
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCapture of volatile iodine by newly prepared and characterized non-porous [CuI]n-based coordination polymers†
Abbas
Tarassoli
*a,
Valiollah
Nobakht
 *a,
Elham
Baladi
a,
Lucia
Carlucci
b and
Davide M.
Proserpio
bc
*a,
Elham
Baladi
a,
Lucia
Carlucci
b and
Davide M.
Proserpio
bc
aDepartment of Chemistry, Faculty of Sciences, Shahid Chamran University of Ahvaz, Ahvaz, Iran. E-mail: tarassoli@scu.ac.ir; v.nobakht@scu.ac.ir; Fax: +98 613 3331042
bDipartimento di Chimica, Università degli Studi di Milano, Via C. Golgi 19, 20133, Milano, Italy
cSamara Center for Theoretical Materials Science (SCTMS), Samara University, Samara 443011, Russia
First published on 11th September 2017
Abstract
Four new non-porous CuI-coordination polymers [Cu2(μ3-I)2(μ-bpb)]n (1a), [Cu(μ2-I)(μ-bpb)]n (1b), [Cu4(μ2-I)4(μ-bpmb)4]n (2), and [CuI(μ-bdb)]n (3) (bpb = 1,4-bis(pyrazolyl)butane; bpmb = 1,4-bis[(pyrazolyl)methyl]benzene; bdb = 1,4-bis[(3,5-dimethylpyrazolyl)methyl]benzene) have been successfully prepared and their structures fully characterized by single-crystal X-ray diffraction, FT-IR spectroscopy, PXRD and elemental analysis. Crystallographic investigation revealed that 1a, 1b, and 2 exhibit two-dimensional (2D) structures; in 1a parallel [Cu2I2]n staircase motifs are cross-linked into two-dimensional sheets by bpb linkers with a fully extended conformation, while in the structures of 1b and 2 Cu2I2 rhomboid dimers are linked by bpb and pbmb ligands, respectively, into two-dimensional sheets with a 44-sql net. Differently, compound 3 shows a one-dimensional (1D) zigzag chain structure with monomeric CuI units. All the four non-porous coordination polymers show the ability to capture volatile iodine in the gas phase. The solid-state photoluminescence properties of 1a, 1b, and 2 have also been investigated. The iodine-adsorbed samples 1a-I2, 1b-I2, and 2-I2 show no fluorescence behavior.
Introduction
Coordination polymers (CPs) and metal–organic frameworks (MOFs) are interesting categories of materials consisting of metal ions or metal clusters connected by organic linker ligands.1 Various metal clusters, especially metal carboxylate aggregates, have been used as secondary building units (SBUs) in the self-assembly of CPs and MOFs.2 Copper(I) iodides are also building blocks in the synthesis of metal–organic materials.3 Intense investigation in the past three decades in this area of research was conducted not only because of the structural diversity of copper iodide based polymers but also because of the uniqueness of the luminescence properties of the copper(I) iodide aggregates.4 Reactions of copper(I) iodide with nitrogen or sulfur donor linkers in some organic solvents convert the 3D structure of bulk CuI into zero-dimensional CunIn (n = 2–12) aggregates or into various one- to three-dimensional motifs which also have different physical and chemical properties. The structural diversity and synthetic routes of copper(I) halide aggregates have been reviewed by R. Peng et al. in detail.3 On the other hand, it should be remembered that the world energy consumption will greatly increase in the near future and nuclear power plants are important energy sources to produce electricity on a large scale. However, the production of radioactive waste and appropriate disposal of such nuclear materials are still unsolved problems.5 Nuclear wastes contain radioisotopes with long half-lives. This means that the radioisotopes stay in the atmosphere and are hazardous to health for thousands of years. Radioactive iodine isotopes, 133I2, 131I2, 129I2, 125I2, are the main components of nuclear wastes. These are volatile hazardous species and are involved in human metabolic processes.6 Among them, radioisotope 129I2 remains in the environment for a long time due to its half-life of 15.7 million years. Due to the harmful effects of radioactive iodine on human health, a safe capture process and subsequent long-term storage by effective adsorbents is extremely important. To find a suitable matrix for capturing radioactive iodine, several different types of adsorbents such as silver-containing zeolite mordenite,7 Hofmann clathrates,8 layered double hydroxides (LDHs),9 porous carbon,10 porous organic frameworks11 and metal–organic frameworks (MOFs)12,13 have already been reported so far. The silver-containing zeolite (AgZ) mordenite has been a benchmark of iodine capture for many years. However, iodine diffusion inside the zeolite is slow and limits the capture efficiency. On the other hand, the sorbent requires silver to bind I2(g) and hence the preparation of AgZ is expensive.14 In particular, MOFs exhibit high iodine sorption efficiency due to their higher porosity compared to zeolite-like materials.13 Various MOFs containing different nodes and linkers have been reported and their tendency to absorb iodine molecules in the vapor or liquid phase has been examined.12 Among the various adsorbent materials, MOFs seem to be one of the most effective candidates for the capture of iodine. Sorption of iodine by MOFs may occur physically or chemically, known as physisorption and chemisorption, respectively. In physisorption, iodine molecules usually diffuse and encapsulate into the channels or voids of porous MOFs, whereas in the chemisorption process, a chemical interaction or chemical reaction occurs between the I2 molecules and the MOF surface. Although distinguishing between chemisorption and physisorption is not simple, Kawano and co-workers clearly visualized physi- and chemisorption of iodine in Cu2I2-based porous MOFs by single crystal X-ray diffraction.12c Crystal-structure analysis confirms the chemisorption of iodine molecules through the formation of an I3− group from each bridging iodide unit with an almost linear geometry for the I3− ion. Thus, CunIn moieties in copper iodide-based metal–organic materials may be versatile groups for the capture of volatile iodine, even by non-porous CPs in the gas phase. With this idea in mind and as a continuation of our previous work on the synthesis of copper(I) based CPs,15 we have prepared four non-porous coordination polymers with flexible bidentate linker ligands (Scheme 1) and investigated their ability to capture volatile iodine in the gas phase. The photoluminescence behavior of the complexes 1a, 1b, and 2 and its relation with iodine capture was also investigated.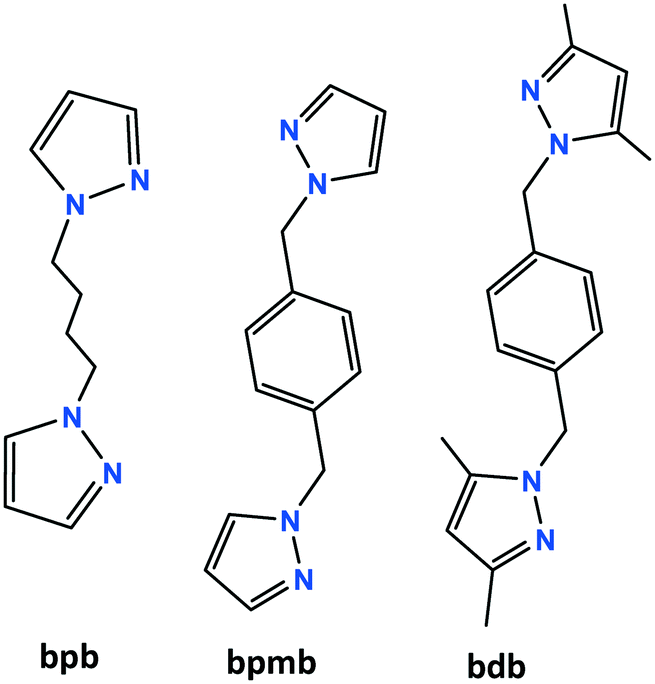 | ||
| Scheme 1 Structure of the ligands used in this work. | ||
Results and discussion
Description of crystal structures
 | ||
| Fig. 1 The structure of [Cu2(μ3-I)2(μ-bpb)]n (1a). (a) Asymmetric unit of 1a with additional atoms to complete the bpb ligand and the coordination of Cu and I. (b) 2D polymeric structure with parallel staircase [Cu2I2]n units and the underlying net bey. (c) View of the ABAB packing modes of three parallel undulating sheets along the [1 0 0] direction. | ||
The pyrazolyl rings of the adjacent bpb ligands in a sheet are parallel by symmetry; however the centroid⋯centroid distance of 4.389 Å implies no significant π⋯π interaction. The [Cu2I2]n ladders run along the crystallographic a axis and the sheets lie parallel to the ac plane. As indicated in Fig. 1c, parallel sheets interdigitate and pack in an ABAB fashion along the b axis. The stacking of the 2D sheets generate weak C–H⋯I interactions in the range 3.18–3.22 Å.
 | ||
| Fig. 2 The structure of [Cu(μ2-I)(μ-bpb)]n (1b). (a) Asymmetric unit of 1b with additional atoms to complete the coordination of Cu. (b) Part of the two-dimensional 44-sql net structure with four equal parallelograms. | ||
 | ||
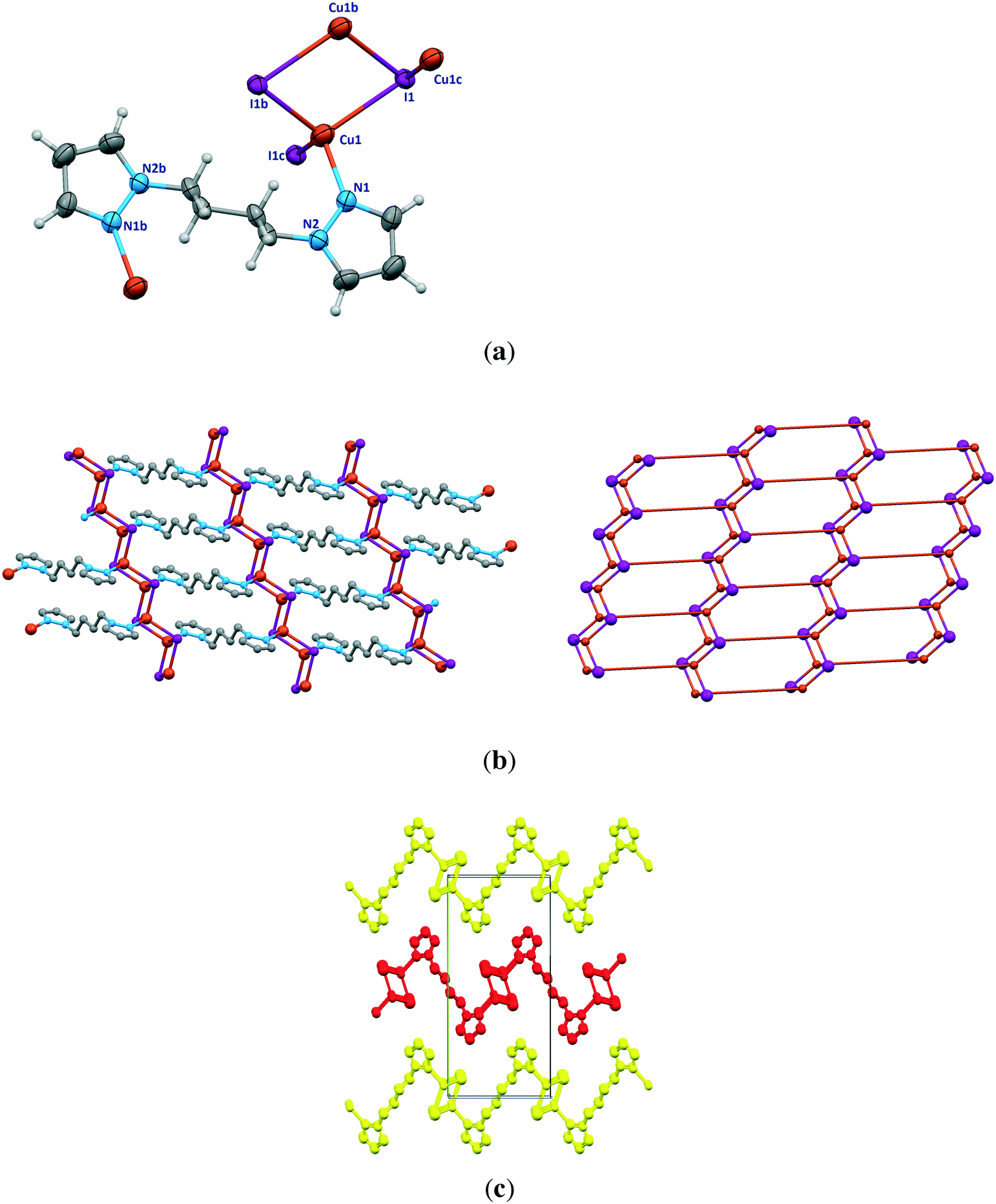
| Fig. 3 The structure of [Cu(μ2-I)(μ-bpmb)]n (2). (a) Asymmetric unit of 2 with labeling scheme for Cu and I. (b) Part of the two-dimensional structure with four crystallographically independent μ-bpmb linkers shown in different colors. | ||
2D sheets lie parallel to the bc plane and show an ABAB packing mode in the crystal. In addition, significant π⋯π stacking interactions occur in 2. These interactions are of three types, two interlayers (brown and green colored rings) and one intralayer (orange colored rings), with centroid⋯centroid distances of 3.633(9), 3.594(9), and 3.846(9) Å and dihedral angles of 7.2(9)°, 18.9(8)°, and 18.9(8)°, respectively. These interactions stabilize the structure and link the discrete 2D layers into a 3D network structure (Fig. 4).
 | ||
| Fig. 4 Three different π⋯π stacking interactions in the structure of 2. | ||
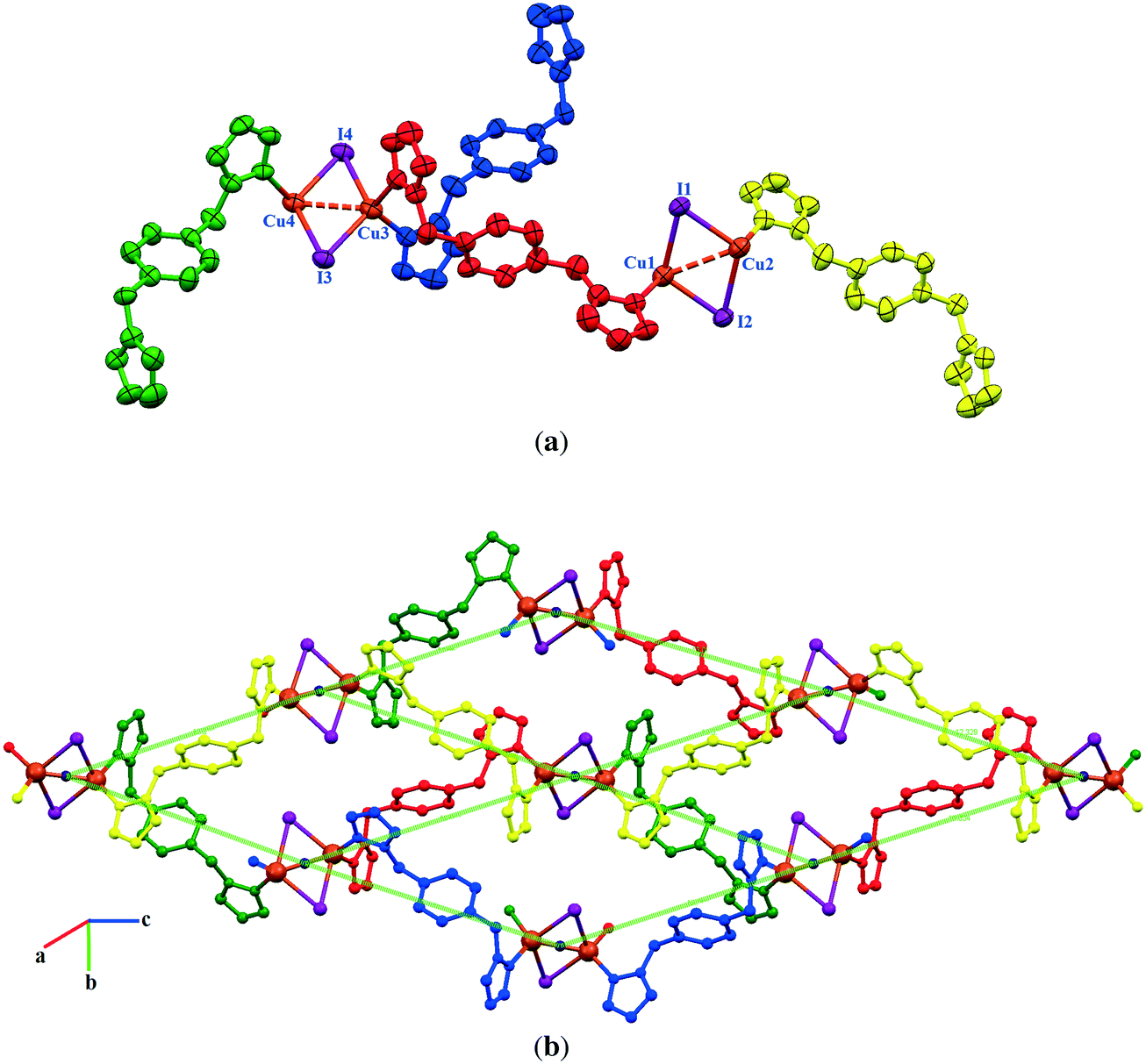
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The asymmetric unit is shown in Fig. 5a. There is a crystallographic inversion center at the midpoint of the phenyl ring of each bdb ligand, so the asymmetric unit consists of one CuI monomer and half each of two bdb ligands. In contrast to the structures of 1a, 1b, and 2, compound 3 contains monomeric CuI units. Copper atoms have a trigonal planar coordination geometry with a slight deviation from the ideal geometry and the angles range from 115.18(7)° to 123.53(5)° (Table S1†). The coordination environment of copper atoms is occupied by a terminal I atom with a Cu–I distance of 2.5206(4) Å and two nitrogen atoms of two crystallographically independent bdb ligands with Cu–N distances of 1.9937(18) and 1.9978(17) Å. The N–Cu–I angles [121.29(6)° and 123.52(5)°] are wider than the N–Cu–N one [115.18(8)°]. As expected, the Cu–I bond length in 3 is much shorter than the bridged ones observed for the other compounds. The structural preference for the trigonal planar arrangement instead of the four-coordinate may be due to the insertion of two methyl substituents on the pyrazolyl rings of bdb, which provide larger steric hindrance around the central copper atoms in 3. The monomeric CuI units are connected by two independent bdb linkers to form a polymeric one-dimensional zigzag chain running along the [1 1 −1] direction (Fig. 5b). As in the structure of 2 there are crystallographically independent μ2-bdb ligands in 3, showing a different pyrazolyl ring rotation around the methylene spacer and consequently different ligand lengths and dihedral and torsion angles. Thus the red colored bdb ligand with a N-to-N distance of 8.73 Å has a greater overall length than the green one (8.20 Å), leading to an alternation of distances between adjacent CuI monomers along the chain, the difference for Cu⋯Cu being close to 0.6 Å (Fig. 5b and c). As both the μ-bdb ligands in the structure of 3 are centrosymmetric, the dihedral angle between two pyrazolyl rings of a ligand are 0° and the nitrogen donor atoms point in exactly opposite directions. This strictly parallel arrangement of rings in the ligands would be better described as antiparallel, with a dihedral angle of 180°. The dihedral angles between the mean plane of the pyrazolyl and phenyl rings of a ligand are 88° (red colored) and 90° (green colored). The 1D zigzag chains pack in a manner such that the pyrazolyl rings of crystallographically equivalent bpb ligands from adjacent chains are strictly parallel and show significant π⋯π interactions. These interactions are of two types with centroid⋯centroid distances of 3.470(2) and 3.871(3) Å and interplanar distances of 3.4141(11) and 3.5787(12) Å between the two rings, respectively. These π⋯π interactions stabilize the structure of 3 and link the discrete 1D chains into a 2D sheet (Fig. 5c).
. The asymmetric unit is shown in Fig. 5a. There is a crystallographic inversion center at the midpoint of the phenyl ring of each bdb ligand, so the asymmetric unit consists of one CuI monomer and half each of two bdb ligands. In contrast to the structures of 1a, 1b, and 2, compound 3 contains monomeric CuI units. Copper atoms have a trigonal planar coordination geometry with a slight deviation from the ideal geometry and the angles range from 115.18(7)° to 123.53(5)° (Table S1†). The coordination environment of copper atoms is occupied by a terminal I atom with a Cu–I distance of 2.5206(4) Å and two nitrogen atoms of two crystallographically independent bdb ligands with Cu–N distances of 1.9937(18) and 1.9978(17) Å. The N–Cu–I angles [121.29(6)° and 123.52(5)°] are wider than the N–Cu–N one [115.18(8)°]. As expected, the Cu–I bond length in 3 is much shorter than the bridged ones observed for the other compounds. The structural preference for the trigonal planar arrangement instead of the four-coordinate may be due to the insertion of two methyl substituents on the pyrazolyl rings of bdb, which provide larger steric hindrance around the central copper atoms in 3. The monomeric CuI units are connected by two independent bdb linkers to form a polymeric one-dimensional zigzag chain running along the [1 1 −1] direction (Fig. 5b). As in the structure of 2 there are crystallographically independent μ2-bdb ligands in 3, showing a different pyrazolyl ring rotation around the methylene spacer and consequently different ligand lengths and dihedral and torsion angles. Thus the red colored bdb ligand with a N-to-N distance of 8.73 Å has a greater overall length than the green one (8.20 Å), leading to an alternation of distances between adjacent CuI monomers along the chain, the difference for Cu⋯Cu being close to 0.6 Å (Fig. 5b and c). As both the μ-bdb ligands in the structure of 3 are centrosymmetric, the dihedral angle between two pyrazolyl rings of a ligand are 0° and the nitrogen donor atoms point in exactly opposite directions. This strictly parallel arrangement of rings in the ligands would be better described as antiparallel, with a dihedral angle of 180°. The dihedral angles between the mean plane of the pyrazolyl and phenyl rings of a ligand are 88° (red colored) and 90° (green colored). The 1D zigzag chains pack in a manner such that the pyrazolyl rings of crystallographically equivalent bpb ligands from adjacent chains are strictly parallel and show significant π⋯π interactions. These interactions are of two types with centroid⋯centroid distances of 3.470(2) and 3.871(3) Å and interplanar distances of 3.4141(11) and 3.5787(12) Å between the two rings, respectively. These π⋯π interactions stabilize the structure of 3 and link the discrete 1D chains into a 2D sheet (Fig. 5c).
 | ||
| Fig. 5 The structure of [CuI(μ-bdb)]n (3). (a) Asymmetric unit of 3 with labeling scheme for non-H and C atoms. (b) 1D zigzag structure of 3 containing a sequence of μ-bdb ligands with two different conformations. (c) π⋯π stacking interactions between the chains giving 2D supramolecular layers. | ||
Spectroscopic characterization
Infrared spectra of the coordination polymers 1–3 are shown in Fig. S1.† The bands in the range of 3020–3130 cm−1 are assigned to the stretching vibration of aromatic C–H bonds of coordinated ligands. Symmetric and asymmetric stretching vibrations of the methylene (–CH2–) and methyl (–CH3) groups of the linkers are observed in the region 2861–2971 cm−1. Medium to strong peaks at 1514 (for 1a), 1517 (for 1b), 1515 (for 2) and 1549 (for 3) cm−1 are also observed, which are attributed to the stretching vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds of the pyrazolyl rings. In compound 3, where the pyrazolyl rings bring two methyl substituents, this vibration is shifted to a higher wavenumber with respect to the values found for compounds 1a, 1b and 2 containing unsubstituted pyrazolyl rings.
N bonds of the pyrazolyl rings. In compound 3, where the pyrazolyl rings bring two methyl substituents, this vibration is shifted to a higher wavenumber with respect to the values found for compounds 1a, 1b and 2 containing unsubstituted pyrazolyl rings.
The reproducibility of the syntheses and the phase purity of the products were investigated by powder X-ray diffraction. PXRD patterns are consistent with the structures obtained by single-crystal X-ray diffraction (Fig. 6, S2 and S3†).
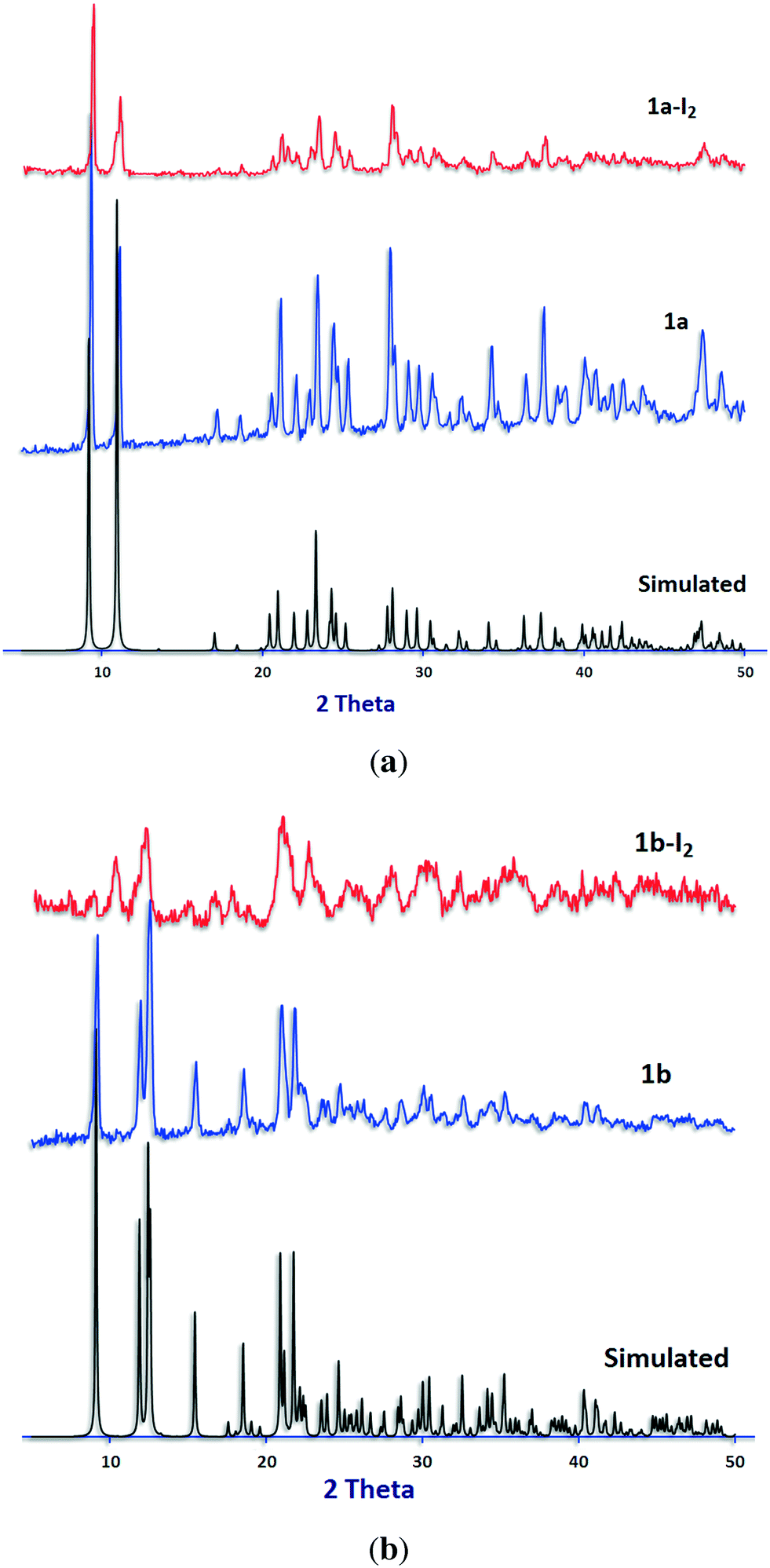 | ||
| Fig. 6 PXRD patterns for (a) 1a and (b) 1b. Calculated from the single-crystal structure (black) before (blue) and after (red) iodine sorption experiments. | ||
Iodine sorption study
Most of the investigations on iodine sorption have been performed using porous coordination polymers (MOFs) but only in solution.12b–g Iodine sorption studies on non-porous coordination polymers in the gas phase are still rare and need to be explored well.22In 2013 Kawano et al. reported on the sorption of iodine in the gas phase by a CuI-based 3D porous coordination network, exhibiting crystallographic evidence for the chemisorption of iodine by the Cu2I2 rhomboid nodes of the structure.12c In this case, each Cu–I moiety converts to a Cu–I3 unit without any change in the charge of the network. In addition, recent reports by Zhao et al.12o and Y.-Z. Zheng et al.12p also reveal halogen-bond interactions between iodine and the Cu–I moieties present in the structures they studied. On the basis of these results we explored the possibility of iodine capture in the gas phase by the four non-porous CunIn-based coordination polymers 1–3. Analysis of the structures using the Platon software23 confirms that there is no accessible void for guest molecules for all.
A fixed iodine vapor pressure strategy was employed, and a vial containing ground colorless crystals of the CPs was kept in a closed system containing crystals of iodine and heated at 55–60 °C at ambient pressure. When the crystals were exposed to iodine vapor, the color of the samples turned to dark brown or black immediately. To evaluate the iodine uptake, the samples were washed with cyclohexane to remove deposited iodine on the surface of the crystals, dried and weighed. Photographs of the samples before and after iodine sorption are shown in Fig. 7. Gravimetric calculations show sorption values of 26.0, 57.0, 57.9, and 92.7 wt% for 1a, 1b, 2, and 3, respectively.
 | ||
| Fig. 7 Photographs of the samples 1–3 before and after iodine sorption. | ||
Iodine uptake in the gas phase degrades the crystallinity of the samples. Several attempts to obtain suitable iodine adsorbed crystals for X-ray crystallography were unsuccessful. Photos of the crystals taken under an optical microscope during gaseous iodine sorption are shown in Fig. S4.†
The FT-IR spectra of compounds 1–3 before and after iodine sorption are almost the same and only the intensity of the vibration bands decreased (Fig. S1†). No other significant difference was observed.
PXRD patterns of the samples after iodine sorption were recorded and are shown in Fig. 6, S2 and S3.† The results show similar PXRD patterns for the iodine-adsorbed samples with respect to their pristine compounds before iodine sorption except for compound 2.
In order to check the thermal stability of the Cu(I) CPs and to get a better insight into the iodine sorption behavior of the samples, thermal gravimetric analysis (TGA) of 1–3 before and after I2 sorption was performed under a nitrogen atmosphere. As shown in Fig. S5,† all the structures are stable up to about 210 °C and start to collapse at higher temperatures. Comparison between the thermal gravimetric behavior of 1a and its related iodine-adsorbed 1a-I2 shows that iodine release occurs at a temperature close to but lower than the decomposition point of 1a. For compound 1b-I2, iodine release happens exactly at the decomposition point of 1b. The results may be a consequence of a strong interaction between iodine molecules and the structures of 1a and 1b. The thermal release of the adsorbed iodine from samples 2-I2 and 3-I2 occurs at ca. 160 °C which is lower than the decomposition point of compounds 2 and 3 (about 240 °C), implying a weaker interaction of iodine molecules with the structures of 2 and 3 with respect to those of 1a and 1b. Release of physisorbed iodine from the pores or surface of the MOFs usually occurs at temperatures lower than those of the chemisorbed samples12b,g,n due to the weaker interaction between the iodine molecules and the surface of the sorbent at the physisorption process. Hence, the release of iodine molecules at a high temperature of 200 °C, close to the decomposition point of compounds 1a and 1b, may be a reason for the chemisorption of iodine. On the other hand, iodine release from the other structures 2 and 3 at lower temperatures may suggest a physisorption process even if the exact interaction mode of iodine with the four structures remains unknown.
To get more insights into the iodine capture process and to confirm the gravimetric iodine uptake amounts in compounds 1–3, the I2 removal process was also investigated.
Iodine release was investigated in non-polar and polar solvents such as cyclohexane, CCl4, EtOH and DMF. The results show a quick iodine release in DMF, a very slow release in ethanol and CCl4 and no release in cyclohexane. On the contrary, heating the samples under vacuum to release adsorbed iodine was unsuccessful. Results of the iodine release test for 1a in the four solvents at different times are shown in Fig. S6.† Based on the above results, the iodine content for samples (1–3)-I2 were determined by release of the adsorbed iodine in DMF (Fig. S7†) and consequent determination by UV/vis spectroscopy at 368 nm. UV/vis measurements give iodine contents of 23.0, 57.7, 56.6, and 93.8 wt% for 1a, 1b, 2, and 3, respectively (Fig. S8 and Table S3†). These results are consistent with the gravimetric amounts and also comparable to the values reported for porous coordination networks.12b,c,g
Luminescence property
The solid-state photoluminescence properties of 1a, 1b, and 2 have been studied at room temperature. The maxima of the emission bands of 1a, 1b, and 2 were observed at 556.2, 548.6, and 558.9 nm (λex = 300 nm), respectively (Fig. 8). The observed photoluminescence could be attributed to MLCT, triplet cluster-centered excited states, a combination of iodine-to-metal charge transfer (IMCT) and d–s transitions by Cu(I)–Cu(I) interaction.4c,24 An intense yellow emission of 1a observed visually by the naked eye under UV irradiation (Fig. 8b) is probably due to the presence of staircase [Cu2I2]n SBUs in the structure of 1a. Interestingly, as iodine species are known as fluorescence quenchers, the fluorescence of the samples is quenched with iodine sorption. The dependence of fluorescence quenching on iodine sorption is an interesting feature observed in this series of CuI-based coordination polymers. This phenomenon has already been observed in a Cd(II)–triazole MOF.12b The iodine-release sample 1a-I2 shows an intense photoluminescence emission again under UV irradiation (Fig. S9†). Such an ON–OFF switching of the photoluminescence emission induced by adsorption/release of iodine in samples 1a/1a-I2 has been shown to be reversible.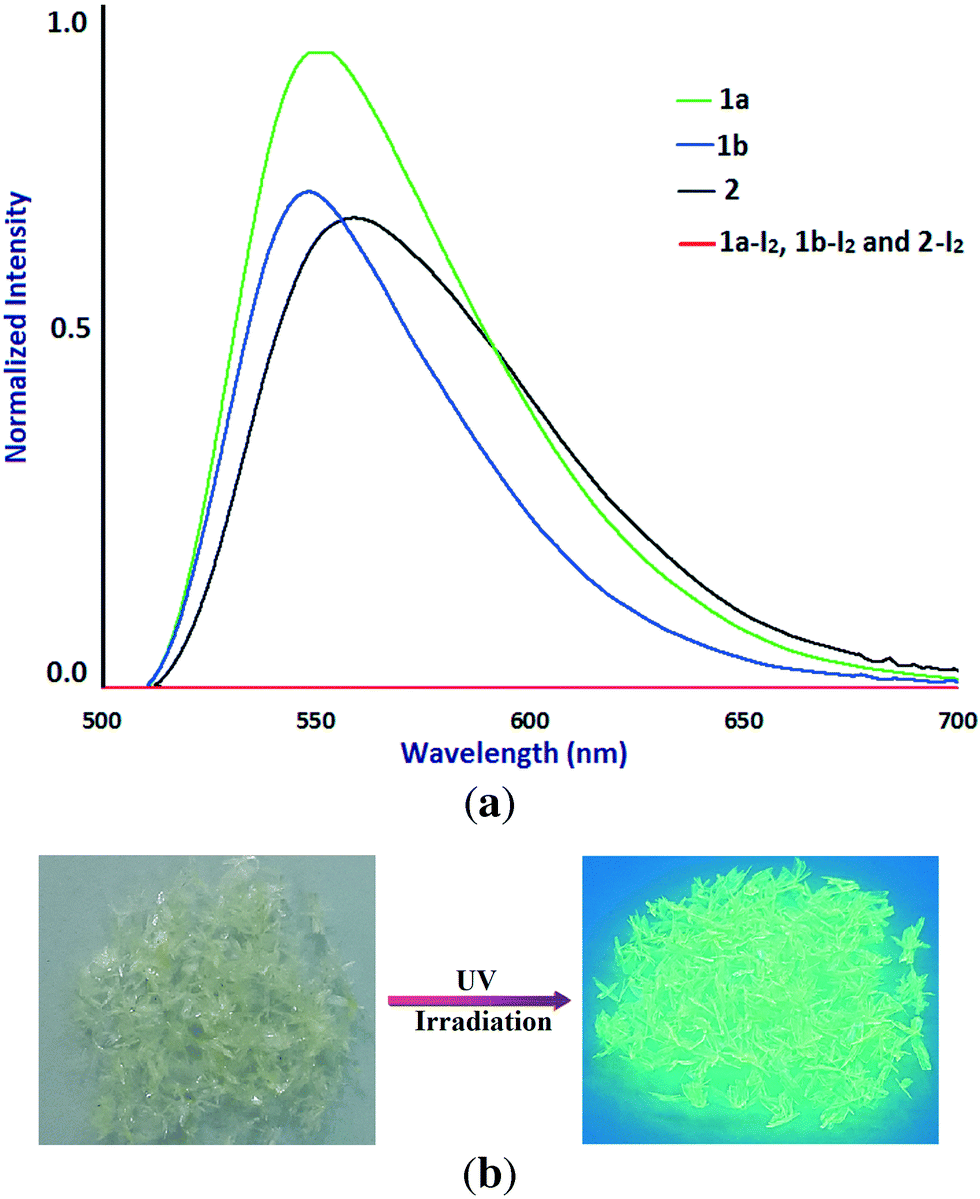 | ||
| Fig. 8 (a) Solid-state luminescence spectra of 1a (green), 1b (blue) and 2 (black) at room temperature (λex = 300 nm) before and after iodine sorption (red). (b) Crystals of 1a under UV irradiation at 254 nm at room temperature. | ||
Experimental
Materials and physical measurements
All experiments were carried out in air. The starting materials were purchased from commercial sources and used without further purification. Infrared spectra (4000–400 cm−1) were recorded as KBr disks on a BOMEN MB102 FT-IR spectrometer. Elemental analyses for C, H and N were performed on a CHNSO Elementar Vario EL III apparatus. X-ray powder diffraction patterns were recorded on a Philips X'Pert Pro diffractometer (Cu Kα radiation, λ = 1.54184 Å) in the 2θ range 5–50°. The simulated XRD powder pattern based on single crystal data was prepared using Mercury software.25 Thermal analyses were carried out on a TGA-DTA Mettler-Toledo TGA/SDTA 851 thermal analyzer between 50 and 600 °C under a dinitrogen atmosphere. Solid-state fluorescence spectra were obtained on a Shimadzu RF-540 spectrofluorometer in the range of 700–500 nm. UV-vis spectra were recorded on a Jenway 6715 spectrophotometer in DMF solution and covered the range 700–200 nm.Synthetic procedures
Direct synthesis of 1a. A solution of CuI (0.1 g, 0.52 mmol) and I2 (0.13 g, 0.51 mmol) in DMF (10 mL) and a solution of bpb (0.05 g, 0.26 mmol) in DMF (5 mL) were mixed in a Teflon-lined stainless steel autoclave and heated at 120 °C for 24 h. The vessel was gradually cooled to room temperature over 24 h. The yellowish solution was transferred into a Petri dish, and needle-shaped single crystals of 1a suitable for single crystal X-ray diffraction were obtained after 1 day which were collected by filtration, washed with DMF, EtOH, and Et2O, and dried in air (0.09 g, 63% yield based on Cu).
Conventional heating. CuI (0.1 g, 0.52 mmol) was added to a solution of bdb (0.308 g, 1.04 mmol) in DMF (30 mL) and the light green mixture was heated at 90 °C for 6 h. The resulting pale yellow solution was filtered. Pale green crystals of 3 were obtained by leaving the filtrate to stand at room temperature for a week; they were collected and washed consecutively with small amounts of DMF, EtOH, and Et2O, and dried in air (0.23 g, 92.0% yield based on Cu). Anal. calcd for C18H22CuIN4: C 44.59, H 4.57, N 11.56; found: C 44.17, H 4.57, N 11.51.
Diffusion method. A solution of CuI (0.01 g) in 3 mL CH3CN was gently layered on the top of a solution of bdb (0.03 g) in 3 mL CH3CN in a test tube. Leaf-shaped crystals of 3 suitable for X-ray crystallography were obtained after a week. They were collected and washed with small amounts of CH3CN and dried in air.
Iodine sorption study
Certain amounts of 1a–3 crystals (30.0 mg) and solid iodine (ca. 30 mg) were added separately to small vials and the vials were placed in a large vessel and sealed. After sublimation of iodine at 55–60 °C, the color of the crystals immediately changed to deep brown or black. To ensure the completion of the process, the samples were exposed to iodine vapor at 55–60 °C for 7 h. The iodine-encapsulated samples were collected, washed with cyclohexane, dried in air, and weighed (37.8, 47.1, 47.4, and 57.8 mg for 1a-I2, 1b-I2, 2-I2, and 3-I2, respectively).Iodine content determination
Iodine-adsorbed 1a-I2 (9.2 mg), 1b-I2 (7.2 mg), 2-I2 (7.8 mg), and 3-I2 (7.9 mg) samples were added to 5 ml DMF and stirred for 2 min. The resulting orange iodine solutions were filtered and diluted in a 10 ml volumetric flask. The four iodine solutions were diluted to the desired concentration and the corresponding I2 contents were determined by UV/vis spectroscopy at 368 nm.Single crystal X-ray crystallographic studies
X-ray data were collected on a Bruker Apex II diffractometer using MoKα radiation. The structures were solved using direct methods and refined using a full-matrix least squares procedure based on F2 using all data.30 Hydrogen atoms were placed at geometrically estimated positions. Details relating to the crystals and the structural refinements are presented in Table 1. Full details of crystal data and the structure refinements in CIF format are available as ESI† (CCDC reference numbers 1556162–1556165).| Compound | 1a | 1b | 2 | 3 |
|---|---|---|---|---|
| Formula | C10H14Cu2I2N4 | C10H14CuIN4 | C56H56Cu4I4N16 | C18H22CuIN4 |
| Molecular weight | 571.13 | 380.69 | 1714.92 | 484.85 |
| T (K) | 296 | 296 | 296 | 296 |
| Cryst syst | Monoclinic | Monoclinic | Monoclinic | Triclinic |
| Space group | P21/c | P21/n | P21/c |
P |
| a (Å) | 4.3887(7) | 8.2449(3) | 17.421(7) | 8.8660(12) |
| b (Å) | 19.267(3) | 19.4227(8) | 15.653(6) | 9.0619(12) |
| c (Å) | 8.9217(15) | 8.8011(3) | 23.368(9) | 12.1365(16) |
| α (°) | 90 | 90 | 90 | 91.656(2) |
| β (°) | 91.730(3) | 113.635(1) | 105.588(5) | 97.796(2) |
| γ (°) | 90 | 90 | 90 | 101.320(2) |
| V (Å3) | 754.0(2) | 1291.17(8) | 6138(4) | 945.7(2) |
| Z | 2 | 4 | 4 | 2 |
| D calcd (g cm−3) | 2.515 | 1.958 | 1.856 | 1.703 |
| Data collected | 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 205 205 |
30671 |
125324 |
22405 |
| Unique data (Rint) | 2489 (0.037) | 4193 (0.024) | 15604 (0.050) |
5946 (0.019) |
| Data/restraints/parameters | 2489/0/82 | 4193/0/145 | 15604/0/721 |
5946/0/221 |
| R 1 [I > 2σ(I)] | 0.0418 | 0.0271 | 0.0313 | 0.0283 |
| wR2 (all data) | 0.0935 | 0.0611 | 0.0715 | 0.0810 |
Conclusion
In summary, four new non-porous copper(I) iodide coordination polymers with diverse bidentate pyrazolyl ligands have been successfully prepared and characterized. The results show that the bispyrazolyl linkers with different spacer groups, lengths, flexibility and steric hindrance on the pyrazolyl rings induce significant effects on the coordination number of copper atoms and dimensionality of the resulting structures. The results confirm that even non-porous CuI coordination polymers show a versatile tendency to capture volatile iodine. Moreover, as the color of the samples immediately turns black when exposed to iodine vapor, these systems could show potential application for iodine sensing purposes. Due to the presence of [Cu2I2]n and Cu2I2 moieties in the structures of 1a, 1b, and 2, the compounds also show photoluminescence behavior that is quenched with iodine sorption.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Shahid Chamran University of Ahvaz (Grant No. 31400) and the Università degli Studi di Milano (Piano di Sviluppo di Ateneo, azione B, progetti di interesse interdisciplinare PSR2015-1716FDEMA_07) for financial support. DMP acknowledges the Ministry of Education and Science of Russia (Grant 14.B25.31.0005). The authors also thank M. Keshavarzi for designing the graphical abstract.References
- (a) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 974 CrossRef CAS PubMed; (b) H.-C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415 RSC; (c) W. Lu, Z. Wei, Z. Gu, T. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H. Zhou, Chem. Soc. Rev., 2014, 43, 5561 RSC; (d) W. L. Leong and J. J. Vittal, Chem. Rev., 2011, 111, 688 CrossRef CAS PubMed.
- (a) V. Guillerm, D. Kim, J. F. Eubank, R. Luebke, X. Liu, K. Adil, M. S. Lah and M. Eddaoudi, Chem. Soc. Rev., 2014, 43, 6141 RSC; (b) D. J. Tranchemontagne, J. L. Mendoza-Cortes, M. O'Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257 RSC; (c) J. J. Perry, J. A. Perman and M. J. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400 RSC.
- R. Peng, M. Li and D. Li, Coord. Chem. Rev., 2010, 254, 1 CrossRef CAS.
- (a) G. Zeng, S. Xing, X. Han, B. Xin, Y. Yang, X. Wang, G. Li, Z. Shi and S. Feng, RSC Adv., 2015, 5, 40792 RSC; (b) M. Knorr, A. Khatyr, A. D. Aleo, A. E. Yaagoubi, C. Strohmann, M. M. Kubicki, Y. Rousselin, S. M. Aly, D. Fortin, A. Lapprand and P. D. Harvey, Cryst. Growth Des., 2014, 14, 5373 CrossRef CAS; (c) F. Wu, H. Tong, Z. Li, W. Lei, L. Liu, W.-Y. Wong, W.-K. Wong and X. Zhu, Dalton Trans., 2014, 43, 12463 RSC; (d) Q. Benito, X. F. L. Goff, G. Nocton, A. Fargues, A. Garcia, A. Berhault, S. Kahlal, J. Saillard, C. Martineau, J. Trebosc, T. Gacoin, J. Boilot and S. Perruchas, Inorg. Chem., 2015, 54, 4483 CrossRef CAS PubMed; (e) Q. Benito, X. F. L. Goff, S. Maron, A. Fargues, A. Garcia, C. Martineau, F. Taulelle, S. Kahlal, T. Gacoin, J. Boilot and S. Perruchas, J. Am. Chem. Soc., 2014, 136, 11311 CrossRef CAS PubMed; (f) P. M. Graham, R. D. Pike, M. Sabat, R. D. Bailey and W. T. Pennington, Inorg. Chem., 2000, 39, 5121 CrossRef CAS PubMed; (g) B. Xin, G. Zeng, L. Gao, Y. Li, S. Xing, J. Hua, G. Li, Z. Shi and S. Feng, Dalton Trans., 2013, 42, 7562 RSC.
- (a) E. Kintisch, Science, 2005, 310, 1406 CrossRef CAS PubMed; (b) R. C. Ewing and F. N. von Hippel, Science, 2009, 325, 151 CrossRef CAS PubMed.
- (a) N. R. Soelberg, T. G. Garn, M. R. Greenlagh, J. D. Law, R. Jubin, D. M. Strachan and P. K. Thallapally, Sci. Technol. Nucl. Install., 2013, 12, 702496 Search PubMed; (b) A. Saiz-Lopez, J. M. C. Plane, A. R. Baker, L. J. Carpenter, R. von Glasow, L. C. G. Martin, G. McFiggans and R. W. Saunders, Chem. Rev., 2012, 112, 1773 CrossRef CAS PubMed; (c) J. E. T. Hoeve and M. Z. Jacobson, Energy Environ. Sci., 2012, 5, 8743 RSC; (d) E. Barea, C. Montoro and J. A. R. Navarro, Chem. Soc. Rev., 2014, 43, 5419 RSC.
- K. W. Chapman, P. J. Chupas and T. M. Nenoff, J. Am. Chem. Soc., 2010, 132, 8897 CrossRef CAS PubMed.
- (a) G. Massasso, J. Long, J. Haines, S. Devautour-Vinot, G. Maurin, A. Grandjean, B. Onida, B. Donnadieu, J. Larionova, C. Guérin and Y. Guari, Inorg. Chem., 2014, 53, 4269 CrossRef CAS PubMed; (b) G. Massasso, M. Rodríguez-Castillo, J. Long, J. Haines, S. Devautour-Vinot, G. Maurin, A. Grandjean, B. Onida, B. Donnadieu, J. Larionova, C. Guérina and Y. Guaria, Dalton Trans., 2015, 44, 19357 RSC; (c) G. Massasso, J. Long, C. Guerin, A. Grandjean, B. Onida, Y. Guari, J. Larionova, G. Maurin and S. Devautour-Vinot, J. Phys. Chem. C, 2015, 119, 9395 CrossRef CAS.
- S. Ma, S. M. Islam, Y. Shim, Q. Gu, P. Wang, H. Li, G. Sun, X. Yang and M. G. Kanatzidis, Chem. Mater., 2014, 26, 7114 CrossRef CAS.
- H. Sun, P. La, Z. Zhu, W. Liang, B. Yang and A. Li, J. Mater. Sci., 2015, 50, 7326 CrossRef CAS.
- (a) C. Pei, T. Ben, S. Xua and S. Qiu, J. Mater. Chem. A, 2014, 2, 7179 RSC; (b) Y. Zhuojun, Y. Ye, T. Yuyang, Z. Daming and Z. Guangshan, Angew. Chem., Int. Ed., 2015, 54, 12733 CrossRef PubMed.
- (a) D. F. Sava, M. A. Rodriguez, K. W. Chapman, P. J. Chupas, J. A. Greathouse, P. S. Crozier and T. M. Nenoff, J. Am. Chem. Soc., 2011, 133, 12398 CrossRef CAS PubMed; (b) Q. K. Liu, J. P. Ma and Y. B. Dong, Chem. Commun., 2011, 47, 7185 RSC; (c) H. Kitagawa, H. Ohtsu and M. Kawano, Angew. Chem., Int. Ed., 2013, 52, 12395 CrossRef CAS PubMed; (d) V. Safarifard and A. Morsali, CrystEngComm, 2014, 16, 8660 RSC; (e) L. Hashemi and A. Morsali, CrystEngComm, 2014, 16, 4955 RSC; (f) S. Parshamoni, S. Sanda, H. S. Jena and S. Konar, Chem. – Asian J., 2015, 10, 653 CrossRef CAS PubMed; (g) W. W. He, S. L. Li, G. S. Yang, Y. Q. Lan, Z. M. Su and Q. Fu, Chem. Commun., 2012, 48, 10001 RSC; (h) J. Wang, J. Luo, X. Luo, J. Zhao, D. Li, G. Li, Q. Huo and Y. Liu, Cryst. Growth Des., 2015, 15, 915 CrossRef CAS; (i) J. He, J. Duan, H. Shi, J. Huang, J. Huang, L. Yu, M. Zeller, A. D. Hunter and Z. Xu, Inorg. Chem., 2014, 53, 6837 CrossRef CAS PubMed; (j) Z. Yin, Q.-Z. Wang and M.-H. Zeng, J. Am. Chem. Soc., 2012, 134, 4857 CrossRef CAS PubMed; (k) J. T. Hughes, D. F. Sava, T. M. Nenoff and A. Navrotsky, J. Am. Chem. Soc., 2013, 135, 16256 CrossRef CAS PubMed; (l) D. F. Sava, K. W. Chapman, M. A. Rodriguez, J. A. Greathouse, P. S. Crozier, H. Zhao, P. J. Chupas and T. M. Nenoff, Chem. Mater., 2013, 25, 2591 CrossRef CAS; (m) M. Zeng, Q. Wang, Y. Tan, S. Hu, H. Zhao, L. Long and M. Kurmoo, J. Am. Chem. Soc., 2010, 132, 2561 CrossRef CAS PubMed; (n) A. K. Chaudhari, S. Mukherjee, S. S. Nagarkar, B. Joarder and S. K. Ghosh, CrystEngComm, 2013, 15, 9465 RSC; (o) S. S. Zhao, L. Chen, X. Zheng, L. Wang and Z. Xie, Chem. – Asian J., 2017, 12, 615 CrossRef CAS PubMed; (p) Y.-Q. Hu, M.-Q. Li, Y. Wang, T. Zhang, P.-Q. Liao, Z. Zheng, X.-M. Chen and Y.-Z. Zheng, Chem. – Eur. J., 2017, 23, 8409 CrossRef CAS PubMed.
- C. Falaise, C. Volkringer, J. Facqueur, T. Bousquet, L. Gasnotb and T. Loiseaua, Chem. Commun., 2013, 49, 10320 RSC.
- J. Liu, B. P. McGrail, D. M. Strachan, J. Liu, J. Tian and P. K. Thallapally, Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd., 2014, p. 1, DOI:10.1002/9781119951438.eibc2198.
- (a) A. Beheshti, W. Clegg, V. Nobakht and R. W. Harrington, Polyhedron, 2014, 81, 256 CrossRef CAS; (b) A. Beheshti, W. Clegg, V. Nobakht and R. W. Harrington, Cryst. Growth Des., 2013, 13, 1023 CrossRef CAS; (c) A. Beheshti, V. Nobakht, L. Carlucci, D. M. Proserpio and C. T. Abrahams, J. Mol. Struct., 2013, 1037, 236 CrossRef CAS.
- T. G. Mitina and V. A. Blatov, Cryst. Growth Des., 2013, 13, 1655 CAS.
- (a) A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS; (b) I. M. C. van Amsterdam, M. Ubbink, G. W. Canters and M. Huber, Angew. Chem., Int. Ed., 2003, 42, 62 CrossRef CAS.
- (a) Z.-P. Deng, H.-L. Qi, L.-H. Huo, S. W. Ng, H. Zhao and S. Gao, Dalton Trans., 2010, 39, 10038 RSC; (b) D. Braga, F. Grepioni, L. Maini, P. P. Mazzeo and B. Ventura, New J. Chem., 2011, 35, 339 RSC.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955 RSC.
- A. Beheshti, W. Clegg, S. A. MousaviFard, R. W. Harrington, V. Nobakht and L. Russo, Inorg. Chim. Acta, 2011, 376, 310 CrossRef CAS.
- V. Nobakht, A. Beheshti, D. M. Proserpio, L. Carlucci and C. T. Abrahams, Inorg. Chim. Acta, 2014, 414, 217 CrossRef CAS.
- K. Miyao, A. Funabiki, K. Takahashi, T. Mochida and M. Uruichi, New J. Chem., 2014, 38, 739 RSC.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- (a) D. Sun, S. Yuan, H. Wang, H. F. Lu, S. Y. Feng and D. F. Sun, Chem. Commun., 2013, 49, 6152 RSC; (b) E. Cariati, X. H. Bu and P. C. Ford, Chem. Mater., 2000, 12, 3385 CrossRef CAS; (c) Y. Song, R. Fan, P. Wang, X. Wang, S. Gao, X. Du, Y. Yang and T. Luanb, J. Mater. Chem. C, 2015, 3, 6249 RSC.
- Mercury 3.0, Copyright Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, CB2 1EZ, UK, 2012 Search PubMed.
- Y. J. Huang, Y. L. Song, Y. Chen, H. X. Li, Y. Zhang and J. P. Lang, Dalton Trans., 2009, 1411 RSC.
- J. F. Ma, J. F. Liu, Y. Xing, H. Q. Jia and Y. H. Lin, J. Chem. Soc., Dalton Trans., 2000, 2403 RSC.
- X. Y. Wang, S. Q. Liu, C. Y. Zhang, G. Song, F. Y. Bai, Y. H. Xing and Z. Shi, Polyhedron, 2012, 47, 151 CrossRef CAS.
- A. S. Potapov, G. A. Domina, A. I. Khlebnikov and V. D. Ogorodnikov, Eur. J. Org. Chem., 2007, 5112 CrossRef CAS.
- G. M. Sheldrick, SHELX97-Programs for Crystal Structure Analysis, release 97–2, Institut fur Anorganische Chemie der Universitat Gottingen, Gottingen, Germany, 1998 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1556162–1556165. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ce01193h |
| This journal is © The Royal Society of Chemistry 2017 |