rate distribution. This allows us to look at the system in terms of nonlinear filtering of noise. The Hill kinetics of the binding of the TF to the operator of the target gene acts as a noise filter that converts the distribution of TF numbers into a different distribution of transcription rates from the target gene. We have shown that the numbers and positions of the minima and maxima of the transcription rate distribution can be found using a geometric construction even without the explicit knowledge of the distribution itself, analogously to the results for a cascade without feedback.1 However, differently from that in the latter case, the present construction contains information not only about the transfer function of the downstream gene (defined by the Hill kinetics of TF–target binding) but also about the transfer function of the upstream gene (describing TF autoregulation). The graphical method demonstrates how the upstream transfer function can “interfere” with the downstream transfer function, amplifying its sensitivity. It also shows whether the uni/bimodality of the upstream gene expression translates itself into the uni/bimodality of the downstream gene expression.
As an example, we have studied a cascade with positive autoregulation of TFs, such that the response of the upstream gene to a signal was graded and the response of the cascade with feedback removed was also graded. We have shown that the shape of the distribution of TF numbers is not simply transferred downstream. The response of the downstream gene can be binary or graded, which depends on whether the sensitivity regions of both transfer functions overlap. And therefore, one upstream gene, simultaneously regulating several downstream genes, can induce not only early or late responses in different downstream genes, but also binary responses in some of them and graded responses in others, depending on their sensitivity to TFs. Interestingly, these response patterns do not depend on whether the regulation of the downstream gene is positive or negative. The recent experimental paper by Rossi and Dunlop34 analysed, in a similar spirit, the diversity of the responses of different genes in E. coli (micF and inaA) to the same TF (MarA). However, their upstream gene was not self-regulated. They confirmed the intuitively expected fact that the noise transmitted to the downstream gene is the highest when the expression of the regulator falls on the sensitive region of its transfer function. The distribution shapes in response to signals were not studied in detail in ref. 34, but the micF response was visibly graded.
The same problem can be alternatively interpreted as a description of a single cascade, where the strength of the target promoter is changed by a mutation. Our model shows that such a mutation may change the pattern of the response of the target from binary to graded (or vice versa). Since cascades with self-regulating TFs are frequent network motifs, a question arises as to whether such a lack of robustness of the response shape to mutations is common in nature, and if so, whether it could itself be an adaptation to different environmental conditions: in some environments, the binary response may be more preferred, whereas other environments may select for graded responses. (However, an adaptation of the response shape by a mutation in promoter–TF affinity would come here at the expense of a change in the signal level at which the response occurs.) This problem is somewhat similar to that explored by Kuwahara et al.35 They have shown that gene systems tend to evolve in such a way that, on one hand, they favour an increased phenotypic diversity, and, on the other hand, their phenotypes undergo strong changes under small genotypic variations. In our model, mutations in promoter–TF affinity may cause significant changes in the pattern (binary/graded) of the gene response to a signal.
Within the tested parameter range, our model can work when the lifetimes of both the TF and target proteins are the same (as in most cases in bacteria).
Using the concept of intrinsic noise representation, we have proposed the relative distribution width as a new measure of the precision of a gene's response to a signal, which compares the distribution width to the range of possible responses. In this way, we demonstrated how the intrinsic noise of the target gene affects the response precision. If the regulatory gene has a low precision of response due to large leakiness, then the target gene with low leakage can increase that precision. The lower the affinity of the target promoter to the TF, the more precise the target gene's response.
4.2 Relevance to known gene systems
Some gene expression models, which have been used to interpret experimental data, assume that the bimodal expression pattern of a self-regulating upstream gene should propagate downstream to the regulated gene (e.g., in ref. 36 the self-regulating TF, GlnG, activates the LacZ reporter and the bimodal response of the regulator is assumed to be mirrored by a bimodal response of the target). However, we have shown that this may not always be the case: the bimodal distribution produced by the upstream gene expression can be converted into a unimodal downstream gene expression and vice versa.There are many studies that analyse the conversion of graded inputs into binary outputs due to feedback37–42 but, to our knowledge, none deals with the possibility of diverse binary/graded response patterns of different targets to the same regulator. Recent evolutionary models43 suggest that if a single TF regulates multiple targets, then TF mutations are less probable than the mutations of TF binding sites. According to these models, a substantial variability in promoter–TF affinities may arise at intermediate selection pressure. Therefore, assuming our model to be valid, the diversity of responses of different target genes to the same TF may be a widespread phenomenon.
Although the concept of early and late responses to mild and acute stress (weak and strong signals) by different target promoters, which have different sensitivities to TFs, is widely known, there are no systematic studies, to our knowledge, of whether these responses are binary or graded. For example, in ref. 44 steeper and milder responses were observed in different GATA-1 targets in G1E cells but no histograms of gene expression variability in a population were recorded and the study presents only dose–response curves from mean expression. However, the binary or graded shape of the gene expression may be of relevance in the context of the cellular response to stress: the large heterogeneity of the response (binary or wide-graded) seems to be an advantage at low-stress levels, when the cost of a possible untimely engagement of cellular machinery over-weighs the threat to the cell population. A narrowly-graded response may, on the other hand, be needed when higher stress levels force all cells to respond unambiguously.45
We have not found systematic experimental studies of the interdependence between the binary and graded responses of a regulatory gene and its multiple targets, except for the analysis of type III secretion in Salmonella by Temme et al.11 However, ref. 11 described the time-dependent response to a single level of inducer. Moreover, this system is much more complex than our model because of co-regulation of genes by multiple transcription factors and self-regulation of targets. The topmost regulator HilD was shown to have a graded response to an environmental signal. HilC, a self-regulating TF, which itself is regulated by HilD and which also responds directly to the signal, showed a graded response as well, but, at the same time, the responses of the downstream genes PrgH and SicA, indirectly or directly regulated by HilC and HilD, were binary.
Among the existing studies, there are examples of self-activating TFs which differentially regulate target genes and which could perhaps be candidates for the validity check of our model: the spo0A system in Bacillus subtilis41 has a somewhat similar topology to that of our model and seems to show a qualitatively similar behaviour. An autoregulated TF, spo0A, when phosphorylated, controls downstream genes (we note, however, that the system is more complex because TF self-regulation is both direct and indirect41,46). Narula et al.41 proposed a model to explain why the TF responded to starvation in a graded manner despite the feedback, but its target, σF, showed a binary response. Their model suggests that this type of response is possible if the target is additionally regulated by other proteins via a post-translational feedforward loop. On the other hand, the model we present here suggests a theoretical possibility that, in a network of a similar topology, the target gene can show a binary response even if the regulator's response is graded, without any post-translational interactions.
Another TF that exhibits a different response pattern from its targets is the eukaryotic gene p53, self-regulating via many indirect loops.47 In ref. 48, it has been shown that p53 target genes display different response types under genotoxic stress: in mouse embryonic fibroblast cells, endogenous Waf1 showed a graded response but artificial promoters with the same TF binding sites responded in a binary manner. In human breast cancer cells, endogenous Waf1 showed a binary response, whereas the level of p53 varied gradually. However, the diversity of the responses of p53 targets may originate from slower or faster rates of recruitment and decomposition of transcriptional apparatus (and not only TF binding).49–51 But if these rates influence the ratio of the on- to off-rates of promoter activity, and hence the K constant in the Hill function, then perhaps our model could be applicable as long as the on/off switching is sufficiently fast to assume Hill kinetics.
In S. cerevisiae, a self-activating TF, Zap1, responds to a range of zinc levels. Among tens of target genes, some are activated under conditions of mild zinc deficiency, and some others are activated at zinc starvation, depending on the strength or number of their Zap1 binding sites.52 In bacteria of the genus Bordetella, the regulatory gene bvgAS responds to the deficiency of MgSO4 or nicotinic acid. Phosphorylated TF BvgA binds to several classes of target promoters,12 which, due to different affinities to the TF, are activated at low, intermediate, or high BvgA∼P levels. The latter gene class is only activated when the positive feedback in the regulatory gene enters the induced regime and drastically increases the BvgA∼P level.53 In bacteria, the NRI transcription factor is encoded by the glnG Gene and autoactivates its transcription. It reacts by phosphorylation to ammonia deprivation. Its target promoters activate in a sequential manner depending on their affinity to the TF and the number of TF binding sites. The first line of response to nitrogen deficiency is activation of the glnALG operon, i.e. autoactivation of the regulatory gene. Higher levels of phosphorylated NRI activate other operons regulated by this TF.54 A recent review on self-regulation in two-component systems12 provides a longer list of possible candidate genes for which our model could be tested. However, Groisman12 notes that bimodal distributions of protein levels have not been observed in three well-characterized two-component systems: PhoB/PhoR in E. coli, PhoP/PhoQ in Salmonella, and BvgA/BvgS in Bordetella.
In Section S16 (ESI†) we discuss possible methods of measurement of transfer functions in an experimental realisation of our model, based on the idea of feedback opening, proposed by Hsu et al.27
Finally, we note that a different, but somewhat related, mechanism of conversion between the graded and binary responses of gene systems is also possible: the process of activation of the TF protein by a signal may itself be strongly nonlinear, due to the Hill kinetics of effector binding, and thus it may create a threshold that converts a unimodal distribution of signals into a bimodally distributed output.55 In our model, we did not take into account the details of TF activation/deactivation.
Authors' contributions
AOM conceived and designed the study, carried out analytical calculations and simulations, and wrote the manuscript. MT proposed the idea of studying a gene cascade with self-regulation, participated in the study conception and design, and revised critically the manuscript. JJ carried out analytical calculations (q(h2) form with leakage, geometric construction with leakage, normalisation of the p1(R) distribution) and participated in study design.Funding
A. O. M. and J. J. were supported by the Ministry of Science and Higher Education Grant No. 0501/IP1/2013/72 (Iuventus Plus).Acknowledgements
We thank Maciej Dobrzyński for literature suggestions on typical protein abundance in E. coli.References
- A. Ochab-Marcinek and M. Tabaka, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 22096–22101 CrossRef CAS PubMed.
- M. S. Sherman, K. Lorenz, M. H. Lanier and B. A. Cohen, Cell Syst., 2015, 1, 315–325 CrossRef CAS PubMed.
- K. H. Kim and H. M. Sauro, BMC Biol., 2012, 10, 89 CrossRef PubMed.
- M. R. Birtwistle, J. Rauch, A. Kiyatkin, E. Aksamitiene, M. Dobrzyński, J. B. Hoek, W. Kolch, B. A. Ogunnaike and B. N. Kholodenko, BMC Syst. Biol., 2012, 6, 109 CrossRef CAS PubMed.
- M. Dobrzyński, L. K. Nguyen, M. R. Birtwistle, A. V. Kriegsheim, A. Blanco, A. Cheong, W. Kolch, B. N. Kholodenko, A. Von and A. B. Ferna, J. R. Soc., Interface, 2014, 11, 20140383 CrossRef PubMed.
- L. A. Sepúlveda, H. Xu, J. Zhang, M. Wang and I. Golding, Science, 2016, 351, 1218–1222 CrossRef PubMed.
- M. Thattai and A. Van Oudenaarden, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8614–8619 CrossRef CAS PubMed.
- M. Thattai and A. van Oudenaarden, Biophys. J., 2002, 82, 2943–2950 CrossRef CAS PubMed.
- S. R. Biggar and G. R. Crabtree, EMBO J., 2001, 20, 3167–3176 CrossRef CAS PubMed.
- S. Takahashi and P. M. Pryciak, Curr. Biol., 2008, 18, 1184–1191 CrossRef CAS PubMed.
- K. Temme, H. Salis, D. Tullman-Ercek, A. Levskaya, S. H. Hong and C. A. Voigt, J. Mol. Biol., 2008, 377, 47–61 CrossRef CAS PubMed.
- E. A. Groisman, Annu. Rev. Microbiol., 2016, 70, 103–124 CrossRef CAS PubMed.
- A. Ochab-Marcinek and M. Tabaka, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2015, 91, 012704 CrossRef PubMed.
- U. Alon, An introduction to systems biology: design principles of biological circuits, Chapman & Hall/CRC, Boca Raton, FL, 2006 Search PubMed.
- E. Altszyler, A. Ventura, A. Colman-Lerner and A. Chernomoretz, Phys. Biol., 2014, 11, 066003 CrossRef PubMed.
- V. Shahrezaei and P. S. Swain, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17256–17261 CrossRef CAS PubMed.
- L. Cai, N. Friedman and X. Xie, Nature, 2006, 440, 358–362 CrossRef CAS PubMed.
- N. Friedman, L. Cai and X. S. Xie, Phys. Rev. Lett., 2006, 97, 168302 CrossRef PubMed.
- M. Gibson and J. Bruck, J. Phys. Chem. A, 2000, 104, 1876–1889 CrossRef CAS.
- Y. Ishihama, T. Schmidt, J. Rappsilber, M. Mann, F. U. Hartl, M. J. Kerner and D. Frishman, BMC Genomics, 2008, 9, 102 CrossRef PubMed.
- A. Ishihama, A. Kori, E. Koshio, K. Yamada, H. Maeda, T. Shimada, H. Makinoshima, A. Iwata and N. Fujita, J. Bacteriol., 2014, 196, 2718–2727 CrossRef PubMed.
- M. R. Maurizi, Experientia, 1992, 48, 178–201 CrossRef CAS PubMed.
- I. Yanai, J. O. Korbel, S. Boue, S. K. McWeeney, P. Bork and M. J. Lercher, Trends Genet., 2006, 22, 132–138 CrossRef CAS PubMed.
- N. T. Ingolia and A. W. Murray, Curr. Biol., 2007, 17, 668–677 CrossRef CAS PubMed.
- M. Slattery, T. Zhou, L. Yang, A. C. Dantas Machado, R. Gordân and R. Rohs, Trends Biochem. Sci., 2014, 39, 381–399 CrossRef CAS PubMed.
- M. Levo, E. Zalckvar, E. Sharon, A. C. Dantas Machado, Y. Kalma, M. Lotam-Pompan, A. Weinberger, Z. Yakhini, R. Rohs and E. Segal, Genome Res., 2015, 25, 1018–1029 CrossRef CAS PubMed.
- C. Hsu, V. Jaquet, F. Maleki and A. Becskei, J. Mol. Biol., 2016, 428, 4115–4128 CrossRef CAS PubMed.
- M. Pájaro, A. A. Alonso and C. Vázquez, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2015, 92, 032712 CrossRef PubMed.
- M. C. Mackey, M. Santillán, M. Tyran-Kamińska and E. S. Zeron, In Silico Biol., 2015, 12, 23–53 CrossRef PubMed.
- H. Qian, Annu. Rev. Biophys., 2012, 41, 179–204 CrossRef CAS PubMed.
- S. Iyer-Biswas, F. Hayot and C. Jayaprakash, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2009, 79, 031911 CrossRef PubMed.
- S. Iyer-Biswas and C. Jayaprakash, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2014, 90, 052712 CrossRef PubMed.
- P. Thomas, N. Popović and R. Grima, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6994–6999 CrossRef CAS PubMed.
- N. A. Rossi and M. J. Dunlop, PLoS Comput. Biol., 2017, 13, e1005310 Search PubMed.
- H. Kuwahara and O. S. Soyer, Mol. Syst. Biol., 2012, 8, 1–11 CrossRef PubMed.
- D.-E. Chang, S. Leung, M. R. Atkinson, A. Reifler, D. Forger and A. J. Ninfa, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 175–180 CrossRef CAS PubMed.
- A. Becskei, B. Séraphin and L. Serrano, EMBO J., 2001, 20, 2528–2535 CrossRef CAS PubMed.
- X. Wang, N. Hao, H. G. Dohlman and T. C. Elston, Biophys. J., 2006, 90, 1961–1978 CrossRef CAS PubMed.
- O. A. Igoshin, R. Alves and M. A. Savageau, Mol. Microbiol., 2008, 68, 1196–1215 CrossRef CAS PubMed.
- S. Palani and C. A. Sarkar, Mol. Syst. Biol., 2011, 7, 480 CrossRef PubMed.
- J. Narula, S. N. Devi, M. Fujita and O. A. Igoshin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E3513–E3522 CrossRef CAS PubMed.
- Y. Shindo, K. Iwamoto, K. Mouri, K. Hibino, M. Tomita, H. Kosako, Y. Sako and K. Takahashi, Nat. Commun., 2016, 7, 10458 CrossRef PubMed.
- M. Lynch and K. Hagner, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E30–E38 CrossRef CAS PubMed.
- K. D. Johnson, S. I. Kim and E. H. Bresnick, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15939–15944 CrossRef CAS PubMed.
- S. Paliwal, P. A. Iglesias, K. Campbell, Z. Hilioti, A. Groisman and A. Levchenko, Nature, 2007, 446, 46–51 CrossRef CAS PubMed.
- D. Schultz, BioEssays, 2016, 38, 440–445 CrossRef PubMed.
- X. Lu, Cold Spring Harbor Perspect. Biol., 2010, 2, a000984 Search PubMed.
- A. Jõers, V. Jaks, J. Kase and T. Maimets, Oncogene, 2004, 23, 6175–6185 CrossRef PubMed.
- N. P. Gomes and J. M. Espinosa, Genes Dev., 2010, 24, 111–114 CrossRef CAS PubMed.
- N. P. Gomes and J. M. Espinosa, Cell Cycle, 2010, 9, 3428–3437 CrossRef CAS PubMed.
- J. M. Morachis, C. M. Murawsky and B. M. Emerson, Genes Dev., 2010, 24, 135–147 CrossRef CAS PubMed.
- C.-Y. Wu, A. J. Bird, L. M. Chung, M. A. Newton, D. R. Winge and D. J. Eide, BMC Genomics, 2008, 9, 370 CrossRef PubMed.
- P. A. Cotter and V. J. DiRita, Annu. Rev. Microbiol., 2000, 54, 519–565 CrossRef CAS PubMed.
- M. Amouyal, C. R. Biol., 2005, 328, 1–9 CrossRef CAS PubMed.
- M. Podtschaske, U. Benary, S. Zwinger, T. Höfer, A. Radbruch and R. Baumgrass, PLoS One, 2007, 2, e935 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: casauto_pccp_supp.pdf. See DOI: 10.1039/c7cp00743d |
| ‡ Current address: Broad Institute of MIT and Harvard, 415 Main Street, Cambridge, MA 02142, USA. |
| This journal is © the Owner Societies 2017 |