
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spatial quenching of a molecular charge-transfer process in a quantum fluid: the Csx–C60 reaction in superfluid helium nanodroplets†
Andreas W.
Hauser
*a and
María Pilar
de Lara-Castells
*b
aGraz University of Technology, Institute of Experimental Physics, Petersgasse 16, 8010 Graz, Austria. E-mail: andreas.w.hauser@gmail.com
bInstituto de Física Fundamental, CSIC, Serrano 123, 28006 Madrid, Spain. E-mail: Pilar.deLara.Castells@csic.es
First published on 28th November 2016
Abstract
A recent experimental study [Renzler et al., J. Chem. Phys., 2016, 145, 181101] on superfluid helium nanodroplets reported different reactivities for Cs atoms and Cs2 dimers with C60 fullerenes inside helium droplets. Alkali metal atoms and clusters are heliophobic, therefore typically residing on the droplet surface, while fullerenes are fully immersed into the droplet. In this theoretical study, which combines standard methods of computational chemistry with orbital-free helium density functional theory, we show that the experimental findings can be interpreted in the light of a quenched electron-transfer reaction between the fullerene and the alkali dopant, which is additionally hindered by a reaction barrier stemming from the necessary extrusion of helium upon approach of the two reactants.
1 Introduction
He-nanodroplets, a common tool for spectroscopy of atoms and molecules in a weakly perturbing matrix,1–4 have been suggested as nanolabs for the study of reactions at lowest temperatures. Most molecules submerge into these droplets after collision due to the stronger interactions between the dopant and He than between the He atoms themselves. However, alkali metal atoms and their smallest clusters tend to stay on the droplet surface since their diffuse electronic distribution would displace too much helium, making a complete submersion energetically unfeasible.5,6Recently, the interaction between a heliophilic C60 molecule, known to reside inside a He droplet, with a heliophobic Cs atom or a Cs2 dimer has been studied experimentally.7 These experiments indicate that the subsequential doping of a C60-doped He-droplet with a single Cs atom does not to lead to a reaction between the two dopants, while a reaction between the heliophilic and the heliophobic dopants takes place in cases where Cs2 is formed on the droplet beforehand. In other words, only if the doping rate with alkali atoms in the experiment is high enough for dimerization a direct reaction on the droplet can be observed.
In this theoretical study, we describe this phenomenon observed by our colleagues via a combination of electronic structure calculations and orbital-free, bosonic helium density functional theory. One-dimensional potential energy scans are generated as approximations to the reaction pathway describing the interaction between a single Cs atom or an alkali metal dimer and a C60 fullerene. These curves, calculated for the free-gas situation, are then corrected for the interaction with the surrounding helium. A similar study on the interaction of a heliophilic and a heliophobic dopant has been performed for the Xe–Rb system.8 However, in the current case, the very high polarizability of the fullerene, together with its high electron affinity, gives rise to two new features: first, the dissociation energies for Cs–C60 and Cs–C60 lie in the range of about 2 eV due to the ionic character of the interaction. Therefore, strong attractive interaction is to be expected between a submerged, heliophilic fullerene and the surface-residing, heliophobic Cs atoms. Second, the high polarizability of C60 leads to a strong van der Waals interaction between the fullerene and its surrounding helium. As a consequence, the helium density, spherically distributed around the C60 molecule in the droplet center, will have a highly peaked radial maximum near the cavity surface. The aim of this article is to show that the extrusion of helium from these areas of high density, together with a reduced mobility due to the He environment, causes an energy penalty high enough to form reaction barriers which can explain the recent experimental findings.
2 Computational methods
2.1 Ab initio calculations
The interaction of a C60 molecule with a single Cs atom and a Cs dimer in its singlet and triplet spin states is studied using (electronic) density functional theory. For carbon we used the Def2-SVP basis set.9 For cesium, the Def2-QZVP basis set of the same family10 was combined with the ECP46MDF effective core potential of the Stuttgart/Köln group.11 We further chose the cost-efficient B97M-V functional, a recent development of the Head-Gordon group,12 which combines a combinatorially-optimized semi-local meta-GGA exchange–correlation functional13 with the VV10 nonlocal correlation functional.14 This density functional approach has been thoroughly tested on several standard databases and shows remarkable accuracy for the prediction of non-bonded interactions and atomization energies at minimal computational cost, which makes it a straight-forward choice for the given task. Although higher levels of theory and larger basis sets would be applicable to the moderately sized, isolated Cs dimer in principle, DFT calculations using the same functional and the same quadruple-zeta basis set were performed for the sake of internal consistency. All Csx–C60 curves have been corrected for basis superposition errors due to the significant difference in the basis set size on each fragment.15 The correction gives rise to geometry shifts in the range of about 0.05 Å. For improved SCF convergence, the pseudo-fractional occupation number method of Rabuck and Scuseria has been applied in all cases.16In order to study the impact of the helium environment on charge mobility we further employ the constrained-DFT method of Wu and Van Voorhis17 to analyze covalent and ionic contributions as well as two-state, wave function-based, multi-configurational SCF calculations (MCSCF),18,19 aided by long-range dispersion corrections. The two-state MCSCF calculations provided both the electronic ground state, asymptotically correlated to C60− + Csx+ fragments, and an excited electronic state which, in the long-range potential region, asymptotically correlated to neutral C60 + Csx species. The necessary dispersion–correction corrections for the neutral state are extracted from the application of the CCSD(T) approach20 to small model (Csx/benzene) systems. The MCSCF and CCSD(T) calculations comprise a slightly altered basis set for the carbon atoms and a fitting of long-range correlation energies for improved accuracy. The details are given in both the Appendix and the ESI.†
All calculations are performed using the Q-Chem program package21 with the exception of the MCSCF and preliminary CCSD(T) calculations, for which the Molpro suite of programs22 is used.
2.2 He density functional theory
Free energies of doped He nanodroplets (HeN) are obtained via helium density functional theory (He-DFT) based on a slightly modified version of the Orsay–Trento density functional.29,30 Note that in contrast to common DFT approaches such as the one discussed above, this functional maps the helium density onto the energy and not the electron density. The free energy of a doped He droplet is minimized with respect to a given arrangement of the dopants within the droplet and on its surface. The free energy F[ρ], a functional of the helium density ρ, can be written as| F[ρ] = E[ρ] + Uext[ρ] − μN[ρ] − F·R[ρ], | (1) |
 | (2) |
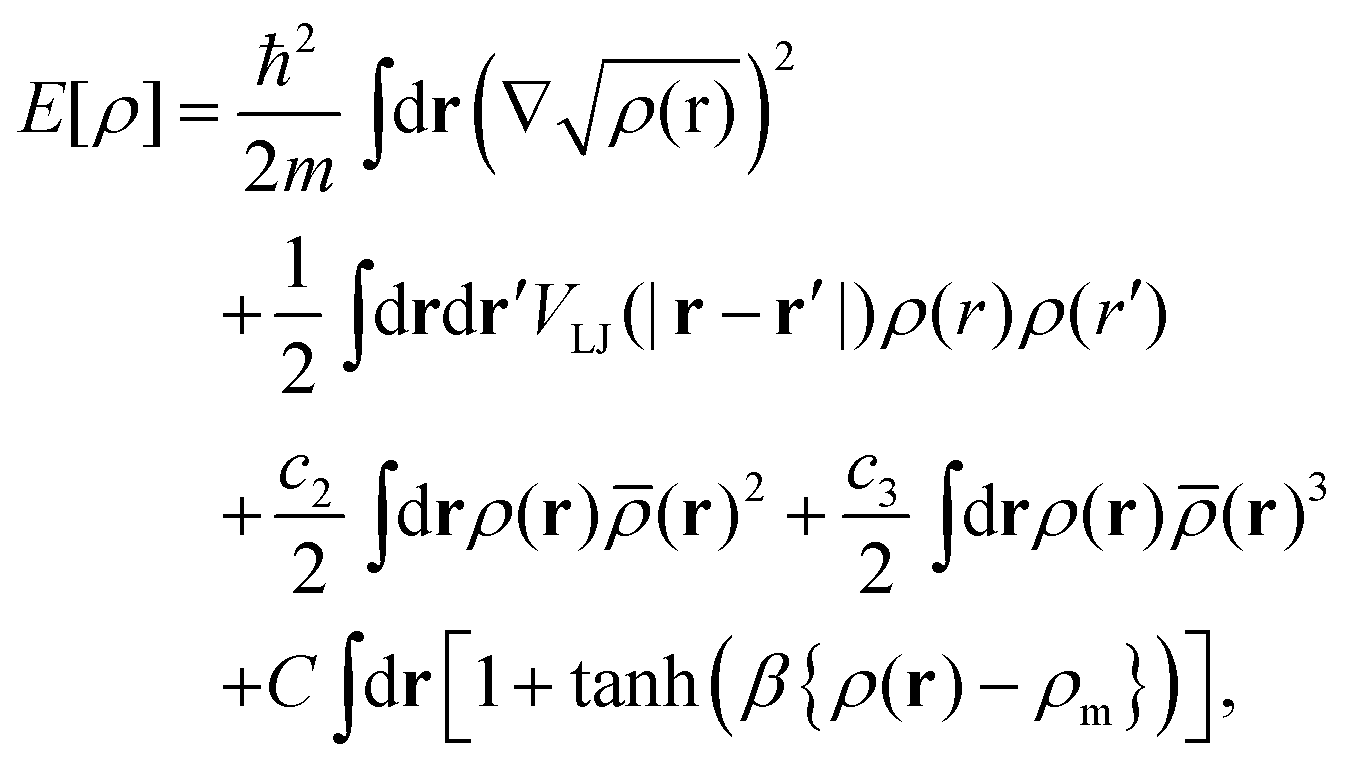 | (3) |
![[small rho, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d1.gif) , a locally averaged density for a given sphere of radius
, a locally averaged density for a given sphere of radius ![[h with combining macron]](https://www.rsc.org/images/entities/i_char_0068_0304.gif) , and finally, a penalty term which forbids an extra pile-up of He density as soon as the density exceeds a threshold value ρm. Note that an additional, nonlocal kinetic energy term which appears in the original formulation (proportional to ∇ρ(r)ρ(r′)) has been dropped here for stability reasons. For details, we refer to ref. 30 and 31, where this type of functional has been used to study the freezing transition of superfluid helium at high pressure.
, and finally, a penalty term which forbids an extra pile-up of He density as soon as the density exceeds a threshold value ρm. Note that an additional, nonlocal kinetic energy term which appears in the original formulation (proportional to ∇ρ(r)ρ(r′)) has been dropped here for stability reasons. For details, we refer to ref. 30 and 31, where this type of functional has been used to study the freezing transition of superfluid helium at high pressure.
3 Results and discussion
3.1 C60 interacting with Cs2
In a first step, the geometry of the Cs dimer was optimized in the singlet and triplet spin states using the chosen density functional. The resulting binding energies and equilibrium bond lengths show a reasonable agreement with the experimental data and previous ab initio studies performed at a higher level of theory (see Table 1). Note, however, that our results are obtained at a fraction of the typical computational cost, which allows us to extend our study towards the interaction with a C60 fullerene. We approach the dimer with a single C60 molecule32 on a straight trajectory perpendicular to the internuclear axis of the dimer and along the C3 axis of the fullerene. This choice of relative positioning (referred to as T-shaped) reflects the typical adsorption geometry for high-spin alkali dimers on the surface of helium droplets.33,34 We calculate the energy of the system as a function of the distance between both centers of mass. The one-dimensional energy surfaces plotted in Fig. 1 can be interpreted as a first approximation to the reaction pathways for the interaction of Cs2 with a single C60 molecule. They are calculated for both spin manifolds, with the Cs–Cs distance kept at the corresponding minimum energy value. The binding energies and equilibrium distances are listed in Table 2. | ||
| Fig. 1 Potential curves for the Cs–C60 (doublet) and the Cs2–C60 systems (singlet and triplet, in a T-shape configuration, i.e. with the Cs2 internuclear axis perpendicular to the C3 axis of the fullerene). Energies are plotted as a function of the distance between both centers of mass. The dashed lines correspond to blue potential energy surfaces (PESs) which are corrected for the spatial hindrance in superfluid helium; see Section 3.6 for details. | ||
| Interaction | State | r min (Å) | E (cm−1) |
|---|---|---|---|
| Cs–C60 | 2S1/2 | 6.27 | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 087 087 |
| Cs2–C60 | X1Σg+ (T-shaped) | 6.12 | 12716 |
| Cs2–C60 | a3Σu+ (T-shaped) | 5.57 | 17320 |
| Cs2–C60 | X1Σg+ (collinear) | 8.73 | 8822 |
| Cs2–C60 | a3Σu+ (collinear) | 9.53 | 11176 |
Our findings are in qualitative agreement with previous studies on the related Na2–C60 system,35 which also shows a deeper minimum for the triplet state. As a consequence, both spin states must cross at a certain distance, since for a free Cs2 molecule the singlet state PES has a deeper minimum than the triplet state (see Table 1). From our current results, the crossing takes place in the potential minimum region of the singlet state (ca. 6 Å). This can be seen if the triplet state of Cs2 in Fig. 1 is mentally shifted to higher energies by the singlet–triplet energy difference of the free gas dimer (4662 cm−1). The Cs–C60 system seems to approach its asymptotic value slightly faster, which indicates a less pronounced interaction in the long range. The different behaviors of the dispersion-dominated tails are easily understood by considering that the value of the average polarizability for triplet Cs2 (ca. 868 a.u. from ref. 36) is almost twice as large as that of atomic Cs (ca. 401 a.u. from ref. 37).
For the sake of completeness, we repeated our scans also for a collinear arrangement of the Cs2 molecule in both spin states, with the internuclear axis parallel to the C3 axis of the fullerene. The corresponding one-dimensional energy surfaces are plotted in Fig. 2. For this geometry, the binding energies are reduced significantly, in particular for the triplet dimer. Again, the distance is measured from the fullerene center to the center of mass of the Cs2 dimer. Note that the different dimer bond lengths of the X1Σg+ ground state and the a3Σu+ state lead to minima at significantly different intermolecular distances in this geometry arrangement.
 | ||
| Fig. 2 Potential curves for the Cs–C60 (doublet) and the Cs2–C60 systems (singlet and triplet, with the Cs2 internuclear axis parallel to the C3 axis of the fullerene). Energies are plotted as a function of the distance between both centers of mass. The dashed lines correspond to PESs which are corrected for the spatial hindrance in superfluid helium; see Section 3.6 for details. | ||
3.2 C60 interacting with a single Cs atom
We further compare both spin manifolds of the dimer (and both relative arrangements, i.e. T-shaped and collinear) to the interaction of C60 with a single alkali atom in its doublet ground state. The corresponding potential depth and minimum position differ considerably from that of the triplet dimer resulting in a T-shaped arrangement, showing about half the binding energy at a much larger distance (see Fig. 1 and 2). The singlet dimer in the T-shaped configuration is also stronger bound than a single atom, while the singlet dimer in a collinear configuration shows an even smaller binding energy than that of the single Cs atom.For the T-shaped configuration, the curvature of the Cs–C60 PES becomes similar to the curvatures of Cs2–C60 in the mid- to long-range. However, the comparably shallow potential minimum and the lack of sufficiently attractive forces at larger distances are already pointing towards an explanation for the quenched reactivity of a single Cs atom when compared to the triplet Cs2 dimer.
3.3 Ionization energies and Mulliken charges
The high electron affinity of the fullerene makes the interaction with the Cs atom clearly ionic in character. For Cs–C60, we obtain a dipole moment of 17.5 D, which is in reasonably good agreement with the experimental value of 21.5 ± 2 D reported in ref. 38. An overview of Mulliken charges and ionization energies (IEs) estimated from the DFT calculations is given in Table 3. For a single Cs atom we observe a positive charge of +0.80e at the Cs–C60 equilibrium position. In the case of the Cs2 molecule in the T-shaped configuration, a transfer of −1.5e onto the fullerene could be observed in the triplet state, while a reduced transfer of only −1.04e occurs in the case of the singlet dimer. The ionization energies of 3.21 and 2.83 eV are significantly higher than those of the corresponding free Cs2 molecules, for which we obtain 2.37 eV for the singlet and 1.96 eV for the triplet state, using the same computational approach. For the collinear geometry arrangement we also find higher ionization energies than for the free molecules (see Table 3). The Mulliken charges in this configuration are particularly interesting: while the total charge transfer in both spin states (about −0.87e on average) is only slightly lower than for the T-shaped geometry, we find an extremely polarized Cs2 molecule for the singlet state.| Interaction | State | Charge on Cs/Cs2 (e) | IEa (eV) |
|---|---|---|---|
| a Estimation based on the energy of the highest occupied orbitals. b The first value refers to the Cs atom which is closer to the fullerene. | |||
| Cs–C60 | 2S1/2 | 0.80 | 3.56 |
| Cs2–C60 | X1Σg+ (T-shaped) | 1.04 | 3.21 |
| Cs2–C60 | a3Σu+ (T-shaped) | 1.50 | 2.83 |
| Cs2–C60 | X1Σg+ (collinear) | 2.03, −1.20 | 3.32b |
| Cs2–C60 | a3Σu+ (collinear) | 0.83, 0.07 | 3.18b |
3.4 Csx–C60 curves without charge transfer
The previous sections revealed that the interaction of a single Cs atom or a Cs2 molecule with a fullerene shows a strong ionic character. However, since both reactants are spatially separated by superfluid helium, a quenched electron mobility is to be expected. In this section, we approximate this feature by constraining both reactants to stay fully neutral upon approaching each other. To do this, we employ constrained-DFT,17 (CDFT) which allows introducing arbitrary constraints to the electron density during the self-consisted-field iterations of the DFT calculations. To each of these constraints corresponds a Lagrange multiplier Vc with the physical meaning of a fictitious external potential, which acts on the density in such a way that the neutrality of both reactants (Csx and the C60) is assured. As a consequence, the additional potential expression occurs in the Kohn–Sham equations, and the corresponding orbitals are evaluated, together with Vc, in a self-consistent manner.In our case, we enforce charge neutrality on both reactants, but also enforce a total spin of 1/2 to the Cs atom (or a total spin 1 to Cs2 in the triplet state) in order to improve the convergence. Unfortunately, the so-obtained PES does not show bound states, neither for Cs nor for Cs2. Since this will be of relevance for the future discussion of long-range dispersion interactions captured via DFT, we included the fully repulsive curves in the ESI.† For comparison, we repeated the CDFT calculations using the B97-D functional,39 which contains an empirical ad hoc correction for van der Waals interactions, and the ωB97X-V, a hybrid functional40 which is closely related to our original choice of B97M-V. None of them shows a bound state. The assumption of an attractive interaction which is difficult to retrieve is supported by a series of benchmark CDFT calculations on the C6H6–Cs model system, which is also not bound at the B97M-V level of theory if charge-neutrality on cesium and the benzene ring is enforced, but which shows a bound state in the CCSD(T) calculations, i.e. coupled cluster singles and doubles excitations with perturbative triples.41
Since the current system is too large for a constrained CCSD(T) calculation we fall back on the multi-configurational SCF (MCSCF) approach18,19 but aided by the necessary long-range dispersion-type corrections in the neutral state. A minimal active space is chosen for the sake of computational feasibility. The long-range energy corrections have been extracted from CCSD(T) studies of the C6H6–Cs and C6H6–Cs2 systems; details are given in the Appendix. Using this technique we obtained a more accurate PES for the two most intriguing systems of our study, i.e. a fullerene interacting with a triplet Cs2 molecule in a T-shaped arrangement (by far the strongest interaction) or a single Cs atom (the weakest interaction). The results of these PES scans are plotted in Fig. 3. As can be seen, the exclusion of any charge transfer has an extreme quenching effect on attractive interactions. In fact, for a single Cs atom, the PES is still fully repulsive. For triplet Cs2, on the other hand, we find a very shallow minimum of about 160 cm−1 at an equilibrium distance of 9 Å. Although this is only a small fraction of the original binding energy, it plays a big role in the overall reaction since it is comparable to the barrier created by spatial hindrance due to the helium environment. We will discuss the consequence of these findings in a later section.
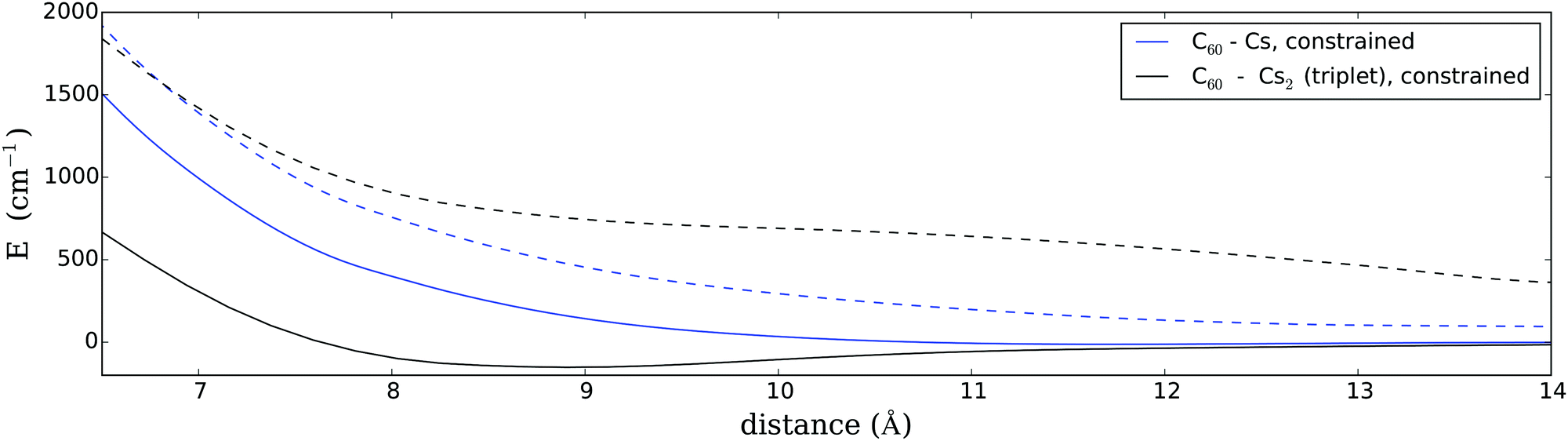 | ||
| Fig. 3 Fully covalent curves for Cs and Cs2 interacting with C60, maintaining charge neutrality on both reactants upon formation. The dashed lines correspond to PESs which are corrected for the spatial hindrance in superfluid helium; see Section 3.6 for details. | ||
3.5 Interactions between dopants and He
To obtain the last ingredients for the follow-up He-DFT calculation, the C60 fullerene is approached with a single He atom on a straight trajectory parallel to the C3 axis. Here, we fall back on the results reported by Hesselmann and Korona,42 who calculated the potential energy as a function of the He–C60 distance via symmetry-adapted perturbation theory with monomers described by density functional theory (DFT-SAPT).43,44To obtain an accurate description of the medium to long-range part, which is crucial for our approach of pair-potential summation in the He-DFT code, we have fitted the DFT-SAPT energies to our new pairwise additive potential model for atom/C60 interactions. The basic outline of this approach can be found in the Appendix. A similar ansatz has been used by us recently to study the submersion of carbon nanotubes in superfluid helium.45
The resulting spherically averaged curve, which will be used for the generation of the total interaction potential of C60 and a helium droplet, is plotted in Fig. 4. It shows a minimum of 57 cm−1 at an equilibrium distance of 6.76 Å. From this comparably strong interaction with a single He atom (a value of 5 cm−1 is typically assumed for the binding energy per He atom in droplets29) we can derive already that a fullerene will not just fully immerse into the He droplet, but will be surrounded by a helium shell of high density, giving rise to a local phase transition from the liquid to a nonspherical, crystalline bulk structure. This well-known phenomenon has been termed ‘snowball formation’ in the He droplet community. However, the He–C60 interaction potential can be assumed to be spherically symmetric due to the high symmetry of the C60 fullerene, and a single curve documenting the radial dependence can be used for the generation of a HeN–C60 interaction potential via summation over pair potentials.
 | ||
| Fig. 4 One-dimensional plot of the pair potential for the interaction of He and a C60 fullerene. A spherically averaged model potential (see Appendix), fit to data points of ref. 42. | ||
In order to obtain a reliable description of the weak interaction between Cs and the He environment, we use the analytical He–Cs potential provided in ref. 46. The corresponding curve is plotted in Fig. 5 for comparison. It shows a minimum of less than 1 cm−1 at a distance of 7.73 Å.
 | ||
| Fig. 5 One-dimensional plots of the pair potentials used for the generation of Uext[ρ] for the He-DFT calculations. For the Cs2–He interaction the two extreme cases of a collinear (ϕ = 0°) and a T-shaped geometry (ϕ = 90°) are plotted. | ||
For the slightly more complex interaction between Cs2 and the He droplet we fall back on the study of Prosmiti et al.,47 which contains a very detailed analysis of the intermolecular potential energy surface for the He–Cs2 system in the triplet state based on high level ab initio calculations. The authors have provided an analytical description of the surface as a function of r, the distance between the centers of mass of the two fragments (He and Cs2), and the angle ϕ, measured between the internuclear axis of the Cs2 molecule and the distance vector between the two centers of mass.
The two angles ϕ = 0° and ϕ = 90° have been selected for plots of the He–Cs2 interaction energies as a function of the distance in Fig. 5. Note that the equilibrium distance for ϕ = 90°, which corresponds to the T-shaped, perpendicular structure, represents the global minimum of the PES. For this geometry, a binding energy of about 2 cm−1 is reported in ref. 47. The characteristics of all relevant curves are summarized in Table 4.
Following the diatomics-in-molecules (DIM) approach,48,49 a three-dimensional estimate of the He–Cs2 pair potential can be obtained from the cuts provided in Fig. 5via an angular-dependent mixing of both curves, either by
| U(r,ϕ) = UT(r)cos2ϕ + ½[Ucoll(r) + UT(r)]sin2ϕ, | (4) |
| U(r,ϕ) = UT(r)sin2ϕ + Ucoll(r)cos2ϕ | (5) |
3.6 He droplet simulations
The aim of this section is the evaluation of a reaction pathway for the interaction of Cs or Cs2 with C60 in the environment of superfluid helium at a temperature of 0.38 K. We simulate a situation which has been realized recently in He droplet experiments of Renzler et al.,7 in which a sequential pickup of C60 fullerenes and Cs atoms was studied by electron ionization mass spectrometry. While heliophilic fullerenes immerse into the droplet completely, the heliophobic alkali metal atoms are known to reside on the droplet surface due to their diffuse valence electron density. This spatial separation, which occurs in cases where C60 pickup takes place before Cs pickup, together with the hindered mobility of the dopants on and in the droplet,57 gives rise to the question whether a reaction between the fullerene and the alkali metal atom can take place or not. An interesting observation of this recent experiment is that Cs2 seems to react with the fullerene, while a single Cs atom does not.From the potential curves shown in Fig. 1 and 2 it can be seen that the interaction of a single Cs atom with C60 is much weaker than the interaction of Cs2 with C60. Assuming a full quenching of charge transfer due to the helium environment we obtained the curves shown in Fig. 3, in which the spin-parallel configuration of Cs2, which is expected to be the dominating spin state on He droplets,58,59 still shows a bound state, while the atomic curve does not. However, this picture is yet incomplete as we have not taken the direct impact of sterical hindrance into consideration, an effect which occurs due to embedding in superfluid helium. The aim of this last computational study is to evaluate this reaction barrier by calculating and comparing the total energies of the systems HeN–Cs–C60 and HeN–Cs2–C60 as a function of the distance between the two dopants. In order to do that we proceed as follows:
First, we create three-dimensional interaction potentials for all dopants from the potential curves given in Fig. 4 and 5. In the case of the ‘spherical’ particles (Cs and C60) the potentials can be generated via a simple pair summation over the corresponding potential curves for the interaction with a single He atom. In the case of Cs2, we use the analytical potential U(r,ϕ) introduced above. In a second step, we evaluate the total energy of a multiply-doped He droplet consisting of N = 2000 He atoms via He-DFT as a function of the distance between the heliophilic and the heliophobic dopant. Three example density distributions for a distance of 20 Å are shown in Fig. 6 as contour plots of planar cuts through the system. Note the helium shell of exceptionally high density which surrounds the fullerene. By repeating the He-DFT energy evaluations for intermolecular distances from 5 to 35 Å we obtain the barriers plotted in Fig. 7. We find a barrier of approximately 400 cm−1 for a single atom, while the barriers for the Cs2 dimer are about three times higher due to the larger perturbation. The maximum of the barrier for the T-shaped configuration is not fully captured by the given scanning range but irrelevant since the Cs2–C60 interaction potential itself is already fully repulsive at a short distance of 5 Å. After setting these energies to zero at the asymptote of infinite distance between Csx and the fullerene inside the He-droplet, this energy correction is added to the interaction energies between the two dopants evaluated in the previous sections.
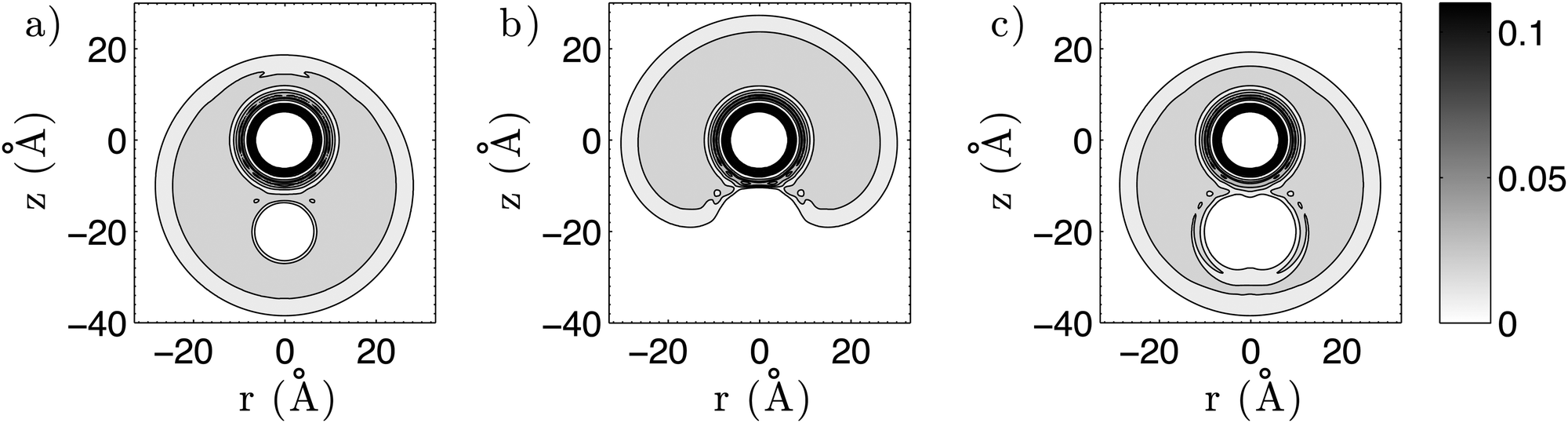 | ||
| Fig. 6 Contour plots of the helium density for a distance of 20 Å between the fullerene and (a) a single Cs atom, (b) the triplet Cs2 dimer in a collinear configuration, (c) the triplet Cs2 dimer in a T-shaped configuration. The density is plotted in units of Å−3. Note that the bulk value for the density of liquid helium is 0.02185 atoms per Å3. | ||
 | ||
| Fig. 7 Estimations of the energy barriers which occur due to sterical hindrance during the reaction of Cs or Cs2 with C60 inside a helium droplet formed by 2000 He atoms, evaluated using orbital-free helium density functional theory. | ||
If we naively assume that the interaction between the fullerene and Csx is not quenched at all by the surrounding helium, we can perform this correction by a simple pointwise summation of the He-DFT results and the free-gas curves of Fig. 1 and 2. The results of this first assumption are already plotted in the same figures as dashed lines. As can be seen, the corrections are marginal compared to binding energies in the range of thousands of wavenumbers and cannot explain any preference for the Cs2–C60 reaction over the Cs–C60 reaction. On the other hand, if we make the assumption of a fully quenched charge transfer, we have to apply our pointwise correction to the potential curves plotted in Fig. 3. Here, the situation is completely different due to the similar magnitude of the remaining weak attractive interaction between triplet Cs2 and the barrier due to the sterical hindrance: the helium environment has a huge impact on the reaction. After this final correction, the weakly bound state disappears even for triplet Cs2. Note that this outcome is sensitive to the actual depth of the charge-constrained potential curve, which might still deviate from the exact curve by a few hundred wavenumbers due to lack of correlation. Indeed, preliminary calculations of the Cs2/C60 dispersion interaction at the DFT-SAPT level indicate that the well-depth could be underestimated by up to 200 cm−1. However, since the helium-induced correction caused by sterical hindrance lies in the range of about 1200 cm−1, it is unlikely that an improved, more accurate description of the charge-constrained state will alter this result. From this, and the experimentally proven fact that the Cs2–C60 reaction takes place, it can be concluded that an electron transfer process must be involved in the overall reaction, despite the dense helium environment. Although our studies do not include any direct description of how or to which extend this transfer is affected by the helium environment, the two extremes described above can be interpreted as boundaries for the actual process. In the light of these two extremes, and given the recent findings of our experimental colleagues, reality seems to be closer to the second case, where charge transfer is significantly quenched but still determines whether the reaction takes place or not. This links our current efforts to much earlier studies on the concept of a ‘harpoon mechanism’, where long-range electron transfer is postulated near crossing points of neutral van der Waals and ionic potential energy curves.60 It remains an open question if this transfer of charge needs to be described via exciplex formation involving He atoms, similar to electron hopping processes between dopants in a rare gas matrix, for example induced by cooperative photoadsorption,61,62 or if a vibrational coupling of the neutral and the ionic states of Csx–C60 through the helium environment is sufficient. In any case, future studies on this subject are needed which also capture the dynamics of this fascinating phenomenon.
4 Conclusions
The interaction of a single Cs atom or a Cs2 dimer with a C60 fullerene has been studied in gas phase as well as by embedding in superfluid helium via a combination of quantum chemistry methods (DFT and MCSCF) and orbital-free bosonic helium density functional theory. This study was triggered by the recent experimental finding that a single Cs atom, which is heliophobic, does not seem to react with a heliophilic fullerene embedded in helium nanodroplets, while a Cs2 dimer, on the other hand, seems to be able to reach the fullerene inside the droplet. We found that the a3Σu+ state of Cs2 is bound to a fullerene with a dissociation energy of more than 17300 cm−1, which is about 4600 cm−1 higher than the dissociation energy of the X1Σg+ ground state, while a single Cs atom, on the other hand, is only bound by about 11100 cm−1. In order to estimate the effect of helium embedding on the reaction pathways, which we approximated by a linear approximation along the C3 axis of the fullerene, we corrected the obtained curves in two ways.
The first correction accounted for a quenching of charge transfer. Since we do not know the impact of helium embedding on the mobility of electrons we made the two extreme assumptions of an unquenched and a fully quenched electron transfer. The first scenario is realized by simple gas-phase computations, while the second is computed via charge-constrained DFT calculations, which enforced the charge neutrality of both fragments at all times, or via two-state MCSCF calculations including long-range dispersion corrections. In the latter case, we obtained a weakly bound state (160 cm−1) of triplet multiplicity for C60–Cs2 in a T-shaped arrangement, while a single Cs atom shows a fully repulsive interaction.
The second correction accounted for the spatial hindrance due to the extrusion of helium upon approach of the two reactants. Based on careful fits of ab initio data for the interactions of helium with both dopants we modeled external potentials for the helium-DFT calculation of total energies for the systems He2000–C60–Cs and He2000–C60–Cs2, and used them for energy corrections of the free-gas and charge-transfer-quenched curves. These corrections are of the order of a few hundred wavenumbers, which turned out to be fully negligible in the case of the free-gas curves, but crucial for the constrained curves: after inclusion the charge-neutral C60–Cs2 state also becomes fully repulsive. From this finding and the experimental fact that the C60–Cs2 reaction takes place we conclude that a minimal charge transfer must occur despite embedding in superfluid helium. We further assume that such an electron transfer process is considerably quenched, but it tips the scale regarding the overall reactivity: it lets the dimer react but prevents the atom from approaching the fullerene.
Appendix
A pairwise potential model for the He/C60 interaction
An additive pairwise potential model has been designed to fit the dispersionless and dispersion contributions to the accurate DFT-SAPT He–C60 interaction energies calculated by Hesselmann and Korona.42 Our potential model can be viewed as an extended version of that previously developed to account for anti-corrugation effects in the interaction of He atoms with metallic surfaces.63 It also extends the Lennard-Jones functional developed by Carlos and Cole,64,65 to account for corrugation effects in the adsorption of noble-gases onto the graphite surface.Our pairwise potential model exploits the fact that DFT-SAPT interaction energies can be decomposed in dispersionless (Edisp-lessint) and dispersion contributions (Edispint). The dispersion energies can be very well fitted by means of the Das functional of Szalewicz and collaborators,66–68 but modulated by a corrugation scaling amplitude:
 | (6) |
When noble-gas atoms are adsorbed onto non-metallic surfaces (see, e.g., ref. 70 and 71), dispersionless energy contributions are repulsive in the short-range and scale exponentially as the distance between the interacting species decrease. They can be fitted to the pairwise additive functional:
 | (7) |
θC adopting a value close to unity. This is translated in positive γR values. The opposite holds when the interaction energy becomes less repulsive for the noble-gas atoms adsorbed on “hollow” sites (e.g., the centers of the pentagons and hexagons in C60). This is the case for the He–C60 interaction (γR = −0.8).
Dispersion-corrected MCSCF calculations of neutral states
Using the Molpro package,22 we have performed additional Hartree–Fock and two-state multi-configurational self-consistent-field (MCSCF)18,19 calculations to characterize the interaction potential between Cs and Cs2(aΣu+) and C60 neutral fragments, with the Cs–Cs axis oriented perpendicular to the C3 axis. For cesium atoms we used the same basis set as in the B97M-V calculations (vide supra). The polarized correlation-consistent double-ζ basis of Woon and Dunning, Jr72 (cc-pVDZ) was adopted for fullerene carbon atoms instead. Test calculations in computing the Cs2/benzene interaction potential showed that the enlargement of the carbon basis set from cc-pVDZ to the polarized correlation-consistent triple-ζ basis increases the well-depth by ca. 10%. A minimal active space was used in the two-state-MCSCF calculations to account for the main non-dynamical correlation effects, consisting of the 5s orbitals from the Cs and Cs2 species along with frontier π-type molecular orbitals of the C60 system.To estimate the dynamical correlation contribution to the interaction, identified as dispersion, we adopted the following strategy: first, coupled-cluster singles, doubles and non-iterative triples [CCSD(T)] calculations20 were performed to calculate the Cs/benzene and Cs2(aΣu+)/benzene interactions. In the relevant potential region, it was checked via a Mulliken population analysis that, for a perpendicular orientation of the Cs–Cs internuclear distance to the C6 axis, the Cs2(aΣu+) and Cs fragments are kept neutral (see the ESI† for details). Next, following a similar strategy as was presented in previous studies (see ref. 73–75), correlation energy contributions in the long-range potential region were fitted to the effective interatomic function Das (the same as in eqn (6), but excluding the corrugation factor). Finally, the dispersion parameters extracted from the benzene model system were used to calculate the dispersion contributions to the Cs/C60 and Cs2(aΣu+)/C60 interactions. These contributions were added to the MCSCF interaction energies.
Acknowledgements
This work has been supported by the COST Action CM1405 Molecules in Motion (MOLIM). AWH thanks Martí Pi for his stimulating, enlightening comments at the MOLIM WG3 meeting in Bratislava, Slovakia. MPdLC is greatly thankful to Alexander O. Mitrushchenkov for very helpful discussions and to MINECO (Spain), under Grant No. MAT2016-75354-P, as well as to the CTI (CSIC) and CESGA supercomputing facilities for the resources provided. Both authors are grateful to Paul Scheier and Andrew M. Ellis for getting in contact and sharing experimental data prior to publication.References
- J. P. Toennies and A. F. Vilesov, Angew. Chem., Int. Ed., 2004, 43, 2622–2648 CrossRef CAS PubMed.
- J. Tiggesbäumker and F. Stienkemeier, Phys. Chem. Chem. Phys., 2007, 9, 4748–4770 RSC.
- C. Callegari and W. E. Ernst, in Handbook of High Resolution Spectroscopy, ed. F. Merkt and M. Quack, John Wiley & Sons, Chichester, 2011 Search PubMed.
- M. Mudrich and F. Stienkemeier, Int. Rev. Phys. Chem., 2014, 33, 301–339 CrossRef CAS.
- J. Nagl, G. Auböck, A. W. Hauser, O. Allard, C. Callegari and W. E. Ernst, Phys. Rev. Lett., 2008, 100, 063001 CrossRef PubMed.
- J. Nagl, G. Auböck, A. W. Hauser, O. Allard, C. Callegari and W. E. Ernst, J. Chem. Phys., 2008, 128, 154320 CrossRef PubMed.
- M. Renzler, M. Daxner, L. Kranabetter, A. Kaiser, A. W. Hauser, W. E. Ernst, A. Lindinger, R. Zillich, P. Scheier and A. M. Ellis, J. Chem. Phys., 2016, 145, 181101 CrossRef PubMed.
- J. Poms, A. W. Hauser and W. E. Ernst, Phys. Chem. Chem. Phys., 2012, 14, 15158–15165 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed.
- T. Leininger, A. Nicklass, W. Küchle, H. Stoll, M. Dolg and A. Bergner, Chem. Phys. Lett., 1996, 255, 274–280 CrossRef CAS.
- N. Mardirossian and M. Head-Gordon, J. Chem. Phys., 2014, 140, 18A527 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1997, 107, 8554 CrossRef CAS.
- O. A. Vydrov and T. Van Voorhis, J. Chem. Phys., 2010, 133, 244103 CrossRef PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- A. D. Rabuck and G. E. Scuseria, J. Chem. Phys., 1999, 110, 695 CrossRef CAS.
- Q. Wu and T. Van Voorhis, Direct Calculation of Electron Transfer Parameters through Constrained Density Functional Theory, American Chemical Society, 2006, vol. 110 Search PubMed.
- H.-J. Werner and W. Meyer, J. Chem. Phys., 1980, 73, 2342 CrossRef CAS.
- P. J. Knowles and H.-J. Werner, Chem. Phys. Lett., 1985, 115, 219 CrossRef.
- J. D. Watts, J. Gauss and R. J. Bartlett, J. Chem. Phys., 1993, 98, 8718–8733 CrossRef CAS.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kuś, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. a. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. a. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C.-M. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. a. DiStasio, H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. Hanson-Heine, P. H. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T.-C. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. a. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S.-P. Mao, N. Mardirossian, A. V. Marenich, S. a. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. O'Neill, J. a. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. i. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stück, Y.-C. Su, A. J. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. a. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z.-Q. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. Chan, D. M. Chipman, C. J. Cramer, W. a. Goddard, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. a. Besley, J.-D. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C.-P. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. a. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. Van Voorhis, J. M. Herbert, A. I. Krylov, P. M. Gill and M. Head-Gordon, Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- H. J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, T. Korona, R. Lindh, A. O. Mitrushchenkov, G. Rauhut, et al., MOLPRO, version 2012.1, a package of ab initio programs, see http://www.molpro.net.
- G. Höning, M. Czajkowski, M. Stock and W. Demtröder, J. Chem. Phys., 1979, 71, 2138–2149 CrossRef.
- M. Raab, H. Weickenmeier and W. Demtröder, Chem. Phys. Lett., 1982, 88, 377–383 CrossRef CAS.
- B. Assadollahzadeh, C. Thierfelder and P. Schwerdtfeger, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 245423 CrossRef.
- I. Moullet, W. Andreoni and P. Giannozzi, J. Chem. Phys., 1989, 90, 7306–7312 CrossRef CAS.
- D. Li, F. Xie, L. Li, S. Magnier, V. Sovkov and V. Ivanov, Chem. Phys. Lett., 2007, 441, 39–42 CrossRef CAS.
- A. W. Hauser, G. Auböck and W. E. Ernst, Vibronic Interactions and the Jahn–Teller Effect, Springer Netherlands, Dordrecht, 2012, pp. 301–316 Search PubMed.
- F. Dalfovo, A. Lastri, L. Pricaupenko, S. Stringari and J. Treiner, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 1193 CrossRef CAS.
- F. Ancilotto, M. Barranco, F. Caupin, R. Mayol and M. Pi, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 214522 CrossRef.
- F. Caupin, F. Ancilotto, M. Barranco, R. Mayol and M. Pi, J. Low Temp. Phys., 2007, 148, 731–736 CrossRef CAS.
- The geometry of the C60 molecule was optimized at the same level of theory.
- G. Auböck, J. Nagl, C. Callegari and W. E. Ernst, J. Phys. Chem. A, 2007, 111, 7404–7410 CrossRef PubMed.
- R. Pérez de Tudela, D. López-Durán, T. González-Lezana, G. Delgado-Barrio, P. Villarreal, F. A. Gianturco and E. Yurtsever, J. Phys. Chem. A, 2011, 115, 6892–6902 CrossRef PubMed.
- A. S. Hira and A. K. Ray, Phys. Rev. A: At., Mol., Opt. Phys., 1996, 54, 2205–2215 CrossRef CAS.
- J. Deigmayr, M. Aymar, R. Wster, M. Weidemüller and O. Dulieu, J. Chem. Phys., 2008, 129, 064309 CrossRef PubMed.
- M. D. Gregorie, I. Hromada, W. F. Holmgren, R. Trubko and A. D. Cronin, Phys. Rev. A: At., Mol., Opt. Phys., 2008, 92, 052513 CrossRef.
- R. Antoine, D. Rayane, E. Benichou, P. Dugourd and M. Broyer, Eur. Phys. J. D, 2000, 12, 147–151 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- N. Mardirossian and M. Head-Gordon, Phys. Chem. Chem. Phys., 2014, 16, 9904–9924 RSC.
- J. D. Watts, J. Gauss and R. J. Bartlett, J. Chem. Phys., 1993, 98, 8718–8733 CrossRef CAS.
- A. Hesselmann and T. Korona, Phys. Chem. Chem. Phys., 2010, 13, 732–743 RSC.
- A. J. Misquitta, B. Jeziorski and K. Szalewicz, Phys. Rev. Lett., 2003, 91, 033201 CrossRef PubMed.
- A. Hesselmann and G. Jansen, Chem. Phys. Lett., 2003, 367, 778 CrossRef CAS.
- A. W. Hauser and M. P. de Lara-Castells, J. Phys. Chem. Lett., 2016, 7, 4929–4935 CrossRef CAS PubMed.
- S. H. Patil, J. Chem. Phys., 1991, 94, 8089–8095 CrossRef CAS.
- R. Prosmiti, G. Delgado-Barrio, P. Villarreal, E. Yurtsever, E. Coccia and F. A. Gianturco, J. Phys. Chem. A, 2009, 113, 14718–14729 CrossRef CAS PubMed.
- F. O. Ellison, J. Chem. Phys., 1983, 78, 5024–5030 CrossRef CAS.
- F. O. Ellison, J. Am. Chem. Soc., 1963, 85, 3540–3544 CrossRef CAS.
- M. Ratschek, J. V. Pototschnig, A. W. Hauser and W. E. Ernst, J. Phys. Chem. A, 2014, 118, 6622–6631 CrossRef CAS PubMed.
- D. Mateo, A. Hernando, M. Barranco, E. Loginov, M. Drabbels and M. Pi, Phys. Chem. Chem. Phys., 2013, 15, 18388–18400 RSC.
- M. Leino, A. Viel and R. E. Zillich, J. Chem. Phys., 2011, 134, 024316 CrossRef PubMed.
- A. Hernando, M. Barranco, R. Mayol, M. Pi and M. Krosnicki, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 024513 CrossRef.
- A. Hernando, M. Barranco, R. Mayol, M. Pi and F. Ancilotto, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 184515 CrossRef.
- O. Bunermann, G. Droppelmann, A. Hernando, R. Mayol and F. Stienkemeier, J. Phys. Chem. A, 2007, 111, 12684–12694 CrossRef PubMed.
- M. Mella, M. C. Colombo and G. Morosi, J. Chem. Phys., 2002, 117, 9695–9702 CrossRef CAS.
- A. W. Hauser, A. Volk, P. Thaler and W. E. Ernst, Phys. Chem. Chem. Phys., 2015, 17, 10805–10812 RSC.
- F. Stienkemeier, W. E. Ernst, J. Higgins and G. Scoles, J. Chem. Phys., 1995, 102, 615–617 CrossRef CAS.
- W. E. Ernst, R. Huber, S. Jiang, R. Beuc, M. Movre and G. Pichler, J. Chem. Phys., 2006, 124, 024313 CrossRef CAS PubMed.
- J. L. Magee, J. Chem. Phys., 1940, 8, 687–698 CrossRef CAS.
- M. E. Fajardo and V. A. Apkarian, J. Chem. Phys., 1986, 85, 5660–5681 CrossRef CAS.
- M. E. Fajardo and V. A. Apkarian, J. Chem. Phys., 1988, 89, 4102–4123 CrossRef CAS.
- M. P. de Lara-Castells, R. Fernández-Perea, F. Madzharova and E. Voloshina, J. Chem. Phys., 2016, 144, 244707 CrossRef PubMed.
- L. W. Brunch, M. C. Cole and E. Zaremba, Physical Adsorption: Forces and Phenomena, Clarendon Press, Oxford, 1997 Search PubMed.
- W. E. Carlos and M. W. Cole, Surf. Sci., 1980, 91, 339–357 CrossRef CAS.
- K. Pernal, R. Podeszwa, K. Patkowski and K. Szalewicz, Phys. Rev. Lett., 2009, 103, 263201 CrossRef PubMed.
- R. Podeszwa and K. Szalewicz, J. Chem. Phys., 2012, 136, 161102 CrossRef PubMed.
- R. Podeszwa, K. Pernal, K. Patkowski and K. Szalewicz, J. Phys. Chem. Lett., 2010, 1, 550–555 CrossRef CAS.
- K. T. Tang and J. P. Toennies, J. Chem. Phys., 1984, 80, 3726–3741 CrossRef CAS.
- M. P. de Lara-Castells, M. Bartolomei, A. O. Mitrushchenkov and H. Stoll, J. Chem. Phys., 2015, 143, 194701 CrossRef PubMed.
- M. P. de Lara-Castells, H. Stoll and A. O. Mitrushchenkov, J. Phys. Chem. A, 2014, 118, 6367–6384 CrossRef CAS PubMed.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1994, 100, 2975–2988 CrossRef CAS.
- M. P. de Lara-Castells, H. Stoll, B. Civalleri, M. Causà, E. Voloshina, A. O. Mitrushchenkov and M. Pi, J. Chem. Phys., 2014, 141, 151102 Search PubMed.
- M. P. de Lara-Castells, N. F. Aguirre, H. Stoll, A. O. Mitrushchenkov, D. Mateo and M. Pi, J. Chem. Phys., 2015, 142, 131101 Search PubMed.
- M. P. de Lara-Castells, A. O. Mitrushchenkov and H. Stoll, J. Chem. Phys., 2015, 143, 102804 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A series of additional He density graphs, details of the MCSCF approach, fully repulsive CDFT curves, a table with the parameters of the pairwise potential model and figures documenting the quality of the fitting approach. See DOI: 10.1039/c6cp06858h |
| This journal is © the Owner Societies 2017 |