
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Base free transfer hydrogenation using a covalent triazine framework based catalyst†
A. V.
Bavykina
a,
H.-H.
Mautscke
b,
M.
Makkee
a,
F.
Kapteijn
 c,
J.
Gascon
*a and
F. X.
Llabrés i Xamena
*b
c,
J.
Gascon
*a and
F. X.
Llabrés i Xamena
*b
aCatalysis Engineering, Delft University of Technology, Delft, Netherlands. E-mail: j.gascon@tudelft.nl
bInstituto de Tecnologia Quimica CSIC-UPV, Universidad Politecnica de Valencia, Consejo Superior de Investigaciones Científicas, Valencia, Spain. E-mail: fllabres@itq.upv.es
cChemical Engineering, TUDelft, Delft, Netherlands
First published on 12th May 2017
Abstract
Isomerisation of allylic alcohols to saturated ketones can be efficiently catalysed by a heterogeneous molecular system resulting from IrIIICp* anchoring to a covalent triazine framework. The obtained catalysts are active, selective, and fully recyclable.
Transfer hydrogenation (TH) reaction – the addition of hydrogen to an unsaturated group of an organic molecule from a source other than H2 – has been gaining a lot of attention as it is an appealing alternative to direct hydrogenation.1 The reasoning behind it is the elimination of pressurised hydrogen and high pressure equipment use. Besides, a conventional hydrogenation catalyst is rarely selective – any present unsaturated or oxidised functional group is exposed to reduction, resulting in, most of the time, a fully hydrogenated product or a mixture of products. Exceptions for highly chemoselective hydrogenation catalysts exist, such as noble metal nanoparticles supported on metal oxides or encapsulated inside metal–organic frameworks.2–4 In contrast with conventional hydrogenation, TH allows the reaction to be performed selectively, aiming for a specific unsaturated bond and leaving the rest of the original molecule intact.
A conventional TH catalyst is a transition metal complex; among different metals, iridium is the most active one. Ir complexes involving a N-heterocycle carbene ligand,5–16 or half-sandwich complexes with a Cp* ligand17–22 are typical examples. Moving from soluble organometallic compounds to materials that remain solid under the reaction conditions, facilitates separation and enables recycling. There exist a number of works on heterogeneous TH systems, including magnetic nanoparticle-,23–26 polymer-,27–33 graphene-,34,35 carbon nanotube-,36–38 silica-,39–45 zeolite-46–49 or oxide-supported50–55 catalysts.
Recently, Porous Organic Frameworks (POFs) have been gaining attention within catalytic and other applications.56,57 Covalent Triazine Frameworks (CTFs), a subclass of POFs, are highly porous and stable solids made by trimerisation of aromatic nitriles.58,59 CTFs are rich in nitrogen functionalities, and their distribution can be varied by using different building blocks. Starting from a pyridine containing building unit, quasi-bipyridine moieties become available in the final material. Bipyridine is a widely-applied ligand in organometallic chemistry; its presence within a framework enables anchoring of a transition-metal complex. Following this approach, a number of molecular heterogeneous catalysts were developed for a range of different catalytic reactions by several research groups.59–67
Recently, we developed a CTF based catalyst which is highly active in the reversible formic acid (FA) dehydrogenation reaction.60,61 Applying different conditions, the same catalyst is active in both hydrogen production from FA and hydrogen storage by reaction with carbon dioxide. Both these reactions proceed via iridium–hydride bond formation. In this work, we report the use of a similar Ir@CTF material as a base free catalyst for the transfer hydrogenation (TH) reaction (Fig. 1).
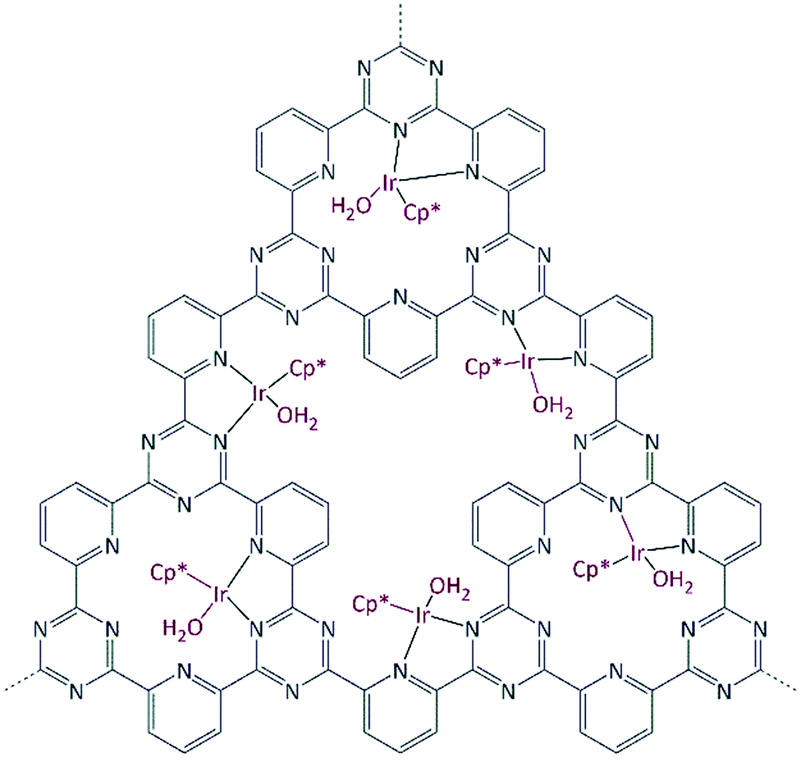 | ||
| Fig. 1 Idealized representation of the Ir@CTF catalyst. Ir is in the oxidation state 3+; charges and (OTf−) weakly coordinated anions are omitted in the figure for clarity. | ||
The commercially available iridium precursor [IrCp*Cl2]2 is coordinated to bipyridine moieties within the framework, resulting in an air- and moisture-stable Ir@CTF catalyst. The inert atmosphere was required only during the catalyst preparation. Once IrCp* was coordinated, the catalyst can be stored at an ambient atmosphere without losing activity. In order to coordinate iridium to the framework, an aqueous solution of [IrCp*Cl2]2 was prepared, in which CTF powder was suspended and stirred. The conventional approach of removing the poisoning chlorine ions by precipitating them with silver cations was ruled out to avoid the precipitation of AgCl within the pores of the material. Instead, HCl was complexed with dimethylformamide, and subsequently washed out.68 The SEM/EDX analysis confirmed that no chlorine was present in the final catalyst. Triflic acid was added during the washing steps to enable the charge balance between Ir3+ and weakly coordinated (OTf−) anions. Due to the insoluble nature and the black color of the CTF solids, catalyst characterization was limited to its structural properties, TGA (see Fig. S1†), SEM (see Fig. S4†), XPS and elemental analysis. The iridium content determined by ICP-OES was 2.4 wt% Ir in the catalyst. Table 1 summarizes the results of elemental analysis and the BET surface area of CTF and Ir@CTF. The framework employed was constructed solely from pyridinedicarbonitrile, resulting in the fully microporous material. Though the BET surface area decreased after introducing the bulky complex, it was still large enough to be accessible to physisorbed N2. XPS analysis (Fig. 2) indicated that Ir was in the oxidation state 3+,69 and the measurements on the spent catalyst confirmed that the oxidation state remained unchanged after several catalytic runs (see Fig. S3†).
| S BET [m2 g−1] | Elemental composition [% C/% N/% H/% Ir] | |
|---|---|---|
| CTF | 930 | 69.7/27.3/3.0/0 |
| Ir@CTF | 500 | 67.9/25.9/3.0/2.4 |
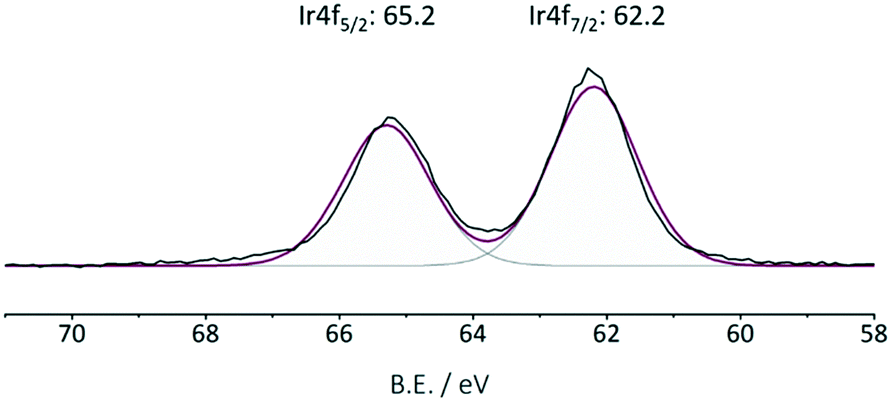 | ||
| Fig. 2 Ir XPS of the fresh Ir@CTF catalyst. The oxidation state of iridium did not change after multiple catalytic runs (see Fig. S3†). | ||
In order to evaluate the catalytic activity of Ir@CTF as a TH catalyst, we considered the redox isomerisation of an allylic alcohol into the corresponding saturated carbonyl compound. Conversion of allylic alcohols into saturated ketones is usually carried out in two steps, involving sequential hydrogenation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and dehydrogenation of the alcohol, which usually requires further protection and deprotection steps. Therefore, the one-pot isomerisation represents an attractive alternative (see Scheme 1).70 This reaction can be considered as an intramolecular TH reaction, in which hydrogen is transferred from the alcohol moiety to the CC bond. Various metals from groups 8, 9, and 10 (including Ir) are known to catalyse this reaction, mostly in the form of homogeneous catalysts.70 However, a few examples exist for transition metal complexes immobilised on suitable supports, such as Ru(OH)x supported on alumina,71 a cationic rhodium complex supported on mesoporous silica72 and, very recently, an iridium N-heterocyclic carbene (NHC) introduced into a metal–organic framework.73 Isomerisation of allylic alcohols is usually carried out in the presence of additives, such as bases or hydrogen acceptors, to promote the reaction.
C bonds and dehydrogenation of the alcohol, which usually requires further protection and deprotection steps. Therefore, the one-pot isomerisation represents an attractive alternative (see Scheme 1).70 This reaction can be considered as an intramolecular TH reaction, in which hydrogen is transferred from the alcohol moiety to the CC bond. Various metals from groups 8, 9, and 10 (including Ir) are known to catalyse this reaction, mostly in the form of homogeneous catalysts.70 However, a few examples exist for transition metal complexes immobilised on suitable supports, such as Ru(OH)x supported on alumina,71 a cationic rhodium complex supported on mesoporous silica72 and, very recently, an iridium N-heterocyclic carbene (NHC) introduced into a metal–organic framework.73 Isomerisation of allylic alcohols is usually carried out in the presence of additives, such as bases or hydrogen acceptors, to promote the reaction.
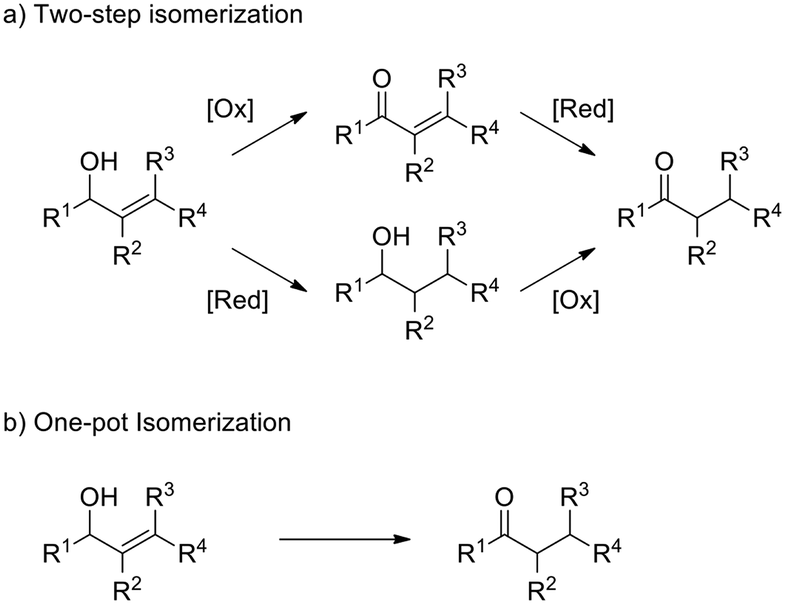 | ||
| Scheme 1 Two step and one step isomerization of allylic alcohols. | ||
In the present study, we have selected 1-octene-3-ol as a model allylic alcohol. Then, we first brought into contact the alcohol (40 mg, 0.31 mmol) with the Ir@CTF material (10 mg, corresponding to 0.40 mol% Ir with respect to the substrate) in toluene (1 mL) at 120 °C under an inert atmosphere (N2, 2 bar). No further additives were used in the reaction. Under these conditions, a conversion of 83% of the alcohol was obtained after a reaction of 48 h with a 78% selectivity to the saturated ketone (final yield of 65%). Besides 3-octanone, another product formed (in 13% yield), which most likely corresponds to the intermediate enone (1-octen-3-one).70Table 2 compares the performance of the Ir@CTF catalysts with those of other catalysts recently reported in the literature.
| Catalyst | Ir (mol%) | Solvent | Temp (°C) | t (h) | Yield (mol%) | TOF (min−1) | Ref. |
|---|---|---|---|---|---|---|---|
| a TOF cannot be calculated from the data provided in the original paper. b Time and yield values are estimated from the plot included in the original publication. | |||||||
| Ir@CTF | 0.4 | Toluene | 120 | 48 | 65 | 0.5 | This work |
| Ir@CTF | 0.4 | Toluene | 120 | 23 | 82 | 24 | This work |
| Ir-UiO-68 | 4 | Toluene | 100 | 48 | 65 | —a | 73 |
| IrCl3·3H2O | 2 | H2O/toluene | 80 | 1.5b | 85b | 14 | 76 |
| K3[IrCl6] | 2 | H2O/toluene | 80 | 1.5b | 85b | 10 | 76 |
In order to improve the catalytic performance of our Ir@CTF material, we carried out a screening of various solvents. In particular, considerably better results were obtained when toluene was replaced by 2-propanol under identical conditions. Fig. 3 shows the corresponding time–conversion plot obtained.
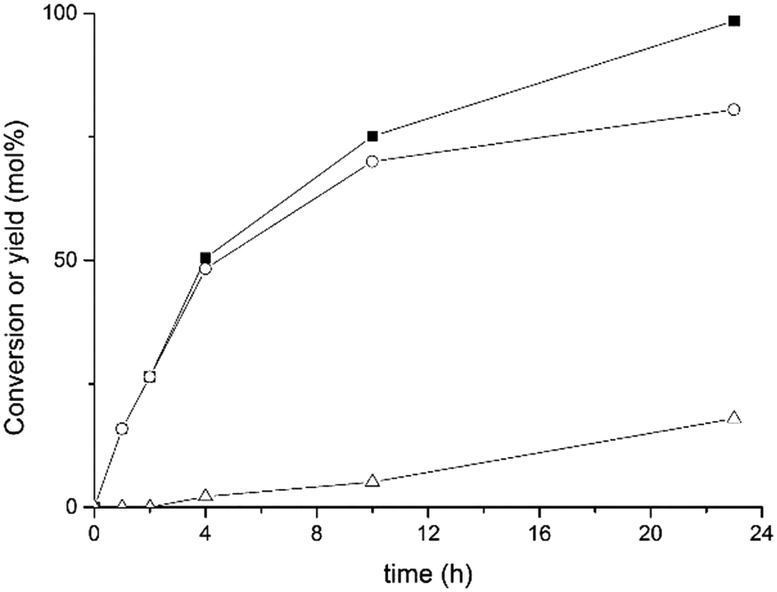 | ||
| Fig. 3 Conversion of 1-octen-3-ol over Ir@CTF (squares) and evolution of products: 3-octanone (circles) and 3-octanol (triangles). The reaction was carried out at 120 °C under an inert atmosphere (N2, 2 bar) using isopropanol as solvent. | ||
The reaction was much faster when isopropanol was used as solvent, attaining a 98% conversion for 1-octen-3-ol after only 23 h, with 82% yield of the target 3-octanone, resulting in a turnover frequency (TOF) of 24 min−1. As the reaction time increased, a secondary product was also observed to develop, which corresponds to the saturated alcohol, 3-octanol. This most likely originated from the further reduction of the saturated ketone through a Meerwein–Ponndorf–Verley TH catalysed by Ir in which isopropanol acted as the hydrogen source to form acetone. As a consequence, the overall selectivity to 3-octanone progressively decreased from >99% at a short reaction time to 83% at the maximum conversion, which was still above the maximum selectivity obtained when toluene was used as solvent (i.e., 78%). Note that the major by-product observed when toluene was used as solvent was the corresponding enone intermediate (1-octen-3-one), which is responsible for the relatively low selectivity attained (78%). When isopropanol was used as solvent, accumulation of the enone in the reaction medium was no longer observed, since it was rapidly converted into the final product, 3-octanone. This may indicate that isopropanol can act as a hydrogen donor solvent, thus assisting the fast conversion of the intermediate enone into the final 3-octanone product, resulting in an overall increment of the selectivity (83%). Given the better performance observed, all further catalytic studies were carried out in isopropanol as solvent.
According to the data shown in Fig. 3, the turnover frequency (TOF) of the Ir@CTF catalyst (calculated at a short reaction time) was 24 min−1. Although this value is far from the performance attained with various Ir(I) transition metal complexes,74,75 Ir@CTF clearly outperformed other catalysts containing Ir(III). Thus, for instance, Sasson et al. reported the conversion of the same allylic alcohol over IrCl3 and K3[IrCl6] at 80 °C in a biphasic H2O/toluene system, reaching TOFs of 14 and 10 min−1, respectively.76 In a recent report by Martín-Matute and co-workers,73 a maximum yield of 65% 3-octanone was obtained after 48 h over an Ir(III)–NHC complex supported in a metal–organic framework (toluene, 100 °C and 4 mol% Ir). The catalytic results obtained with this Ir–NHC-MOF improved upon addition of bases (NaHCO3 or K2CO3) but the amount of Ir that leached into the solution increased considerably and the crystallinity of the MOF support decreased significantly.
It is important to stress that Ir@CTF can catalyse the redox isomerisation of allylic alcohols without the need for additives. Most likely, CTF is not just an inert support to disperse the IrIIICp* metal complexes. Rather, the presence of the pyridine molecules of the support (pKb = 7.8) will play an active role as co-catalysts in the isomerisation reaction, by assisting in the initial alcohol deprotonation and coordination to the Ir centres to form the initial metal enolate.
Reusability studies of the Ir@CTF material using isopropanol (i-PrOH) as solvent were performed for up to six consecutive runs, and the results are shown in Fig. 4. Between two consecutive catalytic runs, the solid was recovered by filtration, washed with i-PrOH, and dried at room temperature. Only a slight decrease of activity was observed over the first three runs (the maximum conversion after 22 h dropped from 99% to 97%, Fig. 4), while a more pronounced decrease of activity was observed starting from the fourth cycle. According to XPS analysis of the material recovered after the reactions, the 3+ oxidation state of Ir was maintained. Analysis of the filtrate after the first cycle revealed that some Ir leaches from the solid into the solution, which amounts to 3.6% of the total Ir used. This was most likely due to the additional washing during the reaction of some loosely bound Ir species in the Ir@CTF material. However, no significant amounts of Ir were detected in the filtrate after the second catalytic run (<1% of the total Ir). Therefore, the catalyst deactivation observed upon reuse was (mainly) attributed to a build-up of adsorbed products on the catalyst surface, progressively blocking the active sites. Meanwhile, the selectivity to the ketone was maintained (or slightly increased) upon consecutive reuses.
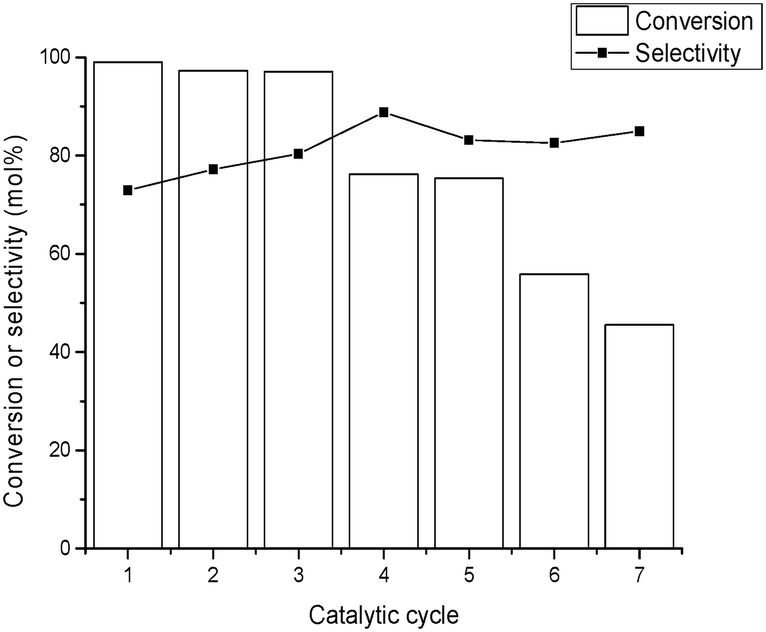 | ||
| Fig. 4 Conversion and selectivity to 3-octanone for six consecutive catalytic runs. The reaction was carried out at 120 °C under an inert atmosphere (N2, 2 bar) using isopropanol as solvent. | ||
Conclusions
A nitrogen rich covalent framework constructed from pyridinedicarbonitrile building block allows a molecular heterogeneous catalyst to be obtained, where CTF plays the role of the scaffold and the base. The Ir@CTF catalyst allows straightforward handling and recycling under ambient conditions.Acknowledgements
Financial support from the Generalitat Valenciana (projects Consolider-Ingenio MULTICAT and AICO/2015/065), the Spanish Ministry of Economy and Competitiveness (MINECO) (program Severo Ochoa SEV20120267), and the Spanish Ministry of Science and Innovation (MICINN) (project MAT2014-52085-C2-1-P) are gratefully acknowledged. This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 641887 (project acronym: DEFNET). Also, financial support from the European Union Seventh Framework Programme (FP7/2007-2013) under the grant agreement number 309701, project Eco2CO2 is acknowledged.References
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- F. G. Cirujano, A. Leyva-Pérez, A. Corma and F. X. Llabrés i Xamena, ChemCatChem, 2013, 5, 538–549 CrossRef CAS.
- H. Khajavi, H. A. Stil, H. P. C. E. Kuipers, J. Gascon and F. Kapteijn, ACS Catal., 2013, 3, 2617–2626 CrossRef CAS.
- A. Corma and P. Serna, Science, 2006, 313, 332 CrossRef CAS PubMed.
- A. Azua, J. A. Mata and E. Peris, Organometallics, 2011, 30, 5532–5536 CrossRef CAS.
- A. Azua, J. A. Mata, E. Peris, F. Lamaty, J. Martinez and E. Colacino, Organometallics, 2012, 31, 3911–3919 CrossRef CAS.
- A. Azua, S. Sanz and E. Peris, Chem. – Eur. J., 2011, 17, 3963–3967 CrossRef CAS PubMed.
- A. Binobaid, M. Iglesias, D. Beetstra, A. Dervisi, I. Fallis and K. J. Cavell, Eur. J. Inorg. Chem., 2010, 2010, 5426–5431 CrossRef.
- S. K. Furfari, M. R. Gyton, D. Twycross and M. L. Cole, Chem. Commun., 2015, 51, 74–76 RSC.
- X. Gong, H. Zhang and X. Li, Tetrahedron Lett., 2011, 52, 5596–5600 CrossRef CAS.
- D. Gulcemal, A. G. Gokce, S. Gulcemal and B. Cetinkaya, RSC Adv., 2014, 4, 26222–26230 RSC.
- S. Gulcemal, A. G. Gokce and B. Cetinkaya, Dalton Trans., 2013, 42, 7305–7311 RSC.
- S. Gülcemal, A. G. Gökçe and B. Çetinkaya, Inorg. Chem., 2013, 52, 10601–10609 CrossRef PubMed.
- M. V. Jiménez, J. Fernández-Tornos, J. J. Pérez-Torrente, F. J. Modrego, P. Garcııa-Orduña and L. A. Oro, Organometallics, 2015, 34, 926–940 CrossRef.
- M. V. Jiménez, J. Fernández-Tornos, J. J. Pérez-Torrente, F. J. Modrego, S. Winterle, C. Cunchillos, F. J. Lahoz and L. A. Oro, Organometallics, 2011, 30, 5493–5508 CrossRef.
- X.-H. Zhu, L.-H. Cai, C.-X. Wang, Y.-N. Wang, X.-Q. Guo and X.-F. Hou, J. Mol. Catal. A: Chem., 2014, 393, 134–141 CrossRef CAS.
- R. Maity, S. Hohloch, C.-Y. Su, M. van der Meer and B. Sarkar, Chem. – Eur. J., 2014, 20, 9952–9961 CrossRef CAS PubMed.
- A. Ruff, C. Kirby, B. C. Chan and A. R. O'Connor, Organometallics, 2016, 35, 327–335 CrossRef CAS.
- H. Vázquez-Villa, S. Reber, M. A. Ariger and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 8979–8981 CrossRef PubMed.
- C. Wang, A. Pettman, J. Bacsa and J. Xiao, Angew. Chem., Int. Ed., 2010, 49, 7548–7552 CrossRef CAS PubMed.
- Y. Wei, D. Xue, Q. Lei, C. Wang and J. Xiao, Green Chem., 2013, 15, 629–634 RSC.
- X. Wu, X. Li, A. Zanotti-Gerosa, A. Pettman, J. Liu, A. J. Mills and J. Xiao, Chem. – Eur. J., 2008, 14, 2209–2222 CrossRef CAS PubMed.
- M. B. Gawande, H. Guo, A. K. Rathi, P. S. Branco, Y. Chen, R. S. Varma and D.-L. Peng, RSC Adv., 2013, 3, 1050–1054 RSC.
- G. Liu, H. Gu, Y. Sun, J. Long, Y. Xu and H. Li, Adv. Synth. Catal., 2011, 353, 1317–1324 CrossRef CAS.
- R. B. Nasir Baig and R. S. Varma, ACS Sustainable Chem. Eng., 2013, 1, 805–809 CrossRef CAS.
- Y. Sun, G. Liu, H. Gu, T. Huang, Y. Zhang and H. Li, Chem. Commun., 2011, 47, 2583–2585 RSC.
- N. Haraguchi, A. Nishiyama and S. Itsuno, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3340–3349 CrossRef CAS.
- R. Marcos, C. Jimeno and M. A. Pericàs, Adv. Synth. Catal., 2011, 353, 1345–1352 CrossRef CAS.
- R. A. Molla, A. S. Roy, K. Ghosh, N. Salam, M. A. Iqubal, K. Tuhina and S. M. Islam, J. Organomet. Chem., 2015, 776, 170–179 CrossRef CAS.
- H. Sugie, Y. Hashimoto, N. Haraguchi and S. Itsuno, J. Organomet. Chem., 2014, 751, 711–716 CrossRef CAS.
- R. Wang, J. Wan, X. Ma, X. Xu and L. Liu, Dalton Trans., 2013, 42, 6513–6522 RSC.
- X. Xu, R. Wang, J. Wan, X. Ma and J. Peng, RSC Adv., 2013, 3, 6747–6751 RSC.
- Z. Zhou and Q. Ma, Appl. Organomet. Chem., 2011, 25, 233–237 CAS.
- M. Gopiraman, S. G. Babu, Z. Khatri, K. Wei, M. Endo, R. Karvembu and I. S. Kim, Catal. Sci. Technol., 2013, 3, 1485–1489 CAS.
- M. Gopiraman, S. Ganesh Babu, Z. Khatri, W. Kai, Y. A. Kim, M. Endo, R. Karvembu and I. S. Kim, J. Phys. Chem. C, 2013, 117, 23582–23596 CAS.
- B. Barati, M. Moghadam, A. Rahmati, V. Mirkhani, S. Tangestaninejad and I. Mohammadpoor-Baltork, J. Organomet. Chem., 2013, 724, 32–39 CrossRef CAS.
- M. Blanco, P. Álvarez, C. Blanco, M. V. Jiménez, J. Fernández-Tornos, J. J. Pérez-Torrente, L. A. Oro and R. Menéndez, ACS Catal., 2013, 3, 1307–1317 CrossRef CAS.
- M. Gopiraman, S. G. Babu, R. Karvembu and I. S. Kim, Appl. Catal., A, 2014, 484, 84–96 CrossRef CAS.
- S.-J. Chen, H.-X. You, G. Vo-Thanh and Y. Liu, Monatsh. Chem., 2013, 144, 851–858 CrossRef CAS.
- T. Cheng, J. Long, X. Liang, R. Liu and G. Liu, Mater. Res. Bull., 2014, 53, 1–6 CrossRef CAS.
- P. N. Liu, P. M. Gu, F. Wang and Y. Q. Tu, Org. Lett., 2004, 6, 169–172 CrossRef CAS PubMed.
- L.-L. Lou, H. Du, Y. Shen, K. Yu, W. Yu, Q. Chen and S. Liu, Microporous Mesoporous Mater., 2014, 187, 94–99 CrossRef CAS.
- S. M. Sarkar, M. M. Yusoff and M. L. Rahman, J. Chin. Chem. Soc., 2015, 62, 177–181 CrossRef CAS.
- Y. Shen, Q. Chen, L.-L. Lou, K. Yu, F. Ding and S. Liu, Catal. Lett., 2010, 137, 104–109 CrossRef CAS.
- D. Zhang, J. Xu, Q. Zhao, T. Cheng and G. Liu, ChemCatChem, 2014, 6, 2998–3003 CrossRef CAS.
- M. L. Kantam, B. P. C. Rao, B. M. Choudary and B. Sreedhar, Adv. Synth. Catal., 2006, 348, 1970–1976 CrossRef CAS.
- A. Corma, M. E. Domine and S. Valencia, J. Catal., 2003, 215, 294–304 CrossRef CAS.
- Y. Zhu, G. Chuah and S. Jaenicke, J. Catal., 2004, 227, 1–10 CrossRef CAS.
- A. Corma, F. X. Llabrés i Xamena, C. Prestipino, M. Renz and S. Valencia, J. Phys. Chem. C, 2009, 113, 11306–11315 CAS.
- Y. Gao, S. Jaenicke and G.-K. Chuah, Appl. Catal., A, 2014, 484, 51–58 CrossRef CAS.
- A. M. Hengne, A. V. Malawadkar, N. S. Biradar and C. V. Rode, RSC Adv., 2014, 4, 9730–9736 RSC.
- S. Muratsugu, Z. Weng, H. Nakai, K. Isobe, Y. Kushida, T. Sasaki and M. Tada, Phys. Chem. Chem. Phys., 2012, 14, 16023–16031 RSC.
- K. Pupovac and R. Palkovits, ChemSusChem, 2013, 6, 2103–2110 CrossRef CAS PubMed.
- K. Shimura and K.-I. Shimizu, Green Chem., 2012, 14, 2983–2985 RSC.
- X. Tang, L. Hu, Y. Sun, G. Zhao, W. Hao and L. Lin, RSC Adv., 2013, 3, 10277–10284 RSC.
- S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed.
- D.-S. Zhang, Z. Chang, Y.-B. Lv, T.-L. Hu and X.-H. Bu, RSC Adv., 2012, 2, 408–410 RSC.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed.
- R. Palkovits, M. Antonietti, P. Kuhn, A. Thomas and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6909–6912 CrossRef CAS PubMed.
- A. V. Bavykina, M. G. Goesten, F. Kapteijn, M. Makkee and J. Gascon, ChemSusChem, 2015, 8, 809–812 CrossRef CAS PubMed.
- A. V. Bavykina, E. Rozhko, M. G. Goesten, T. Wezendonk, B. Seoane, F. Kapteijn, M. Makkee and J. Gascon, ChemCatChem, 2016, 8, 2217–2221 CrossRef CAS.
- G. Gunniya Hariyanandam, D. Hyun, P. Natarajan, K.-D. Jung and S. Yoon, Catal. Today, 2016, 265, 52–55 CrossRef CAS.
- P. J. C. Hausoul, C. Broicher, R. Vegliante, C. Göb and R. Palkovits, Angew. Chem., Int. Ed., 2016, 55, 5597–5601 CrossRef CAS PubMed.
- K. Park, G. H. Gunasekar, N. Prakash, K.-D. Jung and S. Yoon, ChemSusChem, 2015, 8, 3410–3413 CrossRef CAS PubMed.
- M. Pilaski, J. Artz, H.-U. Islam, A. M. Beale and R. Palkovits, Microporous Mesoporous Mater., 2016, 227, 219–227 CrossRef CAS.
- E. Rozhko, A. Bavykina, D. Osadchii, M. Makkee and J. Gascon, J. Catal., 2017, 345, 270–280 CrossRef CAS.
- W.-H. Wang, M. Z. Ertem, S. Xu, N. Onishi, Y. Manaka, Y. Suna, H. Kambayashi, J. T. Muckerman, E. Fujita and Y. Himeda, ACS Catal., 2015, 5, 5496–5504 CrossRef CAS.
- M. G. Goesten, P. C. M. M. Magusin, E. A. Pidko, B. Mezari, E. J. M. Hensen, F. Kapteijn and J. Gascon, Inorg. Chem., 2014, 53, 882–887 CrossRef CAS PubMed.
- F. Angersbach-Bludau, C. Schulz, J. Schoffel and P. Burger, Chem. Commun., 2014, 50, 8735–8738 RSC.
- N. Ahlsten, A. Bartoszewicz and B. Martin-Matute, Dalton Trans., 2012, 41, 1660–1670 RSC.
- K. Yamaguchi, T. Koike, M. Kotani, M. Matsushita, S. Shinachi and N. Mizuno, Chem. – Eur. J., 2005, 11, 6574–6582 CrossRef CAS PubMed.
- S. Sahoo, H. Lundberg, M. Edén, N. Ahlsten, W. Wan, X. Zou and B. Martín-Matute, ChemCatChem, 2012, 4, 243–250 CrossRef CAS.
- F. Carson, E. Martinez-Castro, R. Marcos, G. G. Miera, K. Jansson, X. Zou and B. Martín-Matute, Chem. Commun., 2015, 51, 10864–10867 RSC.
- R. C. van der Drift, E. Bouwman and E. Drent, J. Organomet. Chem., 2002, 650, 1–24 CrossRef CAS.
- R. Uma, C. Crévisy and R. Grée, Chem. Rev., 2003, 103, 27–52 CrossRef CAS PubMed.
- Y. Sasson, A. Zoran and J. Blum, J. Mol. Catal., 1981, 11, 293–300 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ce00561j |
| This journal is © The Royal Society of Chemistry 2017 |