
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evidence for photosensitised hydrogen production from water in the absence of precious metals, redox-mediators and co-catalysts†
S.
Salzl
,
M.
Ertl
and
G.
Knör
 *
*
Johannes Kepler University Linz (JKU), Institute of Inorganic Chemistry, Altenbergerstr. 69, A-4040 Linz, Austria. E-mail: guenther.knoer@jku.at
First published on 2nd February 2017
Abstract
The water-soluble zinc porphyrin complex Zn(TPPS)4− with TPPS = tetrakis-(4-sulfonatophenyl)porphyrin surprisingly was found to produce significant amounts of hydrogen from aqueous sulfite or amine solutions under visible-light exposure without requiring any other components such as electron relays or additional proton reduction catalysts. Although the production rates and total amounts of chemically stored fuel obtained under these conditions are still much too low to be relevant for practical applications, the background of this unprecedented observation was further studied in its own right. Since the central metal zinc is unlikely to be involved in proton-coupled electron transfer steps upon long-wavelength irradiation and the process does not seem to be much affected by variations of the electron donor added, the mechanism of photocatalytic H2 release is suggested to involve previously neglected redox features of the in situ generated hydroporphyrin ligand system in aqueous solution.
Photocatalytic H2 generation enjoys great interest as a research topic, considering possible future applications of hydrogen as a renewable fuel.1–3 To this extent many different systems using visible light and therefore eventually ambient sunlight as a clean energy source for proton reduction have been found. The two main directions of research are heterogeneous and homogeneous photocatalysis, with the former often focusing on nanostructures like semiconductor photoelectrodes or functionalized quantum dots.4 These systems are considered to be more promising due to their ability to photogenerate larger amounts of hydrogen gas already today. However, fabrication and fine-tuning of the materials involved can be difficult, as well as fully understanding the elementary processes involved. Molecular photocatalysts in contrast can offer the advantage of being able to study the effects of different factors influencing the hydrogen production mechanisms more directly, and this deeper understanding could lead to entirely new sustainable energy concepts in the long term.
Several catalytic systems have been found that rely on photoredox-active metal complexes in solution for generating hydrogen gas upon irradiation. One of the first successful examples reported in the literature combined an electron donor, a visible-light absorbing photosensitizer, optionally an additional redox-active compound acting as an electron relay and a heterogeneous catalyst capable of evolving hydrogen when supplied with electrons.5 This prototype system could already operate in purely aqueous medium, but the ruthenium and rhodium complexes employed, as well as the colloidal platinum catalyst required consist of rare and expensive noble metals, which cannot be considered a sustainable solution. Since then, many attempts have been made to replace parts of such a solar fuel generating system with more abundant components, including molecular photosensitizers6–8 as well as catalysts9,10 or even both.11 A combination of all these desirable features in terms of practicability, however, is quite difficult to be achieved, and also the latter example from the Eisenberg group11 still has the disadvantage of not operating in a purely aqueous medium. It requires a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of water and acetonitrile instead for solubility reasons related to the cobalt catalyst [CoIIICl(dmgH)2(py)] used, which is furthermore unstable when dissolved in water, thus limiting reaction times.
:1 mixture of water and acetonitrile instead for solubility reasons related to the cobalt catalyst [CoIIICl(dmgH)2(py)] used, which is furthermore unstable when dissolved in water, thus limiting reaction times.
More recently, a molecular all-abundant element system has been described by Kurz and Probst et al. that utilized a water-soluble tin porphyrin and the same type of cobalt catalyst to produce hydrogen in aqueous solution with a turnover number (TON) barely exceeding unity, just enough to indicate some catalytic activity.12 Here, we wish to report our own results with a further simplified homogeneous photocatalytic system based on earth-abundant components producing hydrogen gas from neat aqueous solution. The system is operating in the presence of a readily available molecular photosensitizer and different sacrificial electron donors including simple inorganic salts, but notably without the requirement of further mediators or catalysts.
Results and discussion
The water-soluble complex zinc(II)-tetrakis(4-sulfonato-phenyl)porphyrin, Zn(TPPS)4− was investigated as a potential sensitizer for a stepwise photoredox reaction sequence with a molecular cobalt-hydride forming catalyst13–15 according to the following simplified scheme (Fig. 1).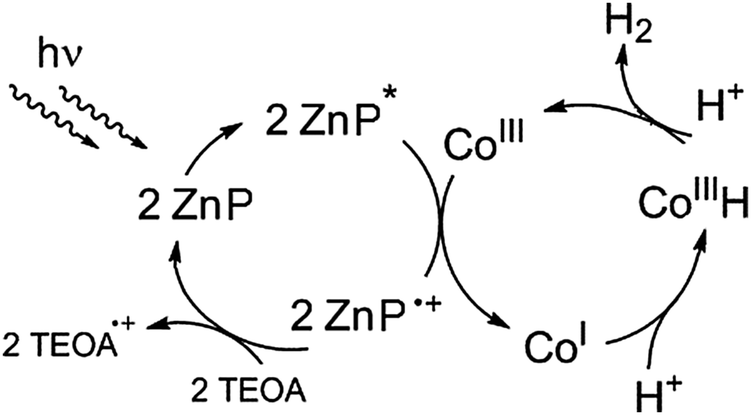 | ||
| Fig. 1 Envisioned homogeneous photocatalytic reaction system for visible-light driven hydrogen formation with a zinc porphyrin sensitizer (ZnP) and a cobalt-based proton reduction catalyst. | ||
During a control experiment using the electron donor triethanolamine (TEOA) at pH 7 in the absence of the cobalt catalyst, unexpectedly some H2 evolution was detected after overnight irradiation (visible range of 100 W/2 HBO mercury short-arc lamp, 400 nm longpass filter). It is not surprising that many redox catalysts, being chromophores themselves, can evolve some H2 without added sensitizer. This amount is then usually substracted from the total amount of H2 measured with the full system (catalyst, sensitizer, donor) in order to give the appropriately corrected yield of H2 gained only from the sensitized cycle. It is however quite unusual for a conventional photosensitizer to evolve H2 on its own in the absence of any additional catalyst. In our case, initially no more than one tenth of a turnover with regard to the amount of Zn(TPPS)4− was detected, but this unprecedented observation warranted further investigation and optimization.
To ensure that the observed H2 evolution was due to the donor-sensitizer interaction, experiments without added TEOA were also performed and monitored via UV-vis spectroscopy. This clearly led to the evolution of a primary photoproduct with a disrupted macrocyclic π-conjugation showing a broad new absorption feature with a maximum at around 740 nm (Fig. 2), which by comparison with characteristic optical properties of similar photolysis products reported in the literature16,17 could either be a metallophlorin-anion (deprotonated 5,24-dihydroporphyrin) derivative (ZnPH−) or an oxygenation primary product with a sp3-saturated meso-carbon site. In the latter case an attack of singlet oxygen at the macrocyclic ligand (generated from remaining trace amounts of O2) has to be postulated. Under such oxidative conditions, closely related metalloporphyrin sensitizers usually undergo bleaching and ring-opening as an irreversible secondary process.17
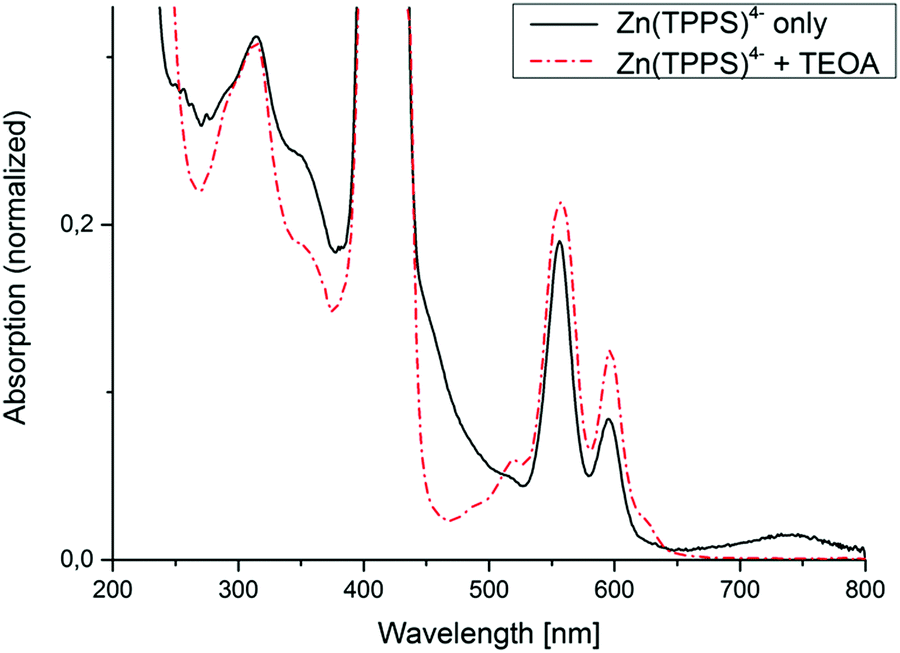 | ||
| Fig. 2 Electronic spectra of Ar-saturated aqueous solutions of Zn(TPPS)4− recorded after visible-light irradiation in the absence and presence of an electron donor (TEOA). | ||
In contrast, the sample irradiated with added TEOA donor upon photolysis clearly showed different spectral changes with gradual rise of a narrow band at 624 nm characteristic for the formation of a chlorin or 2,3-dihydroporphyrin complex (ZnC) without any accumulation of the far-red light absorbing phlorin-type products (Fig. 2). Photochemical conversion of metalloporphyrin complexes to chlorins in the presence of an electron donor is a well-known reaction, for example with ascorbic acid as the reductant and an added amine base18 (TEOA in the present case being both). So far, however, to the best of our knowledge, hydrogen evolution under these conditions has never been mentioned in literature.
Since the photoreaction was initially performed also including blue light excitation (>400 nm filter), it could not be neglected that H2 evolution might arise from an inefficient S2-excited state process requiring irradiation of the porphyrin Soret band, as has been observed before in a few other photoredox reactions.19,20 To find out more about the excited state reactions responsible for triggering H2 production, the photolysis was therefore also carried out with longer-wavelength light (>500 nm longpass filter). Formation of the chlorin derivative ZnC still occurred upon excitation in the porphyrin Q-band range, as well as H2 formation, however only with a turnover number of TON(ZnP) = 0.02 after an overnight reaction. Nevertheless, this result strongly indicates that hydrogen formation is linked to a porphyrin ligand photoreduction process involving the energetically lowest-lying states of ZnP, and not for example due to a reaction sequence requiring the population of higher excited singlet states of the sensitizer.
As already mentioned above, only a very small amount of H2-gas could be detected in the headspace after a long reaction time, which could be partially due to the experimental parameters. The cuvette contained 3 mL of aqueous solution, and hydrogen gas is well soluble in water (Henry volatility of KpcH = 1/Hcp = 128205 Pa m3 mol−1).21 As no efforts were taken to remove all of the evolved H2 from solution, some amount of it (especially in comparison to an overhead space of only 1.35 mL) is not accounted for in the GC measurements, with levels only rising above detection limit when enough had been formed to saturate the solution. To deal with this problem, one could for example freeze or heat the solution for every measurement to try and drive out all of the evolved gas, a complicated process which could easily lead to less reproducible data. This was not attempted, and it is therefore to be assumed that every TON reported here should actually be higher due to some undetected H2 remaining in solution. Instead, conditions were varied to try and arrive at a system still containing only Zn(TPPS)4− as a sensitizer and a sacrificial electron donor, while producing enough H2 to clearly achieve photocatalytic levels (i.e. a TON >1 based on the headspace gas detection only), which indeed could be achieved. These experiments carried out under visible-light irradiation in Ar- or N2-saturated water are summarized in Table 1.
| Entry |
Zn(TPPS)4−
(H2 yield) |
Donor |
Amount of donor
(excess) |
pHa | Irradiat. timeb (h) | TON H2/ZnP |
|---|---|---|---|---|---|---|
| a pH not adjusted unless otherwise stated. b Irradiated >400 nm. c Nitrogen instead of argon used to remove O2. d Self-buffered system. e Irradiated >500 nm. f Buffered. g Calc. TON-value in H2/ZnP corrected for the amount of dissolved hydrogen according to Henry′s law.21 | ||||||
| 1c |
2.5 μmol
(75 nmol) |
TEOA |
10 vol%
(900-fold) |
7d | 70e | 0.03 |
| 2c |
2.7 μmol
(108 nmol) |
TEOA |
10 vol%
(840-fold) |
11.5 | 29 | 0.04 |
| 3 |
5.7 μmol
(171 nmol) |
Na2SO3 |
0.6 mmol
(100-fold) |
8 | 21 | 0.03 |
| 4 |
0.2 μmol
(22 nmol) |
Na2SO3 |
2.4 mmol
(12 |
8 | 29 | 0.11 |
| 5 |
0.7 μmol
(805 nmol) |
TEA |
2 vol%
(610-fold) |
10f | 21 |
1.15
(1.2)g |
Compared to the first experiment described above (entry 1), raising the pH value to above 10 (entry 2) improved the TON. A more drastic change was the formation of a significantly larger amount of chlorin in the photostationary mixtures, which will be discussed in detail later. This improvement in photochemical activity was taken as an indication that increasing the pH (although H2 evolution from water should require less energy in acidic solution) was the correct choice for enhancing the yield of hydrogen with the photosensitizer system used. Still no proof for a photcatalytic reaction cycle was found when changing the donor to sodium sulfite (entry 3). Lowering the concentration of Zn(TPPS)4− did not linearly decrease the TON (entry 4). Finally, however, using triethylamine (TEA) as an alternative donor with a low concentration of Zn(TPPS)4− (entry 5) gave results significantly exceeding stoichiometric product formation, thus proving that the reaction could indeed be driven as a photocatalytic process.
This is, of course, a surprising result considering that no additional catalyst has been added. To discuss the possible processes leading to H2 formation, it is necessary to take a look at the spectral changes measured during the reactions in more detail. The experiment corresponding to entry 2 in Table 1 (TEOA, pH 11.5) will serve as a representative example (Fig. 3). Full spectra over reaction time for each discussed reaction can be found in the ESI.†
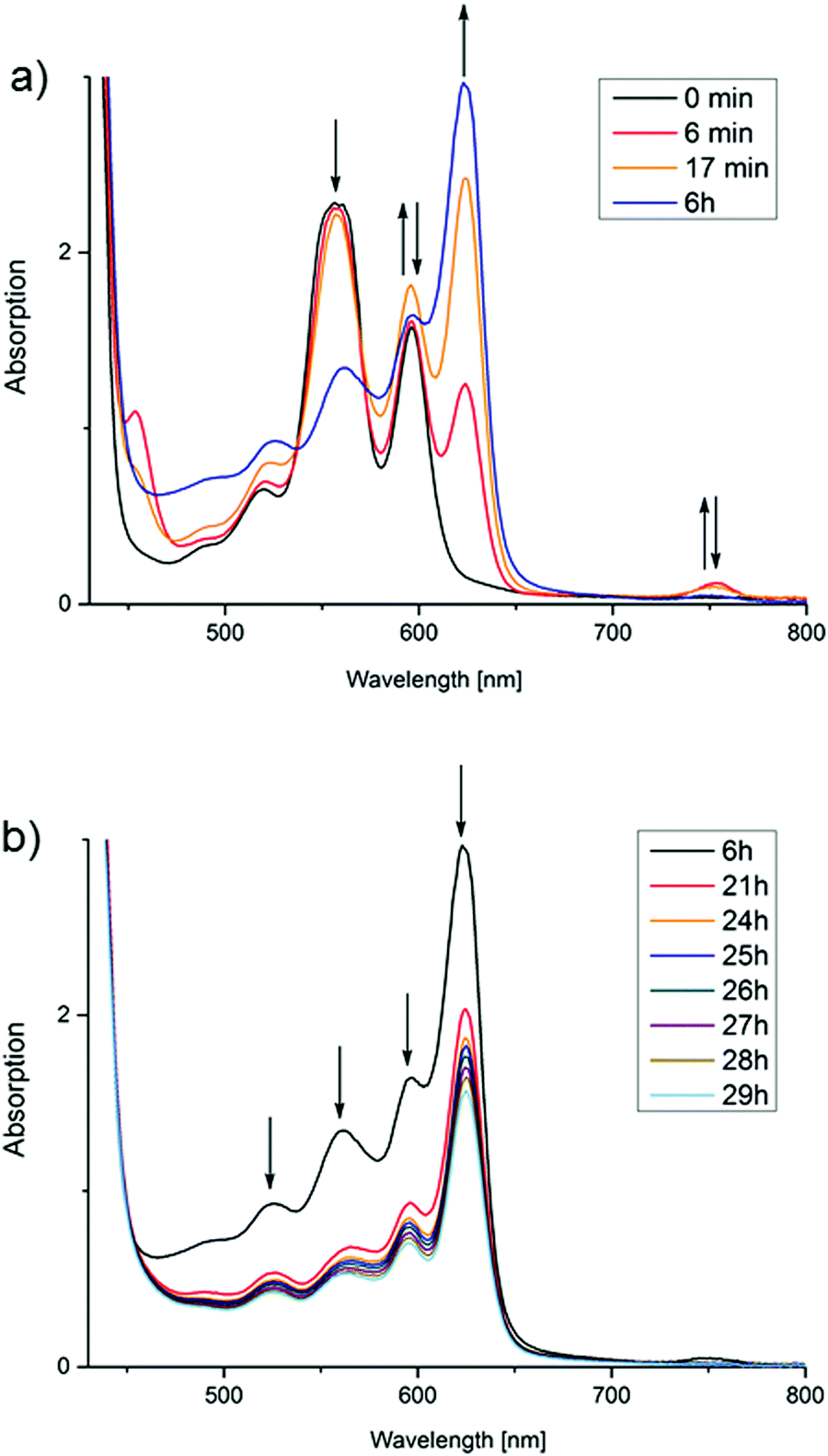 | ||
| Fig. 3 Spectral changes in the course of Zn(TPPS)4− photolysis in water at pH = 11.5 (see entry 2 in Table 1). (a) Accumulation of the zinc chlorin derivative (ZnC) and decrease of the porphyrin (ZnP) within the first hours of irradiation: (b) bleaching of all species upon long-term light exposure (>400 nm). | ||
Taking a look at the Q bands in particular, the starting porphyrin species ZnP shows three characteristic maxima at 520, 557 and 596 nm (Fig. 3). Within a few minutes of irradiation, two new species are formed: the chlorin derivative ZnC with the characteristic maximum at 624 nm, and some bacteriochlorin (2,3,12,13-tetrahydroporphyrin) ZnBC with a narrow peak at 754 nm corresponding to literature.18 The latter signal quickly vanishes, and it could not in fact be detected at any point during most other reactions. This suggests that it is not of importance for the dominant hydrogen evolving process and will therefore not be discussed further.
As already reported by Seely et al.,18 the amount of chlorin formed in the course of the photoreduction process of zinc porphin is highly dependent on the reaction conditions like type of donor used, pH value and initial porphin concentration.
As displayed in Fig. 3, after a certain time, each photolysis experiment reaches a steady-state concentration of chlorin and porphyrin, after which all components apparently bleach. The time needed for this maximum concentration to be reached greatly varies (Fig. 4). It does, however, not correspond directly with the variations in H2 concentration measured in the headspace; at the time of chlorin saturation, no H2 could be detected at any point. It should be mentioned that in the dark, chlorin does not react back into porphyrin, and no additional H2 is produced. Also, in other reactions performed to test the suitability of Zn(TPPS)4− as a sensitizer in combined reactions involving cobalt co-catalysts, no chlorin was formed at any point, regardless if hydrogen was produced or not.
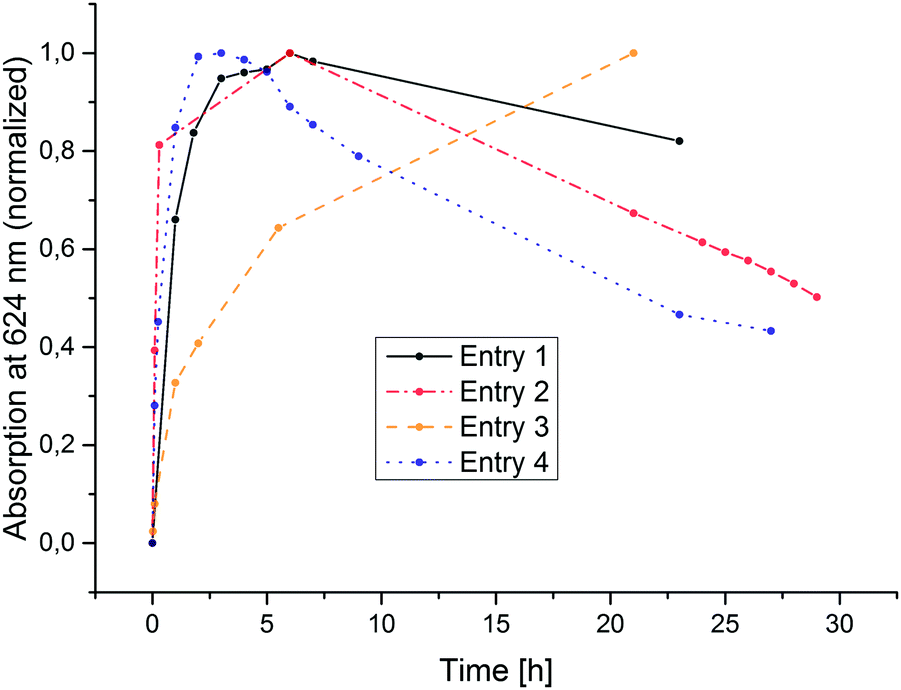 | ||
| Fig. 4 Characteristic rise and fall of peak maxima at 624 nm over irradiation time corresponding to variations in ZnC concentration. Connecting lines added for visual clarity. | ||
The 624 nm signal variation for entry 5 (TEA as donor) is not included in the plot of Fig. 4, because in this reaction, besides the chlorin also another new species strongly absorbing around 600–650 nm was formed. Fig. 5 shows the relevant part of the spectrum for a typical reaction mixture with TEA as donor. This differing behaviour of TEA is known. Seely et al. speculate that it could possibly result from TEA simply being a better donor as well as its ability to coordinate to the central zinc atom.18 The new maximum at 609 nm corresponds well with the reported spectral features of an iso-bacteriochlorin (2,3,7,8-tetrahydroporphyrin) species ZniBC, and while its emergence still does not correspond with hydrogen evolution, it does occur reliably in the TEA-based reaction mixtures giving catalytic TON-values.
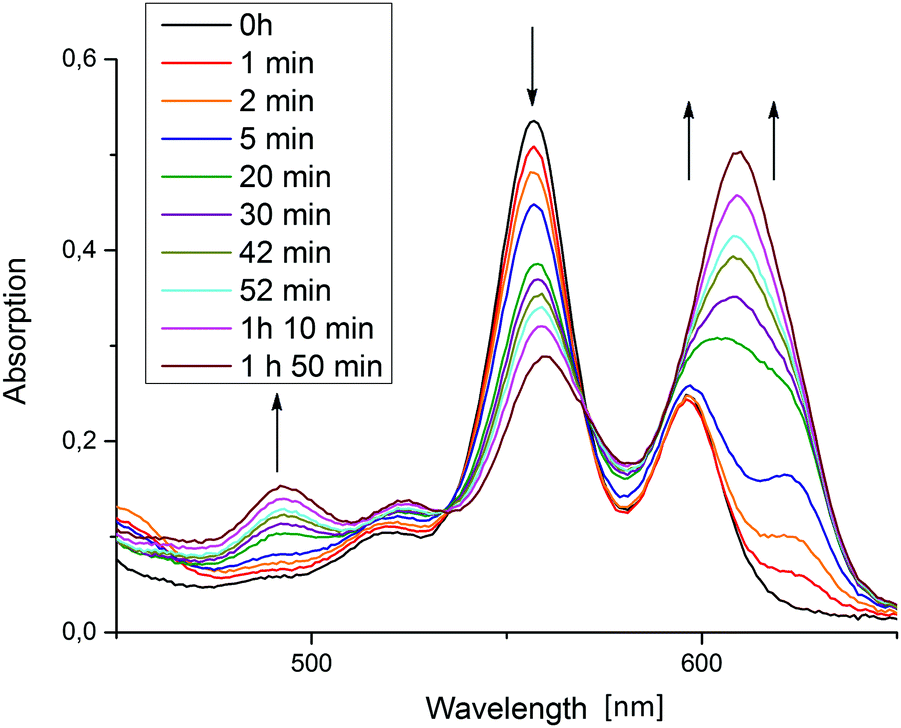 | ||
| Fig. 5 Absorption spectral changes in the porphyrin Q-band region measured in the initial phase of photocatalytic reactions using TEA as donor. | ||
Taking together these spectroscopic results, it has to be concluded that photocatalytic hydrogen generation in anaerobic aqueous solutions of Zn(TPPS)4− in the absence of additional redox mediators or co-catalysts is only occurring after a certain time period of primary photolysis, which corresponds to the initial accumulation of the two-electron reduced chlorin species ZnC (Fig. 4). This is also corroborated by 1H-NMR spectral variations (ESI†).
It should be pointed out that in the field of molecular catalysis the appearance of such a lag-phase with delayed product formation must be handled with care, since gradual decomposition of the starting compounds into metal oxide nanoparticles (and thus actually into a heterogeneous catalyst) may be responsible for all of the effects observed. In the present case, the photogeneration of ZnO nanoparticles via demetallation of the sensitizer upon long-term visible light exposure also cannot be fully excluded. However, since the bandgap of ZnO (3.3 eV) would require an excitation with photons in the UV-range, and successful hydrogen production is also observed upon Q-band irradiation (>500 nm, Table 1), we are convinced that in our system a molecular species derived from irradiated ZnP is indeed operating as a homogeneous H2-producing photocatalyst.
Furthermore, unlike initially anticipated with zinc porphyrins typically representing excellent excited state electron donors (Fig. 1), the production of hydrogen in the present case (without any electron acceptors available other than water) clearly has to be connected to a sequence of reductive excited state quenching steps involving the ZnP sensitizer and its hydroporphyrin photoproducts.
Since the observed hydrogen generation process is directly coupled to the second phase of the photoreaction, where the concentration of the initially accumulated species ZnC decreases again upon further irradiation (Fig. 3b), it can be concluded that reductive quenching of the photoexcited zinc chlorin complex must be the key reaction for triggering H2 evolution, and that ZnC is representing the main sensitizer.
It has been discovered only recently that chlorin complexes may act as efficient photoredox catalysts. By this work carried out in our own group, it could be shown that reductive quenching of a water-soluble tin chlorin based photosystem irradiated in the presence of suitable electron donors may directly lead to a so-called chlorin–phlorin anion complex22,23 (a non-aromatic porphyrin derivative carrying a protonated four-electron reduced ligand with one saturated meso-carbon position, which is characterised by a rather featureless broad absorption pattern in the visible spectral region, lacking the otherwise typical Soret- and Q-bands).
The chlorin–phlorin derivatives turned out to be excellent two-electron reductants, which under anaerobic conditions can reversibly transfer hydride reducing equivalents to other compounds.3,23 Based on these results and considering all of the present findings, we tentatively suggest that visible-light driven hydrogen photogeneration with Zn(TPPS)4− in anaerobic aqueous solution initially requires the formation of the chlorin complex ZnC (Fig. 3a) as the active photocatalyst, which is then converted (in a sequence of steps involving the sacrificial electron donor present)23 into a chlorin–phlorin anion species ZnCH− (Fig. 6) according to eqn (1):
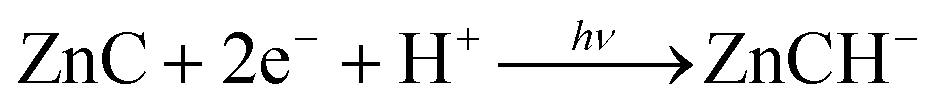 | (1) |
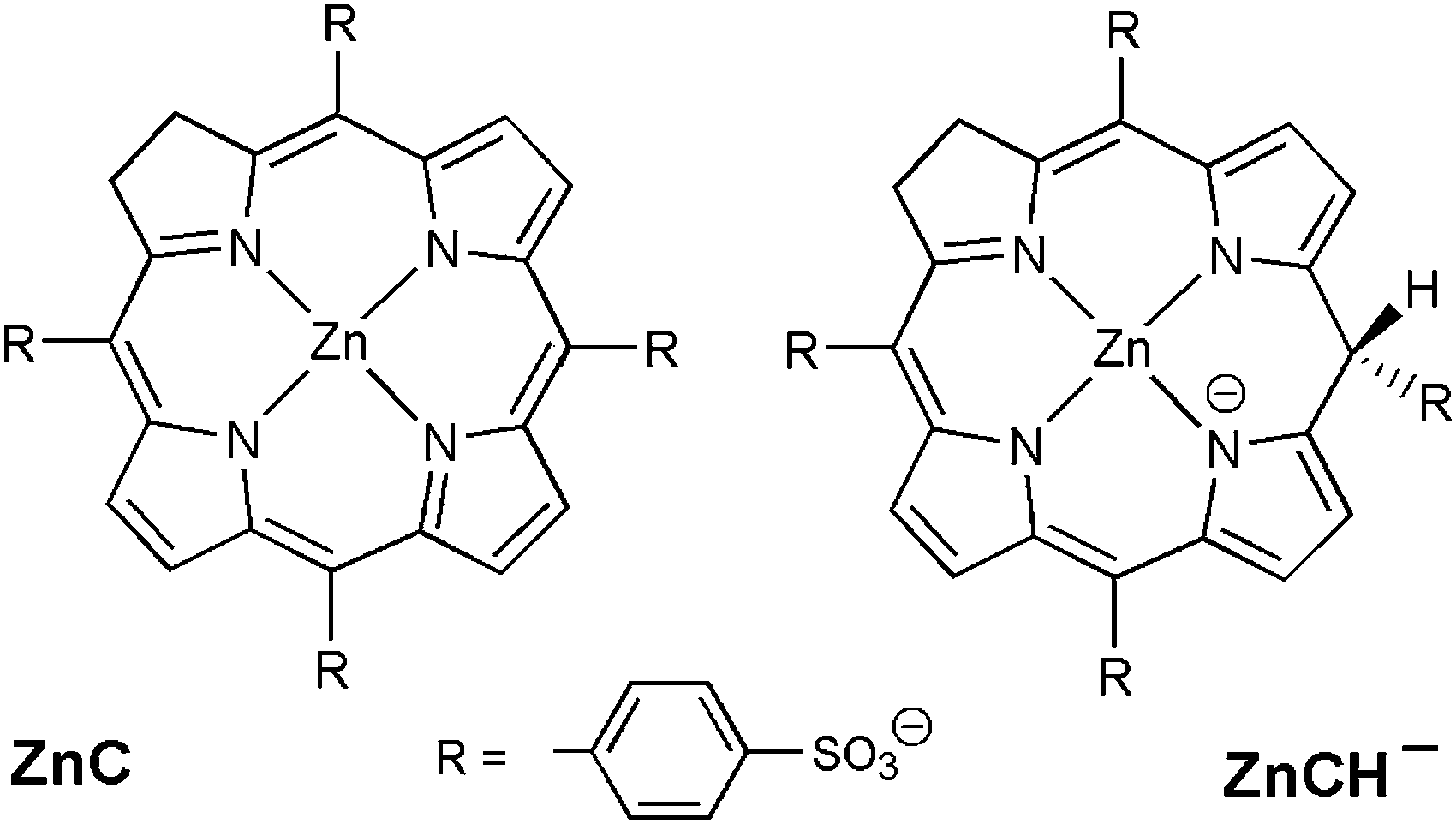 | ||
| Fig. 6 Structural representations of the zinc chlorin complex ZnC and the chlorin–phlorin anion photoreduction product zinc(II)-2,3,10-trihydro-5,10,15,20-tetrakis-(4-sulfonatophenyl)-porphyrin. | ||
Since chlorin–phlorin derivatives are generally expected to be oxygen-sensitive,22,23 in the starting period of photolysis the remaining traces of O2 in the inert-gas saturated systems will be completely consumed in a re-oxidation side-reaction forming back the chlorin complex ZnC according to eqn (2):
| ZnCH− + O2 → ZnC + OOH− | (2) |
After completion of this process, the photoreaction in the absence of co-catalysts can lead to some extent of chlorin–phlorin anion accumulation (eqn (1)), which is then accompanied by an apparent bleaching of the sample and a gradual loss of the initial Soret- and Q-band features (Fig. 3b and 4). Furthermore, depending on the specific reaction conditions, especially on variations of donor type and pH of the samples (Table 1), the additional formation of bacteriochlorin or iso-bacteriochlorin derivatives ZnBC (Fig. 3) or ZniBC (Fig. 5) can also be observed. This well-known reactivity pattern of non-aromatic phlorin-type species22–24 may be explained by subsequent protonation and prototropic tautomerization steps, leading to the formation of compounds carrying less reactive resonance-stabilized tetrahydroporphyrin ligands with intense absorption bands including characteristic Soret- and Q-band features (eqn (3)):
| ZnCH− + H+ → ZnBC/ZniBC | (3) |
In addition to these inevitable side-reactions influencing the fate of chlorin–phlorin type intermediates in aqueous solution, the reduction equivalents (H+, 2e−) reversibly stored in the ligand periphery of a chlorin–phlorin anion metal complex have already been successfully transferred to a metal-hydride forming co-catalyst and could also be very efficiently exploited for the light-mediated two-electron reduction of organic and biological redox cofactors including NAD+.3,23
Our present results reported here indicate that obviously in competition with the ZnCH−-consuming steps described above (eqn (2) and (3)), the protonation of the photogenerated chlorin–phlorin anion species to some extent may also lead to the release of molecular hydrogen according to eqn 4:
| ZnCH− + H+ → ZnC + H2 | (4) |
This important and unprecedented reactivity occuring in the absence of further co-catalysts and redox-mediators opens the possibility to close a very simple homogeneous photocatalytic two-electron redox cycle for solar hydrogen production with the reversibly photoreduced and re-oxidized chlorin species (ZnC) able to act as a light-harvesting photosensitizer for multiple turnovers (Fig. 7).
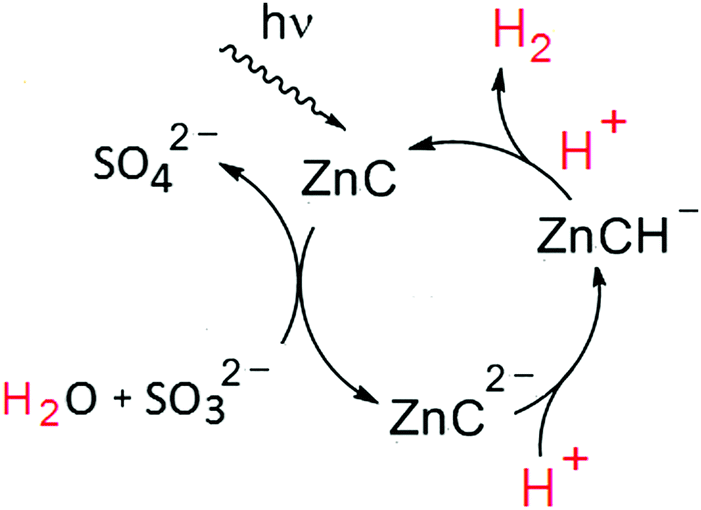 | ||
| Fig. 7 Proposed homogeneous photocatalytic system for visible-light driven water splitting based on a zinc chlorin sensitizer (ZnC) reversibly forming a chlorin–phlorin anion intermediate (ZnCH−) acting as a hydride supplying catalyst. | ||
Besides sulfite, acting as a net two-electron reductant and formally taking up the remaining oxygen core of the split water molecule as shown in Fig. 7, other sacrificial donors such as amines in a stepwise fashion can also provide the electrons required for the ZnC sensitized hydrogen formation (Table 1). Note that compared to conventional systems for H2-photoproduction based on molecular components, in our case ZnC is not only acting as a light-harvesting chromophore, but apparently can also substitute the function of a co-catalyst (e.g. by replacing the required CoIII–H active site with ZnCH− in the hydrogen evolving key-steps, Fig. 1).
Although quite remarkable, this type of all-earth abundant photochemical systems at the moment is not yet able to produce much more hydrogen than is needed to prove that it is indeed catalytic. Nevertheless it still yields more H2 than similar molecular systems shown to operate in pure water.12
It should also be pointed out that the reaction conditions reported here because of the initially unexpected findings were not at all optimized for a possible scenario of H2-production with the chlorin complex ZnC instead of ZnP as the actual photocatalyst (Fig. 1 and 7), and the light-source applied (HBO 100 W/2 mercury lamp equipped with longpass filters) offers only a minor spectral output in the 600–650 nm region. Further studies using for example solar radiation or actinic LED-light better suitable for chlorin excitation, as well as the systematic application of other diamagnetic metallochlorin systems based on earth-abundant components and optimized substituent patterns are therefore expected to enable considerable improvments in TON and hydrogen yield.26
Conclusions
It has been shown for the first time that hydrogen can be produced in aqueous solution, using only a donor and a noble-metal-free sensitizer in the absence of additional redox-mediators and co-catalysts.Moreover, it has also been demonstrated that simple inorganic salts such as sodium sulfite can replace the usually required amines as sacrificial electron donors for visible-light driven hydrogen formation. Thus the two-electron reduction of water for solar fuel production (Fig. 7) could easily be coupled to environmentally relevant redox processes such as flue gas desulfurisation for reducing atmospheric SO2 emissions.
More research needs to be carried out to completely elucidate and confirm the possible mechanisms of hydrogen evolution in the system presented here, as well as to much further raise the turnover numbers measured.
The results obtained indicate that a four-electron reduced (chlorin–phlorin type)22,23 hydroporphyrin derivative with an apparently “bleached out” absorption pattern lacking the typical Soret- and Q-bands of porphyrin derivatives could be involved as a redox catalyst. Such a highly reducing species generated by a reductive quenching mechanism of the photoexcited chlorin sensitizer might be the crucial hydride carrying species triggering catalytic H2 generation upon subsequent protonation. A similar reaction sequence for the photocatalytic accumulation of hydrogen equivalents upon red-light irradiation of a metallochlorin complex has been discovered recently.3,23
The results clearly point out the important role of control experiments when studying multicomponent samples, and how crucial it is to elucidate the functional role of the individual components added. Especially, apparent bleaching of photostationary mixtures in the case of porphyrin-based sensitizers under catalytic performance deserves a more detailed analysis. Thus, our present findings should be useful for other groups working on basic research aspects in the field of solar hydrogen photogeneration.
Experimental
All chemicals and solvents were purchased from commercial sources in reagent quality. Water was purified with a Milli-Q system (Millipore, Bedford, MA, USA). Synthesis of Zn(TPPS)4− was performed according to a procedure described by Amao et al.,25 see ESI† for more details. Hydrogen evolution reactions were performed in rectangular 1 cm quartz cuvettes with gas tight septum, filled with 3 mL of reaction solution and leaving a headspace of 1.35 mL. The samples were bubbled through with N2 or Argon, and afterwards irradiated on an optical bench with a HBO 100 W/2 mercury lamp equipped with a lamp housing and a >400 nm longpass filter (Schott GG400) unless otherwise stated. Absorption spectra were measured directly from the reaction cuvette on a Varian Cary 300 Bio UV-Vis spectrophotometer. H2 detection was carried out after photolysis by removal of 0.5 mL of the headspace gas through septum and injection into a Fisons GC 8000 gas chromatograph. Septa were resealed with polyester resin (nail polish) to prevent H2 from escaping. Further details are given in the ESI.†Acknowledgements
This research was financially supported by the Austrian research promotion agency and the climate and energy funds (FFG project 841186 “Artificial Photosynthesis”).References
- A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022–4047 CrossRef CAS PubMed.
- N. Armaroli and V. Balzani, ChemSusChem, 2010, 4, 21–36 CrossRef PubMed.
- G. Knör, Coord. Chem. Rev., 2015, 304–305, 102–108 CrossRef.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC.
- M. Kirch, J.-M. Lehn and J.-P. Sauvage, Helv. Chim. Acta, 1979, 62, 1345–1384 CrossRef CAS.
- W. Krüger and J.-H. Fuhrhop, Angew. Chem., 1982, 94, 132–133 CrossRef.
- J. R. Darwent, P. Douglas, A. Harriman, G. Porter and M.-C. Richoux, Coord. Chem. Rev., 1982, 44, 83–126 CrossRef CAS.
- A.-M. Manke, K. Geisel, A. Fetzer and P. Kurz, Phys. Chem. Chem. Phys., 2014, 16, 12029–12042 RSC.
- J. I. Goldsmith, W. R. Hudson, M. S. Lowry, T. H. Anderson and S. Bernhard, J. Am. Chem. Soc., 2005, 127, 7502–7510 CrossRef CAS PubMed.
- P. Du, K. Knowles and R. Eisenberg, J. Am. Chem. Soc., 2008, 130, 12576–12577 CrossRef CAS PubMed.
- T. Lazarides, T. McCormick, P. Du, G. Luo, B. Lindley and R. Eisenberg, J. Am. Chem. Soc., 2009, 131, 9192–9194 CrossRef CAS PubMed.
- L. Mintrop, J. Windisch, C. Gotzmann, R. Alberto, B. Probst and P. Kurz, J. Phys. Chem. B, 2015, 119, 13698–13706 CrossRef CAS PubMed.
- T. Lazarides, M. Delor, I. V. Sazanovich, T. M. McCormick, I. Georgakaki, G. Charalambidis, J. A. Weinstein and A. G. Coutsolelos, Chem. Commun., 2014, 50, 521–523 RSC.
- E. S. Wiedner and R. Morris Bullock, J. Am. Chem. Soc., 2016, 138, 8309–8318 CrossRef CAS PubMed.
- N. Kaeffer, J. Massin, C. Lebrun, O. Renault, M. Chavarot-Kerlidou and V. Artero, J. Am. Chem. Soc., 2016, 138, 12308–12311 CrossRef CAS PubMed.
- F. R. Hopf and D. G. Whitten, in The Porphyrins, ed. D. Dolphin, Academic Press, New York, 1978, vol. II, p. 161 Search PubMed.
- K. M. Smith, S. B. Brown, R. F. Troxler and I.-J. Lai, Photochem. Photobiol., 1982, 36, 147–152 CrossRef CAS PubMed.
- G. R. Seely and K. Talmadge, Photochem. Photobiol., 1964, 3, 195–206 CrossRef CAS.
- D. Le Gourriérec, M. Andersson, J. Davidsson, E. Mukhtar, L. Sun and L. Hammarström, J. Phys. Chem. A, 1999, 103, 557–559 CrossRef.
- K. Kiyosawa, N. Shiraishi, T. Shimada, D. Masui, H. Tachibana, S. Takagi, O. Ishitani, D. A. Tryk and H. Inoue, J. Phys. Chem. C, 2009, 113, 11667–11673 CAS.
- R. Sander, Atmos. Chem. Phys., 2015, 15, 4399–4981 CAS.
- H. H. Inhoffen, P. Jäger, R. Mälhop and C.-D. Mengler, Liebigs Ann. Chem., 1967, 704, 188–207 CrossRef CAS.
- K. T. Oppelt, E. Wöβ, M. Stiftinger, W. Schöfberger, W. Buchberger and G. Knör, Inorg. Chem., 2013, 52, 11910–11922 CrossRef CAS PubMed.
- M. Taniguchi and J. S. Lindsey, Chem. Rev., 2017, 117, 344–535 CrossRef CAS PubMed.
- Y. Amao and T. Watanabe, Appl. Catal., B, 2009, 86, 109–113 CrossRef CAS.
- Taking into account the present findings, meaningful turnover numbers for hydrogen production should then be reported relative to the amount of the active zinc chlorin photocatalyst in the system as TON H2/ZnC (instead of H2/ZnP).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional data. See DOI: 10.1039/c6cp07725k |
| This journal is © the Owner Societies 2017 |