
Dual-target cancer theranostic for glutathione S-transferase and hypoxia-inducible factor-1α inhibition†
Zhirong
Geng  *a
and
Zhilin
Wang
*a
*a
and
Zhilin
Wang
*a
*a
and
Zhilin
Wang
*a
Abstract
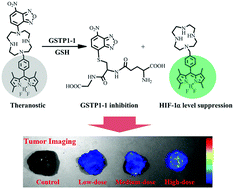
We developed a dual-target theranostic F671, which could exhibit synergetic anticancer effects for inhibiting the activities of glutathione S-transferase and the accumulation of hypoxia inducible factor-1α. F671 undergoes self-immolative cleavage when exposed to GSTP1-1 in live cancer cells, facilitating the visualization of molecule release and distribution, as well as confirming the autophagy-induced apoptosis.
 Please wait while we load your content...
Please wait while we load your content...

