
Asymmetric cross-coupling of alkyl, alkenyl and (hetero)aryl nucleophiles with racemic allyl halides

Abstract
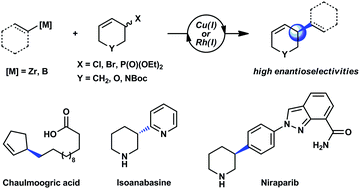
Single enantiomer molecules are important for the pharmaceutical and agrochemical industries and increasingly so in materials science. Most strategies to obtain enantiomerically enriched molecules rely on either generating new stereogenic centres from prochiral substrates or resolving racemic mixtures of enantiomers. Dynamic asymmetric processes are powerful methods that use racemic mixtures of chiral substrates as starting material. This Feature Article focuses on asymmetric additions to racemic substrates using non-stabilized sp2- and sp3-hybridized nucleophiles. These reactions bear considerable resemblance to traditional sp2–sp2 cross-coupling reactions in terms of the starting materials used and the products obtained, but the reaction mechanisms are necessarily different.
 Please wait while we load your content...
Please wait while we load your content...

