
NH3-assisted chloride flux-coating method for direct fabrication of visible-light-responsive SrNbO2N crystal layers†
 ab
Mirabbos
Hojamberdiev,
a
C. Buddie
Mullins,
b
Kazunari
Domen
d
and
Katsuya
Teshima
*ae
ab
Mirabbos
Hojamberdiev,
a
C. Buddie
Mullins,
b
Kazunari
Domen
d
and
Katsuya
Teshima
*ae
Abstract
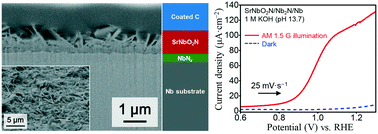
Perovskite-type SrNbO2N crystal layers were prepared on niobium substrates by using an NH3-assisted chloride flux-coating method. The optimization of synthesis parameters (holding temperature and strontium source : flux molar ratio) was performed using a NaCl–KCl flux. By choosing the optimal synthesis conditions, platelet SrNbO2N crystals were grown over the entire substrate surface, and each SrNbO2N platelet has a single-crystalline structure in a cubic symmetry. The optimal crystal layer possesses a well-adhered SrNbO2N/NbNx/Nb structure and absorbed photons with wavelengths up to 680 nm. In addition, the optimal SrNbO2N/NbNx photoelectrode was used for photoelectrochemical water oxidation (2H2O → 4H+ + 4e− + O2↑), as one half of the water splitting reaction. A photocurrent density of 113 μA cm−2 at 1.23 VRHE was recorded on the SrNbO2N/NbNx electrode without any additional co-catalyst loading and treatment under simulated sunlight, due to the higher crystallinity of SrNbO2N and higher interface-adhesion of the SrNbO2N/NbNx/Nb structure, which suppress the recombination of photogenerated electrons and holes at the defects and lead to an increase of the photogenerated electron collection efficiency in a niobium substrate, respectively. This study is the first to address the fabrication of quaternary oxynitride crystal layers on a conductive substrate using an NH3-assisted flux-coating method.
 Please wait while we load your content...
Please wait while we load your content...

