
Flexibility in metal–organic frameworks derived from positional and electronic effects of functional groups†
Hyeonbin
Ha‡
a,
Hyungwoo
Hahm‡
a,
Dong Gyun
Jwa
a,
Kwangho
Yoo
a,
Myung Hwan
Park
 b,
Minyoung
Yoon
c,
Youngjo
Kim
*a and
Min
Kim
*a
b,
Minyoung
Yoon
c,
Youngjo
Kim
*a and
Min
Kim
*a
aDepartment of Chemistry and BK21Plus Research Team, Chungbuk National University, 1 Chungdae-ro, Seowon-gu, Cheongju, 28644, Republic of Korea. E-mail: ykim@chungbuk.ac.kr; minkim@chungbuk.ac.kr
bDepartment of Chemistry Education, Chungbuk National University, Cheongju, 28644, Republic of Korea
cDepartment of Nanochemistry, College of Bionano, Gachon University, Sungnam, 13120, Republic of Korea
First published on 11th August 2017
Abstract
The position of identical functional groups and the subsequent electron density of structural benzene rings in a zinc-based metal–organic framework (MOF) have been controlled to reveal flexibility (or breathing behavior) differences. Both ortho- and para-positioned bi-functional benzene-1,4-dicarboxylic acid (BDC) ligands were synthesized with amino-, chloro-, methoxy-, and nitro groups. Additionally, two tri-functionalized, dimethoxy-amino and dimethoxy-nitro BDCs were prepared. All bi- and tri-functionalized BDCs were successfully incorporated into DABCO MOFs (DMOFs), except two diamino BDCs which were insoluble and thermally unstable. Among the eight bi-/tri-functionalized DMOFs, only para-dimethoxy exhibited flexibility in its framework after evacuation in preparation for N2 isotherm measurement. Since the dimethoxy combination has the most electron-rich environment in the benzene ring of the BDC in this series, this indicates that electron density plays a role in the flexibility changes of identically bi-functionalized DMOFs. However, the electron density alone could not fully explain the flexibility changes suggesting that the position of the functional groups is also important. These findings have been corroborated through the synthesis of two tri-functionalized DMOFs with identical functional group locations but opposite electronic environments.
Introduction
Metal–organic frameworks (MOFs) are hybrid porous materials consisting of metal clusters (or ions) and organic linkers. Three dimensional pores are constructed within MOFs by repeating coordination bonds between these metal clusters (i.e., secondary building units, SBUs) and various binding groups on the organic ligands (e.g., carboxylic acid and pyridine).1 The major advantage of MOFs comes from their high porosity and chemical tunability. The relative ease of functionalization and structural tuning can rapidly create libraries of various sets of MOFs thus creating an ideal platform for evaluating and maximizing various applications such as molecular storage, separation and catalysis.2 The functional groups installed onto the organic linkers of MOFs can be tuned for a particular application, but also, these functional groups may alter the structural features of MOFs.Although many MOFs have rigid structures and permanent porosity, sometimes they exhibit flexibility in their structures and pore sizes (i.e., gate-opening or breathing behavior of MOFs).3,4 In flexible MOFs, the porosity of MOFs is changed by external stimuli such as evacuation, guest molecule adsorption and chemical transformations. For example, CO2 can increase the pore size through strong intermolecular forces whereas N2 or CH4 may not induce the same pore-opening effect under 1 atm (Fig. 1).5–7
 | ||
| Fig. 1 Flexibility changes by external stimuli and ligand controls in DMOFs. | ||
This structural flexibility is a unique feature that MOFs have in comparison with other porous materials (e.g., zeolite, mesoporous silica, activated carbon, etc.). It has been realized that the functional group on an organic linker plays a significant role in the flexibility changes of MOFs. The intramolecular interactions, such as hydrogen bonding between functional groups and guest molecules, are highly related to potential structural transformations and flexibility.8,9 In the case of dialkoxy-functionalized DMOFs (DMOF = DABCO MOF) by Fischer and co-workers, the length of the side chain, polarity, and degree of saturation of the alkyl group affected the flexibility capability of the MOFs (Fig. 1).10,11 From the above cases, it was hypothesized that the role of the functional group is derived from potential intermolecular interactions. However, it was also reported that not only the intermolecular interactions, but also intramolecular changes (e.g., electron-donating or electron-withdrawing effects of functional groups) could affect the framework.
Recently, the relationship between ‘regioisomeric controls of two different functional groups’ and ‘the flexibility of MOFs’ has been revealed.12,13 While the combination of NH2–halogen and NH2–OMe produced flexible DMOF frameworks with para-positioning of the two functional groups, other combinations such as NH2–NO2, NO2–OMe, NO2–Cl, and OMe–Cl formed rigid/inflexible frameworks in relative para-positions (Scheme 1). On the other hand, ortho-isomers, for all combinations (6 examples), showed rigid frameworks after evacuation preparation for gas adsorption. The flexibility changes were verified via N2 adsorption after evacuation and powder X-ray diffraction (PXRD) experiments. Even though all as-synthesized bi-functionalized DMOFs have very similar PXRD patterns, the flexible DMOFs (e.g., DMOF-2,5-NH2Cl and DMOF-2,5-NH2OMe) adsorbed a relatively small amount of N2 in comparison with the inflexible DMOFs (e.g., DMOF-2,3-NH2Cl and DMOF-2,3-NH2OMe). However, the PXRD pattern for flexible, para-positioned DMOF-2,5-NH2Cl changed after evacuation, whereas inflexible ortho-positioned DMOF-2,3-NH2Cl showed no changes after evacuation.12,13
 | ||
| Scheme 1 Flexibility changes by two different functional groups and their positions in zinc-based DMOFs. | ||
The differing electronic environments due to the different substituents were proposed as a major factor for the flexibility changes. The combination of NH2–halogen and NH2–OMe creates electron-rich environments in the benzene ring of the ligand because the amino group is strongly electron-donating. The other combinations, in comparison, are relatively electron-deficient.13 However, the series of regioisomeric-electronic control ligands were somewhat incomplete. In this contribution, two identical organic functional groups were controlled for their regioisomeric positions on the benzene ring and three functional groups have been installed onto the benzene ring to modulate the electronic environment. Using this series of bi-functional and tri-functional ligands, their flexibility changes in a MOF structure were rigorously studied.
Results and discussion
Positional and electronic controls in bi-functionalized DMOFs
First, the synthesis of ortho-(2,3-) and para-(2,5-)di-substituted BDCs was attempted with amino, chloro, methoxy, and nitro groups. Dichloro BDCs (BDC-2,3-Cl2 and BDC-2,5-Cl2) were obtained from Sandmeyer reactions from their respective 2,3- and 2,5-NH2Cl BDC starting ligands in the methyl ester form. BDCE-2,3-NH2Cl and BDCE-2,5-NH2Cl (BDCE = benzene-1,4-dicarboxylic methyl ester) were prepared by chlorination of BDCE-NH2 and separated by silica gel column chromatography (see Scheme 2 and the ESI† for details).12 | ||
| Scheme 2 Preparation of bi- and tri-functional ligands for MOF synthesis. | ||
Next, the dimethoxy-substituted BDCs were obtained by methylation of 2,3-dihydroxy BDC or 2,5-dihydroxy BDC ligands after esterification. The hydrolysis of the methyl ester formed two regioisomeric dimethoxy -substituted isomers suitable for the MOF synthesis (see Scheme 2 and the ESI† for details). Previously, the regioisomeric pairs featuring alkoxy groups were reported by Fischer et al.10,11 By increasing the alkyl chain length, the flexibility was controlled, and it was revealed that the steric bulk between substituents across the pores (or frameworks) plays a key role in flexibility differences. It was initially proposed that the intermolecular interaction between two struts in a MOF pore was important to understand the properties of MOFs and their synthesis. Herein, we propose that the electronic environment of the strut is also a key factor in not only the flexibility but also properties of MOFs as a whole.
Lastly, the dinitro BDCs and diamino BDCs were obtained from nitration of NH2-BDC and the subsequent oxidation or reduction. The nitration was performed with the acetyl protected ester from BDCE-NHAc, followed by column chromatography, giving one major isomer, BDCE-2,5-NHAcNO2. The amino group on BDCE-2,5-NHAcNO2 was successfully oxidized after H2O2 treatment, and BDC-2,5-(NO2)2 was obtained by hydrolysis of the ester under basic conditions. Separately, BDC-2,3-(NO2)2 was prepared through double nitration and oxidation from p-xylene. (see Scheme 2 and the ESI† for details). BDC-2,5-(NH2)2 was purchased from commercial sources and BDC-2,3-(NH2)2 was obtained by reduction of BDC-2,3-(NO2)2 in the ester form (see the ESI† for details).
Using the obtained BDCs, the synthesis of DMOFs was carried out. DMOF-2,3-(OMe)2 and DMOF-2,5-(OMe)2 were successfully synthesized by following a typical DMOF synthesis procedure. Of note, the improved crystallinity of DMOF-2,3-(OMe)2 was achieved by a lower temperature synthesis (see the ESI† for details). The dichloro-functionalized DMOF-2,3-Cl2 and DMOF-2,5-Cl2 were also synthesized under the typical synthetic solvothermal conditions of DMOF (120 °C). Although para-functionalized DMOF-2,5-Cl2 was previously reported by Uemura et al. to see ligand substitution effects,14 this is the first example of an ortho-dihalogen substituted DMOF series. Dinitro-functionalized DMOF-2,3-(NO2)2 and DMOF-2,5-(NO2)2 were carefully monitored for appropriate solvothermal synthetic conditions since multi-nitro compounds can be explosive. These dinitro BDC ligands are thermally stable up to 300 °C as evidenced by thermogravimetric analysis (TGA, Fig. S3†). Both DMOF-2,3-(NO2)2 and DMOF-2,5-(NO2)2 were obtained through solvothermal synthesis at 105 °C. Once again, both mono-nitro-functionalized DMOF-NO2 and para-dinitro-functionalized DMOF-2,5-(NO2)2 were previously reported by Uemura et al.,15 but this is the first example of ortho-dinitro-functionalized MOFs. Finally, the use of diamino functionalized BDCs, BDC-2,3-(NH2)2 and BDC-2,5-(NH2)2, was attempted in DMOF synthesis but we could not obtain crystalline DMOF materials from either ligand. We found two possible reasons for the unsuccessful DMOF synthesis. First, both diamino-functionalized BDC ligands were insoluble in polar aprotic solvents (mainly in DMF). We speculate that the two intramolecular hydrogen bonds, between the ortho-positioned carboxylic acid–amino groups, contribute to this insolubility. Secondly, these ligands are very thermally unstable. Even with low concentrations of BDC-2,3-(NH2)2 or BDC-2,5-(NH2)2 in DMF solution, while originally it is bright yellow, it very easily changes to a dark black-brown solution after thermal treatments.
The formation of DMOF structures was confirmed by PXRD patterns (Fig. 2). The six obtained DMOFs showed similar PXRD patterns, and all major peaks matched well with previously reported non-functionalized or mono-functionalized DMOF structures in the as-synthesized structure (Fig. S2†).12,16 In addition, the unit cell parameter of the functionalized DMOF, determined by PXRD pattern indexing, was similar to the single crystal unit cell parameter of the non-functionalized DMOF (Table S1†). Therefore, the major framework structure is not significantly differentiated from the original DMOF structure. The indexed peak position, intensity and hkl values are summarized in the ESI.†
 | ||
| Fig. 2 PXRD patterns and N2 full isotherms (77 K) of the obtained bi-functional DMOFs. a) DMOF-2,3-Cl2/DMOF-2,5-Cl2 b) DMOF-2,3-(NO2)2/DMOF-2,5-(NO2)2 c) DMOF-2,3-(OMe)2/DMOF-2,5-(OMe)2. | ||
1H NMR analysis, after acid digestion, additionally confirmed that all synthesized, regioisomeric ligands retained their functionality during the DMOF framework synthesis and no decomposition or isomerization of ligands was observed. The ligand ratio between BDCs and DABCO was confirmed to be 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. S1†). The TGA shows that these DMOFs are thermally stable up to 300 °C (Fig. S3†).
:1 (Fig. S1†). The TGA shows that these DMOFs are thermally stable up to 300 °C (Fig. S3†).
The full N2 isotherms for the obtained six symmetrically bi-functionalized DMOFs were carefully monitored to observe the flexibility changes of frameworks by regioisomeric ligand installation of functional groups. While both dichloro- and dinitro-systems (i.e., DMOF-2,3-Cl2 and DMOF-2,5-Cl2/DMOF-2,3-(NO2)2 and DMOF-2,5-(NO2)2) showed porous structures for N2 adsorption after evacuation, only the dimethoxy-functionalized DMOFs displayed flexibility differences based on functional group positional changes (Fig. 2, S4, and S5†). Although both DMOF-2,3-(OMe)2 and DMOF-2,5-(OMe)2 showed almost the same PXRD patterns while synthesizing, the N2 adsorption profile and BET surface area were quite different (Fig. 2 and S6†). DMOF-2,3-(OMe)2 was highly porous to N2 (quantity adsorbed ∼375 cm3 g−1 STP at 1 atm, and BET surface area = 1554 m2 g−1) whereas, DMOF-2,5-(OMe)2 was practically non-porous to N2 (quantity adsorbed <25 cm3 g−1 STP at 1 atm, and BET surface area = 45 m2 g−1). This finding is also consistent with the N2 adsorption profile of 2,5-alkoxy functionalized DMOFs reported by the Fischer group,11 and the 2,5-dichloro functionalized DMOF reported by the Uemura group.14
This is also a similar phenomenon to the previously reported DMOF-2,5-NH2Cl and DMOF-2,5-NH2OMe cases.12,13 In the DMOF-2,5-NH2OMe study, we utilized Hammett's substituent constants to predict the relative electronic environment of the BDC ligands.13 The present results have similar trends to relationships between the framework flexibility and the electronic environments of symmetrical installations of functional groups. Among the dichloro-, dimethoxy-, and dinitro-, only the dimethoxy-functionalized BDCs have an electron-rich benzene ring while the others have relatively electron-deficient benzene rings. Unsurprisingly, only methoxy-functionalized DMOFs (DMOF-2,3-(OMe)2 and DMOF-2,5-(OMe)2) displayed the flexibility changes in N2 adsorption experiments after evacuation (Fig. 2). In several computational studies, it was found that the electron density of the benzene ring could impact the bond strength and length of the metal–carboxylate coordination bonds (i.e., coordination bond between the secondary building unit and the BDC ligand).17,18 We believe that our findings are in line with this hypothesis. It is also clearly revealed that these regioisomeric flexibility changes occur not only in hetero-functional group combinations and hydrogen-bonding combinations (e.g., NH2–Cl and NH2–OMe), but also in homo-functional group combinations (i.e., symmetrical functionalization, OMe–OMe).
Adsorption of CO2 of the obtained bi-functionalized DMOFs generally follows the same trends of the N2 adsorption. DMOF-2,3-Cl2 and DMOF-2,5-Cl2 showed almost identical CO2 adsorption profiles (∼3.0 mmol g−1 CO2 uptake at 760 mmHg), and dinitro-functionalized DMOF-2,3-(NO2)2/DMOF-2,5-(NO2)2 displayed similar CO2 uptake at 760 mmHg. Only dimethoxy-functionalized DMOFs have a significantly different CO2 uptake with ∼3.0 mmol g−1 for DMOF-2,3-(OMe)2versus ∼0.5 mmol g−1 for DMOF-2,5-(OMe)2, respectively. This follows exactly the same trend as for the N2 adsorption for dimethoxy-functionalized DMOFs indicating that, for this series of DMOFs, intermolecular forces between CO2 and the ligands cannot induce the breathing behavior as has been seen in other systems (Fig. S7–S9 and Table S2†).
The pore volume analysis from the N2 adsorption data was used to compare the six DMOFs (Table 1). As expected, even though all six bi-functionalized DMOFs have very similar PXRD patterns, their pore volume showed dramatic differences between DMOF-2,3-(OMe)2 and DMOF-2,5-(OMe)2. Using the t-plot method,19 there was up to 74% difference in the pore volume versus other combinations, like DMOF-2,3-Cl2/DMOF-2,5-Cl2 and DMOF-2,3-(NO2)2/DMOF-2,5-(NO2)2, which showed similar pore sizes.
| DMOF | t-Plot micropore volume |
|---|---|
| DMOF-2,5-Cl2 | 0.471 cm3 g−1 |
| DMOF-2,3-Cl2 | 0.461 cm3 g−1 |
| DMOF-2,5-(NO2)2 | 0.179 cm3 g−1 |
| DMOF-2,3-(NO2)2 | 0.185 cm3 g−1 |
| DMOF-2,5-(OMe)2 | 0.085 cm3 g−1 |
| DMOF-2,3-(OMe)2 | 0.329 cm3 g−1 |
| DMOF-2,5-(OMe)2-3-NO2 | 0.299 cm3 g−1 |
| DMOF-2,5-(OMe)2-3-NH2 | 0.219 cm3 g−1 |
Electronic control in tri-functionalized DMOFs
Based on the flexibility differences from bi-functionalized BDCs, especially BDC-2,5-(OMe)2, tri-functionalized ligands were further examined by introducing NO2 and NH2 groups on the benzene ring as third functional groups since DMOF syntheses with just these functional groups were unsuccessful. Nitration was performed on BDCE-2,5-(OMe)2, and BDCE-2,5-(OMe)2-3-NO2 was selectively collected via column chromatography. The target dimethoxy-nitro BDC was obtained following hydrolysis of the starting methyl ester. For another tri-functionalized BDCE, the amino-dimethoxy ligand was prepared by standard reduction (Pd/C in H2) from BDCE-2,5-(OMe)2-3-NO2. Again, the desired BDC-2,5-(OMe)2-3-NH2 was synthesized by hydrolysis of the methyl ester form. To the best of our knowledge, this is the first example of tri-functionalized BDCs for MOF synthesis. Both dimethoxy-nitro and amino-dimethoxy ligands were confirmed by 1H NMR, 13C NMR, IR and HR-MS. (see the ESI† for detailed procedures)These two tri-functionalized BDC ligands were promptly used in DMOF syntheses. In both cases, a low temperature condition (100 °C) produced a better crystalline material than the standard solvothermal condition (120 °C). The DMOF framework using either DMOF-2,5-(OMe)2-3-NO2 or DMOF-2,5-(OMe)2-3-NH2 was confirmed by PXRD (Fig. 3 and S2†), and 1H NMR analysis, after acid digestion, clearly confirmed that both ligands were intact in each framework (Fig. S1†). However, the TGA trace displays an interesting phenomenon; dimethoxy-nitro functionalized DMOF-2,5-(OMe)2-3-NO2 showed a large weight% drop near 270 °C (Fig. S3†). We believe that, and as we discussed above, this is due to the thermally unstable nitro group within an electron-rich environment (near the methoxy groups).
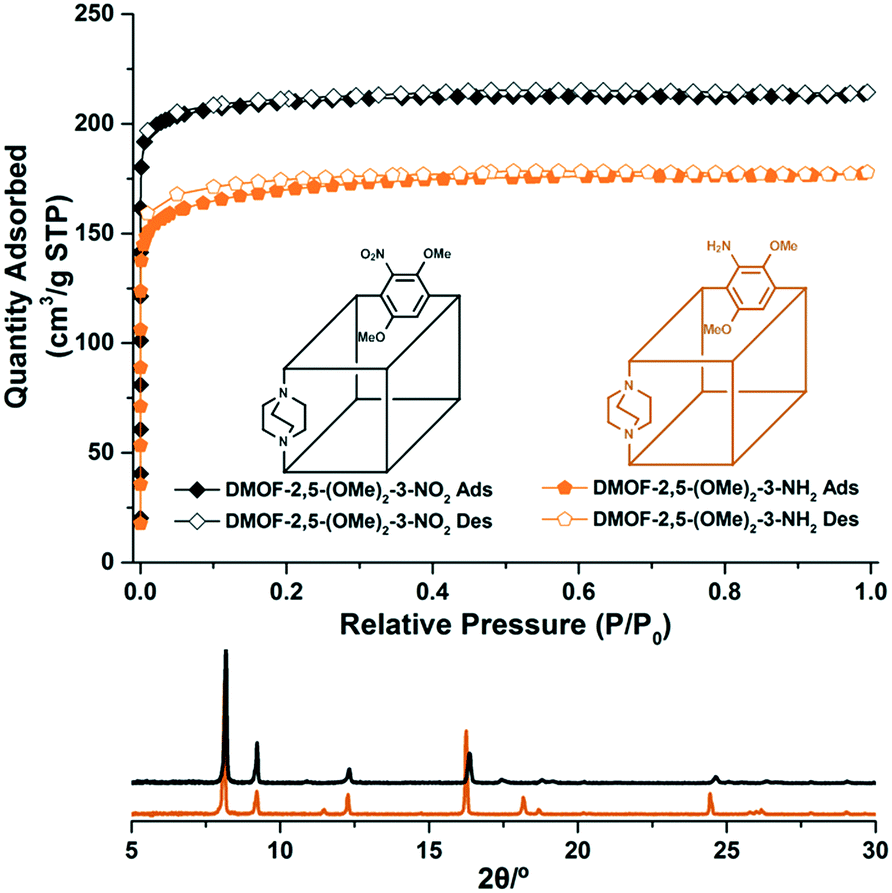 | ||
| Fig. 3 PXRD patterns and N2 full isotherms (77 K) of the tri-functional DMOF-2,5-(OMe)2-3-NO2 and DMOF-2,5-(OMe)2-3-NH2. | ||
The full N2 isotherm experiments for the tri-functionalized DMOFs were performed to monitor the flexibility changes of frameworks by electron density differences with the same regioisomeric positions (Fig. 3). Both DMOF-2,5-(OMe)2-3-NO2 and DMOF-2,5-(OMe)2-3-NH2 displayed porosity to N2, and BET surface areas were measured to be 830 m2 g−1 and 661 m2 g−1, respectively. (Fig. S10†) The pore volume changes between DMOF-2,5-(OMe)2-3-NO2 and DMOF-2,5-(OMe)2-3-NH2 were also correlated with the surface area differences (Table 1).
Lastly, the PXRD patterns for the obtained bi- and tri-functionalized DMOFs after evacuation were compared (samples were re-exposed to air after evacuation under vacuum for measurement). Similar to the full N2 adsorption experiment, only DMOF-2,5-(OMe)2 displayed a peak shift in the PXRD pattern to higher angles, and other DMOFs showed generally retained PXRD patterns (Fig. S2 and S12†). Some decomposition was also measured (i.e., broad backgrounds) since these DMOFs are not stable to moisture including the flexible DMOF-2,5-(OMe)2. Thus, the PXRD data refinement of the changed pattern could not be performed.
Both the N2 isotherm and PXRD pattern changes after evacuation of bi- and tri-functionalized DMOFs suggest that only DMOF-2,5-(OMe)2 demonstrated flexibility in the framework. It is evident that the electron density of the benzene ring alone cannot account for the flexibility changes. The regioisomeric positional variation is an essential factor for the flexibility control in a DMOF system. To date, the ortho-/para-positional changes of two functional groups can alter the flexibility of DMOFs. Among the reported bi-functional DMOFs, only three electron-rich DMOFs (e.g., DMOF-NH2X, DMOF-NH2OMe, DMOF-(OMe)2) have displayed the flexibility changes via regioisomerism. However, electron density control, without different regioisomerism, also had an effect on the flexibility.
Experimental
Ligand synthesis
Dimethyl-2,3-dichloroterephthalate was obtained in comparable yield from a similar procedure using dimethyl-2-amino-3-chloroterephthalate as the starting material.
2,3-Dichloroterephthalic acid was obtained in comparable yield from a similar procedure using dimethyl-2,3-dichloroterephthalate as the starting material.
2,3-Dinitroterephthalic acid was obtained in comparable yield from a similar procedure using dimethyl-2,3-dinitroterephthalate as the starting material.
Dimethyl-2,3-dimethoxyterephthalate was obtained in comparable yield from a similar procedure using dimethyl-2,3-dihydroxyterephthalate as the starting material.
2,3-Dimethoxyterephthalic acid was obtained in comparable yield from a similar procedure using dimethyl-2,3-dihydroxyterephthalate as the starting material.
MOF synthesis
Functionalized BDC (0.5 mmol) and Zn(NO3)2·6H2O (0.5 mmol) were dissolved in DMF (12.5 mL). 1,4-Diazabicyclo[2.2.2]octane (DABCO, 0.8 mmol) was added to this mixture. After the precipitate formed, it was filtered using a fine porosity fritted disc. The clear solution was transferred to a scintillation vial (25 mL size) and heated at a rate of 1–2.5 °C min−1 (detailed conditions for each functionalized DMOF are indicated in the ESI†) from room temperature to 100–120 °C (see the ESI† for details). The temperature was held for 24 h and then cooled to room temperature. The resulting crystals were washed three times with 5 mL DMF. The solvent was exchanged with chloroform (5 mL) over three days, replacing the old chloroform with fresh chloroform every 24 h.Conclusions
In conclusion, we have synthesized a series of bi-functionalized regioisomeric BDC ligands using amino-, chloro-, methoxy-, and nitro-groups with symmetrically controlled positioning along with electron density-controlled tri-functionalized BDC ligands. Among the bi-functionalized ligands, both ortho and para-positioned dichloro-, dimethoxy-, and dinitro-BDCs were successfully incorporated into DMOFs, and two tri-functionalized ligands formed DMOFs with identical PXRD patterns indicating isostructural frameworks. Although all functionalized ligands were perfectly installed into the same DMOF frameworks and eight new bi- or tri-functionalized DMOFs displayed similar structures, the flexibility of the framework (i.e., quantity of N2 adsorbed after evacuation, breathing ability) was modulated by the ligand position from ortho to para in the dimethoxy-functionalized DMOFs. The combination of bi-functionalized 2,3-Cl2, 2,5-Cl2, 2,3-(NO2)2, and 2,5-(NO2)2, and tri-functionalized 2,5-(OMe)2-3-NO2 and 2,5-(OMe)2-3-NH2 showed no significant changes in the N2 adsorption isotherms including pore size analysis, and only the 2,3-/2,5-(OMe)2 combinations displayed highly porous/non-porous framework combinations with respect to N2 and CO2 adsorption. Since the dimethoxy-functionalized BDCs are the only electron-rich benzene rings among the bi-functionalized BDCs, this finding clearly revealed and supported the strong relationship between electron density of the BDC rings and the structural flexibility in two identical functional groups.Among the bi-functionalized DMOFs in this study, only DMOF-2,5-(OMe)2 showed a flexible structure and low N2 adsorption after evacuation. Thus, further pendant group installation of para-dimethoxy functionality was performed. In doing so, a strong electron-withdrawing nitro group or an additionally electron-donating amino group was individually introduced on the benzene ring of the DMOF. Both tri-functionalized DMOFs showed minimal differences in their N2 and CO2 adsorption, even though they have different electronic environments; dimethoxy-nitro has an electron-deficient benzene ring and dimethoxy-amino has an electron-rich benzene ring. Thus, we conclude that the flexibility of DMOFs is not only dependent on the electron density of the benzene rings, but also on the position and local environment of the substituents.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Dr. Corinne A. Allen (Lawrence Berkeley National Laboratory, The Molecular Foundry) for helpful discussions and in preparing this manuscript and Mr. Yoodae Song (Gachon University) for experimental support. This research was supported by the Science Research Center (2016R1A5A1009405) and the Basic Science Research Program (2016R1C1B1011076) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning. The powder X-ray diffraction experiment at the PLS-II, 2D-SMC beamline was supported in part by MSIP and POSTECH.Notes and references
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673 CrossRef CAS PubMed.
- S. M. Cohen, Chem. Rev., 2012, 112, 970 CrossRef CAS PubMed.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062 RSC.
- A. Clearfield, Dalton Trans., 2016, 45, 4100 RSC.
- Z. Wang and S. M. Cohen, J. Am. Chem. Soc., 2009, 131, 16675 CrossRef CAS PubMed.
- K. Nakagawa, D. Tanaka, S. Horike, S. Shimomura, M. Higuchi and S. Kitagawa, Chem. Commun., 2010, 46, 4258 RSC.
- X. Y. Chen, H. Vinh-Thang, D. Rodrigue and S. Kaliaguine, Ind. Eng. Chem. Res., 2012, 51, 6895 CrossRef CAS.
- T. Devic, P. Horcajada, C. Serre, F. Salles, G. Maurin, B. Moulin, D. Heurtaux, G. Clet, A. Vimont, J.-M. Greneche, B. Le Ouay, F. Moreau, E. Magnier, Y. Filinchuk, J. Marrot, J.-C. Lavalley, M. Daturi and G. Ferey, J. Am. Chem. Soc., 2010, 132, 1127 CrossRef CAS PubMed.
- P. Horcajada, F. Salles, S. Wuttke, T. Devic, D. Heurtaux, G. Maurin, A. Vimont, M. Daturi, O. David, E. Magnier, N. Stock, Y. Filinchuk, D. Popov, C. Riekel, G. Ferey and C. Serre, J. Am. Chem. Soc., 2011, 133, 17839 CrossRef CAS PubMed.
- S. Henke, D. C. F. Wieland, M. Meilikhov, M. Paulus, C. Sternemann, K. Yusenko and R. A. Fischer, CrystEngComm, 2011, 13, 6399 RSC.
- S. Henke, A. Schneemann, A. Wutscher and R. A. Fischer, J. Am. Chem. Soc., 2012, 134, 9464 CrossRef CAS PubMed.
- M. Kim, J. A. Boissonnault, P. V. Dau and S. M. Cohen, Angew. Chem., Int. Ed., 2011, 50, 12193 CrossRef CAS PubMed.
- H. Hahm, K. Yoo, H. Ha and M. Kim, Inorg. Chem., 2016, 55, 7576 CrossRef CAS PubMed.
- K. Uemura, Y. Yamasaki, F. Onishi, H. Kita and M. Ebihara, Inorg. Chem., 2010, 49, 10133 CrossRef CAS PubMed.
- K. Uemura, F. Onishi, Y. Yamasaki and H. Kita, J. Solid State Chem., 2009, 182, 2852 CrossRef CAS.
- D. N. Dybtsev, H. Chun and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 5033 CrossRef CAS PubMed.
- A. R. Campanelli, A. Domenicano, F. Ramondo and I. Hargittai, J. Phys. Chem., 2004, 108, 4940 CrossRef CAS.
- T. Soidla, W. P. Oziminski, M. Hoffmann, H. Koroniak and T. M. Krygowski, J. Org. Chem., 2014, 79, 7321 CrossRef PubMed.
- A. Galarneau, F. Villemot, J. Rodriguez, F. Fajula and B. Coasne, Langmuir, 2014, 30, 13266 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and characterization of ligands and materials: Fig. S1–S12, Scheme S1–S5, and Table S1–S2. See DOI: 10.1039/c7ce00971b |
| ‡ H. Ha and H. Hahm equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2017 |