
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Correcting the record: the dimers and trimers of trans-N-methylacetamide†
Thomas
Forsting
 a,
Hannes C.
Gottschalk
a,
Beppo
Hartwig
a,
Michel
Mons
b and
Martin A.
Suhm
*a
a,
Hannes C.
Gottschalk
a,
Beppo
Hartwig
a,
Michel
Mons
b and
Martin A.
Suhm
*a
aInstitut für Physikalische Chemie, Universität Göttingen, Tammannstr. 6, 37077 Göttingen, Germany. E-mail: msuhm@gwdg.de
bLIDYL; CEA, CNRS, Université Paris-Saclay, 91191 Gif-sur-Yvette, France
First published on 3rd January 2017
Abstract
The dimer of trans-N-methylacetamide serves as a simple model for hydrogen bonds in peptides, free of any backbone distortions. Its preferred structures represent benchmark systems for an accurate quantum chemical description of protein interactions. The trimer allows for either two linear or three strained hydrogen bonds, with the former being the only structural motif considered so far in the literature, but the latter winning in energy by a large margin due to London dispersion. A combination of linear Raman and infrared supersonic jet techniques with B3LYP-D3/aug-cc-pVTZ quantum chemical predictions corrects earlier tentative spectroscopic assignments based on a hybrid density functional without dispersion correction. Linear Amide I–III infrared spectra of the jet-cooled monomer are compared to those recently obtained by action spectroscopy.
1 Introduction
The dimer of N-methylacetamide (NMA) offers a good compromise between simplicity and realism when it comes to modelling hydrogen bonds between remotely connected N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups in folded peptides and proteins.1,2 Its monomer contains a full C–(CO)–(NH)–C backbone segment of the peptide bond. The preferred peptide bond conformation is trans (t) for the CO and N–H bonds, which we imply in the following for convenience, unless the less stable cis isomer (c) is explicitly addressed.
O groups in folded peptides and proteins.1,2 Its monomer contains a full C–(CO)–(NH)–C backbone segment of the peptide bond. The preferred peptide bond conformation is trans (t) for the CO and N–H bonds, which we imply in the following for convenience, unless the less stable cis isomer (c) is explicitly addressed.
NMA and its dimer have been investigated extensively, mostly by different levels of molecular mechanics3 and quantum chemistry,4–8 including anharmonic effects in the case of the monomer9–11 and dimer.12 For trimers and longer chain aggregates of NMA, the issue of cooperativity has been discussed.13–17
On the experimental side, vibrational spectroscopy is particularly sensitive to the hydrogen bonds which shape the secondary structure of proteins18 and thus also to NMA aggregation.19,20 The NMA dimer is actually too simple to be amenable to selective UV/IR double resonance techniques,21–23 because it does not provide a favourable electronically excited state for fluorescence or resonant 2-photon ionization studies. In contrast to numerous studies of monomeric amides24,25 and despite a sizeable dipole moment of about 8 D, the tt dimer has also not been studied by microwave spectroscopy, such that most information on the intermolecular N–H⋯OC contact is currently based on low resolution direct absorption FTIR spectroscopy. In inert matrices,1,26 one has to worry about matrix distortion of the spectrum. In the gas phase,27 the monomer spectrum is distorted by thermal excitation and the dimer has negligible abundance. In solution, solvent and thermal effects superimpose.28–32 NMA spectra have indeed been found to be particularly dependent on the environment.2 Therefore, supersonic jet spectroscopy is the method of choice for providing information on cold, isolated, structurally well defined dimers. Indeed, there was a jet FTIR study of NMA dimers and trimers almost a decade ago,33 but its assignments were rather tentative and uncertain at the time due to the low signal-to-noise ratio, the limited variation of expansion conditions and the restricted availability of reliable quantum chemical calculations including a balanced dispersion contribution. At the time, an extension to Raman spectroscopy was already planned33 and will be initiated in the present work. Very recently, while the present work was completed, an IR multiphoton dissociation (IRMPD) + vacuum ultraviolet (VUV) ionization technique was presented for NMA and its clusters.34 It is claimed to be universally applicable, in analogy to the corresponding FTIR approach35 which has been demonstrated between 200 and 8000 cm−1. The results in the range below 1800 cm−1 look promising and the signal-to-noise ratio is impressive. However, the technique is also not conformation-specific, it may suffer from fragmentation of larger clusters to smaller ones (although this issue is not discussed), and there is a systematic wavenumber difference and broadening compared to the previous jet FTIR spectra,33 which may be due to setup calibration and the multi-photon nature of the experiment.
Using predictions based on a large number of aromatic peptide models and the validation of a computationally fast dispersion corrected density functional in combination with harmonic frequency scaling,36 the proposed experimental FTIR assignment of NMA aggregation in ref. 33 was considered doubtful or at least incomplete. Therefore, we come back to this system in the present work and show that the original size assignment indeed needs revision. This conclusion is based on a more systematic FTIR study, hybrid density functional calculations including dispersion corrections, and most importantly linear Raman jet spectroscopy which has become available in the meantime and which complements the IR perspective quite nicely for NMA.2,19 For our purposes, the linear, off-resonant Raman variant has considerable advantages over resonance Raman,37 because it provides robust and predictable scattering intensities for comparison with linear FTIR spectra. The comparison of IR and Raman intensities promises insight into the aggregation topology due to the associated coupling patterns of degenerate monomer vibrations.38 We consider this extensive reevaluation and correction of the scientific record for NMA aggregation necessary and urgent due to the significance of NMA dimer and related systems for benchmarking purposes in the field of peptide interactions.39,40 Microwave spectroscopy should be able to test the structural conclusions drawn in this work, due to a significant polarity (0.4–9 D) predicted for the assigned clusters.
2 Methods
The IR spectra were recorded with a Bruker IFS 66v FTIR spectrometer probing a 600 mm long slit jet expansion of NMA vapor in a variable but large excess of helium gas at room temperature and 0.75 bar.35 The pulsed expansion through the 0.2 mm wide slit into a large vacuum chamber cools the NMA molecules rotationally to about 10 K and vibrationally to a lesser extent. It also leads to the formation of NMA dimers and a minor fraction of NMA trimers on the first few mm of supersonic flow after the nozzle. Although the vacuum chamber has a volume of more than 20 m3, the 150 ms gas flow through the nozzle builds up a background pressure of about 1 hPa, which limits the cross section of the expansion zone to a few cm2, not much larger than the cross section of the mildly lens-focussed infrared beam passing through it. During and after the gas pulse, the vacuum is improved by mechanical pumps at 2500 m3 h−1 for 30–60 s, before the system is ready for the next pulse. This low duty cycle in combination with the large nozzle and vacuum buffer allows for a high signal during the synchronized FTIR scan,35 but the vapor pressure of NMA close to room temperature is so low that more than 1000 pulse-synchronized spectra have to be coadded to achieve an acceptable signal-to-noise ratio at an interferometer resolution of 2 cm−1. The FTIR calibration was verified to be better than 0.5 cm−1 by comparison to water rovibrational transitions. The dilute gas mixtures are prepared by flowing helium through a bed of solid NMA and collecting 67 L in a pre-expansion reservoir. Due to the solid state and the low vapor pressure, the concentration varies somewhat over time despite temperature stabilization of the bed between 293 and 298 K. By sorting batches of 100 scans according to their CH, NH or CO absorbance, an ordering into low, medium and high concentration spectra is still achieved. CaF2 and KBr optics and a LN2-cooled InSb/HgCdTe sandwich detector are used together with a 150 W tungsten lamp or Globar rod, depending on the spectral range of interest. Optical band pass filters restrict the light source to the region of interest (see ESI† for further details about experimental parameters).
The strategy to obtain spontaneous Raman scattering spectra of NMA dimers and trimers in supersonic expansions is somewhat different. Because the feeble Stokes scattering is collected by an f/1.2 photo camera lens and dispersed in a 1 m monochromator equipped with a LN2-cooled CCD camera (Princeton, PyLoN 400B), the slit nozzle can be much shorter (4 mm). To minimize readout noise, the expansion is run continuously over 7–9 min and the photon signals along the center section of the slit nozzle are collected in and binned from 400 pixel rows of the CCD chip during this time, whereas the 1340 pixel columns contain the spectral information at a resolution of about 1 cm−1. Cosmic ray removal is achieved by comparing several such data blocks and calibration of the 532 nm laser and Stokes signal is pushed to about 1 cm−1 by neighboring Ne and Kr discharge lines with known vacuum wavelength. The signal-to-noise ratio is maximized by using as much laser intensity as possible for the scattering, in the present case 20–25 W supplied by a Millenia eV solid state laser from Spectra-Physics and focussed parallel to the slit exit at variable distance with a 50 mm focal length lens. The NMA cluster size is controlled by the NMA/helium ratio, the heating of the tubing and the nozzle as well as the laser-to-nozzle distance. The higher the temperature of the nozzle and the closer the laser to the nozzle, the smaller the expected degree of aggregation for a given NMA concentration. Spectra are shown as Stokes shifts ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) normalized to the same monomer peak intensity after baseline correction, because laser power, alignment, data acquisition time and to some extent sample concentration may vary. The employed diffraction grating performs better for Raman scattering perpendicular to the scattering plane,41 in particular for large Stokes shifts. Therefore, polarized bands (P = I‖/I⊥ ≪ 0.75) have a somewhat higher visibility than depolarized bands (P = 0.75), compared to a balanced grating performance reflected in the calculated Raman cross sections reported in this work.
normalized to the same monomer peak intensity after baseline correction, because laser power, alignment, data acquisition time and to some extent sample concentration may vary. The employed diffraction grating performs better for Raman scattering perpendicular to the scattering plane,41 in particular for large Stokes shifts. Therefore, polarized bands (P = I‖/I⊥ ≪ 0.75) have a somewhat higher visibility than depolarized bands (P = 0.75), compared to a balanced grating performance reflected in the calculated Raman cross sections reported in this work.
Helium (99.996%, Linde), N-methylacetamide (99%, Sigma-Aldrich) and N-methylacetamide-d7 (99%, 98% atom D, Sigma Aldrich) were used as supplied. Traces of methanol(-d4) from partial decomposition of the sample were typically observed in the beginning but quickly vanished in the course of the measurement and had no detectable influence on the N–H or CO stretching spectra.
Quantum chemical calculations (energy minimization, harmonic wavenumbers) were carried out using Gaussian09 Rev. E.01.42 The focus here is on trans dimer and trimer structures and spectra, whereas monomer methyl torsion,43 backbone planarity2 and amide isomerism will be addressed elsewhere. The B3LYP hybrid density functional provides a reasonable entry point for a theoretical description of NMA dimers and trimers. Although dispersion interactions have previously been considered to be minor in such strong hydrogen bonds,33 we use Grimme's pairwise D3 dispersion correction44 with BJ damping for the B3LYP calculations and the aug-cc-pVTZ basis set, now that this correction is available and the large basis set feasible (see ref. 45 for a discussion of lower and higher level approximations in this context and for relevant references). All calculations assume linear restoring force, dipole and polarizability trends with elongation, the so-called double harmonic approximation. Stokes scattering cross sections are calculated assuming a photon-counting detector, low vibrational temperature, laser excitation perpendicular to the scattering plane and non-polarizing diffraction. Tight optimization and a superfine integration grid were used. As will be shown below, our experimental work is in satisfactory agreement with these approximate calculations. It still invites much higher levels of electronic structure and anharmonic vibrational treatment by specialized groups, to further unravel the consequences of aggregation on the complex dynamics of the peptide bond. In contrast, omission of the D3 dispersion correction is shown to lead to serious disagreement for the preferred dimer and trimer structures. Control calculations at B97D level using the TZVPP basis set and D3 correction (ESI†) were carried out using TURBOMOLE.46
3 Amide I–III FTIR spectra
We start with a brief reinvestigation of the CO stretching and peptide bond framework vibrations of monomeric NMA,33 which have recently been measured by a novel IRMPD-VUV scheme,34 but only superficially compared to earlier FTIR spectra at low signal-to-noise ratio.33 The latter had revealed a rather narrow CO stretching fundamental at 1722 cm−1, whereas the IRMPD-detected band center is at 1707 cm−1, i.e. 1% lower and significantly broader. The former deficiency may be attributed to calibration error and the latter to the limited resolution of the free electron laser radiation, but the observed width is actually somewhat larger than the expected 1% laser bandwidth.34 This suggests that the action spectroscopy induces a spectral broadening mechanism, which is not unreasonable for a multiphoton excitation process. To verify this, we have repeated and improved the FTIR CO stretching measurement33 by using a long room temperature slit nozzle and more spectral co-additions. The result shown in Fig. 1 clearly confirms the narrow band width (0.4% of the central wavenumber) and higher wavenumber position (+1%) of the FTIR carbonyl or amide I band. The full width at half maximum of 7 cm−1 is mostly due to residual rotational structure at the ≈10 K rotational expansion temperature and the employed 2 cm−1 spectral resolution, as indicated by the band contour comparison in the ESI.† In line with theoretical predictions, the transition dipole moment is nearly parallel to the CO bond and closely aligns with the b rotational axis, explaining the b-type character of the band. The dip between the jet-cooled P and R branches thus provides the best estimate for the vibrational band center, 1720(1) cm−1. The exact position of the CO stretching band is important to predict the CO overtone for NMA, which may interfere with the N–H stretching fundamental in amides (vide infra). Based on the IRMPD data, it should fall around 3395(10) cm−1, whereas with the FTIR fundamental value, it is expected close to 3423(10) cm−1, if a carbonyl group anharmonicity constant of about −8.5 cm−1 is assumed.47 Comparison to the gas phase spectrum (ESI†) shows good agreement between the estimated FTIR band centers. The CO fundamental has a pronounced P/R rotational structure in the gas phase, which collapses in the jet. The absence of such a structure in the IRMPD spectra confirms that the probed molecules are rotationally cold, ruling out rotational temperature as a broadening factor, as expected. Therefore, the IRMPD broadening must be due to a preferential probing of vibrationally hot molecules or due to another multiphoton effect.
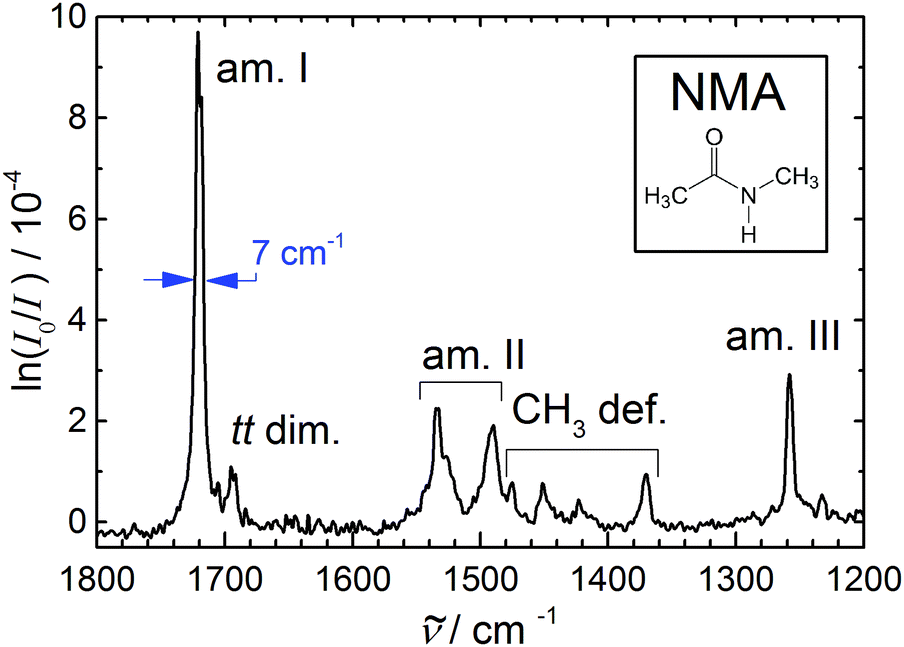 | ||
| Fig. 1 Amide I–III and methyl deformation jet FTIR spectrum of N-methylacetamide (see inset) monomer (with a dimer trace near 1695 cm−1) serving as a reference for anharmonic calculations of this peptide bond prototype9–11 due to the narrowness and reliable position of the bands. | ||
Table 1 summarizes the previous and current findings for the monomer amide I–III and methyl deformation modes. The recent IRMPD data appear to have a somewhat wavenumber-proportional shift with smaller deviations in the central region. One can see that matrix isolation, thermal, and IRMPD or calibration effects are comparable in magnitude, thus rendering the jet FTIR measurement the only currently available reference for highly accurate electronic structure and anharmonic mode-coupling investigations. We refrain from a more detailed analysis of the observed bands and their coupling, as the present contribution focusses on spectral evidence for dimer and trimer structures, we just note that the pronounced splitting of the amide II band has apparently not been captured by available calculations.9–11
Note that the small peak near 1695 cm−1 in Fig. 1 is not due to NMA monomer but rather due to NMA aggregates. It has been found near 1690 cm−1 in much more concentrated expansions, where dimers and trimers contribute significantly.33 Therefore, one may expect the trimer to contribute somewhat below 1690 cm−1, whereas 1695 cm−1 is now the best estimate for the dimer signal. Again, the recent IRMPD values34 of 1681 (dimer) and 1656 cm−1 (trimer) are presumably too low. The fact that the IRMPD shift of the dimer band is very close to the shift of the monomer band points at a calibration origin, rather than a major fragmentation issue in the detection scheme. This suggests that the dimer amide I–III spectrum reported recently34 is indeed due to NMA dimers, and offers a much better signal-to-noise ratio and size resolution than our current FTIR spectra. In Fig. 1, the small relative dimer intensity in the amide I range ensures that dimer contributions to the amide II-III bands will be almost negligible (ESI†). Neither the FTIR nor the IRMPD spectrum indicate any dimer isomerism, as there is only one fairly unstructured CO stretching band (more narrow in the FTIR case). An N–H stretching or amide A spectrum under the same conditions is likely to reveal more details about possible NMA dimers, due to the hydrogen bond-induced intensity enhancement and pronounced shift characteristic for that mode.
4 Amide A FTIR spectra
We thus continue with a reinvestigation of the amide A or N–H stretching infrared spectra, which are currently not accessible to the IRMPD scheme. The upper panel of Fig. 2 shows spectral traces obtained at low, medium and high concentration of NMA in He, from bottom to top. The conditions for the medium concentration correspond roughly to those in the amide I–III spectra in Fig. 1. The strongest signal at 3508 cm−1 is due to the trans monomer, a very weak signal near 3470 cm−1 may arise from the minor cis conformation and another weak bump near 3420–3430 cm−1 tentatively fits the trans NMA CO stretching overtone (see above). Indeed, comparison to the gas phase spectrum (ESI†) shows the presence of a monomer transition near 3425 cm−1, which may or may not profit from Fermi resonance with the N–H stretching fundamental. The rotational band contour of the main N–H band is anomalous and therefore the jet-cooled band center is not easily extracted from this. Possibly, methyl torsional hot bands coupling to the N–H stretching mode are responsible for this distortion and can be efficiently frozen out in the jet. Of interest in the context of NMA aggregation are two peaks at 3395 and 3365 cm−1, which grow relative to the monomer signals with increasing concentration. At the same time, a shoulder of the monomer peak appears to grow in near 3506 cm−1, but is difficult to identify unambiguously on top of the monomer signal. This is a general disadvantage of methods which are not selective to the cluster size. All these spectral contributions are likely due to dimers (the 3506 cm−1 shoulder could also be due to residual methyl torsion hot bands), but the fact that the 3395 cm−1 band grows more quickly at the highest concentration indicates a superimposed trimer contribution. Such a spectral sequence of higher wavenumber trimer and lower wavenumber dimer is quite unusual for hydrogen-bonded hydride stretching modes, as cooperativity normally enhances the hydrogen bond-induced downshift with growing cluster size. Indeed, the tentative assignment proposed before33 was chosen in the opposite way, although the comparison of low and high cluster content spectra (employing a short heated nozzle) should have raised doubts. This already indicates that the expected hydrogen bond cooperativity in the NMA trimer must be counteracted by some effect. In summary, besides offering a better signal-to-noise ratio, the new IR spectra are fully consistent with those reported earlier,33 but the partial trimer assignment is tentatively inverted, because the intensity behaviour with changing concentration is now given higher weight than the wavenumber sequence.
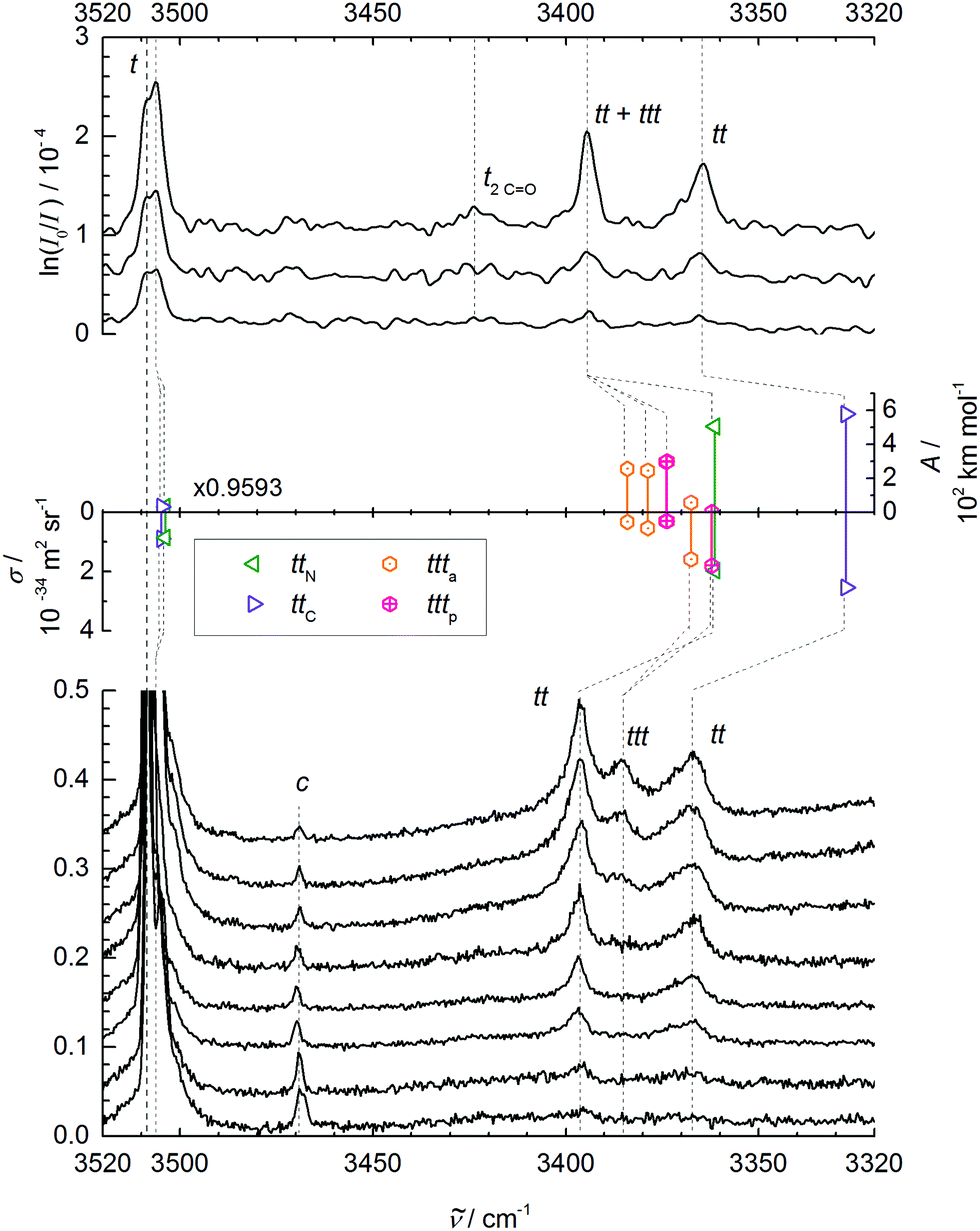 | ||
| Fig. 2 N–H stretching spectra of trans-NMA (t) and its aggregates. Upper panel: FTIR spectra for decreasing NMA concentration in the carrier gas from top to bottom. Middle panel: Monomer-scaled harmonic predictions for trans dimers ttN and ttC and trimers ttta and tttp with IR band strengths (A/(km mol−1), pointing up) and Raman scattering cross sections (σRa/(10−34 m2 sr−1), pointing down). Lower panel: Raman spectra for increasing nozzle temperature (70–175 °C) and thus lower aggregation tendency from top to bottom, recorded at 3 mm nozzle distance and a nominal stagnation pressure of 0.45 bar (helium saturated with NMA vapor at 60 °C), scaled to equal NMA monomer peak intensity (1). Weak bands due to cis-NMA monomer (c) and the trans monomer CO stretching overtone (2CO) are also shown. See Table 2 and ESI† for further details. | ||
At this point, it is helpful to compare the monomer band position with results from other experiments (see Table 2). The dominant jet monomer signal at 3508 cm−1 is upshifted by about 20 cm−1 from the N2 matrix isolation value48 and by about 10 cm−1 from the hot gas phase value as well as from the pH2 value.47 It is thus essential to compare accurate theoretical predictions to the supersonic jet result, rather than to thermal or matrix-embedded measurements.
| Method | FTIR | Raman | Assignment |
|---|---|---|---|
| a This work. | |||
| Jeta | 3508 | 3508 | trans monomer N–H stretch |
| Jet FTIR33 | 3510 | ||
| Exp. vacuum2 | (3545) | Source not clear | |
| Gas phasea | 3494/3509 | ||
| Gas phase27 | 3489/3501 | ||
| H2-matrix47 | 3500 | ||
| N2-matrix48 | 3493/3498 | ||
| CCl4-solution52 | 3475 | ||
| Jeta | (3470) | 3469 | cis monomer N–H stretch |
| H2-matrix47 | 3463 | ||
| N2-matrix48 | 3453/3458 | ||
| CCl4-solution52 | 3431 | ||
| Jeta | ≈3425 |
trans monomer CO overtone |
|
| Gas phasea | 3425 | ||
| Jeta | 3506? | trans dimer free N–H? | |
| Jeta | 3395 | 3396 | trans dimer(+trimer) bound N–H |
| Jet FTIR33 | 3395 | trans dimer bound N–H | |
| CCl4-solution52 | 3391 | ||
| Jeta | (3395) | 3386 | trans cyclic trimer, N–H |
| Jeta | 3365 | 3367 | trans dimer bound N–H |
| Jet FTIR33 | 3365 | trans [chain trimer + dimer], N–H | |
| CCl4-solution52 | 3354 | ||
| CCl4-solution52 | 3332 | trans [chain tetramer], N–H | |
Before the doublet of peaks emerging at 3395 (ttN) and 3365 cm−1 (ttC) is interpreted in terms of two competing tt dimer isomers (with a ttt trimer superposition on the higher wavenumber signal), other possible interpretations have to be ruled out. The cis-NMA content is clearly too low to form significant mixed cis–trans or even the very stable cis–cis dimers in the expansion.33 A tunneling splitting is very unlikely due to the non-equivalence of the two NH bonds, the heavy molecular frames, and the expected weak coupling to facile methyl torsion. However, the closeness of the monomer CO overtone near 3425 cm−1 may induce a Fermi resonance across the hydrogen bond in the dimer, similar to the one observed in a methanol–acetone complex.49 By doubling the amide I downshift of 26 cm−1 observed in Fig. 1, we arrive at about 3368(10) cm−1 for the unperturbed hydrogen bond acceptor CO stretching overtone, which is suspiciously close to the lower of the two bands, but also sufficiently close to the mid-point of the two bands, where a strong resonance partner would be expected to give rise to two more or less equally strong separated bands. A Fermi resonance within the hydrogen bond donor molecule50 would have to build on the donor CO overtone close to 3425 cm−1 and is therefore less likely due to the absence of significant absorption in that region.
To rule out the important possibility of N–H/2CO-Fermi resonance across the hydrogen bond, we have repeated the jet and gas phase experiments with NMA-d7 (ESI†). A comparison of the NMA and NMA-d7 spectra after scaling the latter to the same large dimer–monomer shift and aligning the monomer signals is also provided in the ESI.† Although the concentration and signal-to-noise ratio is lower for the expensive isotopologue, the pattern of NMA and fully deuterated NMA is remarkably similar, with two weak dimer features in the deuterated sample as well and a somewhat stronger cis-monomer peak. Therefore, the Fermi resonance hypothesis can be ruled out. Also, any tunneling effect due to methyl torsion should be attenuated significantly in a –CD3 group. Hence, the two dimer peaks must be due to two different isomers of the tt dimer. We note that for the related indole–NMA complex,51 two different conformations were clearly observed by conformationally selective IR spectroscopy and no evidence for a pronounced Fermi resonance was found. However, the lower wavenumber band at 3307 cm−1 showed some structure, which might point at a weak resonance.
As evidenced by the overproportional growth of the 3395 cm−1 band with concentration, the trimer appears to have only one strong N–H stretching fundamental, instead of the two expected for a chain trimer (besides a weak and weakly shifted free N–H stretch). This was already remarked before33 and tentatively blamed on dimer band overlap. However, given the assignment of the higher wavenumber peak to the main trimer band in this work, such an explanation becomes even less likely. Furthermore, a chain trimer would certainly profit from hydrogen bond cooperativity and thus shift further away from the monomer signal than a dimer. The only plausible explanation is a ring NMA trimer with approximate threefold symmetry of the hydrogen bond pattern and nearly degenerate asymmetric N–H stretching modes underneath the 3395 cm−1 band (due to strained hydrogen bonds), along with a weakly IR active symmetric N–H stretch at lower wavenumber below the detection limit. Such a ring trimer was never discussed in the extensive literature, to the best of our knowledge.
With these two open experimental questions – the structure of the dimer isomers and the hydrogen bond topology of the trimer – it is appropriate to move to a brief quantum chemical analysis of these two systems.
5 Density functional predictions
The trans-NMA dimer tt was predicted to occur in different relative orientations of the monomer planes, some coplanar (β-sheet-like12) like in the solid,1,3,6,12 others more perpendicular,3,7,8,12 like in an α-helix. In some of them, the NH group engages the free electron pair of the carbonyl group pointing to the acetyl side,6–8 in others the CO free electron pair on the amide side.6,8 Although the cis-NMA dimer is actually more stable than the trans dimer, it needs no discussion in this context as the cis monomer abundance is very low33,48 even at high nozzle temperatures, and the interconversion barrier33 is too high to allow for significant trans–cis isomerization during the expansion process. This is consistent with a predicted harmonically zero point corrected energy difference of 9.0 kJ mol−1 between the cis and trans NMA isomers at our standard level of approximation.
An extensive search for possible tt structures at B3LYP/aug-cc-pVTZ level including the pairwise D3 dispersion correction and Becke–Johnson damping revealed two nearly isoenergetic, but spectrally distinct chiral NMA dimers (Fig. 3). One arrangement engages the NH donor to the lone electron pair on the acetyl side (ttC) (with an H⋯OC acceptor angle close to 120°), the other one the lone pair on the amide side (ttN) (with an H⋯OC angle close to 140°). The barrier for lone pair switching is probably not sufficiently high to freeze the corresponding isomers in an early stage of the expansion. We estimate it at <4 kJ mol−1 from preliminary transition state searches. This justifies the use of energy rather than free energy to judge their relative stability (Table 3). The very similar energy of the two isomers leads to a similar population in the jet despite mutual interconversion down to low conformational temperatures, with the ttN isomer having a somewhat more strained and thus weaker hydrogen bond and less spectral visibility. A possible reason for this strain is revealed by a calculation omitting the dispersion correction. The H⋯OC angle increases by about 20° for both dimer conformations and their energy still does not differ by more than 1 kJ mol−1. This suggests that London dispersion stabilizes both dimers to a similar extent and steric hindrance by the N-methyl group is largely responsible for the more open H⋯OC angle in ttN.
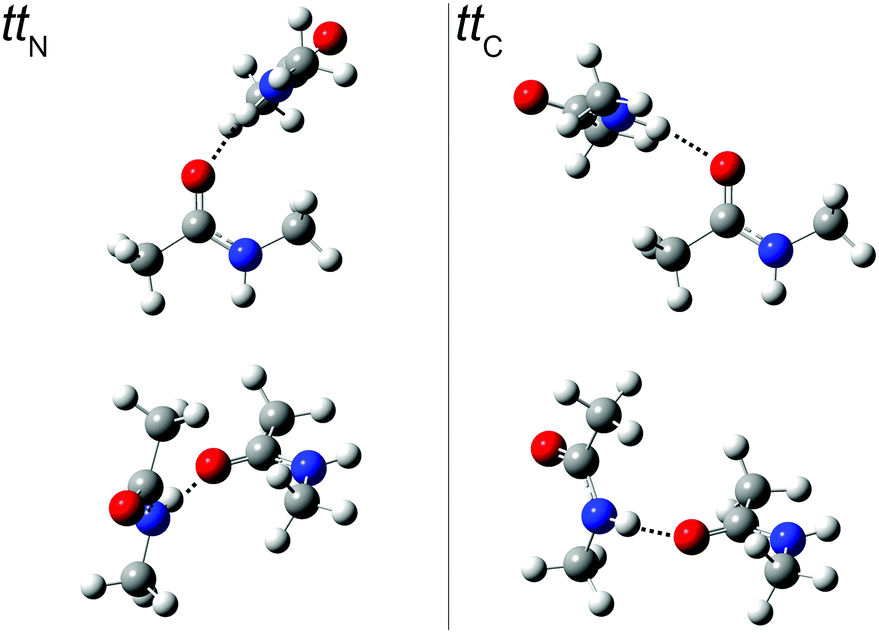 | ||
| Fig. 3
tt
N and ttC dimer structures from two different perspectives. In the top row, the more or less perpendicular orientation of the hydrogen bond donor relative to the plane of lone electron pairs in the acceptor and the (N–)H⋯OC bond angle is visualized (see ESI† for Cartesian coordinates). | ||
O group in N–H hydrogen bonding, at the D3-corrected B3LYP/aug-cc-pVTZ level: relative electronic energies Er,el, harmonically zero point corrected energies Er,0, hydrogen bond distances d(H⋯O), hydrogen bond angles with the CO bond α(H⋯OC), donor N–H stretching wavenumbers (N–H), dipole moments μ/D, infrared intensities AIR and Raman scattering cross sections for perpendicular 532 nm excitation/scattering σRa in the double-harmonic approximation. Results without D3 correction are given in parentheses
| Quantity\isomer | tt C | tt N |
|---|---|---|
| E r,el/kJ mol−1 | 0.0 | 0.0 (1.0) |
| E r,0/kJ mol−1 | 0.0 | 0.1 (0.8) |
| d(H⋯O)/pm | 192 (199) | 193 (200) |
|
α(H⋯OC)/° |
119 (139) | 139 (157) |
|
(N–H)/cm−1 |
3469 (3518) | 3504 (3547) |
| A IR/km mol−1 | 578 (522) | 508 (440) |
| σ Ra/10−35 m2 sr−1 | 25.4 (26.4) | 19.6 (20.5) |
| μ/D | 8 (9) | 8 (9) |
Table 3 lists the computational predictions for the two stable isomers ttC and ttN identified in the calculations. The energetic differences are too small to be significant at this computational level and we invite higher level calculations to determine the exact energy sequence. However, the spectral energy difference in the N–H stretching mode is clear-cut and the 3365 cm−1 band finds straightforward explanation by the ttC isomer, whereas the 3395 cm−1 band must be due to the ttN dimer. In both cases, one observes the typical overestimation of the harmonic shifts relative to the monomer at this computational level (see Fig. 2). Therefore, the two dimer peaks find very satisfactory explanations and reflect the sensitivity of the peptide hydrogen bond on the coordinated lone pair of the CO group. The predictions without dispersion correction show a similar isomer splitting, suggesting that its mechanism is more closely connected to repulsion than to dispersion interaction. The predicted harmonic dimerization shifts now match the experimental shifts perfectly (within 2 cm−1 after scaling of the predicted monomer transition at 3654 cm−1), which is unusual for B3LYP hydrogen bond shifts of smaller molecules. Thus, the dispersion interaction pushes the two monomers together and enhances the hydrogen bond distortion of the N–H bond.
For trans-N-methyl amide trimers ttt, chain conformations are usually proposed, calculated or implied,1,3,4,13–17,19,28–30,32,33,52–54 probably inspired by the solid state chain structure55 and the high dielectric constant of the liquid.28 This even includes the most recent investigation of NMA trimers in vacuum isolation.34
Indeed, we find reasonably stable chain conformations at the dispersion-corrected B3LYP level used for the dimers. However, two types of cyclic trimers are about 17 kJ mol−1 lower in energy than the most stable chain trimers and it is quite surprising that they have not been discovered or discussed before (Fig. 4). One corresponds to three acetyl groups in parallel on the same side (tttp) of a barrel-like arrangement with (non-imposed) C3 symmetry, the other to a partially alternating, less symmetric barrel arrangement (ttta). These two isomers are close in energy (Table 4). Depending on the formation process of the cyclic trimer, one could also look at their free energy difference at some intermediate temperature between the nozzle and final expansion temperatures. As the interconversion barriers will be much higher than in the dimer, even a statistical formation of the two cyclic trimers is conceivable. Note that the NH groups are now pointing at the oxygen end of the π bond, rather than in the direction of the lone electron pairs of the carbonyl group, which represents quite a different type of hydrogen bond.
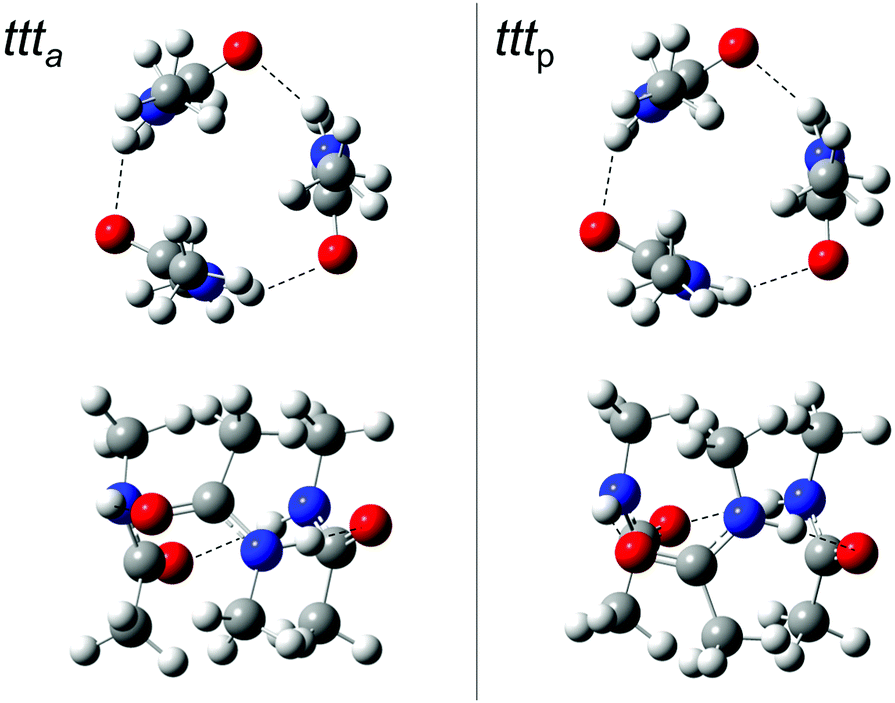 | ||
| Fig. 4 ttt a and tttp trimer structures from two different perspectives. In the top row, the barrel-like arrangement is projected from the top, in the bottom row from the side (see ESI† for Cartesian coordinates). DOI: 10.6084/m9.figshare.4233110.v1. | ||
| Quantity\isomer | ttt a | ttt p | ttt l |
|---|---|---|---|
| E r,el/kJ mol−1 | 0.0 (0.0) | 0.1 (0.3) | 21 (−10) |
| E r,0/kJ mol−1 | 0.0 (0.0) | 0.5 (0.3) | 17 (−12) |
| d(H⋯O)/pm | 212 (226) | 207 (224) | |
| 210 (224) | 207 (224) | 189 (194) | |
| 207 (225) | 207 (224) | 188 (195) | |
|
α(H⋯OC)/° |
104 (110) | 109 (114) | |
| 105 (115) | 109 (114) | 141 (137) | |
| 110 (110) | 109 (114) | 118 (142) | |
|
(N–H)/cm−1 |
3528 (3557) | 3517 (3556) | |
| 3522 (3556) | 3517 (3556) | 3467 (3491) | |
| 3510 (3550) | 3505 (3549) | 3420 (3468) | |
| A IR/km mol−1 | 257 (192) | 299 (199) | |
| 245 (193) | 299 (199) | 547 (400) | |
| 58 (3) | 2 (0) | 801 (986) | |
| σ Ra/10−35 m2 sr−1 | 3.2 (2.3) | 2.9 (2.4) | |
| 5.3 (2.6) | 2.9 (2.4) | 19.0 (20.2) | |
| 15.8 (17.8) | 18.0 (17.5) | 32.4 (45.6) | |
| μ/D | 0.4 (0.3) | 1.2 (0.7) | 11 (15) |
Again it is interesting to switch off the dispersion correction. The cyclic minima still exist, but now, the chain topology which was considered in all previous studies of this trimer wins by a surprisingly large margin (10 kJ mol−1 before and 12 kJ mol−1 after zero point correction). This explains to some extent why the cyclic NMA trimer has remained elusive for such a long time and it provides a rather extreme example of dispersion control in hydrogen bond aggregation.
The two cyclic trimer isomers are not only similar in energy, but also in local structure. The hydrogen bonds are considerably strained, with H⋯OC angles between 104° and 110°, but three of these strained hydrogen bonds are evidently more stable than two nearly unstrained ones, although they do not realize a classical coordination of the lone electron pairs. The strain and non-classical coordination explains why the IR visibility of these structures is lower and why the cyclic cooperativity does not increase the downshift relative to dimers. It even explains why the spectral gap between harmonic theory and experiment is smaller in the trimer than in the dimer (Fig. 2), because the diagonal N–H stretching anharmonicity is certainly attenuated by the strain. This follows from the assumption that the IR-active N–H stretching bands of both isomers somehow overlap with the higher frequency dimer absorption in the upper trace of Fig. 2, thus contributing to its enhanced scaling with NMA concentration. Presence of the chain trimer in the IR spectra can be safely ruled out, not only because of the predicted 17 kJ mol−1 energy handicap, but also because the observed spectral pattern and position is incompatible with the predicted one. As shown in Table 4, the most intense chain trimer band should be further downshifted than any dimer band in the IR spectrum. Thus, the middle panel of Fig. 2 is in complete agreement with the IR spectra in the upper panel if the two nearly degenerate asymmetric trimer modes contribute to the higher wavenumber dimer signal.
Ring closure of the hydrogen bond sequence was previously discussed for less bulky amides than NMA and larger oligomers,4 but only in a planar rather than the barrel fashion proposed here. In the condensed phase, there is also little incentive for such strained barrel structures in competition to longer, unstrained chains, but in the gas phase, three strained cooperative hydrogen bonds are clearly superior to two unstrained ones.
While we have achieved consistency between theory and experiment, the evidence is still somewhat indirect, as it builds on fortuitous band overlap of one dimer and three nearly degenerate trimer bands. It would be interesting to observe the presumably non-overlapping, symmetric stretching fundamental with the lowest trimer N–H stretching wavenumber. Its IR intensity is an order of magnitude weaker than the asymmetric stretch (see Table 4) and thus hidden in the noise of the low concentration FTIR spectra. However, similar to the case of alcohol trimers,38 it should carry most of the Raman activity of the N–H stretching manifold.
6 Temperature-dependent Raman jet spectra
Raman spectroscopy of NMA in condensed phases has been investigated thoroughly and is not without interpretational difficulties. The amide A region involves both Fermi resonance and vibrational Franck–Condon features.19,56 The amide I and II vibrations are only weakly coupled to each other32 but nevertheless fall in a controversially discussed spectral region.19In contrast, the amide A Raman spectra of supersonic expansions, shown in the lower panel of Fig. 2, are rather straightforward to interpret with the knowledge accumulated above from the FTIR spectra and calculations. They are dominated by the t monomer transitions, which are even accompanied by a small fraction of cis isomer (c). The aggregated fraction is controlled by nozzle heating, while keeping the NMA concentration in the stagnant gas mixture more or less constant. The lower the nozzle temperature (from bottom to top), the more extensive is cluster formation. The reduction in relative cis monomer abundance with increasing cluster formation should not be interpreted as preferential cis clustering. It is largely a consequence of the endothermic formation of cis monomers from trans monomers and the lower nozzle temperature in the upper traces, as verified under lower clustering conditions. Two bands growing in simultaneously with nozzle cooling correspond to the dimer isomers identified before. They are broader and skewed towards higher wavenumber, because the expansion is probed close to the nozzle where cooling of internal degrees of freedom is not yet complete. Thus, there is population of thermally excited dimers with reduced hydrogen bond shift. Also, the band maxima are shifted slightly to higher wavenumber due to the thermal population, but the correspondence to the IR spectra is clear. The similar intensity of the two Raman dimer peaks would again be consistent with either a perfect Fermi resonance or similar abundance of two isomers, but the former interpretation has been ruled out in the IR by deuteration (vide supra). Very interesting is the emergence of a third band at the lowest nozzle temperatures, where trimers can form already a few mm downstream the nozzle. With 3386 cm−1, it is close to the position where the Raman-dominant cyclic ttta or tttp trimer band is expected if its IR-dominant bands are to overlap with the ttN dimer. A chain trimer is incompatible with the combined IR/Raman evidence, as it should have a common strong transition further downshifted than any dimer bands (see Tables 3 and 4). Thus, there is strongly supportive Raman evidence for the existence of barrel-like NMA trimers in aggregating supersonic jets. Based on the theoretical Raman scattering cross sections, one can roughly estimate the abundance of clusters in the coldest expansion (uppermost Raman trace), although thermal excitation and polarizing properties of the diffraction grating may introduce additional uncertainties. The total cluster abundance is less than but comparable to the monomer concentration, with similar relative amounts of the two dimers and the trimer and a somewhat larger cumulated contribution from higher oligomers. The latter contribute to a very broad band below 3300 cm−1 (not shown). The trimer is thus just a minor strained intermediate on the way to less strained cyclic or chainlike aggregates.
We have also recorded the amide I–III range of the Raman spectrum at different nozzle temperatures (see ESI† for a comparison to the FTIR spectrum for a diluted expansion). In no case do the predicted spectra suggest a clear separation of the bands from ttN and ttC. This is consistent with the IRMPD + VUV spectrum assigned to the dimer,34 but instead of one relatively broad band, the better resolved Raman spectrum shows several peaks due to the donor and acceptor units in the amide I and amide III case, whereas the amide II region has little Raman activity. This is in contrast to strongly Raman-active CH deformation bands at the lower end of the amide II range. The intensity complementarity between IR and Raman spectra suggests that there is no substantial mixing of these modes. They either show strong Raman activity (when they are CH dominated) or strong IR intensity (when they involve the amide frame). The lower end of the spectrum reveals the amide III vibrations with significant Raman intensity. Two relatively sharp features due to tt dimers appear blue-shifted and have a very weak correspondence in the diluted FTIR spectrum. Further shifted away from the monomer transitions than the sharp dimer peaks, relatively broad peaks emerge in the amide I and III region. They are likely due to cyclic trimers and larger oligomers, as the band maximum shows some evolution with average cluster size. It is interesting to again compare the wavenumbers with those of the size-selective IRMPD spectrum. The amide I dimer transitions at 1704, 1694 and 1685 cm−1 are on average shifted from the IRMPD value of 1688 cm−1 by a smaller amount than the monomer peak. This casts some doubt on a pure calibration interpretation of the shift. For the cyclic trimer, there should be no match between the IRMPD band at 1656 cm−1 and the dominant Raman peak near 1659 cm−1, because the exciton splitting between the IR and Raman active modes is close to 30 cm−1, according to the theoretical predictions. Actually, there is weak evidence for the IR-active (and weakly Raman active) amide I vibration on top of the dominant dimer signal at 1694 cm−1. Such a large discrepancy between the size selected IRMPD trimer spectrum and our linear but overlaid spectra cannot be explained by calibration alone. In the amide III region, the clustering upshifts are almost a mirror image of the amide I clustering downshifts, with clear evidence for two dimer contributions (donor and acceptor) at 1271 and 1285 cm−1. The latter is overlapped by trimer contributions, as evidenced by a stronger scaling with decreasing nozzle temperature. A broader band near 1301 cm−1 appears to be due to cyclic tetramers, according to exploratory theoretical predictions. In summary, the amide I–III region of the Raman spectrum confirms the sequential formation of dimers and cyclic trimers with lower nozzle temperature, without being able to discriminate between the two dimer isomers. Even some of the CH deformation vibrations show weak clustering shifts, which will not be analyzed further. Comparison to IRMPD spectra has to await their recalibration before any firm conclusions can be drawn.
We are not aware of any occurrence of trimeric peptide barrels in proteins, which is quite plausible in view of steric crowding and competition from linear arrangements in a folding peptide chain. Nevertheless, this completely overlooked cyclic intermolecular amide binding motif is valuable for the theoretical description of strained inter-segment peptide hydrogen bonds in general,36 which are often found in proteins. Furthermore, it is a prototypical example for NH coordination perpendicular to the >CO plane. A molecular mechanics or quantum chemical model which is to be trusted in the description of regular and distorted arrangements of peptide hydrogen bonds as well as their spectral signatures should be able to reproduce the energetics and spectroscopy of NMA dimers and trimers, as assigned in the present work for the first time with sufficient confidence.
A first check can be made by using the extensively peptide-validated B97D/TZVPP approach with D3 corrections and empirical linear scaling of harmonic frequencies23 to reproduce the experimental and B3LYP/aug-cc-pVTZ findings with D3 corrections presented here (ESI†). For the tt dimers, the two most stable conformations differ somewhat in structure, which is not surprising given the floppiness of the single hydrogen bond and should serve as a warning not to take the intermolecular structures in Fig. 3 too literally. Rewardingly, their relative energies also fall within a narrow window of less than 0.5 kJ mol−1 with and without zero point energy correction at the B97D level. The two-parameter scaled harmonic wavenumbers are systematically 25 cm−1 lower than the (anharmonic) experiment, but the splitting between ttN and ttC is predicted perfectly. Because intramolecular hydrogen bonds in peptides always tend to have some backbone strain, NMA dimers, deprived of such covalent strains, might represent a useful limiting case to the peptide training set. The two cyclic trimers are predicted similar in energy and ttta is found to be 18 kJ mol−1 more stable than a chain trimer at B97D level. None of the relative trimer stabilities deviates by more than 1.5 kJ mol−1 from the B3LYP predictions, which may be a rough measure for residual errors of our DFT calculations. The two-parameter scaled harmonic trimer NH stretching wavenumbers are finally in perfect agreement with anharmonic experiment. Given the spectral overlap in the trimer IR spectra and the residual thermal excitation in the trimer Raman spectra, it is even hard to put a sign on the theory-experiment discrepancy for ttt NH stretching modes. This nicely confirms that appropriately modified harmonic DFT approaches are able to provide a consistent description of NMA aggregation and vibrational spectroscopy, in agreement with the experimental data.
In a quantum cluster equilibrium treatment of NMA, the high pressure gas phase description16 would possibly profit from inclusion of the cyclic trimer, whereas it should be less important for the condensed phases, as long as sufficiently long chains are included as well.
7 Conclusions
The prevailing view of trans-N-methylacetamide aggregates was that of chain-like dimers tt and trimers tttl. In the present work, it is shown that the dimers occur in two nearly isoenergetic variants engaging the acetyl (ttC) and amide-facing (ttN) lone electron pairs of the acceptor carbonyl group. For trimers, it is shown for the first time that the consistent chain aggregation picture (tttl) presented in more than a dozen publications has to be replaced by an energetically much more favourable barrel-like and thus cyclic pattern, either with alternating (ttta) or parallel (tttp) orientation of the backbones. This cyclic hydrogen bond topology is certainly promoted by the vacuum environment, which is usually also implied in quantum chemical calculations. It is therefore surprising that it has been overlooked for such a long time. This has also contributed to a wrong tentative trimer assignment in our earlier FTIR aggregation study of four different peptide models,33 which we correct in the present work for NMA. A careful comparison of FTIR and very recent IRMPD spectra of NMA and its clusters34 suggests that the latter method must be recalibrated and may induce spectral broadening. Beyond these slight drawbacks, it may turn out to be a sensitive and broadly applicable IR spectroscopy method, certainly complementary to linear FTIR spectroscopy with its limited sensitivity due to the use of conventional light sources but currently still with broader spectral coverage.35 We finally think that there are good prospects to confirm the cyclic NMA trimer structure by microwave spectroscopy, because at least the tttp structure has a sizeable dipole moment (>1 D). Identification of the two dimer isomers ttC and ttN should be even more straightforward due to their high polarity, unless the four methyl rotors cause too much intramolecular dynamics.In conclusion, the switched dimer/trimer assignment and the consistent chain aggregation picture of NMA in the literature were wrong for a right reason, namely underestimated dispersion interactions in hybrid density functionals. This may be slightly more satisfactory than having been right for the wrong reason, but naturally we hope that the present assignments are right for largely the right reasons and will thus remain valid.
Acknowledgements
This contribution is dedicated to the memory of Reinhart Ahlrichs († 12.10.2016), who was often right for the right reasons and whose critical thinking will always remain a role model. We thank Sönke Oswald for valuable help and discussions. The Raman (SU 121/6-1) and FTIR spectroscopy (SU 121/4-1) used in this work is supported by the DFG, as is the general concept that London dispersion interactions have a controlling effect in chemistry (SPP 1807, SU 121/5-1).References
- H. Torii, T. Tatsumi, T. Kanazawa and M. Tasumi, Effects of Intermolecular Hydrogen-Bonding Interactions on the Amide I Mode of N-Methylacetamide: Matrix-Isolation Infrared Studies and ab Initio Molecular Orbital Calculations, J. Phys. Chem. B, 1998, 102, 309–314 CrossRef CAS.
- V. Andrushchenko, P. Matejka, D. T. Anderson, J. Kaminsky, J. Hornicek, L. O. Paulson and P. Bour, Solvent dependence of the N-methylacetamide structure and force field, J. Phys. Chem. A, 2009, 113, 9727–9736 CrossRef CAS PubMed.
- T. W. Whitfield, G. J. Martyna, S. Allison, S. P. Bates, H. Vass and J. Crain, Structure and Hydrogen Bonding in Neat N-Methylacetamide: Classical Molecular Dynamics and Raman Spectroscopy Studies of a Liquid of Peptidic Fragments, J. Phys. Chem. B, 2006, 110, 3624–3637 CrossRef CAS PubMed.
- R. Ludwig, F. Weinhold and T. C. Farrar, Structure of liquid N-methylacetamide: temperature dependence of NMR chemical shifts and quadrupole coupling constants, J. Phys. Chem. A, 1997, 101, 8861–8870 CrossRef CAS.
- R. Vargas, J. Garza, R. A. Friesner, H. Stern, B. P. Hay and D. A. Dixon, Strength of the N–H⋯OC and C–H⋯OC Bonds in Formamide and N-Methylacetamide Dimers, J. Phys. Chem. A, 2001, 105, 4963–4968 CrossRef CAS.
- T. M. Watson and J. D. Hirst, Density Functional Theory Vibrational Frequencies of Amides and Amide Dimers, J. Phys. Chem. A, 2002, 106, 7858–7867 CrossRef CAS.
- G. V. Papamokos and I. N. Demetropoulos, Vibrational Frequencies of Amides and Amide Dimers: The Assessment of PW91XC Functional, J. Phys. Chem. A, 2004, 108, 7291–7300 CrossRef CAS.
- U. Adhikari and S. Scheiner, Preferred configurations of peptide–peptide interactions, J. Phys. Chem. A, 2013, 117, 489–496 CrossRef CAS PubMed.
- S. K. Gregurick, G. M. Chaban and R. B. Gerber, Ab Initio and Improved Empirical Potentials for the Calculation of the Anharmonic Vibrational States and Intramolecular Mode Coupling of N-Methylacetamide, J. Phys. Chem. A, 2002, 106, 8696–8707 CrossRef CAS.
- M. Bounouar and C. Scheurer, Reducing the vibrational coupling network in N-methylacetamide as a model for ab initio infrared spectra computations of peptides, Chem. Phys., 2006, 323, 87–101 CrossRef CAS.
- A. L. Kaledin and J. M. Bowman, Full Dimensional Quantum Calculations of Vibrational Energies of N-Methyl Acetamide, J. Phys. Chem. A, 2007, 111, 5593–5598 CrossRef CAS PubMed.
- S. Ham and M. Cho, Amide I modes in the N-methylacetamide dimer and glycine dipeptide analog: diagonal force constants, J. Chem. Phys., 2003, 118(15), 6915–6922 CrossRef CAS.
- H. Guo and M. Karplus, Ab Initio Studies of Hydrogen Bonding of N-Methylacetamide: Structure, Cooperativity, and Internal Rotational Barriers, J. Phys. Chem., 1992, 96, 7273–7287 CrossRef CAS.
- T. C. Cheam, Ab initio vibrational spectra of hydrogen-bonded N-methylacetamide, J. Mol. Struct.: THEOCHEM, 1992, 257, 57–73 CrossRef.
- L. M. Kuznetsova, V. L. Furer and L. I. Maklakov, Infrared intensities of N-methylacetamide associates, J. Mol. Struct., 1996, 380, 23–29 CrossRef CAS.
- M. Huelsekopf and R. Ludwig, Correlations between structural, NMR and IR spectroscopic properties of N-methylacetamide, Magn. Reson. Chem., 2001, 39, S127–S134 CrossRef CAS.
- X. N. Jiang and C. S. Wang, Evaluation of the individual hydrogen bonding energies in N-methylacetamide chains, Sci. China: Chem., 2010, 53, 1754–1761 CrossRef CAS.
- W. K. Surewicz, H. H. Mantsch and D. Chapman, Determination of Protein Secondary Structure by Fourier Transform Infrared Spectroscopy: A Critical Assessment, Biochemistry, 1993, 32(2), 389–394 CrossRef CAS PubMed.
- W. A. Herrebout, K. Clou and H. O. Desseyn, Vibrational Spectroscopy of N-Methylacetamide Revisited, J. Phys. Chem. A, 2001, 105, 4865–4881 CrossRef CAS.
- M. R. Panman, D. J. Shaw, B. Ensing and S. Woutersen, Local Orientational Order in Liquids Revealed by Resonant Vibrational Energy Transfer, Phys. Rev. Lett., 2014, 113, 207801 CrossRef CAS PubMed.
- M. Gerhards, C. Unterberg, A. Gerlach and A. Jansen, β-Sheet model systems in the gas phase: Structures and vibrations of Ac-Phe-NHMe and its dimer (Ac-Phe-NHMe)2, Phys. Chem. Chem. Phys., 2004, 6, 2682–2690 RSC.
- J. C. Dean, E. G. Buchanan and T. S. Zwier, Mixed 14/16 helices in the gas phase: conformation-specific spectroscopy of Z-(Gly)n, n = 1, 3, 5, J. Am. Chem. Soc., 2012, 134, 17186–17201 CrossRef CAS PubMed.
- E. Gloaguen and M. Mons. Isolated neutral peptides, in Gas-Phase IR Spectroscopy and Structure of Biological Molecules, ed. A. M. Rijs and J. Oomens, Springer, Berlin, 2015, vol. 364, pp. 225–270 Search PubMed.
- Y. Kawashima, T. Usami, N. Ohashi, R. D. Suenram, J. T. Hougen and E. Hirota, Dynamical structure of peptide molecules, Acc. Chem. Res., 2006, 39, 216–220 CrossRef CAS PubMed.
- R. Kannengießer, W. Stahl, H. V. L. Nguyen and I. Kleiner, 14N Nuclear quadrupole coupling and methyl internal rotation in N-tert-butylacetamide as observed by microwave spectroscopy, J. Phys. Chem. A, 2016, 120, 3992–3997 CrossRef PubMed.
- G. Pohl, A. Perczel, E. Vass, G. Magyarfalvi and G. Tarczay, A matrix isolation study on Ac-Gly-NHMe and Ac-L-Ala-NHMe, the simplest chiral and achiral building blocks of peptides and proteins, Phys. Chem. Chem. Phys., 2007, 9, 4698–4708 RSC.
- R. L. Jones, The Infrared Spectra of Some Simple N-Substituted Amides in the Vapor State, J. Mol. Spectrosc., 1963, 11, 411–421 CrossRef CAS.
- K. Pralat, J. Jadzyn and S. Balanicka, Dielectric Properties and Molecular Structure of Amide Solutions. 1. N-Methylacetamide in Carbon Tetrachloride, J. Phys. Chem., 1983, 87, 1385–1390 CrossRef CAS.
- R. Ludwig, O. Reis, R. Winter, F. Weinhold and T. C. Farrar, Quantum Cluster Equilibrium Theory of Liquids: Temperature Dependence of Hydrogen Bonding in Liquid N-Methylacetamide Studied by IR Spectra, J. Phys. Chem. B, 1998, 102, 9312–9318 CrossRef CAS.
- M. Akiyama and H. Torii, Cooperative effect in hydrogen bonding of N-methylacetamide in carbon tetrachloride solution confirmed by NMR and IR spectroscopies, Spectrochim. Acta, Part A, 1999, 56, 137–144 CrossRef.
- M. P. Gaigeot, R. Vuilleumier, M. Sprik and D. Borgis, Infrared Spectroscopy of N-Methylacetamide Revisited by ab Initio Molecular Dynamics Simulation, J. Chem. Theory Comput., 2005, 1, 772–789 CrossRef CAS PubMed.
- L. Piatkowski and H. J. Bakker, Vibrational Relaxation Pathways of AI and AII Modes in N-Methylacetamide Clusters, J. Phys. Chem. A, 2010, 114, 11462–11470 CrossRef CAS PubMed.
- M. Albrecht, C. A. Rice and M. A. Suhm, Elementary peptide motifs in the gas phase: FTIR aggregation study of formamide, acetamide, N-methylformamide, and N-methylacetamide, J. Phys. Chem. A, 2008, 112, 7530–7542 CrossRef CAS PubMed.
- V. Yatsyna, D. J. Bakker, P. Salén, R. Feifel, A. M. Rijs and V. Zhaunerchyk, Infrared action spectroscopy of low-temperature neutral gas-phase molecules of arbitrary structure, Phys. Rev. Lett., 2016, 117, 118101 CrossRef PubMed.
- M. A. Suhm and F. Kollipost, Femtisecond single-mole infrared spectroscopy of molecular clusters, Phys. Chem. Chem. Phys., 2013, 15, 10702–10721 RSC.
- Y. Loquais, E. Gloaguen, S. Habka, V. Vaquero-Vara, V. Brenner, B. Tardivel and M. Mons, Secondary structures in Phe-containing isolated dipeptide chains: laser spectroscopy vs. quantum chemistry, J. Phys. Chem. A, 2015, 119, 5932–5941 CrossRef CAS PubMed.
- L. C. Mayne and B. Hudson, Resonance Raman spectroscopy of N-methylacetamide: overtones and combinations of the C–N stretch (amide II′) and effect of solvation on the CO stretch (amide I) intensity, J. Phys. Chem., 1991, 95, 2962–2967 CrossRef CAS.
- P. Zielke and M. A. Suhm, Concerted proton motion in hydrogen-bonded trimers: a spontaneous Raman scattering perspective, Phys. Chem. Chem. Phys., 2006, 8, 2826–2830 RSC.
- E. G. Buchanan, W. H. James III, S. H. Choi, L. Guo, S. H. Gellman, C. W. Müller and T. S. Zwier, Single-conformation infrared spectra of model peptides in the amide I and II regions: experiment-based determination of local mode frequencies and inter-mode coupling, J. Chem. Phys., 2012, 137, 094301 CrossRef PubMed.
- T. Fornaro, M. Biczysko, J. Bloino and V. Barone, Reliable vibrational wavenumbers for CO and N–H stretchings of isolated and hydrogen-bonded nucleic acid bases, Phys. Chem. Chem. Phys., 2016, 18, 8479–8490 RSC.
- N. O. B. Lüttschwager, Raman Spectroscopy of Conformational Rearrangements at Low Temperatures, Springer, Heidelberg, 2014 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09 Revision E.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- M. Avalos, R. Babiano, J. L. Barneto, J. L. Bravo, P. Cintas, J. L. Jiménez and J. C. Palacios, Can we predict the conformational preference of amides?, J. Org. Chem., 2001, 66, 7275–7282 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. Heger, J. Altnöder, A. Poblotzki and M. A. Suhm, To π or not to π – how does methanol dock onto anisole?, Phys. Chem. Chem. Phys., 2015, 17, 13045–13052 RSC.
- TURBOMOLE V6.3 2011, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- L. O. Paulson and D. T. Anderson, Infrared spectroscopy of the amide I mode of N-methylacetamide in solid hydrogen at 2–4 K, J. Phys. Chem. B, 2011, 115, 13659–13667 CrossRef CAS PubMed.
- S. Ataka, H. Takeuchi and M. Tasumi, Infrared studies of the less stable cis form of N-methylformamide and N-methylacetamide in low-temperature nitrogen matrices and vibrational analyses of the trans and cis forms of these molecules, J. Mol. Struct., 1984, 113, 147–160 CrossRef CAS.
- F. Kollipost, A. V. Domanskaya and M. A. Suhm, Microscopic roots of alcohol–ketone demixing: infrared spectroscopy of methanol–acetone clusters, J. Phys. Chem. A, 2015, 119, 2225–2232 CrossRef CAS PubMed.
- M. Miyazaki, J. Saikawa, H. Ishizuki, T. Taira and M. Fujii, Isomer selective infrared spectroscopy of supersonically cooled cis- and trans-N-phenylamides in the region from the amide band to NH stretching vibration, Phys. Chem. Chem. Phys., 2009, 11, 6098–6106 RSC.
- K. Sakota, Y. Shimazaki and H. Sekiya, Formation of a dual hydrogen bond in the N–H⋯CO moiety in the indole-(N-methylacetamide)1 cluster revealed by IR-dip spectroscopy with natural bond orbital analysis, J. Chem. Phys., 2009, 130, 231105 CrossRef PubMed.
- T. Köddermann and R. Ludwig, N-Methylacetamide/water clusters in a hydrophobic solvent, Phys. Chem. Chem. Phys., 2004, 6, 1867–1873 RSC.
- S. Trabelsi, M. Bahri and S. Nasr, X-ray scattering and density-functional theory calculations to study the presence of hydrogen-bonded clusters in liquid N-methylacetamide, J. Chem. Phys., 2005, 122, 024502 CrossRef PubMed.
- S. Trabelsi, M. Bahri, S. Nasr and M.-C. Bellissent-Funel, Local Order in Fully Deuterated Liquid N-Methylacetamide (C3D7NO) As Studied by Neutron Diffraction and Density Functional Theory Calculations, J. Phys. Chem. B, 2006, 110, 25021–25025 CrossRef CAS PubMed.
- J. Eckert, M. Barthes, W. T. Klooster, A. Albinati, R. Aznar and T. F. Koetzle, No Evidence for Proton Transfer along the N–H⋯O Hydrogen Bond in N-Methylacetamide: Neutron Single Crystal structure at 250 and 276 K, J. Phys. Chem. B, 2001, 105, 19–24 CrossRef CAS.
- J. Edler and P. Hamm, Spectral response of crystalline acetanilide and N-methylacetamide: vibrational self-trapping in hydrogen-bonded crystals, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 214301 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Further NMA gas phase and jet spectra, spectra of the d7 isotopologue, experimental details, tables of B97D results, Cartesian B3LYP-D3 coordinates. See DOI: 10.1039/c6cp07989j |
| This journal is © the Owner Societies 2017 |