DOI:
10.1039/C6CE02383E
(Paper)
CrystEngComm, 2017,
19, 171-178
Preparation of hexagonal prism anatase with high thermal stability from a HTiOF3 precursor†
Received
14th November 2016
, Accepted 29th November 2016
First published on 29th November 2016
Abstract
Anatase hexagonal prism TiO2 with high thermal stability was prepared through the decomposition of precursors (HTiOF3). HTiOF3 was firstly synthesized, using a solvothermal method, from n-octanol, tetrabutyl titanate and hydrofluoric acid. The obtained precipitates were then calcined at different temperatures (200–900 °C) for 2 h. The formation process of the hexagonal prism HTiOF3 precursors included three steps: nuclei growth, dissolution–recrystallization and further growth. The morphologies of the precursors depended strongly on the reaction temperature and the amount of solvent. The precursors were transformed into TiO2 after calcination and the effect of the calcination temperature on the morphology and phase composition of the hexagonal prism TiO2 was investigated. The results indicated that the anatase hexagonal prism TiO2 exhibited a rather high thermal stability, maintaining both the anatase structure and precursor shape up to 800 °C. The sample that was calcined at 550 °C exhibited a superior photocatalytic performance.
Introduction
Titanium dioxide (TiO2) has attracted great attention due to its nontoxicity, environmental compatibility, cheapness and chemical stability. Because of its unique physical and chemical properties, TiO2 materials have been widely used in various fields including solar cells,1,2 gas sensors,3 and photocatalysis.4–6 To date, TiO2 has been synthesized in various shapes such as nanospheres, nanofibers, nanotubes and nanosheets,7–12 by different preparation methods including solvothermal, sol–gel and electrospinning vapour deposition methods, and so on.13–20 Among these preparation methods, the synthesis of anatase TiO2via the decomposition of precursors has recently been regarded as an effective synthetic means to produce novel morphologies. Due to the intrinsic crystal structures and morphologies of the precursors, the shape of the anatase crystals would be sensitively affected by the morphology of the precursors. For example, Wang et al. reported the fabrication of 3D hierarchical TiO2 nanoboxes via topotactic transformation from TiOF2 nanocubes.21 Zhao et al. reported the synthesis of 30% and 80% {001} facet exposed anatase TiO2 through decomposition of two fluorine-containing precursors (TiOXFY).22 Therefore, taking advantage of the decomposition of precursors to obtain unique morphologies of anatase crystals is desired.
On the other hand, the crystalline phase plays an important role in TiO2. It is well known that TiO2 has three main crystalline phases: anatase, rutile, and brookite.23 The phase compositions of TiO2 affect its properties. Among the three major crystalline polymorphs, anatase TiO2 is usually used in photocatalytically active stable titania-coated fields such as self-cleaning glass and bathroom tiles but the processing temperatures are very high. However, pure anatase is metastable and inclined to transform into rutile when the heating temperature is higher than 500 °C owing to it having a higher surface energy than rutile, which limits its suitability for high-temperature applications. Therefore, elevating the thermal stability temperature of anatase TiO2 is vital to broaden its applications. Hitherto, attention has been focused on increasing the thermal stability temperature of anatase TiO2 by doping with metallic or nonmetallic elements. For instance, Song et al. prepared N/Cu-codoped titania nanoparticles with a stability temperature of the anatase structure of up to 700 °C;24 Lv et al. synthesized F-doped titania nanoparticles with a high stability temperature of up to 1000 °C.25 However, doping usually leads to the possible generation of secondary impurity phases which affects the phase purity and thus reduces the photocatalytic activity of titania. Thus, there is an urgent need to increase the stability temperature using an effective method.
Driven by the above consideration, in this paper we extended the method of decomposing a precursor to fabricate complex three dimensional anatase hexagonal prism TiO2 by decomposing precursors of HTiOF3. The precursors were first obtained through a solvothermal method and the experimental parameters, including the solvothermal temperature, reaction time and the volume of solvent, were investigated in detail. Then, the precursors were transformed into TiO2 during the calcination process; the anatase hexagonal prism TiO2 that formed from the HTiOF3 precursor exhibited high thermal stability. The photocatalytic performance of the samples calcined at different temperatures was also discussed.
Experimental section
Materials
Tetrabutyl titanate was purchased from Aladdin Reagent Company and n-octanol was purchased from Shanghai Guoyao Chemical Reagent Co. Ltd., (Shanghai, China). Hydrofluoric acid (40 wt%) was purchased from Laiyang Kant Chemical Co. Ltd. (Laiyang, China). Absolute ethanol was purchased from Tianjin Fuyu Chemical Industry Co. Ltd. (Tianjin, China). All reagents were used directly without further purification.
Synthesis of hexagonal prism TiO2 materials
The hexagonal prism TiO2 was synthesized using a two-step method. Firstly, the precursor was prepared via a solvothermal method. 25 mL of n-octanol was added to a 50 mL Teflon-lined autoclave. 2 mL of titanium titanate was then added and 0.8 mL of hydrofluoric acid (HF, 40 wt%) was dropped slowly into the above solution with stirring for 30 min at room temperature. The solvothermal reaction was carried out at 160 °C for 10 h. After the Teflon-lined autoclave had cooled down to room temperature, the obtained precursors were washed with ethanol several times, then dried at 60 °C overnight. The solvothermal temperature, the reaction time and the volume of solvent of the precursors were investigated in detail. The preparation conditions of the precursors are listed in Table 1. Secondly, the precursors were calcined at 200, 300, 470, 550, 650, 800, 870 or 900 °C for 2 h. The resultant samples calcined at different temperatures were labelled as Tx, where x represents the calcination temperature. For example, T200 °C represents the precursors that were calcined at 200 °C for 2 h in air.
Table 1 Synthetic conditions of the precursor samplesa
| Sample |
n-Octanol (mL) |
Temperature (°C) |
Time |
|
The volumes of titanium titanate and HF added in the experiments were 2 and 0.8 mL, respectively.
|
| 1 |
25 |
120 |
20 h |
| 2 |
25 |
160 |
20 h |
| 3 |
25 |
180 |
20 h |
| 4 |
25 |
160 |
0 h |
| 5 |
25 |
160 |
15 min |
| 6 |
25 |
160 |
10 h |
| 7 |
15 |
160 |
10 h |
| 8 |
25 |
160 |
15 h |
| 9 |
25 |
160 |
5 h |
Characterization
The crystal structures of the as-prepared samples were confirmed using an X-ray diffractometer (XRD, Bruker D8-Advance, Cu Kα target, λ = 1.5406 Å). The morphologies of the samples were observed using a field emission scanning electron microscope (FESEM, FEI, QUANTA FEG 250). Fourier transform infrared (FTIR) spectra were recorded using a Fourier transform infrared spectroscopy instrument (Thermo Electron, Nicolet 380, United States). Raman spectra were obtained using a high-resolution Raman spectrometer (LabRAM HR Evolution, HORIBA JOBIN YVON SAS). Absorption spectra of the samples were obtained using a UV-vis spectrometer (Hitachi U-4100) with a quartz cell.
Photocatalytic investigation
The photocatalytic performance of the as-synthesized samples was evaluated by degrading methyl orange (MO) dye under UV light irradiation and a 500 W Hg lamp (PL-02, Beijing) was used as the ultraviolet light source. In a typical experiment, 10 mg of the sample was dispersed in 25 mL of a MO aqueous solution (10 mg L−1) by vigorously stirring at room temperature. Prior to UV light irradiation, the sample was stirred in the dark for 30 min to establish the adsorption–desorption equilibrium of the solution. 2 mL aliquots of suspension solution were taken out at irradiation time intervals of 30 min and then centrifuged to remove the photocatalyst particles. Finally, the absorbance spectra of the samples were obtained using a UV-vis spectro-photometer.
Results and discussion
Role of the reaction temperature on the morphologies of the precursors
The reaction temperature is regarded as an important factor during the precursor growth process. Fig. 1 shows SEM images with low- and high-magnification of the precursors prepared at different reaction temperatures for 20 h. This result indicates that the samples obtained at 120 °C (Fig. 1(a) and (b)) and 160 °C (Fig. 1(c) and (d)) exhibited similar morphologies except for their sizes. The precursors consisted of four hexagonal prisms closely inserted into each other; eight faces could be seen on the outside, every face displayed a hexagonal shape, and the surface morphology of the hexagonal shape was observed to be smooth. The angle between the adjacent sides in each hexagonal shape was about 120°. In the case of the samples obtained at 120 °C (Fig. 1(a) and (b)), the length of the hexagonal prism was about 950 nm and the width of the top hexagonal shape surface was about 350–450 nm. Upon increasing the temperature to 160 °C, the length of the hexagonal prism increased to 1.2 μm and the width of the top hexagonal shape surface increased to around 450 nm, which suggested that at a higher reaction temperature crystals grew with bigger crystallite sizes due to the faster growth rate, when the reaction time was kept the same for the samples. Upon increasing the reaction temperature to 180 °C, as shown in Fig. 1(e) and (f), it could be observed that the hexagonal prism self-assembled morphology was destroyed; some cubic boxes appeared and the cubic boxes were not uniform.
 |
| | Fig. 1 SEM images of precursors prepared at different reaction temperatures for 20 h: (a and b) 120 °C, (c and d) 160 °C and (e and f) 180 °C. | |
The XRD patterns of the samples prepared at different temperatures are shown in Fig. 2. The XRD patterns of the samples prepared at 120 and 160 °C were completely identical and the samples could be identified as HTiOF3, with the structural and compositional information being similar to that obtained for ammonium oxofluorotitanate, NH4TiOF3.26–28 The detailed diffraction peaks of HTiOF3 have been calculated by Zhao's group,26 and are listed in the ESI† (Table S1). This confirmed that the hexagonal prism self- assembled morphologies shown in Fig. 1(a)–(d) were HTiOF3. However, Fig. 2(c) displayed samples prepared at a temperature of 180 °C which were composed of not only HTiOF3; the diffraction peaks located at 2θ = 33.5, 54.2, 60.0, 69.9 and 74.9 degrees were attributed to TiOF2 (JCPDS no. 08-0060). This revealed that at a higher reaction temperature, HTiOF3 may exhibit a trend of transforming into TiOF2. These results demonstrated that the reaction temperature plays an important role in the growth process, and affects not only the morphologies but also the phase compositions.
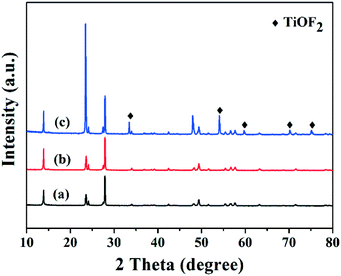 |
| | Fig. 2 XRD patterns of the precursors prepared at different reaction temperatures for 20 h: (a) 120 °C, (b) 160 °C and (c) 180 °C. | |
The role of reaction time on the morphologies of the precursors
In order to investigate the formation of precursors during the solvothermal process, a series of time-dependent samples were prepared at a reaction temperature of 160 °C. Fig. 3 shows the SEM images of the samples prepared at different reaction times. At the initial stage of 0 min (Fig. 3(a)), it was seen that all samples consisted of amorphous nanoparticles. These amorphous nanoparticles then continued to grow. When the reaction time was increased to 15 min (Fig. 3(b)), the nanoparticles had already developed into hexagonal prism self-assembled morphologies. This result indicates that the transformation process from amorphous nanoparticles into hexagonal prism self-assembled precursors under the solvothermal conditions was very rapid. Continuing to increase the reaction time to 5 h (Fig. 3(c)), it was clearly observed that the hexagonal prism morphology was still maintained, but the size of the hexagonal prism became large compared with that of the sample obtained at 15 min and the hexagonal prism morphology appeared more clearly. At a reaction time of 10 h (Fig. 3(d)), the height of the hexagonal prism was up to 1–1.2 μm and the width of the top hexagonal shape surface was about 350–450 nm. On further increasing the reaction time to 15 h (Fig. 3(e)), the size and morphologies of the hexagonal prisms were similar to those of the precursors obtained at 10 h. This result suggested that a reaction time of 10 h was sufficient for the formation of fully developed hexagonal prism self-assembled precursors and longer reaction times would not alter the basic shape. The time-dependent experiment results demonstrated that hexagonal prism precursors form through rapid nuclei growth, dissolution–recrystallization and a further growth process. The formation mechanism of the precursors is illustrated in Scheme 1.
 |
| | Fig. 3 SEM images of the precursors prepared at 160 °C for different times: (a) 0 min; (b) 15 min; (c) 5 h; (d) 10 h; (e) 15 h. | |
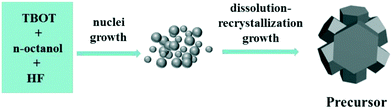 |
| | Scheme 1 Schematic model of the formation of precursors of TiO2. | |
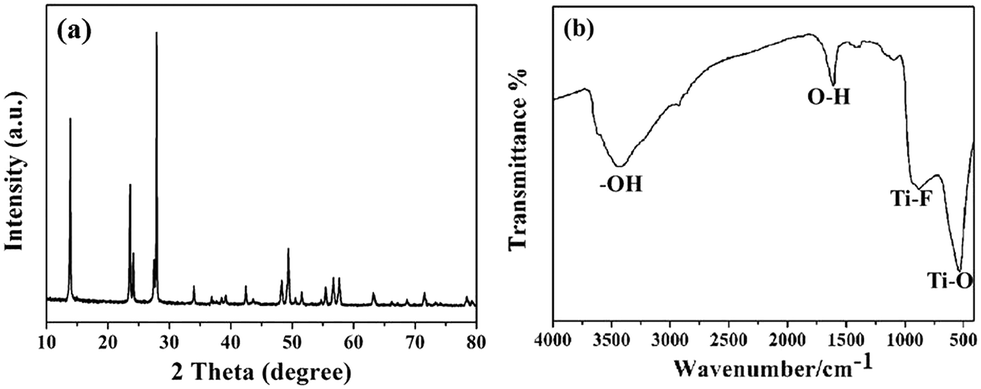
Fig. 4(a) shows the XRD patterns of the sample prepared at 160 °C for 10 h (sample 6); the sample was also identified as HTiOF3. Sample 6 was also measured using FTIR spectroscopy to further confirm the existence of HTiOF3. As shown in Fig. 4(b), the peaks located at around 534 cm−1 and 915 cm−1 were assigned to Ti–O and Ti–F, respectively, and the peaks located at 3400 cm−1 and 1600 cm−1 were assigned to the stretching vibrations of hydroxyl groups.29 The results of the FTIR spectroscopy combined with the XRD patterns were regarded as confirmation that the hexagonal prism self-assembled precursors were HTiOF3.
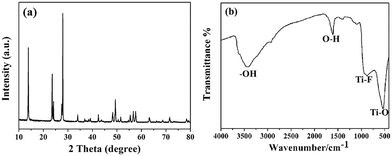 |
| | Fig. 4 (a) XRD pattern of sample 6 (160 °C, 10 h); (b) FTIR spectrum of sample 6 (160 °C, 10 h). | |
The role of the solvent amount on the morphologies of the precursors
The effect of the amount of solvent on the morphologies of the precursors was investigated. Fig. 5(a) and (b) show SEM images at low magnification and high magnification of the precursors prepared using 15 mL of n-octanol. In this case, although the hexagonal prism self-assembled morphologies could be seen in Fig. 5(a) and (b), at the same time many aggregated particles were also observed. Compared with the precursors prepared using 25 mL of n-octanol (Fig. 3(d)), the precursors prepared using 15 mL of n-octanol, under the same reaction temperature and reaction time, were not well developed . Based on the above discussion, it is suggested that the amount of solvent affects the degree of reaction instead of altering the hexagonal prism morphologies; the larger the volume of n-octanol in the limited Teflon-lined autoclave, the larger the pressure in the autoclave, so the more fully the reaction occurs, which contributes to there being more intact morphologies.
 |
| | Fig. 5 (a) Low-magnification and (b) high-magnification SEM images of the hexagonal nanosheet self-assembled precursors prepared using 15 mL n-octanol. | |
The role of the calcination temperature on the hexagonal prism TiO2
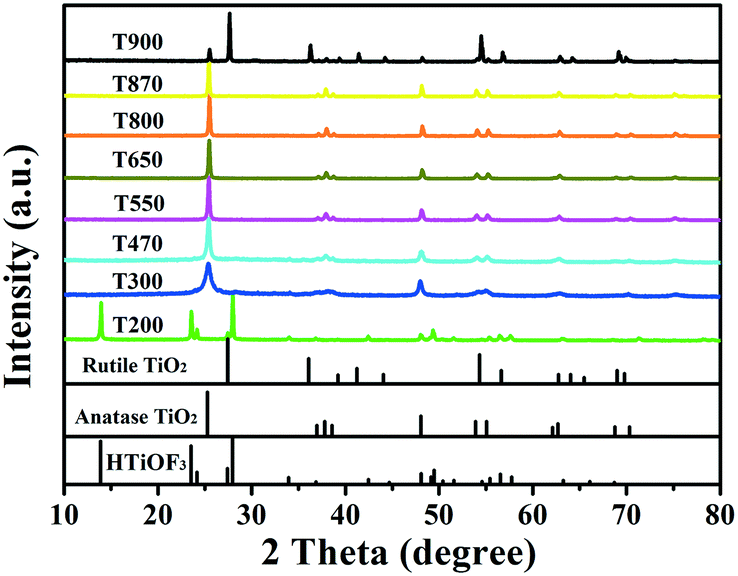
The changes in the phase structures and crystallinity of the samples calcined at different temperatures were investigated using XRD and the results are shown in Fig. 6. For sample T200, all of the diffraction peaks were in accordance with the precursors and no peaks for the anatase or rutile TiO2 phases were observed. This indicates that T200 was still pure HTiOF3 and the precursor had not decomposed at 200 °C. When the calcination temperature was increased to 300 °C, the peak for HTiOF3 disappeared and the characteristic peaks at 2θ values of 25.3, 37.8, 48.0, 53.9, 55.1, 62.7, 68.8, 70.3 and 75.0 degrees could be assigned to the (101), (004), (200), (105), (211), (204), (116), (220) and (215) crystal planes of anatase TiO2 (JCPDS File Card No. 21-1272), which suggested that the precursor transformed into TiO2 when the calcination temperature increased from 200 to 300 °C. Further observation indicates that for the samples calcinated in the range of 400 to 870 °C, all peaks were identified as anatase TiO2 without other phases. When the calcination temperature was increased to 900 °C, rutile TiO2 was observed and sample T900 was a mixed phase of anatase and rutile. Compared with the general phase transformation temperature for TiO2 (500–600 °C),23 the phase transformation temperature of TiO2 obtained from HTiOF3 was far higher. This may be attributed to there being fluoride absorbed on the crystal planes of anatase TiO2, which favours decreasing the energy of anatase and stabilizing the anatase structure.25,30,31
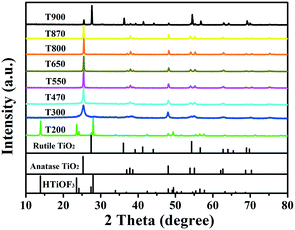 |
| | Fig. 6 XRD patterns of sample 6 calcined at different temperatures. | |
The phase purity of T800 was further confirmed by Raman spectroscopy. Fig. 7 shows the Raman spectrum of the T800 sample; the observed positions of the Raman bands at 142.7, 396.0, 514.8 and 637.9 cm−1 were in good accordance with the Eg(1), B1g(1), A1g(1) + B1g(1) and Eg(2) modes of anatase TiO2 reported in the literature.30 Hence, the phase purity of T800 was further confirmed to be anatase. The morphologies of the samples calcined at 200, 550, 800 and 870 °C were further studied using SEM. Fig. 8(a) shows the SEM images of the precursors calcined at 200 °C; it could be observed that the morphologies of T200 were inherited from the precursors, which is consistent with the results from XRD. Compared with the morphologies of T200, the morphology of T550 has been changed obviously as shown in Fig. 8(b). The morphology of T550 takes the shape of a hollow porous hexagonal prism. It could be inferred that during the calcination process, facets of the hexagonal prism located outside collapsed and the inside of the filler was exposed. As shown in the inset of Fig. 8(b), the filler showed a granular shape which was beneficial to forming the porous structure. The reason why the facets of the hexagonal prism located outside collapsed was that the precursor, HTiOF3, decomposed and released HF during the calcination;26,27 it is because of the overflow of gaseous material that the formation of porous hexagonal prism morphologies occurs. When the calcination temperature was increased to 800 °C, although the porous hexagonal prism morphologies were damaged to a certain extent, hexagonal prism morphologies could still be observed. Continuing to increase the calcination temperature to 870 °C, the porous hexagonal prism morphologies were completely destroyed and some amorphous nanoparticles formed. Fig. 9 shows the TEM images at low magnification and high magnification of T550. Fig. 9(a) shows that T550 indeed has a porous hexagonal prism morphology and the porous characteristics may be caused by overflow of HF during the calcination process. It further could be observed that the angle between two adjacent hexagonal prisms is 60° (Fig. 9(b)).
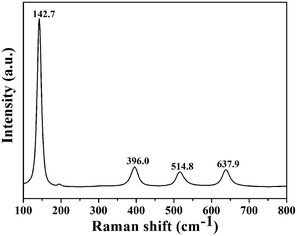 |
| | Fig. 7 Raman spectrum of T800. | |
 |
| | Fig. 8 SEM images of sample 6 calcinated at different temperatures: (a) 200 °C, (b) 550 °C, (c) 800 °C and (d) 870 °C. | |
 |
| | Fig. 9 (a) Low-magnification and (b) high-magnification TEM images of T550. | |
The chemical composition and elemental chemical status of the sample calcined at 550 °C (T550) were characterized by X-ray photoelectron spectroscopy (XPS). The XPS survey spectrum of T550 is illustrated in Fig. 10(a); there can be observed four characteristic peaks of Ti 2p, O 1s, F 1s, and C 1s. The C 1s peak at 285 eV is attributed to contamination from the XPS instrument itself. As illustrated in the Ti 2p high-resolution spectrum (Fig. 10(b)), two main peaks located at about 458.4 and 464.1 eV can be respectively ascribed to both Ti 2p3/2 and Ti 2p1/2, and are in accordance with that of Ti4+ of bulk TiO2.32,33 The peak at 529.6 cm−1 is assigned to O 1s (Fig. 10(c)).34,35 The corresponding high-resolution XPS spectrum of F 1s is shown in Fig. 10(d); it demonstrates that the F species displayed only one peak of binding energy at 684.5 eV. However, the binding energy of F1s for F-doped TiO2 displayed two peaks at around 684.5 and 688.4 eV, which were attributed to the F species adsorbed on the TiO2 surface and those incorporated into the TiO2 lattice, respectively.36–38 Therefore, the F species displayed in Fig. 10(d) could be assigned to F− adsorbed on the TiO2 surface instead of that incorporated into the TiO2 lattice. The F− adsorbed on the TiO2 surface plays an important role in the thermal stability of the anatase phase. Firstly, F− ions adsorbed on the TiO2 surface could reduce the surface energy of anatase in order to stabilize the anatase structure, so the phase transformation of anatase-to-rutile is suppressed by the presence of F− ions.31 Upon further increasing the calcination temperature, the adsorbed F− ions are desorbed from the surface of TiO2, which leads to an increase in the crystal face energy of anatase due to the loss of F− ions, and so finally the anatase-to-rutile phase transformation occurred.30
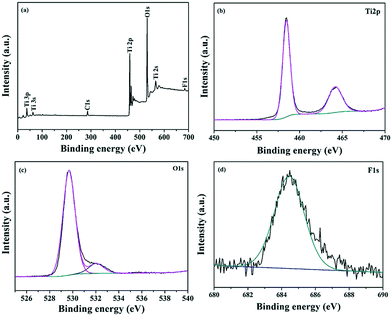 |
| | Fig. 10 The survey (a) and high resolution XPS spectra of (b) Ti 2p (c) O1s and (d) F1s for T550. | |
N2 adsorption–desorption isotherms were investigated to analyze the specific surface area and pore size distribution of samples calcined at different temperatures. All of the samples were type III (Brunauer–Deming–Deming–Teller (BDDT) classification) with a hysteresis loop of type H3, indicating the presence of slit-like pores (Fig. 11). The specific surface areas of samples T200, T300, T470, T550, T650, and T800 were 14.94, 25.56, 28.56, 30.94, 24.10, and 30.69 m2 g−1, respectively. Sample T200 exhibits the smallest surface area; with increased calcination temperature, the surface area increased. When the calcination temperature is increased up to 550 °C, the surface area increased to 30.937 m2 g−1, which is two times larger than that of T200.
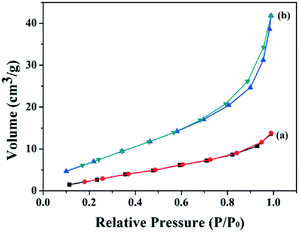 |
| | Fig. 11 (a) N2-adsorption isotherms and pore size distributions of samples calcined at different temperatures: (a) 200 °C; (b) 550 °C. | |
To evaluate the photocatalytic activity of the samples calcined at different temperatures, the photocatalytic degradation of MO was performed under UV light irradiation, the results of which are shown in Fig. 12. T200 exhibited almost no photocatalytic activity due to its relatively low surface area and phase composition. T300 and T800 exhibited similar weak photocatalytic activities, because T300 had low crystallinity and the morphologies of T800 were damaged to a certain degree, both of which could affect their photocatalytic activities, although T800 has a surface area of 30.69 m2 g−1. Compared with T470 and T650, T550 exhibited the highest photocatalytic activity, where 88% of MO dye was degraded in 3 h, which is because its larger surface area and porous morphologies could provide efficient transport pathways to reactant and product molecules. Overall, the calcination temperatures played an important role in the photocatalytic activity. On the one hand, porous TiO2 with a large surface area could be obtained without morphological deformation by controlling the appropriate calcination temperature; abundant open space hexagonal prism morphologies are beneficial to reducing the combination of photo-generated electrons and holes and thus the photocatalytic activity of TiO2 is improved. On the other hand, it is observed that high calcination temperatures could destroy the hexagonal prism morphologies inherited from the precursors and generate amorphous nanoparticles. The photocatalytic activity of the hexagonal prism TiO2 can probably be attributed to its special porous hexagonal prism morphologies as well as their crystallinity and average feature sizes which are affected by calcination temperatures. Although the photocatalytic activities of the samples calcined at different temperatures were not ideal, there existed differences between them and further exploration to improve their photocatalytic activities is desired.
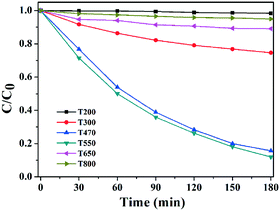 |
| | Fig. 12 Plots of the photocatalytic degradation of MO under UV irradiation. | |
Conclusions
In summary, anatase hexagonal prism TiO2 with high thermal stability was prepared by transformation from HTiOF3 precursors. The effect of the growth conditions, such as the reaction temperature, the reaction time and the amount of solvent, on the morphologies and phase composition of the precursors was also investigated. The reaction temperature played an important role in the formation of the hexagonal prism morphologies. The obtained hexagonal prism TiO2 transformed from HTiOF3 exhibited high thermal stability and the hexagonal prism TiO2 could still retain the anatase phase when the calcination temperature was up to 800 °C. T550 exhibited the highest photocatalytic activity due to its intact poriferous hexagonal prism TiO2 and high surface area. Although the photocatalytic activity of the samples prepared in this study needs to be further optimized, the method to obtain novel morphologies of TiO2 with high thermal stability by the transformation of precursors has provided new insight into the construction of advanced photocatalytic nanostructures with high-temperature stability.
Acknowledgements
This work was supported in part by the project from the National Basic Research Program of China (973 Program, 2013CB632401), the Program for Taishan Scholars and projects from the National Natural Science Foundation of China (51572109, 51501071, 51402123, and 51402124).
Notes and references
- J. T. Jiu, F. M. Wang, S. Isoda and M. Adachi, Chem. Lett., 2005, 34, 1506–1507 CrossRef CAS.
- K. Fujihara, A. Kumar, R. Jose, S. Ramakrishna and S. Uchida, Nanotechnology, 2007, 18, 365709–365713 CrossRef.
- A. P. Caricato, S. Capone, G. Ciccarella, M. Martino, R. Rella, F. Romano, J. Spadavecchia, A. Taurino, T. Tunno and D. Valerini, Appl. Surf. Sci., 2007, 253, 7937–7941 CrossRef CAS.
- M. C. Yan, F. Chen, J. L. Zhang and M. Anpo, J. Phys. Chem. B., 2005, 109, 8673–8678 CrossRef CAS PubMed.
- U. S. Ozkan, Y. P. Cai and M. W. Kumthekar, J. Phys. Chem., 1995, 99, 2363–2371 CrossRef CAS.
- J. Lin, Y. Lin, P. Liu, M. J. Meziani, L. F. Allard and Y. P. Sun, J. Am. Chem. Soc., 2002, 124, 11514–11518 CrossRef CAS PubMed.
- C. Jia, P. Yang, H. S. Chen and J. Wang, CrystEngComm, 2015, 17, 2940–2948 RSC.
- B. Wang, X. Y. Lu, L. K. Yu, J. Xuan, M. K. H. Leung and H. Guo, CrystEngComm, 2014, 16, 10046–10055 RSC.
- N. Watanabe, T. Kaneko, Y. Uchimaru, S. Yanagida, A. Yasumori and Y. Sugahara, CrystEngComm, 2013, 15, 10533–10540 RSC.
- Y. Zheng, Z. Jiang and P. Yang, RSC Adv., 2016, 6, 15485–15491 RSC.
- Z. Bao, H. Xie, Q. Zhu, J. Qian, P. Ruan and X. Zhou, CrystEngComm, 2013, 15, 8972–8978 RSC.
- W. Zhou, X. Liu, J. Cui, D. Liu, J. Li, H. Jiang, J. Wang and H. Liu, CrystEngComm, 2011, 13, 4557–4563 RSC.
- C. Cui, H. Chen, B. Lan, L. Zhang, R. Ma, J. Geng, H. Li and J. Hu, CrystEngComm, 2015, 17, 3763–3767 RSC.
- H. He, C. Jia and P. Yang, Funct. Mater. Lett., 2014, 7, 145001–145005 Search PubMed.
- J. Lu, P. Zhang, A. Li, F. Su, T. Wang, Y. Liu and J. Gong, Chem. Commun, 2013, 49, 5817–5819 RSC.
- E. Stathatos, F. Delmonte and D. Levy, Langmuir, 1997, 13, 4295–4300 CrossRef CAS.
- Z. Xiong, H. Dou, J. Pan, J. Ma, C. Xu and X. S. Zhao, CrystEngComm, 2010, 12, 3455–3457 RSC.
- Y. Cheng, W. Huang, Y. Zhang, L. Zhu, Y. Liu, X. Fan and X. Cao, CrystEngComm, 2010, 12, 2256–2260 RSC.
- X. Liu, Y. Li, D. Deng, N. Chen, X. Xing and Y. Wang, CrystEngComm, 2016, 18, 1964–1975 RSC.
- C. Hu, X. Zhang, W. Li, Y. Yan, G. Xi, H. Yang, J. Li and H. Bai, J. Mater. Chem. A, 2014, 2, 2040–2043 CAS.
- Z. Wang, B. Huang, Y. Dai, X. Zhu, Y. Liu, X. Zhang and X. Qin, CrystEngComm, 2013, 15, 3436–3441 RSC.
- Y. Zhao, Y. Zhang, H. Liu, H. Ji, W. Ma, C. Chen, H. Zhu and J. Zhao, Chem. Mater., 2014, 26, 1014–1018 CrossRef CAS.
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855–874 CrossRef CAS.
- K. Song, J. Zhou, J. Bao and Y. Feng, J. Am. Ceram. Soc., 2008, 91, 1369–1371 CrossRef CAS.
- Y. Lv, L. Yu, H. Huang, H. Liu and Y. Feng, Appl. Surf. Sci., 2009, 255, 9548–9552 CrossRef CAS.
- P. Liu, Y. Wang, H. Zhang, T. An, H. Yang, Z. Tang, W. Cai and H. Zhao, Small, 2012, 8, 3664–3667 CrossRef CAS PubMed.
- T. Wu, G. Wang, X. Zhu, P. Liu, X. Zhang, H. Zhang, Y. Zhang and H. Zhao, Nano Res., 2016, 9, 745–754 CrossRef CAS.
- L. Zhou, D. Smyth-Boyle and P. O'Brien, J. Am. Chem. Soc., 2008, 130, 1309–1320 CrossRef CAS PubMed.
- P. Cozzoli, A. Kornowski and H. Weller, J. Am. Chem. Soc., 2003, 125, 14539–14548 CrossRef CAS PubMed.
- K. Lv, J. Yu, L. Cui, S. Chen and M. Li, J. Alloys Compd., 2011, 509, 4557–4562 CrossRef CAS.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- Q. Shi, Y. Li, E. Zhan, N. Ta and W. Shen, CrystEngComm, 2015, 17, 3376–3382 RSC.
- A. Meng, J. Zhang, D. Xu, B. Cheng and J. Yu, Appl. Catal., B, 2016, 198, 286–294 CrossRef CAS.
- G. Jiang, M. Wei, S. Yuan and Q. Chang, Appl. Surf. Sci., 2016, 362, 418–426 CrossRef CAS.
- G. Li, J. Liu, J. Lan, G. Li, Q. Chen and G. Jiang, CrystEngComm, 2014, 16, 10547–10552 RSC.
- C. Lei, X. Jiang, X. Huang, X. Liu, D. Zeng, Y. Ma, L. Wang and D. Peng, Appl. Surf. Sci., 2015, 359, 860–867 CrossRef CAS.
- E. Samsudin, S. B. A. Hamid, J. C. Juan, W. J. Basirun and Gabriele Centi, Appl. Surf. Sci., 2016, 370, 380–393 CrossRef CAS.
- Q. Xiang, K. Lv and J. Yu, Appl. Catal., B, 2010, 96, 557–564 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ce02383e |
|
| This journal is © The Royal Society of Chemistry 2017 |